
Abstract
Computational modeling and analysis of biomolecular network models annotated with omics data are emerging as a versatile tool for designing personalized therapies. Current endeavors aimed at employing in silico models towards personalized cancer therapeutics remain limited in providing all-in-one approach that ascertains actionable targets, re-positions FDA (Food and Drug Administration) approved drugs, furnishes quantitative cues on therapy responses such as efficacy and cytotoxic effect, and identifies novel drug combinations. Here we propose “CanSeer”—a methodology for developing personalized therapeutics. CanSeer employs patient-specific genetic alterations and RNA-seq data to annotate in silico models followed by dynamical network analyses towards assessment of treatment responses. To exemplify, three use cases involving paired samples, unpaired samples, and cancer samples only, of lung squamous cell carcinoma (LUSC) patients are provided. CanSeer reveals the effectiveness of repositioned drugs along with the identification of several novel LUSC treatment combinations including Afuresertib + Palbociclib, Dinaciclib + Trametinib, Afatinib + Oxaliplatin, Ulixertinib + Olaparib, etc.
Similar content being viewed by others

Introduction
Cancer remains a leading cause of death worldwide despite an ever-increasing repertoire of treatment modalities1,2,3,4. Amongst the salient impediments faced by anticancer regimens are drug resistance5, patient-to-patient variability of therapeutic response6, and cytotoxicity7,8. Towards overcoming these issues, molecular targeting-based therapies, seminally exemplified by imatinib’s success in treating chronic myeloid leukemia in 20019, has paved the way for subsequent breakthroughs. The ensuing research impetus led to the development of several targeted therapies including vemurafenib10,11,12, gilteritinib13,14, temsirolimus15,16, etc. These breakthrough drugs further stimulated efforts aimed at identification of molecular targets in wider varieties of cancer and onward employment in devising targeted therapies17,18,19,20,21.
Towards designing targeted therapies, advancements in high throughput sequencing and multiplex mutational screening have been instrumental in elucidating novel molecular signatures22,23,24,25,26 thus giving rise to the domain of personalized therapeutics27,28,29,30. In an early-harvest project, MD Anderson Cancer Center embarked on a program to screen patient-specific genetic alterations with “matched targeted therapies”31,32,33, leading to momentum in genome-informed precision medicine. However, subsequent clinical trials remained limited to monotherapies (a single target—a single drug combination) which eventually started to lose efficacy and developed resistance. As an example, about 65% of FLT3 mutated refractory acute myeloid leukemia (AML) patients showed resistance to gilteritinib14. Similarly, BRAF targeting single-agent vemurafenib failed to offer benefits during phase-II clinical trials34. This was partially attributable to drug resistance caused by underlying genetic heterogeneity35 and molecular cross-talk between interconnected signaling pathways36, thereby treatment escape37,38. Endeavors at overcoming these undesired outcomes now employ combinations of monotherapies such as trametinib + fluvastatin for treating lung cancer39, trametinib + zoledronate for KRAS-mutated patients of metastatic colorectal cancer40, etc. Results from such combinatorial regimens have demonstrated lowered rates of treatment escape and resistance to therapy41,42.
To assist in predicting effective combinatorial therapies, as well as to better understand the mechanistic underpinnings, dynamical simulations of omics-informed biomolecular network models have gained prominence43,44,45,46,47,48. In particular, mathematical modeling coupled with computational dynamical analysis have advanced our understanding of human biomolecular signaling, along with incorporation of patient-specific multi-omics data. Recent studies, such as those by Beal et al., have used omics profiles to stratify patients and analyze individualized survival49, while Eduati et al. constructed continuous logic models that revealed heterogeneity amongst pancreatic cancer patients50. However, the study remained limited to only a single “apoptotic” readout. Ianevski et al. leveraged machine learning for identifying patient-specific drug combinations, reducing toxicity in leukemic cells51, but lacked mechanistic insights and posed a risk of off-label drug use. In a recent study, we reported a personalized in silico “drosophila patient model” that employed patient-specific somatic mutations and identified personalized combinatorial treatment for colorectal cancer52. The proposed annotation of network models with somatic mutations remained deficient in construing a comprehensive molecular signature of individual patients. Moreover, the coverage of druggable and clinically actionable targets remained uncatered during exhaustive screening for predicting efficacious targets. Later in 2022, Montagud et al. employed PROFILE and enriched the Boolean model of cancer signaling with more interactions, followed by customization for prostate cancer patients53. For developing individualized models, the study considered the presence/absence of somatic mutations and copy number alterations. However, the strategy lacked considering ground knowledge of mutation type and mapping of copy number altered genes with RNA-seq based gene expressions. Also, the in silico drug response approach could not differentiate between the effect of two independent drugs targeting the same gene of patient, one at a time.
To date, a multi-factorial patient data-integrative approach to simulate drug effects using drug scores, followed by quantification of therapy response remains unassessed. In addition, the lack of a systematic all-in-one approach impedes devising personalized therapies. This presents the need of an overarching approach that involves the identification of suitable treatment options, determination of actionable targets, repositioning of FDA (Food and Drug Administration) approved drugs, evaluation of efficacy and cytotoxic effect, and exploration of novel drug combinations. Also, the advantages offered by in silico studies towards personalized cancer therapeutics till date, remain to be fully translated54, thereby, necessitating the development of a computational method for devising translational personalized anticancer therapies.
Towards this aim, we propose a method “CanSeer” to develop in silico models for clinical translation of personalized treatments. The proposed method initiates with the development of literature-derived biomolecular regulatory network models followed by their validation. The validated model is then integrated with patient-specific genomic and transcriptomic data for dynamical analysis, selecting druggable targets and curating drugs to inform personalized treatment decisions based on molecular and clinical considerations. For this purpose, drug activity scores are computed to divulge the effect of different drugs targeting the same gene. Moreover, to decode the varying effect of a drug from patient to patient, CanSeer computes “drug scores” (DS) for patients by employing the drug activity score and normalized gene expressions of each individual cancer patient. DS are subsequently used to annotate the personalized models, followed by the re-analysis of each model. Employing network model’s cell fate outcomes, treatment efficacy and cytotoxic effect are computed, which then elucidates treatment suitability. The method concludes with the comparison of cell fate predictions under tailored treatment of patient cancer models, demonstrating personalized treatment response. The three use cases of CanSeer exemplified with lung squamous cell carcinoma (LUSC) case study demonstrated CanSeer's utility in predicting optimal personalized treatments, suggesting potential repositioned drugs and identifying novel drug combinations like Afuresertib + Palbociclib, Dinaciclib + Trametinib, amongst others.
Taken together, CanSeer provides a next-generation clinical translation framework for personalized cancer therapeutics by combining mechanistic insights from computational modeling of “multi-omics” patient data. As a result, the method helps (i) elicit optimal tailored treatments, (ii) provide mechanistic insights into patient-to-patient variability of therapeutic response, (iii) repositions FDA-approved drugs, (iv) determines and evaluates actionable targets, (v) elucidates treatment efficacy, (vi) reveals cytotoxic effect, and (vii) facilitates the discovery of novel drug combinations. In conclusion, the work provides a translational approach to precision oncology to formulate and assess clinically deployable personalized treatment plans.
Results
CanSeer—a personalized cancer therapeutics framework
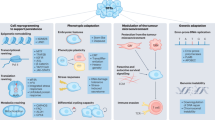
“CanSeer” is a novel methodology for developing in silico biomolecular network models towards designing personalized cancer therapeutics and their clinical evaluation. The proposed method comprises of four salient steps which include: (i) development of literature-derived Boolean models of biomolecular regulations, and their validation, (ii) genetic alterations and expression data acquisition, pre-processing, and normalization, (iii) model annotation with patient-specific omics data comprising of genomic and transcriptomic profiles followed by the dynamical analyses of personalized models, and (iv) therapeutic screening and identification of optimal regimens for cancer patients (Fig. 1).

Algorithmic pipeline for personalized cancer therapeutics: (1) Development of network models from literature and databases with dynamical analysis and validation, (2) Omics data acquisition, pre-processing, and normalization, (3) Patient-specific model personalization and cell fate analysis, and (4) Therapeutic screening for identifying optimal drug regimens.
Below, we provide the exemplification of proposed methodology through a case study on lung squamous cell carcinoma.
