
Abstract
In this study, a series of novel α, β-unsaturated carbonyl compounds (3a–j) and their pyrazoline derivatives (4a–e and 5a–b) were designed and successfully synthesized. All synthesized compounds were characterized using various spectroscopic techniques, including 1H NMR, 13C NMR, and mass spectrometry. The biological activity of these compounds was evaluated against five bacterial strains (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli, Staphylococcus aureus, and Acinetobacter baumannii) and three fungal strains (Candida tropicalis, Candida parapsilosis, and Candida albicans). The results revealed that compound (4c) exhibited potent antifungal activity with a minimum inhibitory concentration (MIC) of 6.25 µg/mL across all tested strains and zone of inhibition (ZOI) against Candida albicans is 27 mm. Furthermore, time kill kinetics of Candida albicans and haemolysis assays also perform in support of their antifungal activity. Additionally, all synthesized compounds were subjected to computational analysis using molecular descriptors, ADMET, molecular docking, and molecular dynamics to find protein-ligand interactions. Molecular docking studies indicated that the most effective antifungal compounds (3h and 4c) exhibited binding energies of -8.76 and −8.44 kcal/mol for DHFR and −7.96 and −8.24 kcal/mol for NMT1, respectively. The obtained results revealed that these compounds exhibit potential interactions with antifungal targets as dual inhibitors. As a result, this study finds an important approach to synthesized compounds with potential antifungal activity.
Similar content being viewed by others
Introduction
The biological potential of heterocyclic compounds is diverse, which makes them important in the field of medicinal chemistry1. In the presence of versatility and diverse biological functions, nitrogen-containing heterocycles have consistently captivated researchers. In the quest for novel compounds with versatile and significant properties, scientists identified pyrazoline as an intriguing member of the heterocyclic family2. Pyrazolines are frequently employed scaffolds in the structures of numerous significant biological and medicinal compounds3. Pyrazolines are found naturally in plants belonging to the Piperaceae family, which includes black pepper (Piper nigrum)4. Recently found that sponges, corals, and other marine organisms display cytotoxic and anti-inflammatory by producing pyrazoline alkaloids5. However, some alkaloids, including sesquiterpene pyrazolines, piperidine alkaloids, and pyrazoline alkaloids, contain pyrazole in their structures. Natural pyrazoline has been interacted with the living organisms by forming the N–N bond6. Therefore, pyrazoline derivatives are the most widely known heterocyclic compounds, with promising biological activities, including HIV reverse transcriptase inhibitors that are non-nucleoside antagonists of neurotensin receptors with analgesic effects and non-steroidal mineralocorticoids7, antiviral8, antitumor9,10, antimicrobial11,12, antitubercular13, antimalarial14, anti-amoebic15, antifungal16, antidiabetic17, anti-inflammatory18, anticancer19. Pyrazoline, also known as a dihydro derivative of pyrazole, can exist in three different forms depending on the position of the double bond, i.e., 1-pyrazoline (1), 2-pyrazoline (2), and 3-pyrazoline (3)20 (Fig. 1).

The general structure of three pyrazoline isomers with their most promising biological activities.
Pyrazoline derivatives and α, β-unsaturated carbonyl compounds exhibit promising potential as antimicrobial agents21. These compounds possess structural motifs that make them effective against pathogens by interfering with essential cellular processes or structures22. α, β-unsaturated carbonyl compounds typically contain conjugated double bonds adjacent to a carbonyl group, which imparts reactivity and biological activity23. They can act as Michael acceptors, readily forming covalent adducts with thiol-containing biomolecules such as cysteine residues in proteins. This mechanism can disrupt key enzymes and cellular processes in fungi, leading to growth inhibition or cell death24,25. Treatment of pathogenic microbial infections depends critically on antimicrobial activity and the way medications interact with their molecular targets26. The rate of antibiotic resistance can rise because of drug interactions that significantly lower microbial resistance. To achieve strong interaction with the target, new and highly active compounds with appropriate active sites are required for the treatment of resistant microbial infections.
Recently, fungal infection also developed as a rising risk to human health27. There are numerous causes for this, but two significant ones are the rise in HIV-positive patients and the number of people receiving chemotherapy for cancer28. This has prompted the search for new molecular targets for antifungal medications. Although a lot of new antifungal medications have been developed in the past five years, some patients are still difficult to treat because of intrinsic or acquired antifungal resistance. Also, serious side effects are a result of many recently developed antifungal medications. Hence, the need for novel antifungal medications that can target fungi specifically without interfering with the host’s biochemical processes is constant. Considering the ever-increasing demand for fungicidal agents and the therapeutic potential of pyrazoline moieties and α, β-unsaturated ketones. The most significant and extensively researched α,β-unsaturated carbonyl-based substances consist of the naturally occurring substances curcumin (4), zerumbone (5), and chalcone (6), along with their derivatives and analogs29 (Fig. 2).

α, β- unsaturated carbonyl and pyrazoline based compounds.
Thus, pyrazolines consequently became a necessary scaffold of various medications with a wide range of biological actions30. Some of the biological activities associated with them include antimicrobial31, antioxidant32, anti-inflamatory33, antitubucular34, anticancer35, cardioprotective, antidiabetic36, antiviral37, anti-ageing38, antiallergic39, and hepatoprotective40. In January 2022, the World Health Organization (WHO) issued a specific report dedicated solely to α,β-unsaturated carbonyl compounds as antibacterial and antifungal agents. However, the WHO regularly monitors and evaluates the safety and efficacy of antimicrobial agents, including those derived from natural sources like α,β-unsaturated carbonyl compounds41. It can be found in a number of commercially available medications across multiple classifications, including ramifenazone (7), antipyrine (8), morazone (9), and famrofazone (10)42,43,44,45, edaravone (amyotrophic lateral sclerosis)46, metamizole (antipyretic and analgesic), muzolimine (diuretic)47, axitinib (the use of VEGFR inhibitors of the second generation to treat metastatic renal cell carcinoma)19 and ibipinabant (as an inverse agonist of the selective cannabinoid CB1 receptor)48 (Fig. 2).
Dihydrofolate reductase (DHFR) is an important enzyme in the folate metabolic pathway. Because it is so important for Candida albicans to be pathogenic and survive, it has been chosen as a key target in the development of antifungal drugs49,50,51. N-myristoyltransferase (NMT) facilitates myristoylation through a sequential ordered Bi–Bi mechanism. It is a saturated fatty acid with 14 carbon atoms that forms an amide bond with the N-terminal glycine residue of a polypeptide during co-translational binding52,53. Recently, NMT has emerged as a novel target for the treatment of fungal diseases, making it a significant focus for antifungal drug design and discovery (DDD).
Our research group is currently working on the design of easy ways to make low molecular weight compounds because they have a potential biological activity. In this study, we synthesized a new series of α, β-unsaturated ketones (3a–j) and their pyrazoline derivatives (4a–f and 5a–b) and in vitro tested their antibacterial and antifungal activities. Furthermore, we also performed in silico molecular descriptors, ADMET, molecular docking, and molecular dynamics analyses to understand their mode of action and drug-like properties. Because of this, we chose two specific proteins, NMT1 (N-myristoyltransferase 1) with the PDB ID: 1IYL and DHFR (dihydrofolate reductase) with the PDB ID: 4HOF, to study their antifungal mechanism. The obtained results were also subjected to comparative analysis between the activity of the standard drug (Fluconazole) as well as reference compounds like R64 (1-Methyl-1 H-imidazol-2-yl)(3-methyl-4-{3-[(pyridin-3-ylmethyl)amino]propoxy}-1-benzofuran-2-yl)methanon) and 18 H (5-[3-(2-methoxy-4-phenylphenyl)but-1-yn-1-yl](-6-methylpyrimidine-2,4-diamine)).
Experimental
Materials and methods
All chemical reagents and solvents were sourced from commercial suppliers and were used without additional purification. The 1H and 13C-NMR experiments were conducted by utilising a Bruker Avance 400 MHz Ultrashield™ spectrometer. About 15 mg of the sample was dissolved in CDCl3 and DMSO-d6. The chemical shift values were calculated in ppm and δ-scale, and the coupling constant, J, was calculated in Hertz (Hz), and their multiplicities expressed as m = multiplet, q = quartet, t = triplet, dd = double doublet, brd = broad doublet, d = doublet, brs = broad singlet, and s = singlet. The ESI-MS spectra were recorded on an ion trap LCQ Advantage Max mass spectrometer (Thermo Electron Corporation). The melting point apparatus (Stuart SMP10) was utilised to obtain the melting points. For the FTIR analysis, a PerkinElmer Nicolet 6700 FTIR spectrometer with attenuated total reflection was used. It could measure frequencies between 150 and 750 cm− 1.
General procedure for the synthesis of α, β-unsaturated carbonyl compounds (3a-j)
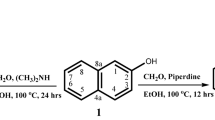
Equimolar quantities of substituted ketone (0.01 mol) and substituted aldehyde (0.01 mol) were dissolved in ethanol (25 mL). Sodium hydroxide solution (0.02 mol) was added slowly, and the mixture stirred for 24 h. The completion of the reaction was monitored by TLC. On completion, the mixture was poured slowly into 400 mL of cold water with constant stirring and 10% HCl was added until pH = 7 was obtained. The precipitate obtained was filtered and washed with distilled water to obtain pure compounds (3a-j) in quantitative yields.
