
Abstract
Given the increasing recognition of frequent drought problems associated with global warming, sunflower (Helianthus annuus L.) has been widely studied as a model plant tolerant to drought and salt stresses. However, there is a lack of information on the systematic identification of the sunflower HaTIFY gene family. In the present study, 21 HaTIFY genes in sunflower were identified and the members of HaTIFY family were divided into four subfamilies, i.e., TIFY, JAZ, ZML and PPD. Gene duplication is a major driver for the expansion of the gene family. Here, three segmental and two tandem duplicated gene pairs were identified via duplication and synteny analysis. Furthermore, five paralogous TIFY gene pairs might have undergone purifying selective pressure during evolution based on Ka/Ks ratio. HaJAZ2/4/5/9/12 from JAZ V subfamily were highly expressed in the majority of tissues. In the analysis of promoter elements of HaTIFYs, more than half of 21 HaTIFY genes contained the drought induction elements. Notably, HaPPD1 and HaPPD4 were significantly upregulated at the early stages of both drought and salt treatments, highlighting their potential roles in enhancing sunflower resistance to abiotic stresses. In conclusion, the HaTIFY gene family plays a crucial role in the positive regulation of sunflower’s response to abiotic stresses, offering key candidate genes for enhancing resistance in sunflower breeding programs.
Similar content being viewed by others


Introduction
Sunflower (Helianthus annuus L.) is the fifth-largest commercial oilseed crop in the world after peanut, soybean, sesame, and rapeseed, and is widely cultured more than 22.9 million hectares on a global scale1. In the context of global warming, more frequent and intense drought and high salinity events in more geographic areas are an alarming issue, resulting in enormous losses in seed yield and oil production2. It has been reported that sunflower is extremely vulnerable to drought and salt stress caused by early spring drought, especially in the young growth stage1. Many reports have shown that sunflowers exposed to abiotic stress have evolved complex signal perception and transduction systems, which is related to the large-scale reprogramming of downstream regulated and co-regulated gene expression by transcription factors (TFs) in response to stress3,4,5. Therefore, the identification of key resistance genes in the sunflower genome and clarifying the potential TFs regulatory mechanisms by bioinformatic and transcriptomic approaches are crucial for enriching resistance gene resources in sunflower to cope with unpredictable and extreme environments.
The plant-specific TIFY proteins were reported to regulate plant development including a highly conserved TIFY domain with the characteristic alpha-alpha–beta fold6,7. The TIFY domain contains 36 amino acids with a conserved motif TIF[F/Y]XG, which is originally known as ZIM (Zinc-finger protein expressed in Inflorescence Meristem). The ZIM domain was initially discovered in the ZIM proteins in Arabidopsis thaliana, and then, to characterize the most conserved amino acid motifs of this family members, Vanholme et al.8 suggested it should be annotated as TIFY. TIFY domain has shuffled or formed new subfamilies by merging with other domains in evolutionary processes7. The TIFY family is typically divided into four clades, i.e., TIFY, ZIM-like (ZML), JAZ (JASMONATE ZIM) and PEAPOD (PPD)8. The members of TIFY subfamily are characterized by one TIFY domain, whereas the other subfamilies possess multiple domains. For instance, the JAZ subfamily proteins contain highly conserved TIFY and Jas (CCT_2) domains9. Previous reports showed that Jas motif is specific of the JAZ subfamily, serving as a negative feedback regulator in the jasmonic acid (JA) signaling pathway via mediating hormone-dependent interactions between JAZ proteins and F-box protein COI1, thereby contributing greatly to environmental adaptation6,9,10. In addition to TIFY domain, the ZML subfamily members are characterized by CCT domain and C2C2-GATA zinc-finger domain10, whereas the PPD subfamily proteins consist of characteristic N-terminal PPD domain and partially Jas domain defecting the conserved Proline-Tyrosine (PY) residue at the C-terminus11,12.
To date, TIFY genes have been identified across the multiple varieties, including 26 in Zea mays12, 18 in Fagopyrum tataricum13, 16 in Dendrobium huoshanense14, and 77 in Brassica napus L.15 and other plants. Indeed, the multiple regulatory effects of the TIFY gene family on plant growth, development16 and abiotic resistance17,18 indicate that this gene family contains a wealth of valuable gene resources. Previous studies across various species suggested that TIFY members have also been showed to engage in increasing plant tolerance to abiotic stresses such as drought, high salinity, or alkali stress15,19. For example, some researchers have highlighted the significant positive role of ClJAZ1/7 genes in watermelon under drought and salinity conditions20. In rice, overexpression of OsJAZ8 has been showed to improve drought resistance21. Additionally, the overexpression of FtJAZ10/12 enhanced the heat stress tolerance in Fagopyrum tataricum13. According to a recent survey, EjJAZ2/4/9 responded to salt stress, which might enhance salinity tolerance through the JA pathway in Eriobotrya japonica18. Moreover, except for the JAZ subfamily proteins, it has been reported that DhTIFY1, PPD1, and ZLM1 had prompt and strong responses in Dendrobium huoshanense14. Previous studies reported that the expression of sunflower HaJAZ gene tended to downregulate under drought and salt stress patterns5. However, extensive studies of other HaTIFY family members and their function involved in drought and salt tolerance are still limited. It is, therefore, hypothesized that the TIFY gene family members in sunflower are involved in regulating the responses to drought and salt stresses.
