
Abstract
Identifying protein–protein interactions (PPIs) is a foundational task in biomedical natural language processing. While specialized models have been developed, the potential of general-domain large language models (LLMs) in PPI extraction, particularly for researchers without computational expertise, remains unexplored. This study evaluates the effectiveness of proprietary LLMs (GPT-3.5, GPT-4, and Google Gemini) in PPI prediction through systematic prompt engineering. We designed six prompting scenarios of increasing complexity, from basic interaction queries to sophisticated entity-tagged formats, and assessed model performance across multiple benchmark datasets (LLL, IEPA, HPRD50, AIMed, BioInfer, and PEDD). Carefully designed prompts effectively guided LLMs in PPI prediction. Gemini 1.5 Pro achieved the highest performance across most datasets, with notable F1-scores in LLL (90.3%), IEPA (68.2%), HPRD50 (67.5%), and PEDD (70.2%). GPT-4 showed competitive performance, particularly in the LLL dataset (87.3%). We identified and addressed a positive prediction bias, demonstrating improved performance after evaluation refinement. While not surpassing specialized models, general-purpose LLMs with appropriate prompting strategies can effectively perform PPI prediction tasks, offering valuable tools for biomedical researchers without extensive computational expertise.
Similar content being viewed by others


Introduction
Protein–protein interactions (PPIs) are crucial in various physiological processes, such as gene expression, signal transduction, and apoptosis, which directly impact health and disease1. Aberrations in PPIs, induced by factors like mutations or infections, are strongly linked to diseases such as cancer2,3. Moreover, PPIs influence a variety of industries, as can be seen in their role in food processing where enzymes like chymotrypsin are essential for breaking down wheat gluten proteins4, and in agriculture where protein interactions affect fruit ripening5. The surge in biomedical literature, exemplified by PubMed’s extensive database, presents a challenge to researchers to efficiently mine and extract actionable insights. To help address this opportunity, advances in Natural Language Processing (NLP), such as Named Entity Recognition (NER), Relation Extraction (RE)6, and Question Answering (QA)7, offer significant potential. The introduction of transformer-based models like BERT8 in 2018 revolutionized this field and have led to specialized versions like BioBERT9, SciBERT10, and Clinical BERT11, enhancing task-specific model performance.
Early methodologies for extracting Protein-Protein Interactions (PPIs) from the literature included pattern-based, co-occurrence, and machine learning strategies. Pattern-based techniques, which involve manually constructing rules to identify protein pairs, provide a foundational approach12,13, while co-occurrence methods leverage occurrences of protein pairs within single sentences to facilitate extraction, albeit with limitations in their capacity to capture complex relationships14. Advanced approaches like machine learning use comprehensive feature sets that combine linguistic and structural elements to enhance PPI extraction, though these methods often face challenges related to structural similarities within the data15,16. Furthermore, kernel-based techniques such as sub-tree17, subset tree18, partial tree19, spectrum tree20, and feature-enriched tree21 kernels effectively use high-dimensional sentence features to optimize the extraction process22,23; and composite kernel methods refine this approach by integrating multiple kernel types to enhance the extraction and analysis of textual information24,25,26. Additionally, the development of the gradient-tree boosting model, LpGBoost, marks a significant advance in computational efficiency through its use of shallow data representations27. Moreover, neural network architectures, particularly convolutional (CNN) and recurrent neural networks (RNN), play a pivotal role by facilitating feature extraction autonomously; this augments kernel methods and improves the overall efficacy of PPI extraction tasks28,29,30. The integration of transformer-based BERT models represents a further evolution in PPI extraction methodologies. These models incorporate deep learning techniques with lexical and syntactic processing to significantly advance the field and enable more precise and effective extraction of protein interactions31,32. However, despite the effectiveness of domain-specific models, their broader application and effective implementation are often limited by the requirement for foundational computer science knowledge. In contrast, the development of large language models (LLMs), such as OpenAI’s ChatGPT introduced in 2022, provides a promising alternative. These models, pre-trained on a hugely wide range of datasets, are capable of generating contextually relevant responses across various domains without the need for task-specific fine-tuning33. Moreover, the LLMs in OpenAI’s Generative Pre-trained Transformer series have evolved from GPT-1 to the more robust and multimodal GPT-4, which demonstrate extensive capabilities in understanding and generating language34,35. These advanced models can seamlessly support a range of applications without requiring domain-specific adjustments36,37.
Compared to open-source language models, which require technical expertise in computational methods for training and prediction, proprietary LLMs such as ChatGPT and Gemini38 enable non-technical users to efficiently obtain domain-specific reference results through well-designed prompts. This capability can significantly accelerate research processes in fields such as pharmaceutical development39. In this study, we use the protein-protein interaction (PPI) prediction task as a case study to evaluate the performance of proprietary LLMs in responding to specifically designed prompts, to simulate the extent of assistance available to biomedical researchers. Our objective is not only to assess the intrinsic capabilities and limitations of these models in specialized tasks but also to explore the extent to which prompt optimization can enhance their performance. In summary, this analysis aims to provide a clearer perspective on the applicability of proprietary LLMs in handling complex biological data, and in so doing, offer valuable insights into the practical deployment of AI technologies in the life sciences.