Step 1—Literature and database-driven development of biomolecular regulatory network models and validation
To exemplify Step 1 in CanSeer (Fig. 2A), a large-scale human signaling network was adopted from Cho et al.55 and analyzed (Fig. 2B). The analysis result along with input file, cell fate file, network rules, and parameters are given in Supplementary Information 1—Supplementary Data 1, while the detailed description of network in provided as Supplementary Information 2—Supplementary Fig. S1 and Supplementary Table S1). The network model comprised of 197 nodes and 744 edges, organized in a mesh topology with 13 input nodes, 8 output nodes, and 176 processing nodes. Topologically, the network followed a scale-free structure, wherein 114 nodes had a high degree (connectivity), playing a critical role in maintaining network robustness. The network contained distinct groups of nodes and edges that programmed five cellular processes including normal proliferation, abnormal proliferation, apoptosis, quiescence and metastasis. Lastly, in terms of structure, the network model had a hierarchical topology with input nodes triggering the downstream processing nodes, which then interact with each other and set the output nodes. Alongside, cell fates involved in oncogenesis such as senescence, and cell cycle arrest were programmed and mapped by updating the published network. Our results from deterministic analysis (DA) of the updated network, under normal input conditions, corroborate with the published outcomes (Fig. 2C; Supplementary Information 1—Supplementary Data 2). To evaluate network robustness, perturbations were introduced as input signals and the network response was scrutinized using DA pipeline56,57. The results exhibited highest variation in normal proliferation (SEM 0.0025) followed by apoptosis (SEM 0.0021) in the network model (Fig. 2D; Supplementary Information 1—Supplementary Data 3). Further, to inspect the distinct input–output relationship, parameter sensitivity analysis was carried out by screening input nodes individually and in combinations (Supplementary Information 1—Supplementary Data 4). Specifically, the analysis assessed how changes in the states of Cho et al.’s input nodes (e.g., switching a node from active to inactive, or 1 to 0 etc.) influenced the overall system behavior i.e. model’s output. Briefly, we first altered the input nodes by switching them between 0 (inactive) and 1 (active), or vice versa, in incremental steps of 0.1. This gradual variation (increase or decrease) in input signal allowed us to observe the incremental effects on the model output, providing insights into how each node contributes to the overall dynamics of the network. The overall process is illustrated as a flowchart in Supplementary Information 2—Supplementary Fig. S2. The results showed that the network was particularly sensitive to the input levels of following nodes “alphailig”, “DNA damage”, “EGF”, “IL1/TNF”, “TGF-β”, and “Wnt”. Noteworthy was that an increase in DNA damage signal led to an increase in apoptosis, along with a decrease in normal proliferation, abnormal proliferation, and metastasis (Fig. 2E). Moreover, the specific input perturbation to cell fate outcomes were classified into 4 categories including: (i) “Apoptosis”, which increased with higher DNA damage signals, while on the other hand, elevated levels of EGF signaling reduced apoptosis by supporting cell growth and survival. (ii) “Normal cell proliferation”, which was observed to increase with Wnt and EGF signaling, both individually and in combination, promoting cell growth. This was in contrast with DNA damage, IL-1/TNF, and TGF-β inhibit normal proliferation, as these signals typically induce stress responses, inflammation, or growth suppression. (iii) “Abnormal cell proliferation” and “Metastasis” decreased with an increase in DNA damage signals or αi ligands expression. Both stimuli act to suppress unchecked cell growth and inhibit the spread of metastatic cells. (iv) Increased IL-1/TNF and TGF-β signaling promoted “Metastasis”, due to their role in inflammation and tumor progression. Summarily, EGF and Wnt signaling promote cell survival by enhancing normal cell proliferation and suppressing apoptosis, supporting growth. On the other hand, DNA damage, IL-1/TNF, TGF-β, and αi ligands lead to anti-survival outcomes by inhibiting proliferation, promoting apoptosis, or suppressing metastasis.

Development and evaluation of biomolecular regulatory network in Step 1 of CanSeer. (A) The workflow involves constructing a biomolecular regulatory network by integrating information from literature and databases. This network is translated into a rules-based or weight-based model, annotated with qualitative input values, and subjected to dynamical analysis (deterministic, probabilistic, or ordinary differential equation (ODE)). Output node propensities from dynamical analysis program cell fate outcomes, which are then compared with literature for validation. Additional validation comprises robustness and sensitivity analyses to confirm biological plausibility and consistency with the existing literature. (B) Cho et al.'s human signaling network was re-analyzed in TISON (Theatre for in silico Systems Oncology) using the deterministic analysis approach. As a result, three cell fates were identified: Normal Proliferation (NP), Abnormal Proliferation (AP), and Metastasis (MT). The propensities for these cell fate outcomes were 0.77, 0.08, and 0.15, respectively, which aligned with the original findings of Cho et al. The bar chart visually compares these propensities with Cho et al.’s results55 wherein the x-axis represents the cell fates i.e. NP, AP and MT, and the y-axis plots their corresponding propensities. (C) The network is modified by tuning the nodes to program other cell fates involved in oncogenesis such as apoptosis, senescence, and cell cycle arrest (. This updated network is re-analyzed and plotted. The x-axis in bar chart shows cell fates and y-axis represents propensities. The cell fates include normal proliferation (NP), abnormal proliferation (AP), metastasis (MT) and apoptosis (Apo). (D) The stability of the model is evaluated by subjecting it to minor input perturbations. The average propensities (mean) of various cell fates are plotted with standard error of means (SEM). The cell fates include normal proliferation (NP), metastasis (MT), apoptosis (Apo), abnormal proliferation (AP) and quiescence (QU). (E) The line graph shows the relationship of input perturbations with the cell fate outcomes in human signaling network. Input-cell fate outcome relationship of DNA damage is only showcased here (Supplementary Information 1—Supplementary Data 4). The line graph indicates the positive relationship between DNA damage and apoptosis (Apo) and negative relationship between DNA damage and the following cell fates: normal proliferation (NP), abnormal proliferation (AP) and metastasis (MT). The x-axis shows increasing stimulus of DNA damage and y-axis indicates propensities of the cell fates.
In addition, we also explored the combined effects of perturbing multiple input nodes, simultaneously. Through this combinatorial screening, we again assessed the system’s sensitivity to complex perturbations. The detailed results are provided in Supplementary Information 1—Supplementary Data 4.
Step 2—Omics data collection, pre-processing, and normalization
In CanSeer’s second step, patient-specific omics data is acquired, pre-processed, and normalized for later use in network model annotation. The detailed workflow of CanSeer’s Step 2 is presented in Fig. 3.

Workflow of Omics Data Acquisition, Pre-processing and Normalization in Step 2 of CanSeer. (A) Acquisition of patient-specific multi-omics data including genetic alterations, expression profiles, and sample sheet from TCGA PanCancer Atlas via GDC (Genomics Data Common) and cBioPortal, ensuring consistency in patient datasets by comparing sample IDs between GDC and cBioPortal. (B) Extraction of RNA-seq data, initially identified by Ensembl IDs and then mapped to gene symbols with BioTools.fr. Gene symbols are filtered based on network nodes, and aliases are sourced from GeneCards or HGNC (HUGO Gene Nomenclature Committee) for unmatched gene names. Using the finalized genes list, RNA-seq based gene expressions are fetched based on the case and sample selection. Patient samples are categorized into three cases based on RNA-seq profiles, which include paired, unpaired and cancer case only. Samples are classified into two types: Normal (N) and Cancer (C). In all three cases, patient’s cancer expressions are integrated with corresponding genetic alterations (copy number variations (CNVs), somatic mutations (SMs) and genomic structural variants (SVs)), and outliers from expression data are removed using statistical methods i.e., 3 × MAD (Median Absolute Deviation) or IQR (interquartile range). Following outlier removal, the expressions from both N and C samples are merged into a combined dataset labeled as "N + CS". (C) In paired and unpaired cases, gene expressions are normalized using the highest expression in N + CS, whereas in cancer only, normalization is done within cancer samples (CS).
Patient data acquisition
To demonstrate Step 2A of CanSeer, we selected normal solid tissue and primary tumor samples of The Cancer Genome Atlas (TCGA) project “Lung Squamous Cell Carcinoma (TCGA-LUSC)” at GDC portal. The samples were then filtered for transcriptomic profiling, mRNA expression quantification, and RNA-seq data in FPKM (Fragments Per Kilobase of transcript per Million mapped reads) format. All openly accessible (551 files) with RNA-seq based expression data for lung cancer samples were downloaded along with the sample sheet (Supplementary Information 1—Supplementary Video 1). In addition, genetic variations including Copy Number Variations (CNVs), Somatic Mutations (SMs) and Genomic Structural Variations (SVs) of the selected samples, were obtained from TCGA PanCancer Atlas study using cBioPortal (Supplementary Information 1—Supplementary Video 2). Patient dataset matching was ensured by comparing sample IDs between GDC and cBioPortal.
Data pre-processing
To describe the data pre-processing step of CanSeer, Ensembl IDs were mapped onto gene symbols (Supplementary Information 1—Supplementary Data 5), gene symbols list was filtered, and aliases were obtained (Supplementary Information 1—Supplementary Data 6). The network nodes list (from Step 1) was converted into gene symbols and the respective RNA-seq based gene expression values were extracted for the designed use-cases (described in Proposed Methodology—Step 2B) in CanSeer (Supplementary Information 1—Supplementary Data 7). On selecting Case 1 and choosing whole dataset, 49 paired normal and cancer samples (Case 1) were obtained. On choosing Case 2 and picking whole dataset, 50 normal and 453 cancer samples were attained. Opting for Case 3 and whole dataset, 453 cancer samples were acquired. Next, to remove the skewed data from normal samples, outliers were removed using interquartile range (IQR). To remove outliers from expression data of cancer samples mapped with CNVs, SMs, and SVs, again IQR was chosen (Supplementary Information 1—Supplementary Data 8). After the removal of outliers from normal and cancer samples, the cleaned data was combined (N + Cs) in Cases 1 and 2. However, the processing remains limited to cancer samples only in Case 3.
Normalization
Subsequent to the data pre-processing step, the process of normalization is undertaken. The dataset pertaining to lung squamous cell carcinoma (LUSC) samples, which had undergone processing in previous step (Step 2B) for all three different cases, was then subjected to normalization (Supplementary Information 1—Supplementary Data 9).
Step 3—Model annotation with patient-specific omics data and dynamical analysis
In the third step, CanSeer employs the normalized gene expression values of cancer samples along with their CNVs, SMs and SVs, and incorporates them into the Boolean logic model for personalizing rules-based biomolecular network models (Fig. 4). A step-by-step description of model annotation with normalized omics data in case of paired (Case 1), unpaired (Case 2), and cancer samples only (Case 3) is given in “Proposed Methodology—Step 3”.

Methodology of model annotation with patient-specific omics data followed by dynamical analysis (Step 3 of CanSeer). (A) The rules-based model is customized with patient-specific omics data including gene expressions, copy number variations (CNVs), somatic mutations (SMs) and genomic structural variants (SVs). The model incorporating patient-specific data is analyzed through dynamical analysis, and the resulting cell fate outcomes are compared between normal and cancer conditions. (B) In normal tissue samples categorized as paired case, normalized gene expression (NGE) values are assigned to the input nodes, and output node propensities are compared to corresponding gene expressions, with network tuning if needed. For unpaired cases, the median NGE values are used for comparison and tuning. (C) In the network model, cancer samples, whether they belong to paired, unpaired, or cancer-only cases, are annotated in the same way. First, patient-specific altered genes (CNVs, SMs and SVs) are searched within the network. Then, the types of SMs are considered for further inclusion and exclusion. SMs such as missense mutation, splice donor variant, splice acceptor variant, splice site variant, frameshift deletion, and non-coding transcript exon variant are included while silent mutation, intron variant, splice region variant, and 5ʹ UTR variant are excluded. Next, the patient-specific altered genes are mapped with the driver genes of same TCGA project and cancer type. The identified patient-specific CNVs, SMs, and SVs are fixed into the model along with the input nodes. These nodes are fixed with the corresponding NGEs of the patient’s cancer sample, to maintain their activity constant throughout the analysis, while other network nodes remain dynamic. The output node propensities from dynamical analysis are compared with the corresponding patient NGEs of cancer sample. If the propensities misalign with the respective expression values, the network is tuned. (D) The cell fates (apoptosis, cell cycle arrest, proliferation, and metastasis) are compared between patient-specific normal and cancer conditions in paired case, and median assigned normal and patient-specific cancer conditions in unpaired case. In cancer only case, only patient-specific cancer fate propensities are plotted. In the bar chart, the x-axis represents cell fates, and the y-axis represents propensities.