In the present study, we identified the TIFY gene family from the sunflower genome and conducted an extensive analysis of the gene structure and conserved motifs, physicochemical properties, conserved domain, promoter cis-elements to shed new light on HaTIFY genes evolutionary relationships and potential functions. Subsequently, the dynamic expression levels of key HaTIFY genes involved in drought and salt stress response were analyzed by RNA-seq and qRT-PCR validation. This study provides a basic foundation for further exploration of the functions of HaTIFY genes in resistance to abiotic stresses and may provide insights into the regulatory mechanisms of sunflower resistance.
Results
Identification and characterization of TIFY genes in Helianthus annuus
A total of 21 HaTIFY genes were identified by screening the sunflower protein database. The HaTIFY proteins can be further categorized into four subfamilies (namely JAZ, PPD, ZML, and TIFY) according to the motifs and domains composition. HaTIFY1 and HaTIFY2 with one TIFY domain belonged to the TIFY subfamily, and the remaining 19 TIFY proteins possessed TIFY and Jas domains. HaJAZ1–HaJAZ12 were grouped as members of the JAZ subfamily; whereas the four proteins without the conserved PY amino acids in the Jas motif were named HaPPD1–HaPPD4. Three protein sequences contained GATA zinc-finger and CCT domains, which is characteristic of ZML subfamily (designated as HaZML1 to HaZML3).
The essential characteristics of the sunflower HaTIFY proteins were subsequently presented in Table 1. Our analysis revealed that the length of CDS exhibited variation, with values spanning from 366 (HaPPD3) to 2254 (HaZML1). Correspondingly, the largest protein (HaZML1) contained 738 amino acids, whereas HaPPD3 was the smallest with 119 amino acids. The molecular weight ranged from 12.95 kDa (HaPPD3) to 81.37 kDa (HaZML1) in these proteins. The fat index of the HaTIFYs were 60.11 to 82.50. Moreover, the average instability index of the HaTIFY proteins was 54.54, indicating that these proteins may be relatively unstable. The values of theoretical pI in 14 out of 21 proteins were more than 7, while the other seven proteins were acidic (pI < 7). All proteins with negative GRVAY values displayed hydrophilic properties because of negative scores. Subcellular localization prediction revealed that nine TIFY proteins localized in the nucleus and chloroplasts, respectively, two in the peroxisomal, and one in the cytoplasm. This result indicated that HaTIFY genes mainly function in the nucleus and chloroplasts, and they may be involved in multiple regulatory pathways. In addition, irregular curling was the main secondary structure mode, the order of proportion was irregular curling > α-helix > extended chain > β-folding. The results of tertiary structures predictions of HaTIFY proteins indicated that the tertiary structures of three pairs of proteins (e.g., HaJAZ3/11, HaPPD1/4 and HaJAZ7/8) were highly similar (Fig. S1).
Phylogenetic analyses and classification of HaTIFY gene family
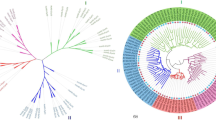
Based on the full length amino acid sequences of 59 TIFY proteins among three species (21 TIFYs in Helianthus annuus, 18 in Arabidopsis thaliana and 20 in Oryza sativa), a phylogenetic tree was constructed using the NJ method (Fig. 1). The result showed that the 59 TIFYs across these species were classified into four subfamilies: JAZ, ZML, TIFY, and PPD. For each group, the number of TIFY proteins varied. Specifically, the JAZ subfamily was the largest group with 37 differential members. This result indicated that the extensive evolution history may be prone to appear in JAZ subfamily to adapt multiple stressors. The JAZ subfamily could be further clustered into five subgroups (JAZ I–JAZ V), in which JAZ I, JAZ II, and JAZ V were universally present in three species. Notably, the JAZ IV subgroup only contained seven OsJAZ proteins, while other species did not include any JAZ proteins in this subgroup. In addition, PPD members only included dicotyledonous plants, indicating that the PPD subgroup might be occurred after differentiation in monocotyledonous plants. The difference in the number of clades members between sunflower and rice was probably that they undergone expansion or contraction since the differentiation from monocots and eudicots.

Phylogenetic analysis of TIFY proteins from Helianthus annuus (Ha), Arabidopsis thaliana (At), Oryza sativa (Os). Neighbor-Joining phylogeny of 59 TIFY proteins of the three species, was determined by MEGA 7.0 program with 1000 bootstrap replicates.
Gene structure and conserved motif analysis of HaTIFY gene family
The exon–intron structure could also be used to provide additional evidence for understanding HaTIFY genes structure evolution to further support phylogenetic grouping. The results revealed that the loss or gain phenomenon of introns or exons was identified in several branches of HaTIFY genes (Fig. 2A). Specifically, HaPPD3 consisted of 4 exons, while others in PPD subfamily contained 7 exons, indicating that exons may be lost during evolution. In addition, HaJAZ2/7/8/9/12 were composed of the same number of exons with two, and HaPPD1/2/4 with 7, and the exon length of these genes displayed almost the same. These TIFY genes might be the product of repeated events. All HaTIFY genes possessed intron. The number of introns belonging to the PPD subfamily members was more than that of the other subfamilies; whereas the members of JAZ V subfamily (only 1–3 introns) were least. Our analysis revealed that there is a strong correlation between the phylogeny of TIFY gene family members and the exon–intron structure, and the genes of the same subfamily usually have similar gene structures.

Phylogenetic relationships, gene structure and motifs of HaTIFYs in Helianthus annuus. (A) The exon–intron structure of HaTIFYs. (B) Distribution of the conserved motifs in HaTIFY proteins. The scale bar indicates 2000 aa. (C) The sequences of TIFY and Jas domains.