Methods
Protein–protein interaction (PPI) task definition
The definition of protein-protein interaction (PPI) can vary depending on the conceptual delineation of entities required for different applications. Since being established in the 1970s40, the interdependence of DNA, RNA, and proteins has been well recognized. However, the gene-centered representation commonly adopted in biomedical literature tends to exacerbate the ambiguity among these molecular entities. To improve the accuracy of biological interaction modeling, many studies have expanded the definition of protein entities to include genes and RNA41,42,43,44. Consequently, PPI datasets differ in their definitions of protein entities based on their intended applications. Moreover, the definition of interactions between proteins spans multiple perspectives, ranging from direct physical contact to broader contextual associations described in textual sources. To mitigate error propagation caused by sequential entity recognition and relation prediction, most PPI datasets provide pre-annotated entities, which facilitate subsequent modeling efforts. In this study, we adhere to the entity annotations provided by each dataset and conduct sentence-level predictions. During the prompt development, the model is presented with a pair of named entities and sentences containing these entities, and it is tasked with predicting whether the protein pairs interact. Sentences containing interacting entity pairs are then classified as positive instances, whereas those without interactions are categorized as negative instances. If a sentence contains multiple entity pairs, each pair’s relationship is assessed separately. For example, if SigK and GerE are determined to have no interaction, the instance is labeled as negative, whereas if SigK and ykvP are identified as interacting within the same sentence, it is labeled as positive. This is illustrated in the examples provided in Supplementary Table 1.
Evaluation dataset and experiment settings
We employed six PPI benchmark datasets to evaluate our prompting strategies: LLL, IEPA, HPRD50, AIMed, BioInfer, and PEDD. The LLL dataset, which originated from the Learning Language in Logic 2005 (LLL05) challenge and was sourced from the Medline database, focuses on extracting protein/gene interactions with only 77 sentences, i.e., a limited quantity of information45. The IEPA dataset comprises 486 sentences extracted from PubMed abstracts46. The HPRD50 dataset includes 50 random abstracts from the Human Protein Reference Database (HPRD), totaling 145 sentences with proteins/genes pre-tagged by ProMiner and interaction relationships annotated by human experts13. The AIMed dataset contains 200 abstracts from PubMed, manually annotated with entities and their interaction relationships47. The BioInfer dataset comprises 1100 sentences and was collected using the Database of Inter-acting Proteins (DIP) to identify PubMed search inputs related to interacting entities; the selected sentences contain more than one pair of interacting entities48. We also used the recently published PEDD dataset, which was derived from the AICUP 2019 competition and focuses on abstracts in PubMed published after 2015 and from journals with impact factors greater than 5 to ensure higher-quality and more recent information49. The original distribution of positive and negative PPI instances across these datasets is presented in the “Raw data” column of Supplementary Table 2. It’s worth noting that since a single sentence may contain both positive and negative instances, the total sentence count is significantly less than the sum of positive and negative instances.
Our experimental workflow followed a two-phase evaluation approach. In the first phase, we selected representative samples from the five benchmark datasets (LLL, IEPA, HPRD50, AIMed, and BioInfer) for prompt design and initial evaluation using GPT-3.5 and GPT-4. In the second phase, we conducted comprehensive performance evaluations using all six datasets (including PEDD) across multiple proprietary LLMs, including GPT-3.5, GPT-4, and Gemini 1.5 (both Flash and Pro versions). All experiments were implemented in Python using the corresponding LLM APIs. To ensure consistent evaluation, we required all LLMs to output their predictions in a standardized JSON format, which helped the systematic calculation of the three standard metrics we employed: recall, precision, and F1-score. Precision measures the accuracy of positive predictions by calculating the proportion of true positive cases among all instances identified as positive; this indicates the model’s ability to avoid false positives. Recall quantifies the proportion of true positives that the system correctly recognizes and thus reflects the model’s ability to capture all relevant interactions. The F1-score provides a balanced measure of the model’s overall performance by combining precision and recall into a single value. To investigate whether providing additional context could improve LLM-based PPI extraction, we experimented with multi-sentence inputs (ranging from 1 to 5 sentences). While adding contextual information might theoretically enhance the model’s ability to infer interactions, it also introduces the possibility of additional noise, potentially affecting prediction reliability. Our results showed that multi-sentence inputs led to performance fluctuations across different models and datasets, indicating that longer inputs do not consistently improve predictive accuracy. To ensure methodological consistency and reproducibility, our primary evaluation was conducted using single-sentence inputs. A detailed analysis of this comparison is provided in the Results section.
Methodological framework overview
This study presents a systematic approach to evaluate the capability of proprietary LLMs in PPI detection through carefully designed prompting strategies. Our methodology comprises three main components: (1) prompt engineering and optimization, (2) context-aware prompt selection, and (3) systematic evaluation across multiple datasets. In the prompt engineering phase, we developed six distinct prompting scenarios, progressively increasing in complexity from basic interaction queries to sophisticated entity-tagged formats. These scenarios were designed to assess how different levels of input structure and contextual information affect model performance. The prompts vary in two key dimensions: the degree of entity tagging (from untagged to comprehensively tagged with numerical identifiers) and the specificity of query statements (from simple interaction queries to structured JSON output requests). The context-aware selection mechanism enables adaptive prompt deployment based on sentence characteristics. This component specifically addresses the challenges posed by various protein entity representations in the biomedical literature, including complicated entities (e.g., "Arp2/3 complex"). The selection process ensures that the most appropriate prompt is applied to each specific context. For systematic evaluation, we implemented a comprehensive workflow that processes PPI datasets sequentially, incorporating pairwise protein labeling and context-specific prompt selection. This structured approach allows for consistent assessment across different datasets while accommodating their unique characteristics. The following sections detail these components, beginning with an in-depth examination of the prompting scenarios, followed by the advanced workflow that integrates these prompts into a coherent PPI detection system.