To exemplify model annotation with patient-specific omics data for Case 1, 7 samples of LUSC were randomly selected from 49 paired normal and cancer samples obtained in Case 1 of Step 2. The network model was annotated for each patient’s normal and cancer samples as described in Case 1 of Proposed Methodology—Step 3. Notably, some input nodes are not directly associated with the gene symbols, and hence were not assigned gene expressions directly. Moreover, input nodes representing biomolecules that contain multiple sub-units were assigned abstracted values computed from downstream nodes’ associated genes. The criteria to assign representative values to network nodes in light of patient’s gene expressions were based on the following: patient mutation (PM), network connectivity (NC), frequency (F), exact match (EM), etc. (Supplementary Information 1—Supplementary Video 3, Supplementary Table S2). The DA of patient-specific cancer models exhibited an overall decrease in apoptosis and senescence with an increase in proliferation as compared to corresponding patient-specific normal models. The other cell fates including cell cycle arrest, quiescence, and metastasis varied from patient to patient (Supplementary Information 1—Supplementary Data 10A). For Case 2, median value of normalized RNA-seq based gene expressions (all 50 normal samples) was assigned to the network model, and DA was performed. The cell fate propensities of proliferation, apoptosis, quiescence, senescence and cell cycle arrest obtained from DA were 0.1370, 0.2709, 0.3608, 0.1214 and 0.0911, respectively. From 453 unpaired cancer samples obtained in Step 2—Case 2, 10 cancer samples were randomly picked for developing personalized cancer models. The 10 personalized cancer models were developed by annotating the network model with patient-specific gene expressions along with SMs, CNVs, and SVs. Next, DA was performed, and the cell fate outcome of each individualized cancer model was compared with the cell fate propensities of median assigned normal model. The comparison revealed decrease in apoptosis and senescence with an increase in quiescence. The patient-to-patient variation in cell fates is shown in Supplementary Information 1—Supplementary Data 10B. For Case 3, the model annotation for cancer samples only was similar to cancer samples in Case 2 of Step 3 (Supplementary Information 1—Supplementary Data 10C).
Step 4—Therapeutic evaluation for personalized cancer treatment
The last step of CanSeer involves the personalization of cancer therapeutics. To demonstrate Step 4, all targetable nodes of the network (either druggable or clinically actionable) were identified (Supplementary Information 1—Supplementary Data 11) and their oncogenic roles i.e. oncogene and tumor suppressor were acquired (Supplementary Table S3). The list of targetable nodes and their corresponding drugs including EGFR—Afatinib, AKT—Ipatasertib, BRAF—Dabrafenib, etc. are listed in Supplementary Information 1—Supplementary Data 12. Next, the genetic alterations of LUSC patients (mentioned in exemplification of Step 3) were mapped with the cancer driver genes of TCGA-LUSC project determined by OncoVar and IntOGen (Supplementary Table S4). The drugs identified to target druggable, or clinically actionable nodes were then queried in GDSC2 to obtain IC50 values. The IC50 values of candidate drugs were extracted for “Lung NSCLC squamous cell carcinoma” specific cell lines. The average IC50 values computed after the removal of outliers for afatinib, afuresertib, APR-246, carmustine, dabrafenib, dinaciclib, ibrutinib, ipatasertib, nutlin-3a, olaparib, osimertinib, oxaliplatin, palbociclib, ribociclib, selumetinib, trametinib, ulixertinib and KU-55933 were 5.06, 17.52, 707.84, 748.64, 203.12, 0.08, 113.09, 74.53, 265.12, 98.49, 4.42, 60.17, 65.97, 63.55, 28.35, 1.23, 18.80 and 208.66, respectively (Supplementary Information 1—Supplementary Data 13). These average IC50 values were then utilized for computing normalized drug activity score (NDAS) e.g., ipatasertib 0.0474, afuresertib 0.0715, osimertinib 0.1377, etc. (Supplementary Information 1—Supplementary Data 13). Next, we computed drug score (DS) for both patient-specific normal and cancer samples (Case 1) whose annotation was exemplified in Step 3. With DS, the personalized models developed in Step 3 were re-analyzed (Supplementary Information 1—Supplementary Data 14A). The cell fates obtained on screening the personalized models with DS reported several efficacious drug combinations along with patient-specific response to various treatment options (Fig. 5A, Supplementary Information 1—Supplementary Data 14A). The cell fate outcomes were employed to compute efficacies and evaluate cytotoxic effects, detailed in the individual patient files in Supplementary Information 1—Supplementary Data 14A. Further, patient-specific therapeutic response index (TRI) for each drug and drug combination was calculated as the difference between efficacy and cytotoxic effect (Supplementary Information 1—Supplementary Data 14A). The drug combination with highest TRI was ranked one. The top ranked treatment options exhibited patient-centric variability (Table 1, Fig. 5B) and their cell fate outcomes were compared against their respective normal and cancer model cell fates (Supplementary Information 1—Supplementary Data 14A). Additionally, the drug combinations were listed in ascending order based on proliferative indices and descending order based on apoptotic indices (Fig. 5C and D, Supplementary Information 1—Supplementary Data 14A). Therapeutic evaluations for cases 2 and 3 are shown in Supplementary Information 1—Supplementary Data 14B and C, respectively.

Therapeutic assessment in patients with lung squamous cell carcinoma (LUSC). The variability in patient-specific response to different drugs and combinations are shown. (A) The therapeutic response indices (TRI) of seven patients are plotted for 50/149 drug combinations in alphabetical order. To demonstrate the response of an individual patient against various drugs and combinations, TRI, proliferative indices and apoptotic indices has been plotted here for only 30 combinations (see patient therapy files in supplementary 14A, B and C for individual responses to 149 drugs/drug combinations). For “patient ID: TCGA-22–5471”, (B) Osimertinib + APR-246 showed highest TRI, (C) Afuresertib + KU-55933 exhibited lowest proliferative index, and (D) Ulixertinib + Ibrutinib indicated highest apoptotic index (For individual patient results, see “Patient Therapy File” in S14A, B and C).
Overall, the LUSC case study revealed four mono-targeted drugs (Afatinib, Osimertinib, Dabrafenib, and Trametinib) and two drug combinations (Osimertinib + Oxaliplatin and Dabrafenib + Trametinib) which have already received FDA (Food and Drug Administration) approval for non-small cell lung cancer (Fig. 6, Supplementary Information 1—Supplementary Data 15). Additionally, our predictions of 26 drugs/drug combinations for LUSC, such as Osimertinib + Selumetinib, Palbociclib + Selumetinib, APR-246 + Olaparib, etc. corroborate with the scientific literature (Fig. 6, Supplementary Information 1—Supplementary Data 15). Moreover, 20 drug combinations predicted by CanSeer require further evaluation in LUSC, including APR-246 + Ibrutinib, Olaparib + Ibrutinib, Ulixertinib + Trametinib, etc. as they are either under investigation or approved for various other cancer types, suggesting potential for repositioning (Fig. 6, Supplementary Information 1—Supplementary Data 15). Finally, CanSeer also identified several novel treatment combinations for LUSC including Afuresertib + Palbociclib, Dinaciclib + Trametinib, Afatinib + Oxaliplatin, Ulixertinib + Olaparib, etc. (Fig. 6, Supplementary Information 1—Supplementary Data 15).

Therapeutic outcome of lung squamous cell carcinoma case study. Employing CanSeer helped identify 4 mono-targeted drugs and 2 drug combinations, which has already been approved by FDA for non-small cell lung cancer (please note that lung squamous cell carcinoma is a sub-type of non-small cell lung cancer). 26 such drugs or combinations were identified that are currently under investigation or reported in various scientific studies for lung cancer especially non-small cell lung cancer. 20 drug combinations predicted for lung squamous cell carcinoma, are previously known, or approved for various other cancer types, suggesting the potential for repositioning. Moreover, CanSeer revealed 97 novel drug combination candidates for lung squamous cell carcinoma (literature validation shown in S15). These four broad categories of drugs or drug combinations are demonstrated through a circos plot.
Discussion
Computational analysis of biomolecular regulatory networks in cancer has become an indispensable tool for elucidating the mechanistic underpinnings of tumorigenesis and tumor progression58,59,60,61. Dynamical simulations of such models have further helped classify patients into clinical sub-groups as well as predict survival46,48,49,62. In this regard, network modeling has also been used to evaluate efficacious drug targets52,63, but lack integration of comprehensive omics data and information on druggable and clinically actionable targets, impeding clinical translation. Incorporating RNA-seq based gene expressions, copy number variations (CNVs), and somatic mutations (SMs) into network models improves mapping of molecular profiles, yet qualitative approaches fall short in developing the clinically employable combinatorial treatments. Additionally, the employment of online drug databases, which also contain clinically disapproved drugs, can lead to off label in silico drug targeting. Consequently, the assistance offered by in silico studies towards personalized cancer care remains rudimentary in the context of clinical adoption. Moreover, the evaluation of drug effects using drug scores coupled with quantitative patient’s omics data to assess therapeutic response, remains unexplored. Lastly, integrative modeling frameworks that facilitate computational investigations into effective treatments, and cytotoxicity towards designing personalized therapies remain lacking. Taken together, there is a need for a systematic method to develop in silico models that provide comprehensive coverage of patient’s molecular profile, as well as facilitate clinical translation of modeling-based personalized therapeutics.