The online website MEME (http://meme-suite.org/) was used to annotate the conserved domain in HaTIFYs. A total of 6 motifs (named motif 1 to motif 6) were identified from the conserved regions in 21 HaTIFYs (Fig. 2B). Although the number of amino acids of TIFY proteins varied, the clustered HaTIFY proteins contained the same number of amino acids and similar conserved domain distribution, such as HaPPD1/4 and HaZML2/3. It was observed that each HaTIFY member contained 1–5 motifs. It is worth mentioning that the members of ZML group generally contained most motifs, while members of TIFY clade only contained one motifs. However, some of which were unique to specific subgroups, such as motif 3 limited to JAZ V, motif 5 distributed only in ZML subfamily; motif 4 was showed in PPD and ZML subgroups. This finding conforms to phylogeny analysis, and they explain together that the members of clustered at the one branch generally possessed a similar conserved sequence motifs. Furthermore, TIFY and Jas domains were performed by drawing their sequence logos. As presented in Fig. 2C, TIFY domain had a conserved TIF[F/Y]XG motif and amino acid sites Val-10 and Val-12. Jas domain was highly conserved, particularly in location sites 7, 10, 11, 14, 17, 18, 19, and 21.
Analysis of cis-acting element in HaTIFY gene family
The cis-elements in the promoter region of TIFY family members were shown in Fig. 3, based on the PlantCARE database. The phytohormone elements were found widely in each HaTIFY gene promoter (Fig. 3A). HaPPD4 had up to 15 phytohormone responsive elements; HaPPD2 and HaJAZ11 had up to 11 and 10 elements, respectively. Further, the relatively abundant cis-elements involved hormone signaling pathways were ABRE and TCA-motif, TGACG-motif. ABRE was present in almost all HaTIFYs promoters (Fig. 3B, C). Another important finding was that more than two-thirds of HaTIFY genes contained TGACG-motif element, which is known to modulate responses to various necrotizing pathogens. This finding underscores the potential involvement of these genes in the JA signaling pathway. The most numerous and widely distributed in stress-responsive elements was ARE, followed by MBS. Interestingly, HaJAZ10, HaPPD4 and HaPPD1 had more drought-induced elements than others, which infers that they may also have the same function in response to drought stress. Conversely, a relatively small number of genes were found to contain TC-rich repeats cis-elements.

Prediction of cis-acting elements in HaTIFYs promoters. (A) Each HaTIFY contains the number of cis-acting elements detected which were divided four types. (B) The number of different elements of each HaTIFY gene in four types of elements. (C) Visualization of four types of elements in HaTIFYs promoters by TBtools, including position, kind and quantity of elements.
Chromosome localization and duplication analysis
The detailed chromosome localization of 21 HaTIFY genes linked to tandem replications events were plotted in Fig. S2. They were mapped irregularly across 12 chromosomes. Among them, Chr4, Chr10, and Chr15 contained the largest number of HaTIFY genes (3 each), followed by Chr6, Chr12, and Chr16 with two HaTIFYs, respectively. Chr1, Chr2, Chr8, Chr11, Chr13, and Chr14 had the least gene distribution, with only one gene each. It is interesting to note that more than half of HaTIFY genes were close to chromosome terminal. Furthermore, the duplication events of the sunflower HaTIFY genes showed that three segmental (HaJAZ2/4, HaJAZ3/11, and HaPPD3/4) and two tandem duplicated gene pairs (HaJAZ12/HaZML1 and HaZML2/3) were identified (Fig. S3). This finding had illustrated the importance of both whole-genome duplication and segmental duplication events in the evolution and expansion of the HaTIFY genes in sunflower. It is worth noting that as a pair of segmental duplicated genes, HaJAZ3/11 exhibited a highly comparable tertiary structure (Fig. S1).
Due to the chromosomal localization lacking of the nature of TIFY genes origin, the gene duplication analysis between sunflower and two model plants (Arabidopsis thaliana and Oryza sativa) were constructed (Fig. S4). Notably, sunflower and Arabidopsis have three pairs of homologous genes, while there are two homologous gene pairs between between sunflower and rice. This is consistent with the fact that both belong to dicotyledonous plants. HaJAZ2 exhibited collinear relationships in both monocotyledons and dicotyledons, indicating that this gene might play crucial roles in evolution. In addition, the selection pressure of homologous TIFY genes was assessed using the ratio of Ka/Ks. The Ka/Ks ratios of the five paralogous pairs were less than 1 (HaJAZ2/AT1G72450.1, HaJAZ8/AT1G72450.1, HaPPD2/AT1G70700.3, HaJAZ2/Os03t0180800-01, and HaJAZ4/Os09t0439200-01) (Table S1). The analysis demonstrated that HaTIFY genes were primarily constrained by purifying selective pressure during their evolutionary development.
Protein–protein interaction (PPI) network
In the study of the interactions between HaTIFY proteins and their potential partners, we constructed protein–protein interaction network based on STRING database (Fig. 4). A total of 17 HaTIFY proteins were presented in the PPI network, including all proteins of the JAZ V subgroup. However, there were only one or two members in other subgroups. Notably, the interaction between all HaJAZ proteins and COI1 could have occurred as a result of JAZ proteins as a transcriptional repressor, by inhibiting downstream gene expression in the JA pathway. Further, the JAZ V subfamily also displayed multiple interactions with MYC2 to inhibit the JA signaling pathway, likely due to the members of JAZ group with the C-terminal jasmonic acid (JA)-related (Jas, CCT_2) domain. Regarding the results, the members of PPD and JAZ V subfamily had more interaction partners, which may play a remarkable role in coordinating hormone signals. The above results reveal the complex network of protein level interactions, and predict their potential functions.