Prompting scenarios
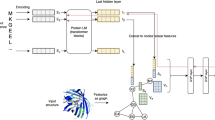
To systematically evaluate PPI extraction, we designed a comprehensive prompting strategy as illustrated in Fig. 1. The strategy categorizes input data into two primary types: Basic Entities and Nested/Complex Entities. For Basic Entities, we developed three distinct entity tagging strategies: (1) No Additional Tagging, where raw text is processed directly; (2) Tag Only One Protein Pair, which focuses on specific protein pairs of interest; and (3) Tag All Protein Entities with Repeated Proteins Assigned Numbers, which provides comprehensive protein markup. These tagging strategies are coupled with progressively complex query statements: from basic interaction verification (Prompt 1), to identifying PPIs without explicit protein information (Prompts 2-3), to full protein-aware PPI identification (Prompts 4-5). For Nested/Complex Entities, we implemented a specialized Pairwise Gene Tagging approach (Prompt 6) to handle compound entities with special characters. More detailed processing methods for these complex cases will be introduced later. All tagged entities use the "<GENE></GENE>" markup schema to standardize the identification of protein entities across different prompting scenarios.

Prompting strategies for PPI identification.
Supplementary Figure 1 showcases examples of input-output pairs for prompts 1 and 3, where ‘USER’ is the designated input and ‘ASSISTANT’ the resulting output from the GPT models. In the sentence 'LLL.d2.s2’, three protein entities are present, which leads to three pairs of input questions generated through permutation and combination. Example (a) in Supplementary Figure 1 illustrates a single prompt pair for prompt 1, while Example (b) demonstrates the application of prompt 3 for the same sentence. Given that prompts 2 and 3 elicit multiple outputs of PPI results from the models, the prompt descriptions have been enhanced to include a request for response in JSON format to simplify subsequent result aggregation and analysis. Descriptions underscored on the input side in the figure show the disparities between prompt 3 and prompt 2. By enumerating all protein entity names in the sentence, the aim is to assess whether the model can enhance its association judgment capability. The most intricate prompt, prompt 5, within the same query framework as prompt 3, entails explicit and comprehensive entity tagging and numbering for the input sentence. Figure 2 illustrates the schematic representation of input sentence tagging.

Schematic representation of tagging in protein entities.
To assess the efficacy of all prompts, we manually selected ten sentences containing protein entities from each of the five PPI benchmark datasets. The selected sentences met the criteria of generating at least 3 to 6 positive/negative instances to maintain a homogeneous distribution in the sample set. The statistical data of the sample dataset are presented in Figure 2 of supplementary materials. The sample dataset was applied to both the GPT-3.5 and GPT-4 models to select the most reliable prompt.
During the data examination, we established a systematic approach to address complex entity scenarios that extend beyond basic predefined prompts. Our analysis revealed three primary patterns requiring specialized handling: symbol-based compound entities, keyword-indicated complexes, and nested entities. First, we implemented symbol-based filtering to identify compound protein entities. Sentences containing protein names with "/" or "-" symbols were flagged as compound protein entities. For example, "Arp2/3 complex" represents two distinct proteins, “Arp2” and "Arp3," functioning as a unified complex. These marked protein names underwent additional analysis to ensure accurate interpretation by the model. Second, we developed keyword-based identification for complex naming entities. Sentences containing specific keywords such as "complex," "subunit," or “component” were identified as potential compound naming entities. For instance, "The Arp2/3 complex binds actin filaments" exemplifies such cases. These sentences received specialized processing by the LLM to prevent prediction errors arising from abbreviations or naming variations. Third, we addressed nested entity scenarios, where both short-form and descriptive long-form protein names appear within the same context. For example, in "... two transmembrane receptors, the p75 neurotrophin receptor and the p140trk (trkA) tyrosine kinase receptor, ...", both “p75” and its longer form "p75 neurotrophin receptor" appear as nested entities. To avoid manual intervention, we employed a gene abbreviation recognition module to detect and process these nested and compound terms automatically. The prompt instructs GPT to consider complete protein names rather than merely abbreviations during PPI identification.
Statistical analysis of the datasets revealed that these complex entity patterns appear exclusively in the AIMed and BioInfer datasets, comprising approximately 23% of instances within each dataset. To address these scenarios, we developed Prompt 6, which builds upon prompt 1 by incorporating comprehensive guidelines for handling compound terms, nested entities, and their variations. The entity tagging strategy requires listing all protein entity names and examining protein pairs systematically, with sentence processing considered complete only after evaluating all possible protein pair combinations. Figure 3 demonstrates this approach using BioInfer dataset examples, with the underlined text highlighting compound entity descriptions and comparing predictions across different LLM models.