Spectral characterization
(E)-1-([1,1′-biphenyl]-4-yl)-3-(2-bromo-5-fluorophenyl) prop-2-en-1-one (3a)
Off white solid, yield 75%, mp 136–137 °C, IR (KBr) νmax : 3068(C-H aromatic), 1659(C = O α, β-unsaturated), 1605(-C = C- aromatic), 1458(C = C) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 6.97–7.02(1 H, m),7.40–7.50(5 H, m), 7.58–7.61(1 H, m), 7.64–7.66(2 H, m), 7.72–7.75(2 H, m),8.06–8.11(3 H, m); 13C-NMR (100 MHz, CDCl3) δc: 114.4(d, JC−F = 2 Hz), 118.4(d, JC−F = 20 Hz), 120.0(d, JC−F = 3 Hz), 125.8, 127.3, 127.8, 128.3, 129.0, 129.3, 134.7(d, JC−F = 10 Hz), 136.3, 136.7(d, JC−F = 10 Hz), 139.8, 141.9(d, JC−F = 2 Hz), 145.9, 160.7(d, JC−F = 250 Hz), 189.3; ES-MS (m/z): 381[M] + and 383[M + 2] +, calculated for C21H14BrFO.
(E)-3-(2-bromo-5-fluorophenyl)-1-(1 H-pyrrol-2-yl) prop-2-en-1-one (3b)
Pale yellow solid, yield 82%, mp 139–140 °C, IR (KBr) νmax : 3076(C-H aromatic), 1648(C = O α, β-unsaturated), 1608(-C = C- aromatic), 1461(C = C), 3332(N-H), 1211(C-N) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 6.35–6.38 (1 H, m),6.95-7.00(1 H, m), 7.07–7.09(1 H, m), 7.13–7.15(1 H, m), 7.25(1 H, d, J = 15.6 Hz),7.44(1 H, dd, J = 3.0, 9.4 Hz), 7.60(1 H, dd, J = 5.3, 8.8 Hz), 8.10(1 H, dd, J = 1.4, 15.6 Hz), 9.75(1 H, s); 13C-NMR (100 MHz, CDCl3) δc: 111.2, 114.3(d, JC−F = 20 Hz), 117.1, 118.1(d, JC−F = 20 Hz), 119.8(d, JC−F = 10 Hz), 125.9, 126.1, 132.8, 134.6(d, JC−F = 10 Hz), 136.8(d, JC−F = 10 Hz), 139.6, 160.7(d, JC−F = 250 Hz), 178.1; ES-MS (m/z): 294 [M] +, 296 [M + 2] +, calculated for C13H9BrFNO.
(E)-3-(2-bromo-5-fluorophenyl)-1-(furan-2-yl) prop-2-en-1-one (3c)
Pale yellow solid, yield 85%, mp 138–139 °C, IR (KBr) νmax : 3081(C-H aromatic), 1658(α, β-unsaturated C = O), 1609(-C = C- aromatic), 1459(C = C), 1101(C-O) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 6.61–6.62 (1 H, m), 7.00(1 H, td, J = 8.2, 2.9 Hz), 7.36(2 H, m), 7.45(1 H, dd, J = 2.9, 9.3 Hz), 7.58–7.61 (1 H, m),7.67(1 H, brs), 8.15(1 H, d, J = 16.4 Hz); 13C-NMR (100 MHz, CDCl3) δc:112.7, 114.4(d, JC−F = 20 Hz), 118.1, 118.5(d, JC−F = 10 Hz), 120.1, 125.0, 134.7(d, JC−F = 10 Hz), 141.7, 146.8, 153.4, 160.7(d, JC−F = 240 Hz), 177.2. ES-MS (m/z): 295[M] +, 297[M + 2]+, calculated for C13H8BrFO2.
(E)-3-(3,5-di-tert-butyl-2-hydroxyphenyl)-1-(furan-2-yl) prop-2-en-1-one (3d)
Bright yellow solid, yield 83%, mp 169–170 °C, IR (KBr) νmax : 3095(C-H aromatic), 1640(C = O α, β-unsaturated), 1587(-C = C- aromatic),1385(C-H),1460(C = C), 3638(O-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.33 (9 H, s), 1.46(9 H, s), 6.25(1 H, s), 6.59(1 H, m), 7.36(1 H, dd, J = 0.6, 3.5 Hz),7.40(1 H, d, J = 2.4 Hz), 7.43(1 H, d, J = 2.4 Hz), 7.45(1 H, d, J = 15.5 Hz) 7.64–7.65(1 H, m), 8.32(1 H, d, J = 15.6 Hz); 13C-NMR (100 MHz, CDCl3) δc:29.9, 31.5, 34.3, 34.9, 112.6, 117.7, 121.5, 122.0, 122.5, 127.2, 136.8, 140.5, 142.6, 146.5, 152.5, 153.7, 178.4. ES-MS (m/z): 327[M + H] +, calculated for C21H26O3.
(E)-3-(3,5-di-tert-butyl-2-hydroxyphenyl)-1-(thiophen-2-yl) prop-2-en-1-one (3e)
Pale yellow solid, yield 78%, mp 170–171 °C, IR (KBr) νmax : 2953(C-H aromatic), 1636(C = O α, β-unsaturated), 1589(-C = C- aromatic),1384(C-H),1413(C = C), 3658(O-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.33 (9 H, s), 1.46(9 H, s), 6.27(1 H, s), 7.16(1 H, dd, J = 3.8, 4.9 Hz), 7.38–7.42(3 H, m), 7.67(1 H, dd, J = 1.0,4.9 Hz), 7.88(1 H, dd, J = 1.0, 3.8 Hz), 8.30(1 H, d, J = 15.3 Hz); 13C-NMR (100 MHz, CDCl3) δc:29.9, 31.5, 34.3, 34.9, 122.2, 122.5, 122.7, 127.1, 128.2, 132.0, 133.9, 136.8, 140.6, 142.8, 145.4, 152.3, 182.5. ES-MS (m/z): 343[M + H] +, calculated for C21H26O2S.
(E)-3-(2-bromo-4,5-dimethoxyphenyl)-1-(furan-2-yl) prop-2-en-1-one (3f)
Yellow solid,, yield 79%, mp 183–184 °C, IR (KBr) νmax : 2994(C-H aromatic), 1650(C = O α, β-unsaturated), 1586(-C = C- aromatic),1389(C-H),1460(C = C), 1191(C-O) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 3.92 (3 H, s), 3.96(3 H, s), 6.60(1 H, dd, J = 1.6, 3.5 Hz), 7.09(1 H, s),7.20(1 H, m), 7.28(1 H, d, J = 15.6 Hz), 7.33–7.34(1 H, m), 7.65–7.66(1 H, m), 8.19(1 H, d, J = 15.7 Hz); 13C-NMR (100 MHz, CDCl3) δc:56.2, 56.3, 109.5, 112.6, 115.8, 117.5, 118.3, 121.8, 126.8, 142.4, 146.4, 148.7, 151.6, 153.7, 177.9. ES-MS (m/z): 337[M] + and 339[M + 2] +, calculated for C15H13BrO4.
(E)-3-(2-bromo-4,5-dimethoxyphenyl)-1-(1 H-pyrrol-2-yl) prop-2-en-1-one (3g)
Pale yellow solid, yield 75%, mp 211–212 °C, IR (KBr) νmax : 3434(N-H), 3092(C-H aromatic), 1647(C = O α, β-unsaturated), 1589(-C = C- aromatic), 1404(C-H), 1265(C-N), 1192(C-O) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 3.91 (3 H, s), 3.95(3 H, s), 6.34–6.36(1 H, m), 7.09–7.07(2 H, m), 7.11–7.13(2 H, m),7.18(1 H, d, J = 6.9 Hz), 7.26(1 H, s), 8.13(1 H, d, J = 15.6 Hz), 9.76(1 H, s); 13C-NMR (100 MHz, CDCl3) δc:56.2, 56.3, 109.5, 111.0, 115.8, 116.0, 116.4, 117.8, 122.8, 125.4, 127.0, 133.0, 140.8, 148.7, 151.3, 178.8. ES-MS (m/z): 336[M] + and 338[M + 2] + calculated for C15H14BrNO3.
(E)-1-([1,1′-biphenyl]-4-yl)-3-(1 H-imidazol-2-yl) prop-2-en-1-one (3h)
Light brown solid, yield 65%, mp 197–198 °C, IR (KBr) νmax: 3434(N-H), 2919(C-H aromatic), 1637(C = O α, β-unsaturated), 1540(-C = C- aromatic), 1637(C = N), 1266(C-N) cm− 1; 1H-NMR (400 MHz, DMSO- d6) δH: 7.43–7.46(2 H, m), 7.51–7.55(4 H, m), 7.80(2 H, d, J = 7.1 Hz), 7.89–7.95(3 H, m), 8.15(2 H, d, J = 8.4 Hz), 12.89(1 H, s); 13C-NMR (100 MHz, DMSO- d6) δc: 121.1, 127.0, 127.1, 128.4, 129.0, 129.0, 131.9, 136.2, 138.8, 143.5, 144.6, 188.1. ES-MS (m/z): 275[M + H] +, calculated for C18H14N2O.