Interaction network of TIFY proteins in sunflower. The green lines represent the interaction between COI1 and HaTIFY proteins; and the blue lines represent the interaction between MYC2 and HaTIFY proteins. The network was constructed using the STRING tool and Cytoscape, and confidence score is 0.4.
Microarray analysis of expression profiles
Based on previous RNA-seq data, the expression patterns of sunflower TIFY gene family in ten main tissues have varying profiles of expression. As showed in Fig. 5, members of the HaTIFY family into four clusters (I to IV) by cluster analysis, according to the varied expression levels. Compared with other groups, HaPPD2 and HaJAZ4/5/12 belonged to the group I showed strong expression in numerous organs. More than one-third of the HaTIFY genes were highly expressed in the majority of tissues, with HaJAZ genes accounting for 75% of them. For example, the expression levels of HaJAZ2/4/5/9/12, members of the JAZ V subfamily, were particularly prominent, in bract, corolla, ligule, ovary, stamen, stem, and style. These results may reveal the important roles of these genes in flower development of sunflower. In contrast, the expression levels of some HaJAZ genes (such as HaJAZ6/10/11) were low or not expressed.

Expression profiles of HaTIFY genes in different tissues and organs. The normalized log2(FPKM) was used to create a heatmap. The indicator ranges in the upper right corner represent the range of the values after normalization. Detailed information is showed in Table S2.
Expression patterns of TIFY genes under abiotic stresses
The transcriptome data was employed to further verify the expression of HaTIFY genes under drought (15% PEG6000) and salt (200 mM NaCl) treatments by extracted the total RNAs from sunflower leaves (Fig. 6). Among them, HaJAZ2/4/5/12, HaPPD1/4, and HaTIFY2 were highly expressed in the control and different stress environments. Moreover, HaJAZ5 and HaPPD1/4 showed a similar up–down-down pattern under both drought and salt stresses, respectively. On the contrary, the expression values of HaJAZ6/10/11, HaPPD2/3, and HaZML1/2/3 were close to or equal to 0. As expected, the homologous genes (such as HaZML2/3 or HaPPD1/4) shared similar expression patterns under the same stress. In general, most of HaTIFY genes with a tendency to express in response to stimuli were from the JAZ V subfamily, which may reveal the potential roles of the JAZ V subfamily in dealing with drought or salt stress.

Expression patterns of HaTIFY genes following 200 mM NaCl and 15% PEG6000 treatments at different times. The normalized log2(FPKM) was used to create a heatmap. The indicator ranges in the upper right corner represent the range of the values after normalization. Detailed information is showed in Table S3.
Based on transcriptome data, six HaTIFY genes (HaJAZ2/5/12, HaPPD1/4 and HaTIFY2) were selected to further validate by qRT-PCR analysis. Overall, the expression levels of the same gene in different stress treatments and stages were found to be highly consistent with transcriptome data (Fig. 7). There were statistically significant upward or downward trends of their expression levels at the initial stage of stresses (P < 0.05). For example, HaJAZ2/5, HaPPD1/4, and HaTIFY2 were significantly induced by drought at 3 h post treatments. Additionally, the expression levels of HaJAZ5, HaPPD1/4, and HaTIFY2 were shown to be significantly up-regulated after 6 h of salt condition compared to the CK group, and then gradually decreased with the increased duration of salt stress.

qRT-PCR analysis of differences in HaTIFYs expression levels under drought (A) and salt (B) stresses. (A) CK, D3, D6 and D9 represent the expression levels of HaTIFYs at 0, 3, 6 and 9 h under drought stress condition in X-axis; (B) CK, S6, S12 and S24 represent in 0, 6, 12 and 24 h exposed to salt treatment. Statistically significant differences were analyzed by Duncan’s multiple-range test. Different lowercase letters above the bars indicate a significant difference between these treatments (p < 0.05).
The expression levels of HaJAZ2 and HaJAZ12 experienced a massive decline during the whole process of salt condition, but the opposite trends appeared in drought condition. Differing from the above two genes, a similar expression pattern with increasing at first and then decreasing was observed the expression levels of other four genes (HaJAZ5, HaPPD1/4 and HaTIFY2) at different periods following the two types of stress. Interestingly, the extent of HaPPD1 and HaPPD4 upregulation is higher compared to other genes, indicating that they have potentially function roles in drought and high salinity tolerance. For instance, the peak expression levels of HaPPD1 and HaPPD4 were significantly upregulated under drought and salt treatments compared to CK. This phenomenon may be related to the presence of numerous drought induction elements in the promoter of HaPPD1 and HaPPD4 genes. In summary, these results provide useful clues for specific mechanism that HaTIFY genes involved in the response to several abiotic stresses tolerance, which is an intriguing issue to deserve further study.
Discussion
Numerous studies have documented that TIFY genes encode plant-specific transcription factors, which not only participate in the regulation of plant growth and development9,22, but also play an important role in abiotic/biotic stress responses23,24,25,26,27,28. With an increasing number of genome sequences being released, TIFY gene family has been widely identified in oilseed crops, including Brassica napus15, Zea mays12, Juglans regia28 and Glycine soja17. Sunflower has been well known for its outstanding resistance to environmental stresses2, while the functional characterizations of TIFY genes in sunflower have not been reported yet. Accordingly, we conducted an identification of the TIFY gene family in the sunflower genome and investigated their structure and expression levels with a focus on identifying sensitive members responsive to abiotic stresses.