Design of Prompt 6 for handling complex protein entities.
A scenario-based prompting framework for PPI detection
Sentences describing PPIs often vary widely due to differences in research topics, author writing styles, and experimental types, which makes them challenging to categorize. To address these challenges, we developed a comprehensive framework as illustrated in Fig. 4, which begins with prompt design and evaluation using sample datasets. The framework initiates from the prompting scenarios (lower left in the figure, with detailed design principles shown in Fig. 1). In this phase, input sentences are categorized based on entity complexity: basic entities and complex entities (including complex, compound, or nested structures). For each category, candidate prompts undergo evaluation using GPT models to determine the most effective prompt for that specific scenario. The result is Prompt 5 for basic entities and Prompt 6 for complex entities. Following prompt assessment, all PPI dataset sentences undergo scenario classification based on their entity complexity. The sentences are then processed using appropriate marking strategies to highlight gene locations: basic entities undergo gene tagging and numbering, while complex entities are processed through pairwise gene tagging. Finally, these processed sentences are combined with their corresponding scenario-specific prompts for performance evaluation using proprietary LLMs including GPT and Gemini models. This systematic approach ensures appropriate handling of various PPI descriptions while optimizing prompt selection based on entity complexity and sentence structure. The performance of the predictions can be comprehensively evaluated through this structured methodology.

PPI prompting framework.
Results
Determination of candidate prompts
The primary objective of the experiment was to ascertain the optimal general prompts (Prompts 1–5) to help the model discern the different PPIs. To achieve this, we employed the PPI sample dataset delineated in Supplementary Figure 2 as the focal point for evaluation and conducted performance assessments of general prompts across both the GPT-3.5 and GPT-4 models. The findings, shown in Table 1, consistently demonstrate that Prompt 5 exhibited superior performance within the sample dataset across both model iterations. According to Supplementary Figure 3, the design strategy of Prompt 5 helps GPT accurately identify multiple proteins with the same name during PPI prediction and subsequently lists the actual interacting groups based on content associations. By subsequently leveraging the efficacy demonstrated by Prompt 5, we investigated the impact of inputting multiple sentences simultaneously on the performance of the GPT models (Table 2). The findings indicate demonstrate not only GPT-4’s superior performance over GPT-3.5 but also a more consistent effect on model performance with the single-sentence input methodology.
To evaluate the performance of Prompt 6, which is specifically designed to handle cases involving nested protein named entities, we created an evaluation sample set. This set consists of 10 sentences each from the AIMed and Bioinfer datasets. In the AIMed dataset, there are 15 positive instances and 27 negative instances, while the Bioinfer dataset contains 42 positive instances and 30 negative instances. This sample composition allows for a balanced assessment of the prompt’s effectiveness in identifying relevant entities. Given their similar query statement structures, Prompt 1 served as the baseline for evaluation. The results presented in Table 3 indicate that Prompt 6 outperforms Prompt 1 in both GPT-3.5 and GPT-4 models. This highlights the beneficial impact of providing compound term information on the enhancement of large language model performance.
Performance evaluation of PPI datasets
Based on the previously established prompt filtering process, we evaluated the performance of GPT-3.5, GPT-4, and Google Gemini 1.5 (both flash and pro versions) across six PPI datasets using the final Prompts 5 and 6. The “Raw data” column of Table 4 presents comparative results across all models. It was notable that Gemini 1.5 pro demonstrated superior performance across most datasets, achieving the highest F1-scores in the LLL (90.3%), IEPA (68.2%), HPRD50 (67.5%), and PEDD (70.2%) datasets. GPT-4 showed competitive performance particularly in the LLL dataset (87.3% F1-score), while generally maintaining stable performance across the other datasets. GPT-3.5 demonstrated consistent but relatively lower performance compared to the other models. The variation in model performance across datasets reflects the inherent challenges in their characteristics. The LLL dataset, despite being the smallest, yielded the highest performance across all models (ranging from 79.2 to 90.3% F1-score), which suggests that its smaller size and potentially more straightforward entity relationships facilitate better model comprehension. In contrast, more complex datasets like AIMed, BioInfer, and PEDD showed lower performance across all models, with F1-scores typically ranging from 37.9 to 70.2% for the raw data. This performance pattern highlights the challenges posed by complex entity features and rich content in these datasets.
Furthermore, we identified a bias in all models towards predicting positive input instances, which led to a diminished discriminatory capability when processing exclusively negative instances. To address this limitation, we excluded sentences containing solely negative instances from the datasets during performance evaluation. The “Refined data” column of Table 4 demonstrates the models’ performance after this modification. The LLL dataset, lacking such instances, maintained its original distribution and was excluded from refined evaluation. In this scenario, all models showed improved precision and F1-scores across the refined datasets. For instance, in the IEPA dataset, Gemini 1.5 pro achieved an F1-score of 89.7%, while GPT-4 and GPT-3.5 reached 86.4% and 84.7% respectively. This consistent improvement across models and datasets further validates our observation regarding the models’ positive instance bias and demonstrates the effectiveness of our refinement strategy.