To address this need, we have proposed “CanSeer”, a method to develop in silico models for clinical translation of personalized therapeutics. CanSeer couples’ dynamical analysis of biomolecular network models with patient-specific genomic and transcriptomic data to assess the individualized therapeutic responses to targeted drugs, chemotherapeutic agents, and their combinations. CanSeer is network model agnostic and supports all major types of biomolecular network models. Once any network model is defined and converted into a computable form (e.g. Boolean rules, weight-based, or differential equations-based models), it can be readily ported for onward dynamical analysis.
Towards a quantitative evaluation of personalized in silico models, CanSeer employs RNA-seq based gene expression data to annotate the network models. Moreover, the patient’s CNVs, SMs, and genomic structural variants (SVs) are incorporated to enable the elucidation of molecular signatures. CanSeer envisages quantitative personalized models in comparison to recent approaches that encoded CNVs (amplified genes and homozygous deletions), and SMs (oncogenes and tumor suppressors) as 0 or 1. For an integrative investigation into patient tumors, statistical outliers detected in gene expression data having CNVs, SMs, and SVs are tagged and stored. Considering the heterogeneous impact of genetic alterations, network nodes are annotated with normalized expressions such that the node activity remains conserved throughout the analysis for genetically altered genes.
For enabling translational pertinence and prioritizing repositioning opportunities, cancer driver genes in patients are identified, and druggable and/or clinically actionable target nodes are selected for therapeutic interventions. As a result, CanSeer can elicit optimal tailored treatment by employing therapeutic screening of druggable and clinically actionable targets, thus enabling clinical translation. The proposed formulation of drug scores assisted in capturing the effect of drug-to-drug variability e.g. Ipatasertib and Afuresertib are both AKT inhibitors with average IC50 of 74.5 and 17.5 for lung NSCLC (non-small-cell lung cancer) squamous cell carcinoma cell lines. Computing drug scores for patients and performing targeted in silico screening on cancer samples allowed identifying patient-specific responses to targeted drugs and their combinations. Likewise, administering chemotherapeutic agents to both normal and cancer samples helped reveal patient-centric responses to chemotherapy drugs and their combinations with targeted drugs. Finally, the patient-specific treatment efficacy together with evaluating cytotoxic effect helped identify and rank optimal tailored treatments.
In terms of modelling approach, CanSeer takes Kauffman’s logical modeling64,65,66 paradigm and employs deterministic analysis pipeline56,57, to enable customization and evaluation of large personalized networks. Earlier, Beal et al. devised personalized models by integrating CNVs, and SMs as discrete data, and expressions (gene/protein) as continuous data into Boolean models49. However, the resultant model’s integrative investigation remained limited as CNVs, and SMs remained unmapped with expression data. CanSeer addresses this by integrating RNA-seq based gene expressions with CNVs, SMs, and SVs for a consolidative investigation into patient’s tumor. The mapping between gene expression and genetic alterations is based on the positive correlation of CNVs, SMs, and SVs with gene expression levels67,68,69,70,71,72,73. In 2022, Montagud et al. applied Beal et al.’s approach on Fumia’s Boolean model to personalize 488 prostate cancer patients and eight cell lines53. Despite having multiple readouts including proliferation, invasion, migration, metastasis, DNA repair, and apoptosis, the study remained limited to eliciting proliferation and apoptosis in patient-centric models. Further, to identify potential treatments, knockouts were performed to increase apoptosis and deplete proliferation. A similar approach was applied in our previous study52 to predict combinatorial treatment for colorectal cancer. However, a caveat in employing discrete knock-out perturbations is that they show maximum effect, and hence cannot be employed as direct targets in wet-lab experiments and clinical trials. In contrast, CanSeer considers druggable and clinically actionable targets and investigates the quantitative patient treatment response by incorporating drugs’ IC50 values to establish clinical relevancy and applicability. Moreover, in our colorectal cancer study, cell-type specific gene expression data of fly gut was integrated with patient-specific mutations to identify personalized drug combinations. In contrast, CanSeer incorporates a comprehensive molecular signature comprising of patient-specific RNA-seq based gene expressions, CNVs, SMs, and SVs. Towards improving clinical translation of computational precision oncology approaches, Ianevski et al. executed a machine learning approach to select patient-specific drug combinations with synergy-efficacy-toxicity balance51. However, some of the identified combinations were not clinically applicable due to the utilization of public drug databases that contain incomplete information. CanSeer employs extensive literature review in addition to drug database utilization to avoid devising inoperable combinations. In addition, CanSeer applies computational modeling approach that provides mechanistic insights into patient tumors, and optimal personalized therapy identification.
CanSeer’s methodology exemplified through three use cases suggest that the proposed methodology is effective in identifying personalized anticancer therapies. However, the published model used for demonstration of CanSeer lacked coverage for certain mutations in some patients, resulting in inconclusive outcomes. Moreover, the study remains limited in prioritizing synergy-based drug combinations. Alongside, the patient-specific sensitivity to drug cocktails revealed by the in silico personalized models require further evaluation by employing tumor-on-chip models that have the potential to reproduce patient’s cancer cells and microenvironment74,75,76. Prospectively, the proposed methodology can be extended by integrating in silico personalized models with microfluidics-based tumor-on-chip models to expedite testing of predicted personalized treatment options and assessing response. In terms of limitations and possibilities for future extension, CanSeer provides a roadmap for developing tailored therapeutic combinations in light of patient genomic and transcriptomic profiles, however, wet lab validation of predicted efficacious drug combinations, and repositioned drugs can enhance CanSeer’s clinical acceptability. Moreover, CanSeer provides a direction for model annotation with patient’s data, however, requires an expansion to cater for panomics data, to avoid under or overfitting of patient’s profile. Pertaining to patient’s tumor profile, it is also important to discover patient-specific cancer driver mutations for which the algorithms77,78,79 can be combined with CanSeer to identify potential driver genes in patient. Likewise, various algorithms identifying CNVs, SMs, and SVs can be integrated to abstract the missing information of a particular patient from whole-exome sequencing data80,81,82,83,84,85,86,87,88.
Furthermore, in view of future extension, CanSeer cues the development of in silico organoids89, and multi-scale modeling pipelines90 that can simulate the spatiotemporal evolution of patient tumors, and the temporal effect of therapy on personalized in silico organoid models. Such automated multi-scale modeling pipelines backed by high-throughput performance computing leveraging massively on parallel graphics processing units (GPUs) can enable real-time analyses for predicting precision medicine.
In terms of clinical feasibility, CanSeer has the potential to assist oncologists in making informed treatment decisions by integrating patient genomic and transcriptomic data, potentiating effective cancer therapies and improved patient survival. The direct use of molecular data enhances the accuracy of individualized treatment options, while the capability to reposition FDA-approved drugs accelerates development of novel therapeutic avenues. In addition, CanSeer’s capacity to furnish quantitative treatment responses empowers clinicians to tailor therapies with precise risk–benefit assessments. Its scalability across different cancer types broadens the framework’s applicability in clinical oncology, and its identification of novel drug combinations may improve efficacy and outcomes possibilities in end-stage cancer cases.
Navigating lengthy regulatory processes for novel drug combinations can delay clinical deployment, rendering an impediment in translation of in silico predictions into practice. This could be confounded by the limited provision of high-quality omics datasets. Additionally, the complexity of personalized dynamic network modeling demands high-performance computing posing scalability challenges in clinical settings. Furthermore, accurately predicting drug synergies and toxicity is complicated by intricate drug interactions, while the successful integration of CanSeer into clinical workflows requires the development of intuitive software solutions.
In conclusion, the proposed methodology demonstrates assessment of response and cytotoxicity of personalized cancer therapeutics. The novel framework thus fills a crucial void in clinical provision of personalized cancer treatments through a comprehensive incorporation of patient’s molecular profile coupled with evaluation of clinically actionable targets.
Materials and methods
Biomolecular network model construction
The process of network model development comprises of two sub-steps, i.e. (a) model assembly, and (b) assembled model’s implementation within the modeling and simulation platform, TISON, for onward analysis. The model assembly process is typically manual and starts with a review of published literature to gather data on gene regulatory interactions, protein–protein interactions, signaling pathways, etc. Once an exhaustive list of interacting partners and the type of interactions is collected, individual Boolean rules are defined for each gene or protein (node) using logic operators. The process of defining Boolean rules is detailed in Supplementary Information 2—Supplementary Method 1. Note that users can also choose to construct networks by retrieving interactions from interaction databases like Pybel, Reactome, BioPAX, or from literature using natural language processing. To implement the assembled model, users can employ TISON’s rules editor (Supplementary Information 2—Supplementary Method 2).
To develop a biomolecular regulatory network model, pathways and interactions were retrieved from existing databases including Kyoto Encyclopedia of Genes and Genomes (KEGG), PathBank, Pathway Interaction Database (PID), and Reactome. These pathways were then integrated, and their crosstalk was incorporated into the network architecture. Rules-based and weight-based formalisms were used for modelling the resultant network architecture towards carrying out network analyses. Case study exemplars were developed around the human signaling network comprising of 197 nodes and their interactions. Boolean logic rules were defined to translate the adopted network into a rules-based model. For developing a weight-based version of rules-based biomolecular regulations, the basal value of each network node was set at 1. Next, interaction weights were computed based on the number of adjacent nodes such that the output of weight-based model becomes comparable to the rules-based model91. The interaction weights were then adjusted iteratively until the results closely matched those of a deterministic analysis of rules-based model. To compare the results from both approaches, the steady state cell fate propensities of the output nodes were compared.
Input and output node setup using fixed node states and cell fate programming
To analyze the human signaling network under normal conditions, values for input nodes were abstracted from published literature (Supplementary Table S5). Each input value was “fixed” to cater for inputs such that the nodes’ state remains unchanged during dynamical analysis. The normalized patient gene expression for each input node was selected to personalize the network model. In the personalized cancer model, patient’s genetic alterations were also fixed as “fixed node states”. A fixed node state refers to a node in a biological network that is “locked” in a specific state—either ON (1) or OFF (0)—and remains unchanged throughout the simulation, regardless of the rules governing the node’s behavior (Supplementary Information 2—Supplementary Method 3). To program the cell fates, associated biomarkers and the associated network nodes’ states were acquired from the published literature (Supplementary Table S6). Output nodes and their associated nodes’ states defining cellular functions such as apoptosis, senescence, cell cycle arrest, etc. were abstracted from literature to further expand the set of cell fates (Supplementary Table S7). The resultant cell fate classification program was tuned in the light of normalized RNA-seq based gene expression data of patients to personalize the models (Supplementary Table S8). Combinations of nodes’ states were created to provide an exhaustive coverage for the cell fate classification scenario.