(E)-3-(1 H-imidazol-2-yl)-1-(1 H-pyrrol-2-yl) prop-2-en-1-one (3i)
Light brown solid, yield 71%, mp 210–211 °C, IR (KBr) νmax : 3401(N-H), 2918(C-H aromatic), 1651(C = O α, β-unsaturated), 1543(-C = C- aromatic), 1651(C = N), 1300(C-N) cm− 1; 1H-NMR (400 MHz, DMSO- d6) δH: 6.29–6.27(1 H, m), 7.12–1.14(1 H, m), 7.17–7.18(1 H, m), 7.26(2 H, brs), 7.43(1 H, d, J = 16.0 Hz), 7.65(1 H, d, J = 15.6 Hz), 12.00(1 H, s), 12.7(1 H, s); 13C-NMR (100 MHz, DMSO- d6) δc: 110.7, 117.2, 123.1, 127.0, 129.3, 133.3, 144.1, 177.8. ES-MS (m/z): 188[M + H] +, calculated for C10H9N3O.
(E)-3-(1 H-imidazol-2-yl)-1-(thiophen-2-yl) prop-2-en-1-one (3j)
Light brown solid, yield 69%, mp 189–190 °C, IR (KBr) νmax : 3435(N-H), 2918(C-H aromatic), 1645(C = O α, β-unsaturated), 1562(-C = C- aromatic), 1645(C = N), 1264(C-N) cm− 1; 1H-NMR (400 MHz, DMSO-d6) δH: 7.32–7.34(2 H, m),7.51(1 H, d, J = 15.2 Hz), 7.79(1 H, d, J = 15.6 Hz), 8.06–8.08(3 H, m), 12.83(1 H, s); 13C-NMR (100 MHz, DMSO- d6) δc: 121.6, 129.5, 131.4, 133.4, 136.0, 143.7, 145.6, 181.6. ES-MS (m/z): 205[M + H] +, calculated for C10H8N2OS.
General procedure for the synthesis of pyrazoline derivatives (4a-e and 5a-b)
To the solution of appropriate chalcone (0.001 mol) in the methanol (10 ml), hydrazine hydrate or thiosemicarbazide (0.002 mol) was added and mixture stirred for 2–3 h (Fig. 3; Table 1). The progress of the reaction was monitored using TLC54. On completion, the crude mixture was poured into ice-cold water, and separated precipitate was filtered off. Obtained solid compounds were washed by distilled water and n-hexane to obtain pure compounds (4a-e and 5a-b).

Scheme for the Synthesis of α,β-unsaturated ketones (3a-j) and their pyrazoline derivatives (4a-e and 5a-b). Reaction conditions: (a) EtOH, NaOH, rt, 24 h; (b) NH2NH2, MeOH, rt, 2–3 h; (c) thiosemicarbazide, EtOH, rt. 2–3 h.
2,4-di-tert-butyl-6-(3-(furan-2-yl)-4,5-dihydro-1 H-pyrazol-5-yl) phenol (4a)
White solid, yield 59%, mp 176–177 °C, IR (KBr) νmax: 3093(C-H aromatic), 1608(C = N), 1300(C-N), 1608(-C = C- aromatic), 1436(-C-H), 3307(O-H), 3575(N-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.29(9 H, s), 1.41(9 H, s), 3.21(1 H, dd, J = 14.4, 16.2 Hz), 3.31(1 H, dd, J = 10.0, 16.2 Hz) 4.87(1 H, dd, J = 10.0, 14.4 Hz), 6.11(1 H, brs), 6.48(1 H, dd, J = 1.8 Hz, J = 3.4 Hz ), 6.64(1 H, d, J = 3.4 Hz), 6.89(1 H, d, J = 2.4 Hz), 7.28(1 H, d, J = 2.4 Hz), 7.52 (1 H, dd, J = 0.6 Hz, J = 1.7 Hz ), 9.32(1 H, brs); 13C-NMR (100 MHz, CDCl3) δc: 29.6, 31.6, 34.1, 35.1, 40.1, 66.5, 111.4, 111.6, 121.4, 123.7, 123.8, 137.5, 141.2, 144.2, 147.4, 150.3, 153.1; ES-MS (m/z): 341[M + H] +, calculated for C21H28N2O2.
2,4-di-tert-butyl-6-(3-(thiophen-2-yl)-4,5-dihydro-1 H-pyrazol-5-yl) phenol (4b)
White solid, yield 46%, mp 219–220 °C, IR (KBr) νmax: 3078(C-H aromatic), 1596(C = N), 1300(C-N), 1596(-C = C- aromatic), 1436(-C-H), 3361(O-H), 3306(N-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.29(9 H, s), 1.42(9 H, s), 3.28(1 H, dd, J = 14.3, 15.9 MHz), 3.38(1 H, dd, J = 10.0, 16.1 MHz), 4.91(1 H, dd, J = 10.0, 14.3 MHz), 6.05(1 H, brs) 6.90(1 H, d, J = 2.4 Hz), 7.05(1 H, dd, J = 3.6, 5.0 Hz), 7.16(1 H, dd, J = 0.9, 3.6 Hz), 7.29(1 H, d, J = 2.4 Hz), 7.38(1 H, dd, J = 0.9, 5.0 Hz ), 9.37(1 H, brs); 13C-NMR (100 MHz, CDCl3) δc:29.7, 31.6, 34.2, 35.2, 41.1, 66.9, 121.6, 123.7, 123.9, 126.1, 127.4, 127.9, 135.8, 137.6, 141.2, 153.1, 154.3 ; ES-MS (m/z): 357[M + H] +, calculated for C21H28N2OS.
3-([1,1′-biphenyl]-4-yl)-5-(1 H-imidazol-2-yl)-4,5-dihydro-1 H-pyrazole (4c)
Pale yellow solid, yield 43%, mp 250 °C above, IR (KBr) νmax: 3023(C-H aromatic), 1671(C = N), 1304(C-N), 1564(-C = C- aromatic), 1401(-C-H), 3312(N-H) cm− 1; 1H-NMR (400 MHz, DMSO- d6) δH: 3.32(1 H, dd, J = 10.5, 16.3 MHz), 3.44(1 H, dd, J = 10.9, 16.3 MHz), 4.91(1 H, t), 6.97(1 H, brs), 7.35–7.39(1 H, m), 7.54–7.44(3 H, m), 7.78–7.70(6 H, m); 13C-NMR (100 MHz, DMSO- d6) δc: 38.3, 57.9, 126.6, 126.9, 127.2, 127.3, 128.0, 129.4, 129.5,132.5, 139.9, 140.2, 148.4, 149.6; ES-MS (m/z): 289[M + H] +, calculated for C18H16N4.
5-(1 H-imidazol-2-yl)-3-(1 H-pyrrol-2-yl)-4,5-dihydro-1 H-pyrazole (4d)
Light brown solid, yield 59%, mp 242–243 °C, IR (KBr) νmax: 3098(C-H aromatic), 1611(C = N), 1293(C-N), 1579(-C = C- aromatic), 1383(-C-H), 3285(N-H) cm− 1; 1H-NMR (400 MHz, DMSO- d6) δH: 3.15(1 H, dd, J = 9.5, 19.7 MHz), 3.44(1 H, dd, J = 11.5, 16.4 MHz), 4.76(1 H, t), 6.07(1 H, m), 6.26(1 H, brs), 6.80(1 H, brs), 6.94(2 H, brs), 11.19(1 H, brs); 13C-NMR (100 MHz, DMSO- d6) δc: 39.1, 57.2, 108.8, 109.6, 120.8, 122.1, 125.9, 145.2, 148.5; ES-MS (m/z): 202[M + H] +, calculated for C10H11N5.
5-(1 H-imidazol-2-yl)-3-(thiophen-2-yl)-4,5-dihydro-1 H-pyrazole (4e)
Light brown solid, yield 63%, mp 188–189 °C, IR (KBr) νmax: 2920(C-H aromatic), 1652(C = N), 1293(C-N), 1436(-C = C- aromatic), 1383(-C-H), 3402(N-H) cm− 1; 1H-NMR (400 MHz, DMSO- d6) δH: 3.22(1 H, dd, J = 10.3, 16.2, MHz), 3.44(1 H, dd, J = 10.8, 16.2 MHz), 4.87(1 H, t), 6.94(2 H, brs ), 7.09(1 H, dd, J = 3.6, 5.0 Hz), 7.20(1 H, dd, J = 1.0, 3.5 Hz), 7.37(1 H, brs), 7.51(1 H, dd, J = 1.0, 5.0 Hz); 13C-NMR (100 MHz, DMSO- d6) δc: 39.1, 40.6, 58.0, 122.3, 126.9, 127.0, 128.0, 137.1, 146.3, 148.1; ES-MS (m/z): 219[M + H] +, calculated for C10H10N4S.