Discussion
The application of LLMs to biological relationship extraction presents both promising opportunities and notable challenges. Our comprehensive evaluation of LLMs in PPI prediction reveals several key insights regarding model performance, limitations, and practical implications for the biomedical research community. In this discussion, we first systematically examine the relationship between model performance and dataset characteristics, followed by a detailed analysis of model-specific results. We then address key limitations and challenges before exploring future directions and potential applications. Our analysis demonstrates varying levels of effectiveness across different models and datasets, with particularly notable performance patterns. The most striking observation is the consistently superior performance achieved on the LLL dataset across all models, with F1-scores ranging from 79.2 to 90.3%, and Gemini 1.5 Pro achieving the highest F1-score of 90.3%. This exceptional performance can be attributed to several factors, including LLL’s smaller size, straightforward annotation scheme, and simpler grammatical structures, which provide a less challenging environment for model comprehension50. These dataset-specific characteristics form the foundation for understanding the broader patterns of model performance across our evaluation suite.
The performance variations across datasets reveal critical insights into model behavior and limitations. Beyond quantitative performance metrics, our linguistic analysis uncovered significant challenges in semantic parsing and referential understanding. We observed that LLMs demonstrate notable susceptibility to misclassification when confronted with complex interaction-related linguistic structures. Two illustrative examples underscore these interpretative challenges. In the first instance, consider the sentence excerpted from BioInfer.d245 "Here we have now identified the reciprocal complementary binding site in alpha-catenin which mediates its interaction with <GENE>beta-catenin</GENE> and <GENE>plakoglobin</GENE>." Lexical cues such as "mediates" and "interaction" precipitated a model-driven misinterpretation, erroneously suggesting a direct PPI relationship between beta-catenin and plakoglobin. Critically, the models overlooked the nuanced reality that alpha-catenin separately interacts with these entities. The referential narrative structure, particularly the pronomial phrase "its interaction," represents a significant interpretative vulnerability for contemporary LLMs. A second example, excerpted from AIMed.d182, further highlights the challenges of referential disambiguation in LLMs. Specifically, the sentence “The MDM2 oncoprotein is a cellular inhibitor of the <GENE>p53</GENE> tumor suppressor in that it can bind the transactivation domain of <GENE>p53</GENE> and downregulate its ability to activate transcription” demonstrates how repeated gene mentions require accurate coreference resolution to preserve semantic clarity. In this case, the two p53 entities plainly lack a protein-protein interaction. The pronoun “it” unambiguously references the sentence’s primary subject, "MDM2 oncoprotein". Nevertheless, the models consistently failed to discern these critical contextual nuances, erroneously predicting an interconnection. The marked contrast between simpler datasets like LLL and more complex ones such as AIMed and BioInfer highlights how sentence complexity and grammatical structure significantly impact extraction accuracy. This is particularly evident in AIMed’s sophisticated sentence constructions and nested entities, which resulted in notably lower precision scores of 26.6% and 23.8% for GPT-3.5 and GPT-4, respectively. The representation of biological entities across different datasets also proves to be a crucial factor, with BioInfer’s complex compound entities and abbreviations significantly challenging model comprehension, as reflected in its lower F1-scores ranging from 45.3 to 55.3% in the raw data. In contrast, datasets featuring more straightforward entity mentions, such as HPRD50, demonstrated more robust performance with F1-scores between 63.6 and 67.5%. Additionally, our analysis uncovered a consistent bias across all models toward predicting positive instances, leading us to refine our evaluation approach by excluding sentences containing solely negative instances. This refinement resulted in substantial performance improvements, as evidenced by IEPA’s F1-scores increasing from 65.6 to 84.7% for GPT-3.5 and from 64.0 to 86.4% for GPT-4; this underscores the significant impact of data distribution on model behavior.
Gemini 1.5 Pro demonstrated superior performance across most datasets, which can be attributed to its advanced model architecture and optimization. This superior performance was particularly evident for the LLL (90.3% F1-score), IEPA (89.7% F1-score in refined data), and HPRD50 (87.1% F1-score in refined data) datasets. GPT-4 showed competitive performance, particularly in the LLL dataset (87.3% F1-score), while maintaining stable performance across other datasets. GPT-3.5, while showing consistent performance, generally achieved lower scores compared to other models. To contextualize these results within the current research landscape, a recent study by Rehana et al.51 evaluated GPT and BERT models on PPI tasks, achieving F1-scores of 86.49% (GPT-4 on LLL), 71.54% (GPT-4 on IEPA), and 65.0% (GPT-4 on HPRD50) using strategies such as protein dictionary normalization and protein masking. While their approach relies on sophisticated preprocessing techniques, our prompt design approach achieves comparable or superior results while focusing solely on sentence structure, and in so doing, offers a more user-friendly approach for non-technical biomedical researchers. Further analysis of the LLMs’ behavior in specific linguistic contexts revealed additional performance patterns. We examined PPI sentences from the PEDD dataset containing inference-related keywords including whether, may, might, could, potential, and other similar terms; we identified 2,570 cases with 282 positive and 2,288 negative instances for evaluation. In these inferential contexts, Gemini 1.5 Pro maintained its superior performance with an F1-score of 0.2, followed by Gemini 1.5 Flash at 0.179 and GPT-3.5 at 0.163, while GPT-4 unexpectedly showed the lowest performance at 0.154. The significantly lower F1-scores in inferential contexts compared to general PPI detection suggest that LLMs struggle to interpret protein interactions when presented with hypothetical or uncertain relationships. Notably, all models exhibited high false positive rates in inferential contexts, with even the best-performing Gemini 1.5 Pro producing 1,550 false positive cases. This observation provides additional evidence of a broader pattern in LLMs’ prediction behavior that warrants further investigation, as we discuss in our analysis of the study’s limitations.