Dynamical deterministic analyses of biomolecular networks
Dynamical assessment of networks was undertaken by performing Deterministic Analysis (DA) using an in-house web-based modelling and simulation platform, Theatre for in silico Systems Oncology (TISON)57. TISON provides a user-friendly and web-based implementation of both deterministic56,57 as well as probabilistic92,93 analysis pipelines within the network’s editor (Supplementary Information 2—Supplementary Method 4). To derive the Boolean rules from Cho et al.55 provided truth tables (Supplementary Information 2—Supplementary Method 5), we employed Kadelka et al.’s logical rules generation toolbox94. Next, for deterministic analysis of the network in TISON, two additional files including fixed node states file (inputs) and the cell fate classification file were adopted from Cho et al. The fixed node file contained initial input conditions to cue the DA. The maximum iterations to search for the steady state were set to 500 and a bootstrap of 256 network states was applied. The maximum number of iterations for finding the steady state and the bootstrap value were estimated through iterative network analysis and simulations (Supplementary Information 2—Supplementary Method 6). The node update policy in DA updates all nodes in the system simultaneously at each time step following predefined rules (Supplementary Information 2—Supplementary Method 7). Finally, output node propensities from DA were used to arrive at the cell fates.
For the input nodes, normalized gene expression data of patients, under normal and cancer conditions, were defined in the fixed node states file. Additionally, the normalized gene expressions of genetically altered genes were set as fixed node states for the patient-specific cancer network. The fixed node states file of the cancer patient was used as-is for therapy. An additional input “exhaustive screening file” was defined for performing DA-based therapeutic evaluation. The patient drug scores were integrated to target the specific nodes using the exhaustive screening file.
Robustness analysis and sensitivity analyses
To determine the susceptibility of the biomolecular network to minor variations, the network inputs were perturbed by up to 10%. The input conditions were taken in combination for network analyses. A random sample of 256 network states was selected along with 10% of fixed node combinations. DA was performed to compute average node propensities, standard deviation, and standard error of mean (SEM) for each mapped cell fate. The average propensities and SEMs of emergent cellular phenotypes were plotted to evaluate the stability of the network. To threshold, a SEM value of 0.05 was used for declaring the network robust.
To analyze the network behavior under large input perturbations, the adopted network with cell fate expansion was exposed to all possible input values (parameters), sequentially. Each input was assessed at uniform increment of 0.1 from 0 to 1. In order to see the influence of each parameter, all other inputs were kept at normal levels. For that, DA for each input was performed with bootstrap of 256 network states. Results from each input node perturbation were compiled to see the overall effect of each input on the network. Biological plausibility was established by validating the interpretation of each input-cell fate relationship against literature (Supplementary Information 2—Supplementary Method 8). Additionally, multiple input perturbations were performed to investigate the synergistic or antagonistic relationship of the input stimuli in the human signaling network. For combinatorial parameter sensitivity assessment, the levels of co-occurring inputs were varied, and the effect on associated cell fates was stored. DA was performed using TISON.
Genomic and transcriptomic data assembly, pre-processing, normalization and model personalization
To acquire patients’ transcriptomic profiles (RNA-seq based gene expression data), The Cancer Genome Atlas (TCGA) PanCancer Atlas studies were accessed using Genomic Data Commons (GDC) portal. For obtaining the genomic data including copy number variations (CNVs), somatic mutations (SMs), and genomic structural variants (SVs), the same TCGA project was queried in cBioPortal. Patient data matching was ensured by comparing sample IDs between GDC and cBioPortal. Patients’ genomic and transcriptomic data were obtained for the TCGA-LUSC project to demonstrate the three cases of proposed method “CanSeer”. MATLAB 2020b95 was used to implement the pre-processing and normalization of genomic, and transcriptomic data.
Implementation of genomic and transcriptomic data pre-processing and normalization
The CanSeer genomic data pre-processing and RNA-seq based gene expression data normalization algorithm was implemented in MATLAB 2020b. The MATLAB script is available in Supplementary Information 1—Supplementary Data 7 and as well as at GitHub (https://github.com/BIRL/CanSeer). The process initiates with filtering of the sample sheet obtained from the TCGA program based on the following keywords: “normal”, “tumor”, “primary”, and “metastatic” to include all types of normal and tumor samples, except ‘recurrent tumors’. Next, specific case amongst Cases 1, 2, and 3 as (i) “paired”, (ii) “unpaired”, and (iii) “cancer samples only” is selected. For case 1, a new sample sheet is assembled for both normal and cancer samples that include patient/sample IDs along with the RNA-seq gene expression file names mapped against normal and cancer samples. For case 2, two separate sample sheets are formed for the normal and cancer case. Each sample sheet contains patient/sample IDs with their corresponding RNA-seq gene expression file names. For case 3, the sample sheet is assembled for cancer samples only. Subsequently, the option of random sample or complete dataset of RNA-seq gene expressions can be opted. Next, the network’s nodes list, comprising of all network nodes, is employed to extract and align RNA-seq gene expressions of patients with the respective genes and patient IDs. In the next step, the outliers are detected in the dataset using MAD (Median Absolute Deviation) or IQR (interquartile range) methods. After detection of statistical outliers, Copy Number Variations (CNVs), Somatic Mutations (SMs) and Genomic Structural Variations (SVs) are selected and processed. From CNVs, deep deletions (-2) and amplification (+ 2) (based on GISTIC processing) are saved. CNVs having low-confidence values of –1 and + 1, obtained using GISTIC processing, are removed. The MATLAB script encodes deep deletions of genes as -2, amplifications as + 2, and remaining data as 0. Next, the filtered SMs (for selected patients and network genes) are transformed into logical arrays, where 0 and 1 represent the absence and presence of mutation, respectively. Similarly, patient-specific SVs are also transformed into logical arrays representing the absence (0) and presence (1) of genomic SVs. Lastly, the detected outliers are super-imposed with the CNVs, SMs, and SVs to retain the highly altered RNA-seq gene expressions resulting from CNVs, SMs, and SVs. The remaining outlier gene expressions are removed after which the normal and cancer samples are combined and normalized between 0 and 1 using the highest gene expression across patients. The normalization strategy for Case 3 is similar to Cases 1 and 2, with the exception of RNA-seq gene expression data availability for only cancer samples, wherein, the RNA-seq gene expressions are normalized by the maximum gene expression from cancer samples only.
The normalized RNA-seq based gene expression data was used to annotate the network model for personalization.
Target identification using literature and drug-target databases and evaluation
Drugs targeting each node (biomolecule) in the network were obtained from published literature, and drug-target databases including PanDrugs, DrugBank, and The Drug Gene Interaction Database (DGIdb). Only druggable and clinically actionable nodes were selected to enable the clinical translation of resultant models. The drugs identified were then queried in Genomics of Drug Sensitivity in Cancer (GDSC2) to obtain inhibitory concentrations (IC50). The IC50 values of candidate drugs were acquired for “Lung NSCLC squamous cell carcinoma” cell lines and their mean were computed after removing the outliers. Next, the log of mean IC50 values of candidate drugs were used to formulate the drug activity score (DAS). DAS was then normalized and utilized to compute a “drug score for patient”, which was then employed to annotate the personalized model. The cell fate outcomes from the resultant model’s DA were then used to calculate the patient-specific responses including treatment efficacy, cytotoxic effect, therapeutic response index (TRI), proliferative index and apoptotic index.
Proposed methodology
The proposed methodology of CanSeer comprises of four steps, which are detailed below:
Step 1—Literature and database-driven development of biomolecular regulatory network models and validation
To abstract biomolecular regulation and develop a cancer-type specific biomolecular network architecture, published literature as well as online databases including KEGG96, PathBank97, PID98, and Reactome99 are employed. The resultant network topology includes input, output, and processing nodes along with their interactions. The input nodes in the network cue the downstream processing nodes which then crosstalk, and signal output nodes. For dynamical analysis of the model, this topology is translated into the rules-based100,101,102 or weight-based103,104 network models. For the analysis of rules-based or weight-based model, normal conditions are abstracted from the literature and are assigned to the input nodes. Next, the dynamical analysis of the network is performed using either deterministic (DA)56,57, probabilistic(PA)92,93, or ordinary differential equation(ODE)105 modalities under the influence of input conditions. DA, PA and ODE modalities are detailed in (Supplementary Information 2—Supplementary Method 9). Note that the case study provided in this work utilized deterministic analysis modality from TISON, only and the resulting outcomes are detailed in the results section. Results from dynamical analyses include output node propensities which are then used to program cell fate outcomes such as quiescence, proliferation, cell cycle arrest, apoptosis, etc. To validate the cell fate outcomes, the trends in node-specific cell fate propensities are tallied with the published literature. To further evaluate the biological plausibility and sensitivity of the validated model, random and systematic perturbations are introduced into the model by varying input conditions termed as robustness106, and parameter sensitivity107,108 analyses. Here again, the resulting cell fate outcomes are matched and validated against the published literature. The workflow of network model development and dynamical evaluation is shown in Fig. 2A.
Step 2—Omics data collection, pre-processing, and normalization
In CanSeer’s second step, patient-specific omics data is acquired, pre-processed, and normalized for later use in network model annotation. The detailed workflow of CanSeer’s Step 2 is presented in Fig. 3.