In the present study, 21 HaTIFY genes unevenly distributed on 12 chromosomes were identified, and HaTIFY proteins were divided into four subfamilies, namely JAZ, PPD, ZML, and TIFY (Fig. 1), which were consistent with the classification methods used in Juglans regia28, Fagopyrum tataricum13, and Solanum lycopersicum12. In agreement with our findings, other published results showed that there is no PPD subfamily in monocotyledonous plants29. It might be linked with the fact that neofunctionalization or loss of function driven by differences in selective constraints between monocots and dicots29. Previous study has provided evidence that the PPD subfamily was formed from the merging of TIFY domain and PPD domain before vascular plant formation30. It was worth noting that HaTIFYs within the same subfamily typically had similar conserved motif and exon–intron structure, as shown by gene structure and phylogenetic analysis (Fig. 1 and Fig. 2). Similar observations were reported in Arabidopsis and other plants, that a strong correlation existed between phylogenetics and exon–intron structure23,31.
Furthermore, the genes from the same subfamily have similar gene structures based on the information contained in expression pattern and phylogenetic analysis (Fig. 1 and Fig. 7). For example, the genes located in the closest phylogenetic branches, such as HaPPD1/4 and HaJAZ2/12 with similar tendency under drought and salt stresses, respectively. Moreover, HaJAZ11 and HaJAZ6 were clustered, and these two genes both tended to be not or less expressed illustrates, presumably due to pseudogenization5. Among groups JAZ I–JAZ V, JAZ V subfamily had the maximum HaJAZs members, 8 HaJAZs were clustered, possibly leading to species-specific adaptation. It has been reported that OsJAZ8 was overexpressed in leaves of seedlings of rice to eventually enhanced salt stress tolerance21. Meanwhile, AtJAZ1 can directly interact with the negative regulator of ABA signaling to regulate the response to salt and drought treatments32. Considering this aspect, OsJAZ8, AtJAZ1 and 8 HaJAZ proteins from one clade might share a similar biological function. As expected, the majority of JAZ V subfamily members (i.e., HaJAZ2/4/5/9/12) were highly expressed in floral tissues, including bract, corolla, ligule, ovary and style (Fig. 5), which indicates that their functions in flower development of sunflower are indispensable33. Furthermore, our findings showed that the expression levels of HaJAZ2/4/5/12 tended to be highly were observed under drought and salt stresses. This result suggests that the genes in JAZ V subfamily might play a more important role than other subfamilies in responding to abiotic stresses.
Tandem and segmental duplications are widely recognized as key mechanisms in plant evolution, gene family expansion, and functional differentiation, particularly in response to abiotic stress34. In our study, three pairs of segmental duplication genes from the JAZ subfamily (HaJAZ2/4, HaJAZ3/11, and HaPPD3/4), as well as two tandem replication events (HaJAZ12/ZML1 and HaZML2/3) (Fig. S3), revealing that these duplication events might contribute to the evolution and expansion of the HaTIFY gene family31. Researchers have showed that the functions and expression patterns of segmentally duplicated genes were similar35. This viewpoint only partially supports by our observation that the expression level of HaJAZ2 and HaJAZ4, which underwent segmental duplications, showed similar up-and-down expression patterns during drought stress (Fig. 6). This result suggested that HaJAZ2 and HaJAZ4 might have similar biological functions. However, the opposite trend was observed in kiwifruit29 and sunflower5 that repeated the expression patterns of segmentally duplicated gene pairs were not completely consistent, resulting in neofunctionalization to regulating abiotic tolerance. Our result showed that HaJAZ genes were conserved in sequence but varied in expression among duplicated genes. Specifically, HaJAZ11 tended to be not expressed in most of tissues, while HaJAZ3 tended to be divergently expressed among tissues or stress treatments (Fig. 5 and Fig. 6). Meanwhile, despite the fact that HaPPD3/4 was duplicated, the expression patterns of them varied in response to drought and salt stresses. Accordingly, we imply that duplicated HaTIFY genes may undergo neofunctionalization to breakdown functional redundancy in the adaptation to abiotic stresses. Furthermore, the inter-species collinearity analysis showed that there were more pairs of homologous genes in sunflowers and Arabidopsis, compared those existed between sunflowers and rice (Fig. S4). It is possible due to the difference between monocotyledonous and dicotyledonous plants33. The evolutionary selection pressure analysis also showed that five duplicated paralogous gene pairs were strongly constrained by purification selection as Ka/Ks ratio was less than 1 (Table S1), which might have resulted in preserved functionality or pseudogenization12,33.
It is important to mention that MYC2 as the direct target of JA receptor COI1 genes, interacts with the C-terminal domain of JAZ protein and then induce a set of JA responsive genes after degradation of JAZ protein36. Increasing evidence supports the argument that JAZ proteins through bind to bHLH TFs repressed the expression of the early JA-responsive genes36,37. Our results obtained from the PPI analysis align with above findings, in which the multiple proteins in JAZ V subfamily interacting with COI1 and MYC2 may play an important role in the JA pathway (Fig. 4). Furthermore, with the increase in time of salinity stress, the expression level of HaJAZ2/12 continuous significant declined (Fig. 7), which can be clearly demonstrated the fact that the JAZ gene as a “repressor” response to environmental stresses in sunflower5. Subsequently, the expression patterns of six differentially expressed genes in the continuous drought or salt environment were quantified by qRT-PCR (Fig. 7). Noteworthy, HaJAZ5, HaPPD1/4 and HaTIFY2 exhibited the similar expression patterns that initially increased and then declined, whether exposed to drought or salt treatment. Interestingly, HaPPD1 and HaPPD4 were significantly upregulated at the early stages of both drought and salt treatments, highlighting their potential role in enhancing sunflower resistance to abiotic stresses.