While these results demonstrate promising capabilities of LLMs in biomedical relationship extraction, several important limitations and challenges emerged from our analysis. The observed bias of LLMs towards positive PPI predictions warrants careful consideration. A comprehensive review discusses how biases can manifest in various natural language processing tasks, including literature mining, highlighting the tendency of LLMs to generate outputs that may favor certain narratives or perspectives52. The ability to accurately identify both the presence and absence of protein interactions is crucial for understanding biological systems and developing therapeutic interventions. Our findings suggest that current LLMs, despite their sophisticated architecture, may exhibit a systematic bias that could lead to false positive predictions in PPI detection tasks. This limitation could be particularly problematic in exploratory research where identifying non-interacting protein pairs is as valuable as detecting interactions. Our analysis also revealed interesting variations in how different LLMs identify potential PPIs within established complexes. For example, when analyzing the sentence "Arp2/3 complex from Acanthamoeba binds profilin and cross-links actin filaments" and subsequently asked whether Arp2 and Arp3 components interact with each other, we observed divergent interpretations: GPT-3.5 identified a positive PPI between Arp2 and Arp3 subunits within the complex, while GPT-4, Gemini 1.5 Flash, and Gemini 1.5 Pro classified it as a negative interaction. This discrepancy highlights a fundamental challenge in how LLMs process relationships between proteins that function as part of established complexes. The Arp2/3 example demonstrates that even when addressing interactions between components of well-characterized molecular assemblies, LLMs can produce inconsistent results. This variation represents an important consideration when employing LLMs for biomedical knowledge extraction, particularly for questions about the internal structure of protein complexes that may not be explicitly stated in the text.
Traditional transformer-based models (BioBERT, PubMedBERT, SciBERT) have demonstrated superior performance across these datasets, with BioBERT achieving F1-scores ranging from 74.95% (HPRD50) to 86.84% (LLL)51. Similarly, previous machine learning approaches have shown competitive results, with sdpCNN achieving 66% and 75.2% on AImed and BioInfer respectively53, and the tree-based DSTK model achieving scores ranging from 71.01% (AImed) to 89.20% (LLL)23. Based on these findings and observed limitations, we recommend that practitioners implementing LLMs for PPI detection should: (1) implement additional validation steps for negative predictions, (2) consider incorporating confidence scores or uncertainty measures in their predictions, and (3) potentially employ ensemble approaches that combine LLM predictions with traditional machine learning methods that have shown robust performance in identifying negative instances. Looking toward future developments, while the system testing results obtained through prompt design may not surpass various state-of-the-art deep learning models, LLMs offer unique advantages. Their ability to adapt prompt suitability continuously without retraining for specific task demands54 and potential to outperform state-of-the-art models when fine-tuned on specific datasets55 makes them particularly valuable for non-computer science experts56. Recent research has shown promising strategies for improving LLM performance in biomedical applications. Few-shot learning approaches have demonstrated comparable results to SOTA models in tasks such as NER, relation extraction, summarization, and QA55. In the Nephrology domain, chain-of-thought (CoT) strategies have successfully guided models in clinical decision support57. Future development of LLMs for biomedical applications should explicitly address the positive prediction bias, perhaps through specialized pre-training strategies or architectural modifications that better balance the model’s ability to identify both positive and negative protein interactions. LLMs have already begun to appear in highly specialized clinical research, such as extracting diagnosis information from cancer examination reports58,59. Their performance on the recent PEDD dataset (achieving 70.2% F1-score compared to BioBERT’s 77.06%)49 demonstrates their ability to handle contemporary biomedical literature effectively, though precise task completion still relies heavily on appropriate prompt guidance60. In an era emphasizing multi-objective applications, the integration of large language models can provide a robust foundation for development and research across various domains. Our findings suggest that when combined with effective prompt engineering strategies and domain-specific considerations, LLMs have the potential to revolutionize biomedical relationship extraction, particularly for researchers without extensive computational expertise.
Conclusion
Verifying interactions in biomedical experiments requires precise conditions and results in complex biomedical literature and PPI datasets. This study shows that general-purpose LLMs like the GPT and Gemini models can reliably predict protein interactions and serve as valuable tools for non-experts. Using progressive prompt designs tailored to PPI dataset specificities, we addressed model biases, such as misclassifying negative instances as positive, by refining data preprocessing and prompt design to significantly enhance performance. Although the GPT and Gemini models currently lag behind traditional state-of-the-art methods, the rapid advancement of LLM technology offers a promising future. Innovative techniques, such as chain-of-thought prompts and ensemble predictions with multiple LLMs, are expected to further improve performance. The continued evolution of LLMs holds significant potential for advancing research across an ever diversifying range of fields.
Data availability
The five benchmark PPI datasets (AImed, Bioinfer, HPRD50, IEPA and LLL) analyzed in this study are publicly available on the GitHub repository at https://github.com/metalrt/ppi-dataset/tree/master/csv_output. Additionally, the latest PPI dataset referenced in this study, PPED, can be accessed via the AI CUP 2019 platform at https://www.aicup.tw/ai-cup-2019.