Patient data acquisition
Patient-specific multi-omics data containing expression profiles and genetic alterations (copy number variations (CNVs), somatic mutations (SMs) and genomic structural variants (SVs)) are acquired from TCGA PanCancer Atlas studies109 using Genomics Data Common (GDC)110 and cBioPortal111,112, respectively. Note that patient information can be employed from other programs and projects as well such as TARGET (Therapeutically Applicable Research to Generate Effective Treatments) program and GENIE-MSK (Genomics, Evidence, Neoplasia, Information and Exchange at Memorial Sloan Kettering Cancer Center) project which are available at GDC and cBioPortal. In addition to the expression profiles, sample sheet containing patients’ clinical information are obtained from TCGA program using GDC. Patient dataset matching is ensured by comparing sample IDs between GDC and cBioPortal.
Data pre-processing
The acquired RNA-seq based expression data is informed by Ensembl IDs which are then also mapped onto gene symbols using BioTools.fr toolbox113. The gene symbols list is filtered based on the nodes (biomolecules) present in the network model. Aliases for biomolecules that do not tally with the list of gene symbols are obtained from GeneCards114 or HUGO Gene Nomenclature Committee (HGNC)115. Onwards, the complete network nodes list is converted into the gene symbols to enable the fetching of corresponding gene expression values. To extract the sample type specific RNA-seq based gene expressions, the sample selection i.e., solid tissue normal and primary tumor is carried out while downloading the expression data from TCGA.
The files required as an input to the script for further processing include (i) gene list, (ii) RNA expression files, (iii) sample sheet, (iv) SM files, (v) CNV files and (vi) SV files. Based on the availability of RNA-seq profiles in TCGA, the patient samples are selected and categorized into three cases: Case 1—paired samples which comprise normal (N) and cancer (C) RNA-seq data from the same patient, Case 2—unpaired samples which contain ‘N’ and ‘C’ RNA-seq data from different patients, and Case 3—cancer samples only that consists of gene expression data from ‘C’ patients. The script further allows the selection of whole dataset and random sampling for the case study, and fetches expressions against finalized genes list. Next, each patient’s cancer expressions are mapped with corresponding SMs, CNVs and SVs. For Cases 1 and 2, patients’ expression data is mapped with the network’s gene symbols for both normal and cancer samples, separately. While in Case 3, patients’ expression data is mapped with the network’s gene symbols for cancer samples only. The patients’ cancer expressions in all three cases are then mapped with the CNVs, SMs, and SVs.
Following the mapping, outliers in expression data are detected. In Cases 1 and 2, outliers from expression data of normal samples are removed using statistical outlier removal methods i.e., 3 × MAD (Median Absolute Deviation)116 or IQR (interquartile range)117 to eliminate skewed data (Supplementary Information 2—Supplementary Method 10). While, for cancer samples, the method detects outliers using MAD or IQR followed by splitting the expression values into “outliers” and “tag & store” (Cs) expression data. If the highly altered expression maps with the CNVs, SMs, or SVs, then the expression value from amongst the outliers is stored. Otherwise, the highly altered expressions are considered statistical outliers and removed. The expressions from normal and cancer samples after the removal of outlier genes are combined (N + Cs). However, Case 3 caters to the situation wherein the availability of RNA-seq data is limited to cancer samples only.
Normalization
In Cases 1 and 2, the RNA-seq based gene expressions are normalized using the maximal expression in N + CS, while in Case 3 the expressions are normalized using the highest expression in Cs only.
Step 3—Model annotation with patient-specific omics data and dynamical analysis
In the third step, CanSeer employs the normalized gene expression values of cancer samples along with their CNVs, SMs and SVs, and incorporates them into the Boolean logic model for personalizing both rules-based and weight-based biomolecular network models (Fig. 4, Supplementary Information 2—Supplementary Method 11). A step-by-step description of model annotation with normalized omics data in case of paired (Case 1), unpaired (Case 2), and cancer samples only (Case 3) is given below.
Case 1: Paired normal and cancer samples
In Case 1, patient-specific normalized gene expression values from paired normal (N) and stored cancer expressions (Cs) are used to annotate the “Normal (N)” and “Cancer (C)” network models, respectively. The normalized gene expression values are assigned to the input nodes in the rules-based networks while in case of weight-based networks all network nodes are set with normalized gene expression data. For weight-based networks, basal values are computed for all network nodes in the light of normalized gene expressions of patients.
Basal value calculation
Basal values are computed using the formula given below:
where, bi is the basal value of the node, xj is the experimental value (e.g., gene expression), and wij is the network’s adjacency matrix.
In addition to the gene expressions, networks are annotated with their genomic alterations including CNVs, SMs, and SVs to develop models of personalized cancers. The types of SMs are considered while incorporating SMs in the model. SMs such as missense mutation, splice donor variant, splice acceptor variant, splice site variant, frameshift deletion, and non-coding transcript exon variant are included while silent mutation, intron variant, splice region variant, and 5’ UTR variant are excluded. In case of both rules-based and weight-based networks, CNVs, SMs, and SVs are incorporated into the model by setting corresponding nodes to normalized gene expressions such that the node activity is maintained at the same level for each time-step of dynamical analysis. Dynamical analysis is carried out for each personalized normal and cancer network model followed by comparing the average node activity of output nodes with the patient-specific normalized gene expressions of respective output genes. A “match” is declared if the average node activity of output nodes is comparable to the normalized gene expressions of corresponding genes within a user-defined tolerance. If the outcome is not comparable, then the network is iteratively fine-tuned for making the model representative of the patient (Supplementary Information 2—Supplementary Fig. S3, Supplementary Information 2—Supplementary Method 12). The approach for tuning the network model includes modifying the logical rules and bootstrapping the edge weights in rules-based and weight-based networks, respectively. Cell fates from the tuned personalized models are compared for normal and cancer case.
Case 2: Unpaired normal and cancer samples
For Case 2, CanSeer computes the median gene expression values of each gene in normalized ‘N’ from N + Cs, which is used to annotate the network model. Normalized median gene expression values are integrated into the network model to represent a normal network. The normalized median gene expression values are assigned to the input nodes in the rules-based networks. On the other hand, for weight-based networks, all the network nodes are annotated with normalized median gene expression data. Basal values are computed in the light of normalized median gene expressions in the weight-based networks.
Next, dynamical analysis is performed for the normal network, and the average node activity of output nodes is compared with the normalized median gene expressions of respective genes. Next, the network is annotated with the patient’s cancer gene expressions along with CNVs, SMs, and SVs to model individual tumors. The method of model annotation for cancer sample is like Step 3—Case 1. Lastly, a comparison is made between cell fate outcomes from dynamical analysis of median assigned normal and patient-specific cancer models.
Case 3: Cancer samples only
For case 3, CanSeer utilizes the normalized patient-specific cancer gene expressions together with the CNVs, SMs, and SVs for the personalization of network model. The integration of omics data into the network and in silico validation of personalized cancer model is carried out as described in Step 3—Case 1. The cell fate outcomes from the tuned personalized cancer models are plotted.
Step 4—Therapeutic evaluation for personalized cancer treatment
The last step of CanSeer involves the personalization of cancer therapeutics (Fig. 7). For that, personalized in silico cancer models (constructed in Step 3) are screened under the effect of different drugs and their combinations. For the clinical translation of models, targetable nodes (either druggable or clinically actionable) of the network are identified. The oncogenic role of each targetable node is obtained from OncoKB118. Drugs targeting each node (biomolecule) in the network are obtained from published literature, and drug-target databases such as PanDrugs, DrugBank119, and DGIdb. To systematically prioritize druggability and repositioning opportunities, cancer driver genes in the selected TCGA project are acquired from OncoVar120, and IntOGen121. The drugs identified to target druggable, or clinically actionable nodes are then queried in Genomics of Drug Sensitivity in Cancer122 (GDSC2) to obtain IC50. The IC50 values of a drug for all tissue specific cell lines are extracted and their mean is computed after removing the outliers. The log of mean IC50 values of candidate drugs is utilized to formulate the potential drug activity. To quantify the potential activity of each drug for a specific tissue type, drug activity scores (DAS) are computed by taking the reciprocal of log IC50 value of the drug, as given in the equation below.

Method of therapeutic evaluation for personalized cancer treatment (Step 4 of CanSeer). The final step of CanSeer involves personalized cancer therapeutics. This includes screening personalized in silico cancer models with different drugs and their combinations. (A) First, druggable genome and clinically actionable target nodes in the network are identified, and their oncogenic roles are obtained from OncoKB. Drugs targeting these nodes are gathered from literature and databases (DrugBank, PanDrugs, The Drug Gene Interaction Database (DGIdb), etc. Moreover, cancer driver genes are acquired from OncoVar and IntOGen to prioritize druggability. (B) Subsequently, drug IC50 values sourced from “Genomics of Drug Sensitivity in Cancer (GDSC2)” are employed to compute drug activity scores (DAS), which are then normalized (NDAS). (C) NDAS values together with the normalized RNA-seq based gene expression values of a patient in cancer (NRGEPC) and maximal efficacy gain induced by a drug (MEGID) per se, are utilized to calculate drug scores for patients (DS). (D) Using DS, personalized cancer models undergo re-annotation and re-analysis. (E and F) Dynamical analysis cell fate outcomes are then utilized to assess patient-specific responses to individual drugs and combinations, considering both efficacy and cytotoxic effect. (G) Next, CanSeer establishes the "therapeutic response index (TRI)" by quantifying the difference between efficacy and cytotoxic effect. (H and I) TRI is then used to prioritize treatment options followed by comparing treatment-induced cell fates with those from normal and cancer models.
Drug activity scores are then normalized (NDAS) using the highest drug activity score, as shown in the equation below.
Next, to predict the patient-specific response to treatment, the maximal efficacy gain induced by a drug (MEGID) per se is calculated using the normalized RNA-seq based gene expression values of a patient in cancer (NRGEPC). For the case of a tumor suppressor, MEGID is computed as follows.