5-(3,5-di-tert-butyl-2-hydroxyphenyl)-3-(furan-2-yl)-4,5-dihydro-1 H-pyrazole-1-carbothioamide (5a)
Off white solid, yield 58%, mp 225–226 °C, IR (KBr) νmax: 3099(C-H aromatic), 1589(C = N), 1221(C-N), 1589(-C = C- aromatic), 1370(-C-H), 3278(O-H), 3145(N-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.18(9 H, s), 1.43(9 H, s), 3.38(1 H, dd, J = 3.2, 18.0 Hz), 3.80(1 H, dd, J = 11.2, 18.0 Hz), 6.10(1 H, dd, J = 3.2, 11.2 Hz ), 6.59(1 H, dd, J = 1.8, 3.4 Hz ), 6.85(1 H, d, J = 2.3 Hz), 6.91(1 H, d, J = 3.3 Hz), 7.27(1 H, d, J = 2.2 Hz), 7.63 (1 H, d, J = 1.2 Hz), 8.06(1 H, s); 13C-NMR (100 MHz, CDCl3) δc: 29.9, 31.4, 34.3, 35.2, 42.9, 57.1, 112.4, 114.6, 120.1, 124.3, 129.5, 139.4, 143.9, 145.8, 145.9, 149.3, 149.6, 174.9; ES-MS (m/z): 400[M + H]+, calculated for C22H29N3O2S.
5-(3,5-di-tert-butyl-2-hydroxyphenyl)-3-(thiophen-2-yl)-4,5-dihydro-1 H-pyrazole-1-carbothioamide (5b)
Off white solid, yield 61%, mp 242–243 °C, IR (KBr) νmax: 3078(C-H aromatic), 1583(C = N), 1230(C-N), 1583(-C = C- aromatic), 1378(-C-H), 3271(O-H), 3148(N-H) cm− 1; 1H-NMR (400 MHz, CDCl3) δH: 1.17(9 H, s), 1.43(9 H, s), 3.44(1 H, dd, J = 3.0, 17.8 Hz), 3.86(1 H, dd, J = 11.1, 17.8 Hz), 6.13(1 H, dd, J = 3.0, 11.0 Hz ), 6.87(1 H, d, J = 2.0 Hz), 7.16(1 H, m), 7.27(1 H, d, J = 2.0 Hz), 7.39(1 H, d, J = 3.12 Hz), 7.54 (1 H, d, J = 4.8 Hz), 8.08(1 H, s); 13C-NMR (100 MHz, CDCl3) δc: 29.9, 31.4, 34.3, 35.1, 43.9, 57.6, 120.1, 124.3, 128.1, 129.5, 130.2, 130.3, 133.6, 139.4, 143.9, 149.7, 153.7, 174.6; ES-MS (m/z): 416[M + H]+, calculated for C22H29N3OS2.
Biological evaluation
In-vitro antibacterial activity
The in vitro anti-bacterial activity of prepared films was evaluated using zone of inhibition and microbial load inhibition. Antibacterial activity was evaluated using a standard minimum inhibitory concentration assay and microbial load inhibition55.
Preparation of bacterial culture for the experiment
Bacteria that had been grown on blood agar plates were moved to 10 mL of 3% w/v tryptic soy broth (TSB) and left there to grow for 12–18 h. To get the test bacteria to the mid-logarithmic phase, 200 µL of the overnight culture was added to 10 mL of fresh 3% w/v tryptic soy broth (TSB). To get 1–2 × 109 CFU/mL of the bacterial suspension, 10 mM Tris, pH 7.4, was added to the pellet, and the bacteria were spun at 10,000 RPM for 10 min. We then discarded the supernatant.
Minimum inhibitory concentration
The antimicrobial activities of the compounds were performed using different approaches in the literature. For the tests, five bacteria were used: Acinetobacter baumannii (ATCC 1605), Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 27853), and Klebsiella pneumoniae (ATCC 27736). The synthesized compounds were mixed with DMSO to make stock solutions. These solutions had a concentration of 5 mg/mL, which is about the same as colistin, levofloxacin, and vancomycin. We grew the bacteria in 3% TSB. Then, we centrifuged the bacteria all night until they reached the mid-logarithmic phase. They were then washed twice in 10 mM Tris at pH 7.4 and put back together in 2X MHB broth (Becton Dickinson, USA) to get 1 × 105 CFU/mL. The amount of peptide in 96 wells of a polypropylene microtiter plate (Cat. No. 3790 from Costar Corp.) was lowered from 100 µg/mL to 0.78 µg/mL. Following this, 50 µL of 2X MHB broth containing 1 × 105 CFU/mL of bacteria was added to each corresponding well. We then incubated the plate at 37 °C for 16 h. The Resazurin method was used to look at the plates and find the minimum inhibitory concentration (MIC). The value that was seen to be higher than the MIC was > 100. We carried out the assay and MIC determination in triplicate56.
In vitro antifungal activity
The in vitro antifungal efficacy of compounds was evaluated against the fungi: Candida albicans (ATCC-90028), Candida tropicalis (ATCC-750), and Candida parapsilosis (ATCC-22019). The standard broth microdilution method was employed for susceptibility testing following the guidelines established by the Clinical and Laboratory Standards Institute (CLSI), utilising RPMI 1640 medium buffered with MOPS [3-(N-morpholino propanesulphonic acid)] for fungal cultures in 96-well plates57. The maximum concentration examined was 50 µg/mL, with each test well containing an inoculum load between 1 and 5 × 10³ cells. The plates were incubated for yeasts for 24 to 48 h and for mycelial fungi for 72 to 96 h at 35 ± 1 °C, followed by visual assessment to determine the MIC55.
Zone of Inhibition (ZOI)
The antifungal effect of the film was tested against (Candida albicans, Candida parapsilosis and Candida tropicalis) and standard. In brief, inoculum sus pension (~ 106CFU/mL) was streaked on a sabouraud dextrose agar (SDA) plate for lawn growth. Then, using sterile forceps formulation, loaded disks were gently pressed onto the surface of SDA plates and incubated for 24 h at 35 ± 0.5℃. The zone of inhibition diameter was determined using a metric ruler55.
Time kill kinetics Candida albicans
The Klepser et al. method was used to perform time kill kinetics in regular RPMI. The isolates were subculture at least twice and cultivated on SDA plates for 24 h at 35 °C before to the experiments. Using spectrophotometry, the inoculum was adjusted to a 0.5 McFarland turbidity standard’s density at 530 nm. To achieve the required concentration of test chemicals, including Fluconazole, the modified inoculum suspension was diluted in RPMI. Compounds were evaluated for antifungal activity at 1/2 X, 1X, and 5X MIC. 30 µL of solution was taken out of the control (drug-free tube) and test compound tube at pre-established intervals of 0, 4, 8, and 24 h, and it was serially diluted in RPMI medium. Four drops of 25 µL were poured on SDA plates and incubated at 35 °C for 24 to 48 h to determine the CFU per millilitre58.
Hemolysis assay
Compound’s (3h and 4c) hemolytic activity was evaluated with 50% whole blood present. In accordance with CSIR-CDRI human ethical authorization, blood was drawn from a healthy volunteer in a vacutainer containing EDTA (CDRI/IEC/2014/A1). An equivalent volume of 1X Phosphate Buffer Saline (PBS) was used to dilute the blood. A prepared blood suspension containing varying concentrations of test compound (50 and 100 µg/mL) was incubated for one hour at 37 °C. PBS and 2% Triton-X were used to achieve 0 and 100% hemolysis, respectively, whereas Fluconazole was utilized as a positive control. The samples were centrifuged at 800 x g for 10 min, and 100 µL of the supernatant was then transferred into a fresh 96-well microtiter plate. The Tecan Infinite M Nano + spectrophotometer was used to measure the absorbance at 540 nm, and the amount of released hemoglobin in the supernatant was used to estimate the hemolysis59,60.
Computational evaluation
Molecular descriptors (MDs) analysis
To calculate all the molecular physicochemical properties of the synthesized compounds, the Molinspiration Property Engine v2022.08 (https://www.molinspiration.com/cgi-bin/properties) was used. Here, absorption (% ABS) was calculated by using the formula % ABS = 109 − (0.345 × TPSA) and calculate their topological polar surface area (TPSA), molecular weight (MW), partition coefficient (log P) rotatable bonds (RB), hydrogen bond acceptor sites (ON), and hydrogen bond donors (OHNH). Recently, a range of MD’s rules are presented to reveal the drug-likeness of a chemical scaffold, with Lipinski’s rule of five (Ro5) being the most widely accepted. According to this rule, drug design and development (DDD) is highly effective in reducing miss-lead2.
Absorption, distribution, metabolism, excretion and toxicity (ADMET) analysis
Here, we employed the admeSAR tool to assess the ADMET characteristics of the synthesized compounds. This server regularly update the available datasets for doing structure-based searches in order to identify ADMET properties61,62.