References
Berggård, T., Linse, S. & James, P. Methods for the detection and analysis of protein–protein interactions. Proteomics 7(16), 2833–2842 (2007).
Garner, A. L. & Janda, K. D. Protein-protein interactions and cancer: targeting the central dogma. Curr. Top. Med. Chem. 11(3), 258–280 (2011).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2), 271–280.e8 (2020).
Agyare, K. K., Addo, K. & Xiong, Y. L. Emulsifying and foaming properties of transglutaminase-treated wheat gluten hydrolysate as influenced by pH, temperature and salt. Food Hydrocolloids 23(1), 72–81 (2009).
Wang, S. et al. Phosphorylation of MdERF17 by MdMPK4 promotes apple fruit peel degreening during light/dark transitions. Plant Cell 34(5), 1980–2000 (2022).
Kim, J.-D., et al. Overview of BioNLP shared task 2011 In Proceedings of the BioNLP Shared Task 2011 Workshop (Association for Computational Linguistics, 2011).
Tsatsaronis, G. et al. An overview of the BIOASQ large-scale biomedical semantic indexing and question answering competition. BMC Bioinform. 16, 1–28 (2015).
Devlin, J., et al. BERT: Pre-training of deep bidirectional transformers for language understanding. (North American Chapter of the Association for Computational Linguistics, 2019).
Lee, J. et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 36(4), 1234–1240 (2020).
Beltagy, I., Lo, K. & Cohan, A. SciBERT: A pretrained language model for scientific text. In Conference on Empirical Methods in Natural Language Processing (2019).
Alsentzer, E. et al. Publicly Available Clinical BERT Embeddings (Association for Computational Linguistics, 2019).
Huang, M. et al. Discovering patterns to extract protein–protein interactions from full texts. Bioinformatics 20(18), 3604–3612 (2004).
Fundel, K., Küffner, R. & Zimmer, R. RelEx—Relation extraction using dependency parse trees. Bioinformatics 23(3), 365–371 (2007).
Bunescu, R., et al. Integrating co-occurrence statistics with information extraction for robust retrieval of protein interactions from Medline. In Proceedings of the hlt-naacl bionlp Workshop on Linking Natural Language and Biology (2006).
Van Landeghem, S., et al. Extracting protein-protein interactions from text using rich feature vectors and feature selection. In 3rd International symposium on Semantic Mining in Biomedicine (SMBM 2008). (Turku Centre for Computer Sciences (TUCS), 2008).
Liu, B., et al. Dependency-driven feature-based learning for extracting protein-protein interactions from biomedical text. In Coling 2010: Posters (2010).
Vishwanathan, S. & Smola, A. J. Fast kernels for string and tree matching. Adv. Neural. Inf. Process. Syst. 15, 569–576 (2003).
Collins, M., Parsing with a single neuron: Convolution kernels for natural language problems. Technical Report, (University of California at Santa Cruz, 2001).
Moschitti, A. Making tree kernels practical for natural language learning. In 11th conference of the European Chapter of the Association for Computational Linguistics (2006).
Kuboyama, T. et al. A spectrum tree kernel. Inf. Media Technol. 2(1), 292–299 (2007).
Sun, L. & Han, X. A feature-enriched tree kernel for relation extraction. In Proceedings of the 52nd Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers) (2014).
Li, L. et al. An approach to improve kernel-based protein–protein interaction extraction by learning from large-scale network data. Methods 83, 44–50 (2015).
Murugesan, G., Abdulkadhar, S. & Natarajan, J. Distributed smoothed tree kernel for protein–protein interaction extraction from the biomedical literature. PLoS ONE 12(11), e0187379 (2017).
Miwa, M. et al. Protein–protein interaction extraction by leveraging multiple kernels and parsers. Int. J. Med. Inform. 78(12), e39–e46 (2009).
Li, L. et al. Integrating semantic information into multiple kernels for protein-protein interaction extraction from biomedical literatures. PLoS ONE 9(3), e91898 (2014).
Chang, Y.-C. et al. PIPE: A protein–protein interaction passage extraction module for BioCreative challenge. Database 2016, baw101 (2016).
Warikoo, N., Chang, Y.-C. & Ma, S.-P. Gradient boosting over linguistic-pattern-structured trees for learning protein–protein interaction in the biomedical literature. Appl. Sci. 12(20), 10199 (2022).
Hsieh, Y.-L., et al. Identifying protein-protein interactions in biomedical literature using recurrent neural networks with long short-term memory. In Proceedings of the eighth international joint conference on natural language processing (volume 2: short papers) (2017).
Peng, Y. & Lu, Z. Deep Learning for Extracting Protein–Protein Interactions from Biomedical literature (Association for Computational Linguistics, 2017).
Yadav, S., et al. Feature assisted bi-directional LSTM model for protein–protein interaction identification from biomedical texts (2018).
Su, P., Peng, Y. & Vijay-Shanker, K. Improving BERT model using contrastive learning for biomedical relation extraction. In Workshop on Biomedical Natural Language Processing (2021).