In summary, 21 HaTIFY genes were identified in sunflower and HaTIFY proteins were divided into four subfamilies, namely JAZ, PPD, ZML, and TIFY. The proteins from the same subfamily were similar in conserved motifs and domains. Segmental and tandem duplication events contributed to the expansion of HaTIFY gene family. Phylogenetic analysis and the synteny comparisons between sunflower and other plant species (Arabidopsis thaliana and Oryza sativa) uncovered the vital evolution information of TIFY gene family. Furthermore, more than half of 21 HaTIFY genes contained the drought induction elements. Subsequently, RNA-seq and qRT-PCR analysis showed that most members of PPD and JAZ V groups had relatively high expression levels. HaPPD1 and HaPPD4 tend to be sensitive to drought and salt stresses. In conclusion, the HaTIFY gene family plays a crucial role in the positive regulation of sunflower’s response to abiotic stresses, offering key candidate genes for enhancing resistance in sunflower breeding programs.
Materials and methods
Identification and analysis of HaTIFY gene family
To identify the members of TIFY gene family in sunflower genome, the whole-genome, protein sequence, and annotation files of sunflower genome were downloaded from the Sunflower Genome Sequence database (http://www.sunflowergenome.org). The Hidden Markov Model (HMM) profile of the conserved functional domain of TIFY (PF06200) was obtained from the Pfam Database v34.0 (http://pfam.xfam.org/). Firstly, the HMM profile was used to screen the sunflower proteomes to obtain the all potential TIFY genes in sunflower using the hmmsearch software in the HMMER package v3.029. After removing redundant and incomplete sequences (E-value < 1e-5), the conserved domain architectures of the acquired sequences were further validated using the online SMART tool (http://smart.embl-heidelberg.de/). The amino acid sequences containing the conservative TIFY domain were identified as the candidate TIFY members, while proteins without the typical functional domain of TIFY were manually excluded31.
Subsequently, the properties of the TIFY proteins, including theoretical isoelectric point (pI), molecular weight (Mw), and grand average of hydropathy (GRAVY), were analyzed using the ExPASy server (https://web.expasy.org/protparam/). The predictive analysis of the subcellular localization of sunflower TIFY protein was predicted by the ProtComp v9.0 tool (http://www.softberry.com/)15. The secondary structures and tertiary structures were performed via SOPMA web-server (https://npsa.lyon.inserm.fr/) and SWISS-MODEL server (https://swissmodel.expasy.org/), respectively. Next, the STRING web-server (https://cn.string-db.org/) and Cytoscape_v3.10.1 were used for protein–protein interaction network analysis.
Phylogenetic analysis, motif recognition, and promoter cis-elements
We compared 59 TIFY proteins including 18 protein sequences from Arabidopsis thaliana, 20 protein sequences from Oryza sativa and 21 protein sequences from Helianthus annuus. A rootless phylogenetic tree was constructed using MEGA 7.0 software with the neighbor-joining (NJ) method and setting bootstrap analysis with 1000 bootstrap replicates38. TIFY protein information in Arabidopsis thaliana (TAIR, https://www.arabidopsis.org/) and Oryza sativa (TIGR, http://rice.plantbiology.msu.edu/) was available at a publicly-accessibly website, respectively4.
To further understand the structural diversity of TIFY genes in sunflower, the Gene Structure Display Server 2.0 (GSDS 2.0) (http://gsds.cbi.pku.edu.cn/) was used to determine their intron–exon organizations. The conserved motifs of full-length TIFY proteins were identified by MEME Suite v5.4.1 (https://meme-suite.org/meme/), using the default parameters and set the conserved motif number to 6. The promoter sequences of HaTIFY genes were obtained in Phytozome v13 database (https://phytozome-next.jgi.doe.gov/pz/portal.html). Further, 2000 bp upstream of CDS was extracted each HaTIFY promoter. The cis-acting regulatory elements of the plant promoter sequence were predicted through PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) and visualized via TBtools26.
Chromosomal distribution, gene duplication, and synteny analysis
The chromosomal location and potential tandem duplications of the HaTIFY genes were analyzed using TBtools39. The entire sunflower genome sequence was retrieved from the Genome Database for Sunflower and all the predicted TIFY genes were mapped onto the corresponding chromosomes to visualize their distribution patterns. Genes were considered to be duplicated when there was more than 85% identity at their nucleotide sequences, which were manually marked on the chromosomal location12. To analyze the duplication events of the TIFY genes, we used MCScanX in TBtools. The selection pressure of the duplicated pairs and dividing of homologous TIFY genes was estimated based on the synonymous (Ks) and non-synonymous (Ka) rate among the gene pairs using the KaKs_Calculator 2.0 software40.
Expression profiles of HaTIFY genes in sunflower tissues and organs
The transcriptome data of sunflower TIFY genes in different tissues and organs were obtained from the Sunflower Genome Database (https://www.sunflowergenome.org/). The expression data of 10 tissues and organs were selected, including bract, corolla, leaf, ligule, ovary, pollen, seed, stamen, stem, and style. All the relative expression levels of HaTIFY genes were normalized as FPKM values (Fragments Per Kilobase of transcript per Million mapped reads). Finally, the heatmaps of the expression patterns of HaTIFYs were created by TBtools based on the normalized log2(FPKM)40.