Molecular docking
The PDB database provided the atomic coordinates for NMT1 (PDB ID: 1IYL) and DHFR (PDB ID: 4HOF). Then, using the Autodock Tool, the protein structures were optimized for molecular docking. Using ChemDraw, the compound (4c) 2D structure was prepared. The OpenBabel GUI version 2.4.1 was used to optimize and convert it into the PDBQT format63. MM2 force field is used in Chem3D to minimize ligand energy. Regarding the docking simulations, specific grid box dimensions were set as follows: X: 13.4, Y: 47.7, Z: -1.0 for NMT1 and X: 0.2, Y: 4.9, Z: 32.7 for DHFR based on the co-crystallized ligand coordinates. Subsequently, Autodock v4.2 was employed to determine the interaction energy and theoretical inhibition constant (Ki). 2D interactions are visualised in Discovery Studio Visualizer 2.5 and 3D interactions are visualized in ChimeraX63,64.
Molecular dynamics
In this study, we utilised Gromacs-2024.1 to perform a molecular dynamics (MD) simulation of the most potent pyrazoline (4c) derivative. After assembling the protein-ligand complex (4c) and prior to system relaxation, we set up the simulation with the following parameters: The TIP3P water model was used as the solvent, the box form was configured to be cubic, and the force constants (fcx, fcy, fcz) were established at 1000. We calibrated 23,601 water molecules (SOL) and four sodium ions (Na+ and Cl−) to neutralise the system. We performed a steepest-down energy minimisation step before initiating the simulation. We conducted the simulation for 100 ns under NPT conditions (constant temperature, pressure, and particle number) at 300 K and 1.01 bar. For electrostatic interactions, we employed the smooth particle mesh Ewald technique. Post-simulation, investigate the H-bond, RMSF, RMSD, and ligand-protein interaction patterns.
Results and discussion
Chemistry
The synthesis of α, β-unsaturated carbonyl compounds was optimized by exploring different reaction conditions, catalysts, and purification methods to improve the yield, selectivity, and efficiency of the reaction (Fig. 3). Finally, compounds (3a-j) were successfully synthesized via Claisen-Schmidt condensation reaction between respective aldehyde and methyl ketone using sodium hydroxide as base in the presence of ethanol at room temperature. The reaction proceeded smoothly and almost completed in 24 h. Desired compounds were obtained in moderate to good yields (69–85%), after purification with water followed by n-hexane. Further, 2-pyrazoline derivatives (4a-e and 5a-b) were synthesized by reacting α, β-unsaturated carbonyl compounds with hydrazine or its derivatives to form the pyrazoline ring using sodium hydroxide in methanol as base, at room temperature (Table 1). All the synthesized compounds were characterized using NMR, MS, UV and FTIR spectral techniques.
Biological evaluation
In vitro antibacterial activity
The synthesized series of compounds (3a-j) was evaluated against five bacterial strains. The results have been tabulated in Table 2. It could be seen that all the tested compounds except (3j) did not show any inhibitory effect up to 100 µg/mL concentration. Compound (3j) had shown moderate activity against S. aureus, E. coli, K. pneumoniae and A. baumannii with MIC of 100 µg/mL. Though, the result was not encouraging but it clearly indicated the selectivity towards three bacterial strains. Also, these three strains are the most heard among human infections and could be a cause of serious concern if left untreated. Selectivity of only one compound towards these pathogens highlights the role of pyrazole and thiophene substituent on the α, β-unsaturated ketones in the activity. Hence, the present series lights and paves path for further development of α, β-unsaturated ketones with heterocyclic substituents as antibacterial agents.
In vitro antifungal activity
Candida is the most common fungal pathogen responsible for mucosal, cutaneous and systemic infections in humans of different age groups. The three well known species of Candida accounting for > 75% of infections are Candida albicans, Candida parapsilosis and Candida Tropicalis. The virulence of these fungal infections and the risk of increasing resistance towards available marketed drugs will always boost the finding of effective treatment. The synthesized α, β-unsaturated carbonyl compounds (3a-j) and their pyrazoline derivatives (4a-e and 5a-b) were subjected to in vitro antifungal assay against three strains of fungus, namely Candida albicans, Candida parapsilosis and Candida tropicalis, utilizing the Standard Broth Micro dilution method CLSI guidelines57. The obtained results were then compared, in terms of MIC (50 µg/mL), with those of the standard drugs Fluconazole (Fluc.). as displayed in Table 3; Fig. 4. Two compounds (3h and 4c) displayed significant antifungal activity with MIC value of 6.25, 12.5, 12.5 µg/mL and 6.25, 6.25, 6.25 µg/mL against C. albicans, C. parapsilosis and C. Tropicalis, respectively. Compounds (3b and 3j) showed moderate activity with MIC value of 25–50 µg/mL while rest of the compound did not show any activity up to 50 µg/mL concentration. Previous reports of the pyrazoline derivatives have not been encouraging as compared to the present series, wherein the activity profile of compound (4c) against all three fungal strains made it an instant hit for further development of antifungal agents.

Bar graph representation of antifungal activity (Candida albicans (ATCC 90028), Candida parapsilosis (ATCC 22019) and Candida tropicalis (ATCC 750)) of α, β-unsaturated ketones (3a-j) and their pyrazoline derivatives (4a-e and 5a-b).
In this study, zone of inhibition (ZOI) also performed to validates the in vitro antifungal activity of the synthesized compounds (3h and 4c) against Candida albicans (ATCC 90028), Candida parapsilosis (ATCC 22019), and Candida tropicalis (ATCC 750) (Fig. 5). Here, used the standard drug Fluconazole solution in DMSO as a positive control. The results showed that the ZOI of the Fluconazole standard and synthesized compounds (3h and 4c) against Candida albicans was 28, 24, and 27 mm after 48 h of incubation. Similarly, we found the ZOI to be 22, 18, and 19 mm against Candida parapsilosis, and 23, 19, and 20 mm against Candida tropicalis respectively (Fig. 5).

(I) Zone of inhibition of Fluconazole standard, compound (3h and 4c) in DMSO solution against Candida albicans (ATCC 90028); (II) Zone of inhibition of Fluconazole standard, compound (3h and 4c) in DMSO solution against Candida parapsilosis (ATCC 22019) and (III) Zone of inhibition of Fluconazole standard, compound (3h and 4c) in DMSO solution against Candida tropicalis (ATCC 750).
Time kill kinetics Candida albicans
Time kill kinetic was performed at different time points i.e., 0, 4, 8, 24 h to check the killing patterns of antifungal compounds. However, no significant log reduction of CFU in Candida albicans ATCC 90,028 was observed at ½X MIC, 1X MIC & 5X MIC after 24 h of treatment in any of above-mentioned compounds including Fluconazole (Fig. 6).

Time kill kinetics for Candida albicans ATCC 90,028 at three different concentrations i.e., ½X MIC, 1X MIC & 5X MIC for compounds (A) 3h (B) 4c, and (C) Fluconazole.
Hemolysis assay
Hemolysis of test compounds was evaluated under physiological conditions i.e., 50% (v/v) suspension of whole blood. Cytotoxicity of compounds against human RBCs was performed at 50 and 100 µg/mL. It was found that all the compounds were showing around 6% hemolysis both at 50 and 100 µg/mL concentration which is less than our positive control Fluconazole. The present study indicates that at the tested concentrations, compounds (3h and 4c) are non-hemolytic compounds and therefore seem to be safe (Fig. 7).

Hemolytic activity of compounds (Hemolytic effects of the compounds on human RBCs were analysed at 50 and 100 µg/mL. Hemoglobin released was measured at 540 nm and is expressed as % of Triton X-100 induced hemolysis. Experiment was performed in triplicate).
In silico drug likeness and ADMET study
A weak pharmacokinetic profile causes most hits to fail in clinical trials. Therefore, it is indisputable that research on ADMET is important for the development of new drugs65. Failure of the hit molecule in the late-stage drug development increases the financial burden on the funding agencies, as well brings wastage of uncompensated time. Hence, the in silico ADMET shows to be cost and time-effective approach in the drug discovery method which eliminates the unfit candidates in the early stages. With this aim, the series of α, β-unsaturated carbonyl compound (3a-j) and their pyrazoline derivatives (4a-e and 5a-b) were subjected to an in silico study using the Molinspiration web tool and the values thus obtained are summarised in Table 3. For most of the compounds the TPSA was between 44.77 and 74.99 Å2 which means that they have good cellular absorption, nRB is ≥ 3 which represented good molecular flexibility, molecular volume (MV) ranged between 200 and 400 showing less steric hinderance and more cellular transportation, logP values which is a measure of molecular hydrophobicity was found < 5 denoted good absorption, bioavailability, hydrophobic drug-receptor interactions. Besides this, Lipinski’s rule was followed in majority of the molecules (Table 4). Thus, from the calculated ADMET parameters, the series of synthetic compounds showed a good absorption profile, which could be interpreted. i.e., 83.13–107.56% with drug-likeness properties (Table 5).