On the other hand, for drug targeting of an oncogene or proto-oncogene, MEGID is set as:
Using MEGID, drug scores are customized for patients termed as “drug score for patient (DS)”. DS is then defined for tumor suppressors as:
For the case of oncogene and proto-oncogenes:
Next, patient-specific drug scores are used to annotate nodes in the personalized network models and the network models are re-analyzed. The cell fate outcomes from dynamical analyses of network models are further employed to calculate the patient-specific response against each drug and drug combination. The patient-specific response includes treatment efficacy and its cytotoxic effect. To calculate efficacy, the ratio of anticancer cell fate propensities to cancer-promoting cell fate propensities is taken. Next, the cytotoxic effect of each drug and drug combination for individual patients is evaluated using the formula given below:
Further, the difference between efficacy and cytotoxic effect is calculated and referred to as “therapeutic response index (TRI)”. Based on TRI, CanSeer ranks the treatment options from highest to lowest. The cell fates under patient-specific treatment options (top 3) are compared with the corresponding normal and cancer model cell fate outcomes. Lastly, in addition to the conclusive TRI based treatment ranking, CanSeer enlists proliferative and apoptotic indices for clinical relevance.
Data availability
All the data and its interpretation needed to evaluate the results of the study are available along with the manuscript as supplementary information and supplementary tables. Supplementary information 1 contains supplementary dataset, supplementary data, and supplementary videos. Supplementary dataset and supplementary data are publicly accessible at https://data.mendeley.com/datasets/rm7vm2kf4s/2. The supplementary videos are available at YouTube (https://www.youtube.com/playlist?list=PLaNVq-kFOn0a6HWmZq_KxeSbY6UpnvvKB). Supplementary information 2 contains supplementary figures and supplementary methods. The MATLAB script is available at GitHub (https://github.com/BIRL/CanSeer).
Change history
26 May 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-03481-y
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 71, 209–249 (2021).
Kinch, M. S. An analysis of FDA-approved drugs for oncology. Drug Discov. Today 19, 1831–1835 (2014).
Blagosklonny, M. V. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell Cycle 3, 1035–1042 (2004).
Abbas, Z. & Rehman, S. An overview of cancer treatment modalities. In Neoplasm (2018).
Holohan, C., Van Schaeybroeck, S., Longley, D. B. & Johnston, P. G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013).
Yang, R., Niepel, M., Mitchison, T. & Sorger, P. Dissecting variability in responses to cancer chemotherapy through systems pharmacology. Clin. Pharmacol. Ther. 88, 34–38 (2010).
Florento, L. et al. Comparison of cytotoxic activity of anticancer drugs against various human tumor cell lines using in vitro cell-based approach. Int. J. Biomed. Sci. 8, 76–80 (2012).
Krishna, I. V., Vanaja, G. R., Kumar, N. S. K. & Suman, G. Cytotoxic studies of anti-neoplastic drugs on human lymphocytes—In vitro studies. Cancer Biomark. 5, 261–272 (2009).
Iqbal, N. & Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 1–9 (2014).
Flaherty, K. T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010).
Chapman, P. B. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011).
Hyman, D. M. et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 373, 726–736 (2015).
Levis, M. & Perl, A. E. Gilteritinib: Potent targeting of FLT3 mutations in AML. Blood Adv. 4, 1178–1191 (2020).
Perl, A. E. et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 381, 1728–1740 (2019).
Rini, B. I. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin. Cancer Res. 14, 1286–1290 (2008).
Hudes, G. et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281 (2009).
Xu, W., Yang, Z. & Lu, N. Molecular targeted therapy for the treatment of gastric cancer. J. Exp. Clin. Cancer Res. 35, 1–11 (2016).
Montenegro, R. C. et al. Identification of molecular targets for the targeted treatment of gastric cancer using dasatinib. Oncotarget 11, 535 (2020).
Zeng, J.-H. et al. Identification of molecular targets for esophageal carcinoma diagnosis using miRNA-seq and RNA-seq data from The Cancer Genome Atlas: A study of 187 cases. Oncotarget 8, 35681 (2017).
Isozaki, Y. et al. Identification of novel molecular targets regulated by tumor suppressive miR-375 induced by histone acetylation in esophageal squamous cell carcinoma. Int. J. Oncol. 41, 985–994 (2012).
Oliff, A., Gibbs, J. B. & Mccormick, F. New molecular targets for cancer therapy. Sci. Am. 275, 144–149 (1996).
Luthra, R. et al. A targeted high-throughput next-generation sequencing panel for clinical screening of mutations, gene amplifications, and fusions in solid tumors. J. Mol. Diagn. 19, 255–264 (2017).
Beadling, C. et al. Multiplex mutation screening by mass spectrometry: Evaluation of 820 cases from a personalized cancer medicine registry. J. Mol. Diagn. 13, 504–513 (2011).
Hansen, A. R. & Bedard, P. L. Clinical application of high-throughput genomic technologies for treatment selection in breast cancer. Breast Cancer Res. 15, R97 (2013).
Matulonis, U. A. et al. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS ONE 6, e24433 (2011).
Chae, Y. K. et al. Path toward precision oncology: Review of targeted therapy studies and tools to aid in defining “actionability” of a molecular lesion and patient management support. Mol. Cancer Ther. 16, 2645–2655 (2017).
Prasad, V., Fojo, T. & Brada, M. Precision oncology: Origins, optimism, and potential. Lancet. Oncol. 17, e81–e86 (2016).
Lassen, U. N. et al. Precision oncology: A clinical and patient perspective. Futur. Oncol. 17, 3995–4009 (2021).
Tsimberidou, A. M., Fountzilas, E., Nikanjam, M. & Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 86, 102019 (2020).
Terry, S. F. Obama’s precision medicine initiative. Genet. Test. Mol. Biomark. 19, 113–114 (2015).
Tsimberidou, A. M. et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center Initiative. Clin. Cancer Res. 18, 6373–6383 (2012).
Tsimberidou, A.-M. et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: Validation and landmark analyses. Clin. Cancer Res. 20, 4827–4836 (2014).
Tsimberidou, A.-M. et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00002 (2017).
Kopetz, S. et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J. Clin. Oncol. 33, 4032–4038 (2015).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Sun, X., Bao, J., You, Z., Chen, X. & Cui, J. Modeling of signaling crosstalk-mediated drug resistance and its implications on drug combination. Oncotarget 7, 63995–64006 (2016).
Santer, F. R., Erb, H. H. H. & McNeill, R. V. Therapy escape mechanisms in the malignant prostate. Semin. Cancer Biol. 35, 133–144 (2015).
Nguyen, C. & Yi, C. YAP/TAZ signaling and resistance to cancer therapy. Trends Cancer 5, 283–296 (2019).
Levine, B. D. & Cagan, R. L. Drosophila lung cancer models identify trametinib plus statin as candidate therapeutic. Cell Rep. 14, 1477–1487 (2016).
Bangi, E. et al. A personalized platform identifies trametinib plus zoledronate for a patient with KRAS-mutant metastatic colorectal cancer. Sci. Adv. 5, eaav6528 (2019).
Chen, S. H. & Lahav, G. Two is better than one; toward a rational design of combinatorial therapy. Curr. Opin. Struct. Biol. 41, 145–150 (2016).
Steinway, S. N. et al. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. npj Syst. Biol. Appl. 1, 1–12 (2015).
Saez-Rodriguez, J., MacNamara, A. & Cook, S. Modeling signaling networks to advance new cancer therapies. Annu. Rev. Biomed. Eng. 17, 143–163 (2015).
Zañudo, J. G. T., Steinway, S. N. & Albert, R. Discrete dynamic network modeling of oncogenic signaling: Mechanistic insights for personalized treatment of cancer. Curr. Opin. Syst. Biol. 9, 1–10 (2018).
Gómez, J., Zañudo, T., Scaltriti, M. & Albert, R. A network modeling approach to elucidate drug resistance mechanisms and predict combinatorial drug treatments in breast cancer. Cancer Converg. 1, 1–25 (2017).
Fey, D. et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci. Signal. 8, ra130 (2015).
Eduati, F. et al. Drug resistance mechanisms in colorectal cancer dissected with cell type-specific dynamic logic models. Cancer Res. 77, 3364–3375 (2017).
Hidalgo, M. R. et al. High throughput estimation of functional cell activities reveals disease mechanisms and predicts relevant clinical outcomes. Oncotarget 8, 5160–5178 (2016).
Béal, J., Montagud, A., Traynard, P., Barillot, E. & Calzone, L. Personalization of logical models with multi-omics data allows clinical stratification of patients. Front. Physiol. 9, 1965 (2019).
Eduati, F. et al. Patient-specific logic models of signaling pathways from screenings on cancer biopsies to prioritize personalized combination therapies. Mol. Syst. Biol. 16, e8664 (2020).
Ianevski, A. et al. Patient-tailored design for selective co-inhibition of leukemic cell subpopulations. Sci. Adv. 7, eabe4038 (2021).
Gondal, M. N. et al. A personalized therapeutics approach using an in silico drosophila patient model reveals optimal chemo- and targeted therapy combinations for colorectal cancer. Front. Oncol. 11, 692592 (2021).
Montagud, A. et al. Patient-specific Boolean models of signalling networks guide personalised treatments. Elife 11, e72626 (2022).
Seyhan, A. A. Lost in translation: the valley of death across preclinical and clinical divide—identification of problems and overcoming obstacles. Transl. Med. Commun. 4, 1–19 (2019).
Cho, S. H., Park, S. M., Lee, H. S., Lee, H. Y. & Cho, K. H. Attractor landscape analysis of colorectal tumorigenesis and its reversion. BMC Syst. Biol. 10, 1–13 (2016).
Shah, O. S. et al. ATLANTIS—attractor landscape analysis toolbox for cell fate discovery and reprogramming. Sci. Rep. 8, 1–11 (2018).
Gondal, M. N. et al. TISON: A next-generation multi-scale modeling theatre for in silico systems oncology. bioRxiv 10, 2021.05.04.442539 (2021).
Enderling, H. & Chaplain, M. Mathematical modeling of tumor growth and treatment. Curr. Pharm. Des. 20, 4934–4940 (2014).
Watanabe, Y., Dahlman, E. L., Leder, K. Z. & Hui, S. K. A mathematical model of tumor growth and its response to single irradiation. Theor. Biol. Med. Model. 13, 1–20 (2016).
Andasari, V. et al. Mathematical modeling of cancer cell invasion of tissue: biological insight from mathematical analysis and computational simulation. J. Math. Biol. 2010(63), 141–171 (2010).