Plant materials, stress treatments and RNA-seq
The sunflower material used in this study was local cultivated variety (NWS-567) in Taiyuan, Shanxi Province, China. This material was identified in 2018 by provincial agricultural regulatory department in accordance with relevant institutional guidelines and regulations. The plants were grown in the mixed substrate of fertile soil and vermiculite (1:1 v/v) in plastic pots (170 × 220 mm) with in a 16 h light/8 h dark greenhouse located in Taiyuan University (Taiyuan, Shaanxi province, China) under a condition of 28℃ and the relative humidity of 65%. Four-leaf-old plantlets were treated with 200 mM NaCl (sampled at 0, 6, 12 and 24 h) and 15% polyethylene glycol (PEG) 6000 (collected at 0, 3, 6 and 9 h). Three biological replicates were used for each sample. Afterwards, the sunflower leaves samples at different durations of stimuli were quick-frozen in liquid nitrogen and stored at -80℃ for RNA extraction and RNA-seq.
Total RNA was extracted using OmniPlant RNA Kit (DNase I) (CW2598S, Kangwei Century Biotechnology Co., Ltd, Beijing, China). Sequencing was performed at the illumina Novseq Xplus (Illumina Biotech Co, Ltd., San Diego, CA, USA). Trim the original paired end readings and quality control were conducted using fastp v0.23.4 software. Then, the clean reads were localized to reference genome with orientation mode using HISAT2 v2.2.1 software5. The mapping results were subjected to String Tie to assemble transcripts in each sample25. The heatmap was generated using the log2 (FPKM) through TBtools.
Quantitative real-time PCR (qRT-PCR) analysis
Select genes with high expression levels and significant changes for qRT-PCR analysis. First-strand cDNA synthesis was performed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Wuhan Servicebio Technology Co, Ltd.). The primer sequences listed in Table S4. qRT-PCR was conducted using Universal Blue SYBR Green qPCR Master Mix, and the parameters were as follows: pre-denaturation at 95℃ for 30 s, 1 cycle; annealing at 95℃ for 15 s, 60℃ extension for 30 s, 40 cycles. The ACT2 gene was used as reference41. Three biological and technical repetitions were conducted for each sample. The relative expression was calculated by 2−△△Ct. The SPSS 12.0 was used to analyze the statistical significance.
Data availability
The datasets of this article are included in the current article and its supplementary files, and they can be available from the corresponding author on reasonable request. The Sunflower Genome Sequence database (http://www.sunflowergenome.org). The Pfam database (http://pfam.xfam.org/). The SMART online tool (http://smart.embl-heidelberg.de/). The ExPASy server (https://web.expasy.org/protparam/). The ProtComp v9.0 tool (http://www.softberry.com). The SOPMA web-server (https://npsa.lyon.inserm.fr/). The SWISS-MODEL server (https://swissmodel.expasy.org/). The Arabidopsis thaliana Information Resource (TAIR) database (https://www.arabidopsis.org/). The Oryza sativa Information Resource (TIGR) database (http://rice.plantbiology.msu.edu/). The GSDS 2.0 (http://gsds.cbi.pku.edu.cn/). The MEME Suite v5.4.1 (https://meme-suite.org/meme/). The Phytozome v13 database (https://phytozome-next.jgi.doe.gov/pz/portal.html). The PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).
References
Li, W. et al. A salt tolerance evaluation method for sunflower (Helianthus annuus L.) at the seed germination stage. Sci. Rep. 10, 10626 (2020).
Khalifani, S. et al. Comparison of influential input variables in the deep learning modeling of sunflower grain yields under normal and drought stress conditions. Field Crops Res. 303, 109145 (2023).
Liu, A. et al. Phylogenetic analysis and transcriptional profiling of WRKY genes in sunflower (Helianthus annuus L.): Genetic diversity and their responses to different biotic and abiotic stresses. Ind. Crops Prod. 148, 112268 (2020).
Li, L. et al. Genome-wide identification of the TIFY family in Salvia miltiorrhiza reveals that SmJAZ3 interacts with SmWD40-170, a relevant protein that modulates secondary metabolism and development. Front. Plant Sci. 18, 630424 (2021).
Song, H. et al. Phylogenetic analysis and expression profiles of jasmonate ZIM-domain gene family provide insight into abiotic stress resistance in sunflower. Front. Plant Sci. 13, 1010404 (2022).
Chung, H. & Howe, G. A critical role for the TIFY motif in repression of jasmonate signaling by a stabilized splice variant of the JASMONATE ZIM-domain protein JAZ10 in Arabidopsis. Plant Cell 21, 131–145 (2009).
Singh, P. & Mukhopadhyay, K. Comprehensive molecular dissection of TIFY transcription factors reveal their dynamic responses to biotic and abiotic stress in wheat (Triticum aestivum L.). Sci. Rep. 11, 9739 (2021).
Vanholme, B. et al. The TIFY family previously known as ZIM. Trends Plant Sci. 12, 239–244 (2007).
Zhou, S. L. et al. Genome-wide identification of JAZ gene family in sugarcane and function analysis of ScJAZ1/2 in drought stress response and flowering regulation. Plant Physiol. Biochem. 210, 108577 (2024).
Song, H. et al. Genome-wide identification, expression pattern and subcellular localization analysis of the JAZ gene family in Toona ciliata. Ind. Crops Prod. 178, 114582 (2022).
Bai, Y. et al. Origin and evolutionary analysis of the plant-specific TIFY transcription factor family. Genome 98, 128–136 (2011).
Heidari, P. et al. New insights into structure and function of TIFY genes in Zea mays and Solanum lycopersicum: A genome-wide comprehensive analysis. Front. Genet. 12, 657970 (2021).
Zhao, Z. et al. Genome-wide identification and functional analysis of the TIFY family genes in response to abiotic stresses and hormone treatments in Tartary Buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 24, 10916 (2023).
Li, G. et al. Genome-wide identification and expression analysis of TIFY genes under MeJA, cold and PEG-induced drought stress treatment in Dendrobium huoshanense. Physiol. Mol. Biol. Plants 30, 527–542 (2024).