Molecular docking studies
In this study, we perform docking studies of the most active compounds (3h and 4c) with the target proteins including Dihydrofolate Reductase (DHFR) and N-myristoyl Transferase 1 (NMT1) from C. albicans to figure out their most favourable target and best binding site. Here, the obtained results are also compared with the standard drug (Fluconazole) and reference compounds (R64 (1-Methyl-1 H-imidazol-2-yl)(3-methyl-4-{3-[(pyridin-3-ylmethyl)amino]propoxy}-1-benzofuran-2-yl)methanon) and 18 H (5-[3-(2-methoxy-4-phenylphenyl)but-1-yn-1-yl](-6-methylpyrimidine-2,4-diamine)) receptivity. Furthermore, the free binding energy of synthesized compounds computed by using the molecular docking analysis against DHFR and NMT1. Compound (4c) potentially interacted with DHFR and NMT1 (C. albicans), they interacted very strongly (-8.44 and −8.76 nM) and their estimated Ki values 646.91 and 381.92 nM, respectively are found. Similarly, compound 3h also had a binding affinity of -7.96 for DHFR and −8.24 for NMT1 (C. albicans), with Ki values of 1.45 µM for DHFR and 915.49 nM for NMT1. Based on the compound’s binding interactions with DHFR and NMT1 (Table 6, and Table S1-S2).
The primary characteristics of these interactions between the ligand and protein are hydrophobic and hydrogen bonding, which play a major role in determining how well a ligand will bind to the protein and stabilization of molecular complexes. Hydrogen bonds stabilise 3D biological entities, including proteins, nucleic acids, and ligands, through specialised and directional interactions. π-π stacking interactions reinforce complex stability by adding non-covalent binding energy. They are essential for stabilising protein-ligand interactions, DNA base pairing, and supramolecular structures. Weak Van der Waals forces contribute greatly to complex stability. They complement stronger interactions like hydrogen bonds and π-π stacking by maintaining tight contact between molecules. Specific ionic (electrostatic) interactions provide high binding energy. They bind ligands to enzyme active sites and generate protein salt bridges while helping proteins retain their structure. Proteins fold and lipid bilayers develop in cell membranes due to hydrophobic interactions. They stabilise complex structures and lower free energy by clustering nonpolar areas away from water. The DHFR-compound (4c) complex (Fig. 8, and Table S1-S2) exhibited one hydrogen bond with catalytic residue THR58 and additional hydrophobic interactions with DHFR residues. We observed hydrophobic interactions with NMT1 residues in the NMT1-compound 4c complex, which forms one hydrogen bond with ASN392 (Fig. 8). Additionally, we found that synthesized compounds display potential interactions against both target proteins as compared to standard drugs. On the other hand, these compounds show slightly lower interaction potentials with reference compounds or physiological ligands. Moreover, these results potentially favour in vitro antifungal activity (Fig. 6; Table S1-S2), which suggests that compounds (3h and 4c) may be showing dual inhibition against both targets, DHFR and NMT1, for their antifungal activity.

(A): 3D View of the DHFR-molecule complex displaying hydrogen bond with THR58 against (C. albicans) fungal strain. (B): 3D View of the NMT1-molecule complex displaying hydrogen bond with HIS227 from (C. albicans) fungal strain synthesized compound (4c).
Molecular dynamics simulation
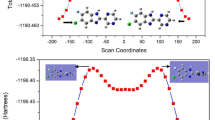
Molecular docking is a fundamental tool in DDD for predicting binding affinity. The process involves numerous approximations, such as treating proteins as rigid entities, simplifying the roles of water and solvents, and making imprecise estimations of entropic and electrical influences66. These simplifications can lead to potential errors. To address these limitations, conducted a MD simulation to study the motion of solvated protein-ligand complexes over a simulated period67. For the MD simulation, selected the combination of the NMT1 protein and the most potent molecule among the synthesized pyrazoline (4c). We calculated the root mean square deviation (RMSD) values for each frame in the trajectory after completing the MD simulation. Generally, RMSD values in simulations indicate the deviation of atomic configurations from their original states. The y-axis (left) depicts the RMSD value of the protein backbone atoms. The RMSD of the protein post-initial stabilization ranged from 0.2 Å to 0.6 Å, remaining comfortably within the 3 Å threshold. This range signifies that the protein maintains its structural integrity during the simulation. The ligand RMSD quantifies ligand stability relative to the protein and its binding pocket. The ligand’s RMSD slightly exceeds that of the protein backbone. However, the RMSD of the ligand and protein remains distinct throughout the simulation, indicating that the ligand remains securely bound to the protein (Fig. 9i). Root mean square fluctuation (RMSF) is important tool for examining subtle variations within the protein chain during simulations. In RMSF graphs, peaks indicate residues exhibiting significant variation throughout the simulation. Typically, terminal and loop sections exhibit greater variability than secondary structures like alpha-helices and beta-strands. Prominent peaks in the graphs indicate regions of the protein experiencing frequent and continuous fluctuations during the simulation. The areas of the protein interacting with the ligand are indicated by green vertical bars in the graph. Figure 9ii shows that residues such as TYR225, HIS227, PHE240, PHE339, LEU350, ILE352, TYR354, VAL390, CYS393, LEU394, LEU415, VAL449, LEU450, and LEU451 exhibit a favourable RMSF profile. Complexes are classified as rigid when the RMSF of their largest residues is below 2.5 Å. Assessing the bonding and structural changes of a biological system requires examining its hydrogen bonds (Fig. 9ii). This study evaluated the stability of the interaction complex through the calculation of its radius of gyration (Rg). The Rg of a frame pertains to the temporal aspect associated with molecular rotation, evaluating the reactivity and folding of the target molecule. Any alteration to the folding conditions of compound 4c typically affects the Rg value (Fig. 9iii). Furthermore, analyses of hydrogen bonds and polar bonds throughout the pathway indicated that hydrogen bonding consistently contributed to the stability of the ligand molecule within the binding pocket (Fig. 9iv). The obtained results display a variety of interactions, including ionic interactions, hydrophobic interactions, hydrogen bonds and water bridges. We discovered that within these varied interactions, ionic interactions were not present, likely because the binding pocket of the protein and the ligand is primarily hydrophobic. The diagram illustrating the protein-ligand interaction from molecular dynamics simulations highlights the significant ways in which the pyrazoline ligand interacts with the protein.

(i) The RMSD plot illustrates the dynamics of proteins and ligand (4c) during a simulation period lasting 100 ns. Protein RMSD is represented in green, while ligand RMSD is shown in maroon, plotted against simulation time (x-axis). (ii) RMSF visualisation of the protein-ligand complex, with green vertical lines denoting interactions between the ligand and protein residues. (iii) Radius of gyration value (Rg). (iv) Hydrogen bonds.
Structure activity relationship (SAR)
Various available aldehydes and methyl ketones were condensed together with hydrazine to form the desired compounds. In vitro antifungal evaluation provided conclusive observations regarding the SAR (Fig. 10). It was found that aldehydes with substituted phenyl rings and methyl ketones with furan, pyrrole, or thiophene showed significant activity. In contrast, the aldehyde with a pyrazole substituent and the ketone with a biphenyl ring exhibited significant inhibitory effects on all three fungal strains. Both the α, β-unsaturated ketone (3h) and its pyrazoline form (4c) demonstrated these effects. The conversion of unsaturated ketone to pyrazoline form increased activity, whereas the methane thioamide substituent on the ‘N’ atom of pyrazoline rings (5a-b) did not enhance activity.

Structure-activity relationship (SAR) analysis of all the synthesized compounds along with the various fragments against Candida albicans.
When pyrazole is present at R1 and biphenyl at R2, α, β-unsaturated ketones exhibit potential antifungal activity against Candida albicans, as measured by a ZOI of 24 mm and a MIC of 6.25 µg/mL. Their conversion to pyrazoline derivatives slightly increased antifungal activity against Candida albicans, with a ZOI of 27 mm and an MIC of 6.25 µg/mL. This finding indicates that incorporating pyrazole and extending resonance not only enhances stability but also improves antifungal potential. We also evaluated the antifungal effects of furan, thiophene, and pyrrole at the R2 position and pyrazole at the R1 position. Among these, only thiophene (3j) showed some activity against Candida albicans, with an MIC of 25 µg/mL. This suggests that pyrazole-based compounds are antifungal but more effective with extended resonance at the R2 position. Various R1 substitutions, including 2-bromo-5-fluorophenyl, 3,5-di-tert-butyl-2-ol-phenyl, and 2-bromo-3,4-dimethoxyphenyl, were also tested with similar R2 substitutions like furan, thiophene, pyrrole, and biphenyl. However, these did not significantly impact antifungal activity. Thus, it can be deduced that pyrazole at R1, biphenyl at R2, and no substitution on the ‘N’ atom of the pyrazoline ring favour antifungal activity.
Additionally, we also compared the obtained results with the existing literature68,69,70 (Fig. 11). To do this, we used MIC and ZOI values to compare the antifungal activity (against Candida albicans) of the synthesized compound (4c) with the some pyrazoline derivatives that were recently reported. Compounds (11) and (12) show ZOIs that are 35 and 32 mm, respectively. They have phenyl rings with NO2 and OCH3 substitutions at the R1 position, phenyl rings only at the R2 position, and N substitutions with (2-methoxyphenoxy)ethan-1-one. Except for these compounds, other reported derivatives (13–16) do not display appropriate antifungal activity against Candida albicans. This again backs up our finding that extending resonance enhance the antifungal activity without N-substitutions.