Al-Huniti, N. et al. Tumor growth dynamic modeling in oncology drug development and regulatory approval: Past, present, and future opportunities. CPT Pharmacometrics Syst. Pharmacol. 9, 419–427 (2020).
Hofree, M., Shen, J. P., Carter, H., Gross, A. & Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115 (2013).
Kim, Y., Choi, S., Shin, D. & Cho, K. H. Quantitative evaluation and reversion analysis of the attractor landscapes of an intracellular regulatory network for colorectal cancer. BMC Syst. Biol. 11, 1–11 (2017).
Kauffman, S. A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 22, 437–467 (1969).
Kauffman, S. Homeostasis and differentiation in random genetic control networks. Nature 224, 177–178 (1969).
Glass, L. & Kauffman, S. A. The logical analysis of continuous, non-linear biochemical control networks. J. Theor. Biol. 39, 103–129 (1973).
Budczies, J. et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274)—associations with gene expression, mutational load, and survival. Genes. Chromosomes Cancer 55, 626–639 (2016).
Kuzyk, A. et al. MYCN overexpression is associated with unbalanced copy number gain, altered nuclear location, and overexpression of chromosome arm 17q genes in neuroblastoma tumors and cell lines. Genes. Chromosomes Cancer 54, 616–628 (2015).
Samulin Erdem, J., Arnoldussen, Y. J., Skaug, V., Haugen, A. & Zienolddiny, S. Copy number variation, increased gene expression, and molecular mechanisms of neurofascin in lung cancer. Mol. Carcinog. 56, 2076–2085 (2017).
Zhou, C. et al. Integrated analysis of copy number variations and gene expression profiling in hepatocellular carcinoma. Sci. Rep. 7, 10570 (2017).
Shao, X. et al. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 20, 1–14 (2019).
Chiang, C. et al. The impact of structural variation on human gene expression. Nat. Genet. 49, 692–699 (2017).
Jia, P. & Zhao, Z. Impacts of somatic mutations on gene expression: An association perspective. Brief. Bioinform. 18, 413–425 (2017).
Subia, B. et al. Breast tumor-on-chip models: From disease modeling to personalized drug screening. J. Control. Release 331, 103–120 (2021).
Liu, X. et al. Tumor-on-a-chip: From bioinspired design to biomedical application. Microsyst. Nanoeng. https://doi.org/10.1038/s41378-021-00277-8 (2021).
Tsai, H. F., Trubelja, A., Shen, A. Q. & Bao, G. Tumour-on-a-chip: microfluidic models of tumour morphology, growth and microenvironment. J. R. Soc. Interface 14, (2017).
Guo, W. F. et al. Discovering personalized driver mutation profiles of single samples in cancer by network control strategy. Bioinformatics 34, 1893–1903 (2018).
Hou, J. P. & Ma, J. DawnRank: Discovering personalized driver genes in cancer. Genome Med. https://doi.org/10.1186/s13073-014-0056-8 (2014).
Erten, C., Houdjedj, A., Kazan, H. & Taleb Bahmed, A. A. PersonaDrive: A method for the identification and prioritization of personalized cancer drivers. Bioinformatics 38, 3407–3414 (2022).
Koboldt, D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Fan, Y. et al. MuSE: Accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17, 178 (2016).
Serin Harmanci, A., Harmanci, A. O. & Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 11, 1–16 (2020).
Flensburg, C., Oshlack, A. & Majewski, I. J. Detecting copy number alterations in RNA-Seq using SuperFreq. Bioinformatics 37, 4023–4032 (2021).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Rausch, T. et al. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Zhang, J., Wang, J. & Wu, Y. An improved approach for accurate and efficient calling of structural variations with low-coverage sequence data. BMC Bioinform. 13, 1–11 (2012).
Bartenhagen, C. & Dugas, M. Robust and exact structural variation detection with paired-end and soft-clipped alignments: SoftSV compared with eight algorithms. Brief. Bioinform. 17, 51–62 (2016).
Rejniak, K. A. et al. In silico organoids: A model for deconvolution of microenvironmental and drug effects on tumor growth [abstract]. In Proceedings of the AACR Special Conference on the Evolving Landscape of Cancer Modeling; 2020 Mar 2–5; San Diego, CA. Philadelphia (PA): Cancer Res. 80, A47 (2020).
Gondal, M. N. & Chaudhary, S. U. Navigating multi-scale cancer systems biology towards model-driven clinical oncology and its applications in personalized therapeutics. Front. Oncol. 11, 4767 (2021).
Kim, Y., Choi, S., Shin, D. & Cho, K. H. Quantitative evaluation and reversion analysis of the attractor landscapes of an intracellular regulatory network for colorectal cancer. BMC Syst. Biol. 11, 45 (2017).
Shmulevich, I., Dougherty, E. R., Kim, S. & Zhang, W. Probabilistic Boolean networks: A rule-based uncertainty model for gene regulatory networks. Bioinformatics 18, 261–274 (2002).
Guo, J., Lin, F., Zhang, X., Tanavde, V. & Zheng, J. NetLand: Quantitative modeling and visualization of Waddington’s epigenetic landscape using probabilistic potential. Bioinformatics 33, 1583–1585 (2017).
Kadelka, C. DesignPrinciplesGeneNetworks. A collection of functions and tools to analyze Boolean gene regulatory networks. https://github.com/ckadelka/DesignPrinciplesGeneNetworks/blob/main/update_rules_models_in_literature_we_randomly_come_across/get_rules_for27765040.py (2023).
MathWorks. MATLAB version (R2020b). (The MathWorks Inc, 2020).
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Wishart, D. S. et al. PathBank: A comprehensive pathway database for model organisms. Nucleic Acids Res. 48, D470–D478 (2020).
Schaefer, C. et al. PID: The pathway interaction database. Nat. Preced. 2008, 1–1. https://doi.org/10.1038/npre.2008.2243.1 (2008).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Bustos, Á., Fuenzalida, I., Santibáñez, R., Pérez-Acle, T. & Martin, A. J. M. Rule-based models and applications in biology. Methods Mol. Biol. 1819, 3–32 (2018).
Maus, C., Rybacki, S. & Uhrmacher, A. M. Rule-based multi-level modeling of cell biological systems. BMC Syst. Biol. 5, 1–20 (2011).
Chylek, L. A. et al. Rule-based modeling: A computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip. Rev. Syst. Biol. Med. 6, 13–36 (2014).
Dimitrova, E. et al. Parameter estimation for Boolean models of biological networks. Theor. Comput. Sci. 412, 2816–2826 (2011).
Pavlopoulos, G. A. et al. Using graph theory to analyze biological networks. BioData Min. 4, 1–27 (2011).
Spencer, S. L., Berryman, M. J., García, J. A. & Abbott, D. An ordinary differential equation model for the multistep transformation to cancer. J. Theor. Biol. 231, 515–524 (2004).
Kwon, Y. K. & Cho, K. H. Quantitative analysis of robustness and fragility in biological networks based on feedback dynamics. Bioinformatics 24, 987–994 (2008).
Zi, Z. Sensitivity analysis approaches applied to systems biology models. IET Syst. Biol. 5, 336–346 (2011).
Cho, K. H., Shin, S. Y., Kolch, W. & Wolkenhauer, O. Experimental design in systems biology, based on parameter sensitivity analysis using a Monte Carlo method: A case study for the TNFα-mediated NF-κ B signal transduction pathway. SIMULATION 79, 726–739 (2016).
Cooper, L. A. D. et al. PanCancer insights from The Cancer Genome Atlas: The pathologist’s perspective. J. Pathol. 244, 512–524 (2018).
Jensen, M. A., Ferretti, V., Grossman, R. L. & Staudt, L. M. The NCI Genomic Data Commons as an engine for precision medicine. Blood 130, 453–459 (2017).
Cerami, E. et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Saurin, A. BioTools.fr: ENSEMBL ID to Gene Symbol Converter - Genomics Biotools. https://www.biotools.fr/human/ensembl_symbol_converter.
Stelzer, G. et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.1-1.30.33 (2016).
Tweedie, S. et al. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 49, D939–D946 (2021).
Leys, C., Ley, C., Klein, O., Bernard, P. & Licata, L. Detecting outliers: Do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Soc. Psychol. 49, 764–766 (2013).
Steven, W. A Review of Statistical Outlier Methods (Pharmaceutical Technology, 2006).
Chakravarty, D. et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00011 (2017).
Wishart, D. S. et al. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36, D901–D906 (2008).
Wang, T. et al. OncoVar: An integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res. 49, D1289–D1301 (2021).
Gonzalez-Perez, A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013(10), 1081–1082 (2013).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, 955–961 (2013).
Acknowledgements
This work was supported by Ignite National Technology Fund [National ICT-R&D Fund (SRG-209 to S.U.)], the Higher Education Commission of Pakistan [Grant No. 20-GCF-521/RGM/R&D/HEC/2020 to S.U., Grant No. TTSF 2022-23-160 to M.S., and S. U.], Health Research Institute, National Institute of Health [Grant No. NHCG-22/R2-91 to M.S., and S. U.], and National Center for Big Data and Cloud Computing [RF-NCBC-015 to S.U.].
Author information
Authors and Affiliations
Contributions
Conceptualization: R.N., M.S. and S.U., investigation: R.N., B.A., M.N., R.H., R.A., S.U., supervision: S.U., analysis: R.N., methodology: R.N. and S.U., interpreted results: R.N., M.S., and S.U., writing—review and editing: R.N., M.S., Z.N., A.A.C., A.F. and S.U., implementation code: R.N., B.A., M.U., Z.B., M.N., S.K. and M.F., and funding acquisition: M.S., S.U., A. A. C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: In the original version of this Article, Asher Alban Channan-Khan was omitted as a co-corresponding author. Correspondence and requests for materials should also be addressed to chanan-khan.asher@mayo.edu.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Butt, R.N., Amina, B., Sultan, M.U. et al. CanSeer: a translational methodology for developing personalized cancer models and therapeutics. Sci Rep 15, 15080 (2025). https://doi.org/10.1038/s41598-025-99219-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99219-x