Warikoo, N., Chang, Y.-C. & Hsu, W.-L. LBERT: Lexically aware transformer-based bidirectional encoder representation model for learning universal bio-entity relations. Bioinformatics 37(3), 404–412 (2021).
Wu, T. et al. A brief overview of ChatGPT: The history, status quo and potential future development. IEEE/CAA J. Autom. Sin. 10(5), 1122–1136 (2023).
Radford, A., et al. Improving language understanding by generative pre-training. OpenAI preprint (2018).
Radford, A. et al. Language models are unsupervised multitask learners. OpenAI blog 1(8), 9 (2019).
Brown, T. et al. Language models are few-shot learners. Adv. Neural. Inf. Process. Syst. 33, 1877–1901 (2020).
Gallifant, J. et al. Peer review of GPT-4 technical report and systems card. PLOS Digital Health 3(1), e0000417 (2024).
Team, G., et al. Gemini: A family of highly capable multimodal models. arXiv preprint arXiv:2312.11805 (2023).
Tripathi, S. et al. Large language models reshaping molecular biology and drug development. Chem. Biol. Drug Des. 103(6), e14568 (2024).
Crick, F. Central dogma of molecular biology. Nature 227(5258), 561–563 (1970).
Jaeger, S. et al. Integrating protein-protein interactions and text mining for protein function prediction. BMC Bioinform. 9, S2 (2008).
Krallinger, M., Valencia, A. & Hirschman, L. Linking genes to literature: text mining, information extraction, and retrieval applications for biology. Genome Biol. 9, 1–14 (2008).
Taha, K. & Yoo, P. D. Predicting the functions of a protein from its ability to associate with other molecules. BMC Bioinform. 17, 1–28 (2016).
Al-Aamri, A. et al. Constructing genetic networks using biomedical literature and rare event classification. Sci. Rep. 7(1), 15784 (2017).
Nédellec, C. Learning language in logic-genic interaction extraction challenge. In Proceedings of the 4th Learning Language in Logic Workshop (LLL05). Citeseer (2005).
Ding, J., et al. Mining MEDLINE: abstracts, sentences, or phrases?, In Biocomputing 2002, 326–337. (World Scientific, 2001).
Bunescu, R. et al. Comparative experiments on learning information extractors for proteins and their interactions. Artif. Intell. Med. 33(2), 139–155 (2005).
Pyysalo, S. et al. BioInfer: a corpus for information extraction in the biomedical domain. BMC Bioinform. 8, 1–24 (2007).
Huang, M.-S. et al. Surveying biomedical relation extraction: a critical examination of current datasets and the proposal of a new resource. Brief. Bioinform. 25(3), bbae132 (2024).
Gajendran, S., Manjula, D. & Sugumaran, V. Character level and word level embedding with bidirectional LSTM–Dynamic recurrent neural network for biomedical named entity recognition from literature. J. Biomed. Inform. 112, 103609 (2020).
Rehana, H. et al. Evaluating GPT and BERT models for protein–protein interaction identification in biomedical text. Bioinform. Adv. 4(1), vbae133 (2024).
Guo, Y., et al. Bias in large language models: Origin, evaluation, and mitigation. arXiv preprint arXiv:2411.10915 (2024).
Hua, L. & Quan, C. A shortest dependency path based convolutional neural network for protein-protein relation extraction. Biomed. Res. Int. 2016(1), 8479587 (2016).
Wang, J. et al. Review of large vision models and visual prompt engineering. Meta-Radiology 1, 100047 (2023).
Jahan, I. et al. A comprehensive evaluation of large language models on benchmark biomedical text processing tasks. Comput. Biol. Med. 171, 108189 (2024).
Wang, J., et al. Prompt engineering for healthcare: Methodologies and applications. arXiv preprint arXiv:2304.14670 (2023).
Miao, J. et al. Chain of thought utilization in large language models and application in nephrology. Medicina 60(1), 148 (2024).
Choi, H. S. et al. Developing prompts from large language model for extracting clinical information from pathology and ultrasound reports in breast cancer. Radiat. Oncol. J. 41(3), 209 (2023).
Huang, J. et al. A critical assessment of using ChatGPT for extracting structured data from clinical notes. npj Digital Med. 7(1), 106 (2024).
White, J., et al. A prompt pattern catalog to enhance prompt engineering with Chatgpt. arXiv preprint arXiv:2302.11382. (2023).
Funding
This research was supported by the National Science and Technology Council of Taiwan, grant number NSTC 113-2221-E-038-019-MY3 and NSTC 113-2627-M-A49-002, as well as from the National Health Research Institutes, under grant number NHRI-12A1-PHCO-1823244. This work was also financially supported by the Higher Education Sprout Project, funded by the Ministry of Education (MOE) in Taiwan (grant number DP2-TMU-114-A-04).
Author information
Authors and Affiliations
Contributions
Yung-Chun Chang designed the architecture and supervised the project. Ming-Siang Huang wrote the main manuscript text and refined the data interpretation. Yi-Hsuan Huang collected the data and conducted the experiments. Yi-Hsuan Lin prepared all the tables and figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, YC., Huang, MS., Huang, YH. et al. The influence of prompt engineering on large language models for protein–protein interaction identification in biomedical literature. Sci Rep 15, 15493 (2025). https://doi.org/10.1038/s41598-025-99290-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99290-4