He, X. et al. Genome-wide identification and functional analysis of the TIFY gene family in the response to multiple stresses in Brassica napus L.. BMC Genom. 21, 1–13 (2020).
Guan, Y. et al. Genome-Wide identification and expression pattern analysis of TIFY family genes reveal their potential roles in Phalaenopsis aphrodite flower opening. Int. J. Mol. Sci. 25, 5422 (2024).
Zhu, D. et al. Identification of wild soybean (Glycine soja) TIFY family genes and their expression profiling analysis under bicarbonate stress. Plant Cell Rep. 32, 263–272 (2013).
Li, X. et al. Genome-wide identification and expression analysis of the Eriobotrya japonica TIFY gene family reveals its functional diversity under abiotic stress conditions. BMC Genom. 25, 468 (2024).
Zhao, C. et al. Overexpression of a TIFY family gene, GsJAZ2, exhibits enhanced tolerance to alkaline stress in soybean. Mol. Breed 40, 1–13 (2020).
Yang, Y. et al. Comprehensive analysis of TIFY transcription factors and their expression profiles under jasmonic acid and abiotic stresses in watermelon. Int. J. Genome 1, 6813086 (2019).
Peethambaran, P. K. et al. Salt-inducible expression of OsJAZ8 improves resilience against salt-stress. BMC Plant Biol. 18, 1–15 (2018).
Hakata, M. et al. Overexpression of TIFY genes promotes plant growth in rice through jasmonate signaling. Biosci. Biotechnol. Biochem. 81, 906–913 (2017).
Dai, Z. et al. Genome-wide identification of TIFY genes and their response to various pathogen infections in cucumber (Cucumis sativus L.). Sci. Hortic. 295, 110814 (2022).
Zhang, X. et al. Genome-wide identification of the TIFY gene family and their expression profiles in response to biotic and abiotic stresses in tea plants (Camellia sinensis). Int. J. Mol. Sci. 21, 8316 (2020).
Lian, C. et al. Genome-wide identification, characterization and expression pattern analysis of TIFY family members in Artemisia argyi. BMC Genom. 25(1), 925–925 (2024).
Zheng, L. et al. Genome-wide identification and expression of TIFY family in cassava (Manihot esculenta Crantz). Front. Plant Sci. 13, 1017840 (2022).
Zhang, C. et al. ZmTIFY16, a novel maize TIFY transcription factor gene, promotes root growth and development and enhances drought and salt tolerance in Arabidopsis and Zea mays. Plant Growth Regul. 100, 149–160 (2023).
Liu, X. et al. Identification of TIFY gene family in walnut and analysis of its expression under abiotic stresses. BMC Genom. 23, 190 (2022).
Tao, J. et al. Genome-wide identification and characterization of the TIFY gene family in kiwifruit. BMC Genom. 23, 179 (2022).
Zhang, H. F. et al. Genome-wide identification of the TIFY gene family in tobacco and expression analysis in response to Ralstonia solanacearum infection. Genomics 116, 110823–110823 (2024).
Liu, K. et al. Pan-Genome analysis of TIFY gene family and functional analysis of CsTIFY genes in Cucumber. Int. J. Mol. Sci. 22, 185 (2023).
Luo, X. et al. ABA signaling is negatively regulated by GbWRKY1 through JAZ1 and ABI1 to affect salt and drought tolerance. Plant Cell Rep. 39, 181–194 (2020).
Sun, F. et al. Genome-wide identification of the TIFY gene family in Brassiceae and its potential association with heavy metal stress in rapeseed. Plants 11, 667 (2022).
Magadum, S. et al. Gene duplication as a major force in evolution. J. Genet. 92, 155–161 (2013).
Faraji, S. et al. The AP2/ERF gene family in Triticum durum: genome-wide identification and expression analysis under drought and salinity stresses. Genes 11, 1464 (2020).
Yan, J. B. et al. The Arabidopsis CORONATINE INSENSITIVE1 protein is a jasmonate receptor. Plant Cell 21, 2220–2236 (2009).
Fernández-Calvo, P. et al. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 23, 701–715 (2011).
Li, Y. et al. New insights into the TIFY gene family of Brassica napus and its involvement in the regulation of shoot branching. Int. J. Mol. Sci. 24, 17114 (2023).
Chen, C. et al. TBtools: An Integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Zuo D. et al.Genome-wide identification and stress response analysis of BcaCPK gene family in amphidiploid Brassica carinata. BMC Plant Biol. 24, 296 (2024).
Li, J. et al. Genome-wide investigation of bHLH genes and expression analysis under different biotic and abiotic stresses in Helianthus annuus L.. Int. J. Biol. Macromol. 189, 72–83 (2021).
Acknowledgements
This work was supported by Fundamental Research Program of Shanxi Province (Project No. 202203021212013, 202303021212358); College Students’ innovation and entrepreneurship training program of Shanxi Province Project (Project No. 20231458, 20241412); Taiyuan University Key Research Project (Project No. 23TYZD03, 23TYZD04).
Author information
Authors and Affiliations
Contributions
Methodology, Y.Z. and J.Z.; Project administration, X.Z.; Software, Y.J. and J.S.; Validation, L.L.; Writing–original draft, K.Z.; Writing–review & editing, Y.Z. and J. G. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, K., Zhou, Y., Jiang, Y. et al. Genome-wide identification of the TIFY gene family in Helianthus annuus and expression analysis in response to drought and salt stresses. Sci Rep 15, 15138 (2025). https://doi.org/10.1038/s41598-025-99315-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99315-y