Conclusion
In this study, we synthesized a group of new α, β-unsaturated carbonyl compounds (3a–j) and their pyrazoline derivatives (4a–e and 5a–b) and evaluated their antibacterial and antifungal activities. Compound (3j) showed selective antibacterial activity against three bacterial strains, while compounds (3b, 3h and 3j) displayed moderate to significant antifungal activity against three pathogenic fungal strains. Notably, compound (4c) was showing potential antifungal activity, with an MIC value of 6.25 µg/mL against Candida albicans, Candida parapsilosis, and Candida tropicalis. We also conducted time kill kinetics of Candida albicans, zone of inhibition, and haemolysis assays in support of their antifungal activity. We further verified their potential antifungal activity against Candida albicans using the obtained results. In addition, in silico drug-likeness and ADMET evaluation showed that these synthesized compounds have potential ADMET properties, making them potential candidates for further analysis. Molecular docking analysis showed that the DHFR and NMT1 proteins from Candida albicans might be better targets for the inhibition process at the molecular level. Docking results revealed that for the most promising antifungal compounds (3h and 4c), the binding energies were −8.76 and −8.44 kcal/mol, as well as -7.96 and −8.24 kcal/mol, respectively. This means that they had potential interactions with the antifungal targets as dual inhibitors. Therefore, the molecular docking results indicate that compound (3h and 4c) have significant antifungal potential and suitable candidates for further studies.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reason-able request.
References
Henary, M. et al. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 10, 14170–14197 (2020).
Azad, I., Nasibullah, M., Khan, T., Hassan, F. & Akhter, Y. Exploring the novel heterocyclic derivatives as lead molecules for design and development of potent anticancer agents. J. Mol. Graph Model. 81, 211–228 (2018).
Ebenezer, O. et al. An overview of the biological evaluation of selected nitrogen-containing heterocycle medicinal chemistry compounds. Int. J. Mol. Sci. 2022. 23, 8117 (2022).
Dhiman, P., Malik, N. & Khatkar, A. Docking-Related survey on Natural-Product-Based new monoamine oxidase inhibitors and their therapeutic potential. Comb. Chem. High. Throughput Screen. 20, (2017).
Cañete-Molina, Á. et al. Bromination-a versatile tool for drugs optimization. Medical-Surgical J. 122, 614–626 (2018).
Yadav, S. Recent advances in the synthesis of pyrazoline derivatives from chalcones as potent pharmacological agents: A comprehensive review. Results Chem. 7, 101326 (2024).
Casimiro-Garcia, A. et al. Identification of (R)-6-(1-(4-cyano-3-methylphenyl)-5-cyclopentyl-4,5- dihydro-1H-pyrazol-3-yl)-2-methoxynicotinic acid, a highly potent and selective nonsteroidal mineralocorticoid receptor antagonist. J. Med. Chem. 57, 4273–4288 (2014).
Mantzanidou, M. & Pontiki, E. & Hadjipavlou-Litina, D. Pyrazoles and pyrazolines as anti-inflammatory agents. Molecules 26, 3439 (2021).
Payne, M. et al. Synthesis and biological evaluation of 3,5-substituted pyrazoles as possible antibacterial agents. Bioorg. Med. Chem. 48, 116401 (2021).
Nurkenov, O. A. et al. Synthesis, structure, and anti-inflammatory activity of functionally substituted chalcones and their derivatives. Russ J. Gen. Chem. 89, 1360–1367 (2019).
Kumar, L. et al. Pyrazoline tethered 1, 2, 3-triazoles: Synthesis, antimicrobial evaluation and in silico studies. Journal of Molecular Structure. 1246, 131154 (2021).
Kumar, L. et al. Synthesis, characterization, α-glucosidase inhibition and molecular modeling studies of some pyrazoline-1H-1, 2, 3-triazole hybrids. Journal of Molecular Structure. 1216, 128253 (2020)
Rasal, N. K., Sonawane, B. & Jagtap, S. V. R. Synthesis, characterization, and biological study of 3-Trifluoromethylpyrazole tethered Chalcone‐Pyrrole and Pyrazoline‐Pyrrole derivatives. Chemistry & Biodiversity. 18, e2100504 (2021).
Nair, A. S. et al. Development of halogenated pyrazolines as selective monoamine oxidase-B inhibitors: Deciphering via molecular dynamics approach. Mol. 2021. 26, 3264 (2021).
Kumar, A. et al. Synthesis, antidiabetic evaluation and bioisosteric modification of quinoline incorporated 2-pyrazoline derivatives. Indian J. Pharm. Educ. Res. 55, 574–580 (2021).
Pharm Sci, P. J. et al. Synthesis, molecular Docking and anti-diabetic studies of novel benzimidazole-pyrazoline hybrid molecules. 33, 847–854 (2020).
Akolkar, H. N. et al. Design, synthesis and biological evaluation of novel furan & thiophene containing pyrazolyl pyrazolines as antimalarial agents. Polycycl. Aromat. Compd. 42, 1959–1971 (2022).
Çelik, G., Arslan, T., Şentürk, M., der Pharmazie, D. E. A. & Şentürk, M. undefined. Synthesis and characterization of some new pyrazolines and their inhibitory potencies against carbonic anhydrases. Archiv der Pharmazie. 353, 1900292 (2020).
Kamogawa, E. & Sueishi, Y. A multiple free-radical scavenging (MULTIS) study on the antioxidant capacity of a neuroprotective drug, Edaravone as compared with uric acid, glutathione, and trolox. Bioorg. Med. Chem. Lett. 24, 1376–1379 (2014).
Beebany, S. et al. Preparation and identification of new 1,4-bis (5,3-substituted-2,3-dihydro-1H-pyrazole-1-yl) Buta-1,4-Dione derivatives with their antibacterial effect evaluation. Chemical Methodologies. 7, 123–136 (2023).
Kumar, H., Bansal, K. K. & Goyal, A. Synthetic methods and antimicrobial perspective of pyrazole derivatives: an insight. Anti-Infective Agents. 18, 207–223 (2019).
Martin, H., Kavanagh, K. & Velasco-Torrijos, T. Targeting adhesion in fungal pathogen Candida albicans. Future Med. Chem. 13, 313–334 (2021).
Schultz, T. W. & Yarbrough, J. W. Trends in structure–toxicity relationships for carbonyl-containing α,β-unsaturated compounds. SAR QSAR Environ. Res. 15, 139–146 (2004).
Susilawati, S., Anwar, C., Saleh, I. & Salni, S. Flavonoid as anti-Candida agents. IJFAC (Indonesian J. Fundam Appl. Chem. 8, 88–97 (2023).
Yadav, C. S. et al. Exploring the therapeutic potential of Chalcones in oncology: A comprehensive review. Curr. Bioact Compd. 20, 12–47 (2024).
Farooq, S. & Ngaini, Z. Synthesis, molecular docking and antimicrobial activity of Α, β-unsaturated ketone exchange moiety for chalcone and pyrazoline derivatives. ChemistrySelect 5, 9974–9979 (2020).
Ramírez–Prada, J. et al. Synthesis of novel quinoline–based 4,5–dihydro–1H–pyrazoles as potential anticancer, antifungal, antibacterial and antiprotozoal agents. Eur. J. Med. Chem. 131, 237–254 (2017).
Kumar, S., Gupta, S., Rani, V. & Sharma, P. Pyrazole containing anti-HIV agents: an update. Med. Chem. (Los Angeles). 18, 831–846 (2022).
Zhao, S., Pi, C., Ye, Y., Zhao, L. & Wei, Y. Recent advances of analogues of curcumin for treatment of cancer. Eur. J. Med. Chem. 180, 524–535 (2019).
Alex, J. & Kumar, R. 4,5-Dihydro-1H-pyrazole: an indispensable scaffold. Journal of enzyme inhibition and medicinal chemistry. 29, 427–442 (2014).
Goodarzi, M., Saeys, W., De Araujo, M. C. U., Galvão, R. K. H. & Vander Heyden, Y. Binary classification of chalcone derivatives with LDA or KNN based on their antileishmanial activity and molecular descriptors selected using the successive projections algorithm feature-selection technique. Eur. J. Pharm. Sci. 51, 189–195 (2014).
Chen, B., Zhu, Z., Chen, M., Dong, W. & Li, Z. Three-dimensional quantitative structure–activity relationship study on antioxidant capacity of curcumin analogues. J. Mol. Struct. 1061, 134–139 (2014).
Du, Z. Y. et al. Anti-proliferative, anti-inflammatory and antioxidant effects of curcumin analogue A2. Arch. Pharm. Res. 36, 1204–1210 (2013).
Bukhari, S., Franzblau, S., Jantan, I. & Jasamai, M. Current prospects of synthetic curcumin analogs and chalcone derivatives against Mycobacterium Tuberculosis. Med. Chem. (Los Angeles). 9, 897–903 (2013).
Qin, H. L. et al. Synthesis of α,β-unsaturated carbonyl-based compounds, oxime and oxime ether analogs as potential anticancer agents for overcoming cancer multidrug resistance by modulation of efflux pumps in tumor cells. J. Med. Chem. 59, 3549–3561 (2016).
Iraji, A. et al. Ugi adducts: design and synthesis of natural-based α-glucosidase inhibitors. Lett. Org. Chem. 19, 1084–1093 (2022).
Ou, J. L. et al. Structure–activity relationship analysis of curcumin analogues on anti-influenza virus activity. FEBS J. 280, 5829–5840 (2013).
Correia-Da-Silva, M. et al. Synthesis and characterization of curcumin-chitosan loaded gold nanoparticles by Oryctes rhinoceros’ chitin for cosmeceutical application. Mol. 2023. 28, 1799 (2023).
Arshad L. et al. Immunosuppressive Effects of Natural α,β-Unsaturated Carbonyl-Based Compounds, and Their Analogs and Derivatives, on Immune Cells: A Review. Front. Pharmacol. 8, 1–22 (2017).
HUO, Z. P., FENG, X. C., WANG, Y., TIAN, Y. T. & QIU, F. Sulfite as the substrate of C-sulfonate metabolism of Α, β-unsaturated carbonyl containing Andrographolide: analysis of sulfite in rats’ intestinal tract and the reaction kinetics of Andrographolide with sulfite. Chin. J. Nat. Med. 19, 706–712 (2021).
Amslinger, S. The tunable functionality of α,β-Unsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem 5, 351–356 (2010).
Dowling, G. & Malone, E. Analytical strategy for the confirmatory analysis of the non-steroidal anti-inflammatory drugs firocoxib, propyphenazone, ramifenazone and piroxicam in bovine plasma by liquid chromatography tandem mass spectrometry. J. Pharm. Biomed. Anal. 56, 359–365 (2011).
Grima, M. et al. Mechanism of action of the diuretic effect of muzolimine. Archives des Maladies du Coeur et des Vaisseaux, 83, 1205–1208 (1990).
Surendra Kumar, R., Arif, I. A., Ahamed, A. & Idhayadhulla, A. Anti-inflammatory and antimicrobial activities of novel pyrazole analogues. Saudi J. Biol. Sci. 23, 614–620 (2016).
Hosamani, K. M. & Chavan, R. R. Microwave-assisted synthesis, computational studies and antibacterial/ anti-inflammatory activities of compounds based on coumarin-pyrazole hybrid. Royal Society open science. 5, 172435 (2018).
Nehra, B. et al. Recent advancements in the development of bioactive pyrazoline derivatives. European Journal of Medicinal Chemistry. 205, 112666 (2020).
Schmidt, A. et al. Influence of muzolimine on arterial wall Elastin. Biochem. Pharmacol. 33, 1915–1921 (1984).
Siddiqui, M. R., AlOthman, Z. A. & Rahman, N. Analytical techniques in pharmaceutical analysis: A review. Arab. J. Chem. 10, S1409–S1421 (2017).
Basak, T., Nath, V., Kumar, V. & Goyal, A. K. Silico identification of antifungal compounds as mutant DHFRase inhibitors: Structure-based approach, molecular dynamics simulation and structural integrity analysis. J. Comput. Biophys. Chem. 20, 589–602 (2021).
Feng, R. et al. Discovery of novel rhizoctonia Solani DHFR inhibitors as fungicides using virtual screening. J. Agric. Food Chem. 71, 19385–19395 (2023).
DeJarnette, C., Luna-Tapia, A., Estredge, L. R. & Palmer, G. E. Dihydrofolate Reductase Is a Valid Target for Antifungal Development in the Human Pathogen Candida albicans. mSphere 5 (2020).
Wang, Y. et al. N-Myristoyltransferase, a potential antifungal candidate drug-target for Aspergillus flavus. Microbiol. Spectr. 11, e04212–e04222 (2022).
Javid, S. et al. Rational design, synthesis, analysis and antifungal activity of novel myristic acid derivatives as N-myristoyltransferase inhibitors. J. Mol. Struct. 1303, 137568 (2024).
Ahmad, A., Husain, A., Khan, S. A., Mujeeb, M. & Bhandari, A. Synthesis, antimicrobial and antitubercular activities of some novel pyrazoline derivatives antimicrobial and antitubercular activities of novel pyrazoline derivatives. J. Saudi Chem. Soc. 20, 577–584 (2016).
Yadav, C. S. et al. Synthesis, characterization, quantum chemical modelling, molecular docking, in silico and in vitro assessment of 3-(2-bromo-5-fluorophenyl))-1-(thiophen-2-yl)prop-2-en-1-one. Sci. Rep. 14, 29838 (2024).
Li, Z. et al. Insights into the antifungal properties of Myxobacteria outer membrane β-1,6-glucanase. J. Agric. Food Chem. 71, 9656–9666 (2023).
Clinical and Laboratory Standards Institute (CLSI). Reference method for broth dilution. In Ref. Method Broth Dilution Antifung Susceptibility Test. Yeasts Approv Stand. 3th ed. 28, 0–13 (2008).
Klepser, M. E. et al. Multi-center evaluation of antifungal time-kill methods. J. Infect. Dis. Pharmacother. 5, 29–41 (2001).
Hasan, M. M. et al. HLPpred-Fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 36, 3350–3356 (2020).
Karasev, D. A., Malakhov, G. S. & Sobolev, B. N. Quantitative prediction of hemolytic activity of peptides. Comput. Toxicol. 32, 100335 (2024).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017 71 7, 1–13 (2017).
Yang, H. et al. AdmetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35, 1067–1069 (2019).
Azad, I., Khan, T., Ahmad, N., Khan, A. R. & Akhter, Y. Updates on drug designing approach through computational strategies: a review. Futur Sci. OA 9, (2023).
Azad, I. Molecular docking in the study of ligand-protein recognition: an overview. Molecular Docking-Recent Advances, IntechOpen. (2023).
Khan, H. et al. Structure based docking and biological evaluation towards exploring potential anti-cancerous and apoptotic activity of 6-gingerol against human prostate carcinoma cells. BMC Complement. Med. Ther. 24, 1–23 (2024).
Agu, P. C. et al. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 13, 13398 (2023).
Pechlaner, M., van Gunsteren, W. F., Hansen, N. & Smith, L. J. Molecular dynamics simulation or structure refinement of proteins: are solvent molecules required? A case study using hen lysozyme. Eur. Biophys. J. 51, 265–282 (2022).
Dhonnar, S. L., Jagdale, B. S., Adole, V. A. & Sadgir, N. V. PEG-mediated synthesis, antibacterial, antifungal and antioxidant studies of some new 1,3,5-trisubstituted 2-pyrazolines. Mol. Divers. 27, 2441–2452 (2023).
Mervat Mohammed, K. & Omar, T. N. A. Synthesis characterization, anti-inflammatory, and antimicrobial evaluation of new 2-pyrazolines derivatives derived from guaiacol. Iraqi J. Pharm. Sci. 32, 254–261 (2023).
Agare, S. U., More, M. P., Tajane, S. P. & Kadre, T. V. Synthesis, characterization and biological activity of novel heterocyclic compounds containing acylated pyrazoline. Orient. J. Chem. 40, 499–505 (2024).
Acknowledgements
The authors acknowledge the Division of Medicinal and Process Chemistry, Microbiology and Laboratory Animal Facility Division CSIR-CDRI, Lucknow, India for providing the facilities to carry out research work and financial support. The manuscript bears communication number IU/R&D/2024-MCN0002614 from Integral University, Lucknow. The authors are also thankful to the Deanship of Scientific Research (DSR) at King Abdulaziz University (KAU), Jeddah, Saudi Arabia, for funding this work under Grant no. (GPIP: 1674-156-2024).
Funding
Rajdeep Guha secured funding, provided consumables and edited the manuscript. This project was funded by the Deanship of Scientific Research (D.S.R.), King Abdulaziz University, Jeddah, Saudi Arabia, under Grant no. (GPIP: 1674-156-2024). The authors, therefore, acknowledge with thanks to D.S.R. technical and financial support.
Author information
Authors and Affiliations
Contributions
ARK, MBL, RG, and IA planned and supervised the experiments. CSY synthesized, characterized compounds and prepare the initial draft of the manuscript, while IA assisted in the manuscript’s writing and review. AK and SC carried out the experiments involving antifungal activities. SPS and IA supervised the experiments in chemistry. JK carried out experiments involving antibacterial activities. AK and JK carried out experiments involving time kill kinetics and hemolysis assay. SA carried out experiments involving cytotoxicity. CSY, IA and KS carried out computational studies and molecular docking analyses. RG, VA and AAA secured funding, provided chemicals and consumables, and edited the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yadav, C.S., Krishna, A., Singh, S.P. et al. Synthesis, characterization and bio-evaluation of novel series of pyrazoline derivatives as potential antifungal agents. Sci Rep 15, 14752 (2025). https://doi.org/10.1038/s41598-025-98645-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98645-1
