
Abstract
All methods to synthesize graphene oxide on a large scale utilize a strongly acidic medium. Graphene oxide (GO) to reduced graphene oxide (rGO) conversion, i.e., obtaining widespread sp2–sp2 hybridized carbon allotropes is a laborious and tedious process. Herein, we report an innovative one-step method, through which we fabricate ‘partially oxidized graphene’ on a gram scale directly from graphite powder, without using any acids. The emergence of the (002) XRD peak, band gap (Eg ~ 1.42 ± 0.01 eV), and carbon-to-oxygen (C/O) ratio of 3.67 validate the direct POG phase formation without any reduction step. Raman (ID/IG ~ 0.80 ± 0.06) and AFM studies reveal the less defective and bi- to few-layer character of POG sheets of thickness ca. 1.59 ± 0.14 nm. The measured conductivity of the POG material (9.22 ± 0.04 S/cm) is found to be higher than the conductivity of the rGO using the standard method (0.30 ± 0.03 S/cm). The zeta potential study suggests a good stability of the POG aqueous dispersion. Furthermore, the proposed method is efficient (2–3 h), safe, and renders a higher yield (77%) than many conventional methods (40–70%) for GO preparation. This offers a promising source for conductive electrodes, carbon paste, and more.
Similar content being viewed by others

Introduction
Synthesis of graphene and its derivatives has received a lot of interest since its discovery in 20041,2. Formation of graphene in derivative forms like graphene oxide (GO), reduced graphene oxide (rGO), and graphene quantum dots (GQD) renders a peculiar change in their characteristics3,4,5. For instance, graphene oxide has an intriguing semiconducting nature, whereas, graphene quantum dots exhibit a high degree of emissivity6,7. Similarly, rGO is akin to few-layer graphene as the band gap shrinks noticeably after the reduction of GO prepared via chemical exfoliation (or the so-called Hummers method)8. Single-layer graphene or few-layer graphene which is 1–2 atoms thick, an idealized sp2 carbon structure with fewer defects offers low sheet resistance with a narrow band gap (0.2–2 eV)9,10,11. Fabrication of few-layer graphene (FLG) via CVD/PVD (chemical or physical vapor deposition) or mechanical exfoliation techniques has proven to be extremely challenging, being non-economical and poor in yield12,13,14. For example, along with the other researchers, María Lozano-Chico et al. also reported (2024) that the yield of the liquid-phase exfoliated graphene is around 10%15,16,17. On the contrary, reduced graphene oxide is particularly interesting because of its easily tunable band-gap and surface applicability for composite formation with an ease of top-down fabrication which could be scaled to large quantities. Reduced graphene oxide (rGO) has been obtained (from GO) via many reduction methods with a claim of properties like FLG, however, among various properties, the electrical properties have also been found to be far from graphene/FLG9,10,11. To bridge the gap between the rGO and FLG properties, researchers have been concerned with reducing the oxidation kinetics in graphene oxide to eliminate the oxygenated groups and defects generated during oxidation.
Conventional processes (Brodie, Staudenmaier, Hummers, Tour etc.) to exfoliate graphite are highly oxidative and render one-few layers of graphene oxide only18,19. The GO reduction is routinely performed employing different chemical reagents (hydrazine, ammonia or ascorbic acid) to retrieve the graphene/FLG character20. However, there is no uniformity in these processes (in terms of quality of the final product, processing time etc.) and rGO obtained is found to be reduced at different scales20. Typically, the reduced graphene oxide obtained so will have abundant pinholes and defects, which will have detrimental effects on its properties21. Generally, it is seen that the characteristic properties of graphene oxide are sensitive to the reaction conditions, specific oxidants and precursors used22. For example, Kang et al. revealed that as the oxidation temperature increases during the exfoliation, the C/O ratio and the average GO size are found to reduce23. The work demonstrates that at higher oxidation temperatures more functional groups and defect sites are produced, while the lower temperature leads to a larger size of GO23. Similarly, Zhang et al. illustrated that the size of the GO sheet varies with a change in oxidation time/oxidant concentration. The size distribution is found to be broadened with the reaction time and increased oxidant concentration24. It has also been reported by Qiang et al. that as the concentration of H2SO4 decreases in water, GO has lesser disruption in basal planes, with a less defective sp2 structure18,19. In terms of properties, it is worth noting that as compared to graphene/FLG (~ 5 to 103 S/cm), the electrical conductivity for rGO is found to be in a range of 10− 3 S/cm to 21 S/cm12,13,14,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39. All these reports25,26,27,28,29,30,31,32,33,34,35,36,37,38,39 are based on varying reduction methods (which include chemical, thermal, hydrothermal, microwave-assisted, sonication, and laser-scribing) including green/ecologically friendly reducing agents. An ideal scenario would be to exfoliate the graphite layers at a large scale without using any acids. This may assist us in having the sp2 structure intact at a larger scale. The presence of larger sp2 domains with mild oxidation may directly yield ‘partially oxidized graphene’ (POG). A benign solvent route that can offer a larger lateral size of graphene layers with a smaller amount of defects may provide a very good quality ‘partially oxidized graphene’. Hence, new synthesis methods and processing must be designed to enhance the qualities of rGO to realize its full potential.
In this study, we have developed a simple direct route to obtain partially oxidized graphene at the gram scale from graphite. The ‘partially oxidized graphene’ formation in an acid-free medium occurring at normal temperature is found to be highly efficient (2–3 h). The as-synthesized ‘partially oxidized graphene’ film (Eg ~ 1.42 ± 0.01 eV) shows conductivity of 9.22 ± 0.04 S/cm, which is higher or in the comparable range of previously reported conductivity values of rGO films of standard methods (considering the fact that we have not used any reduction methods as well as any acids/harmful chemicals to obtain the POG from graphite)25,26,27,28,29,30,31,32,33,34,40,41,42,43. The ID/IG ratio of 0.80 ± 0.06 and the appearance of (002) characteristic XRD peak at 2θ = 21.79°, give a clear indication of direct exfoliation of graphite to ‘partially oxidized graphene’ with fewer structural defects. The high carbon-to-oxygen (C/O) ratio (3.67) and specific surface area (107.23 m2/g) verify the partially oxidized nature of the as-prepared material. We hypothesize the mechanism of the ‘partially oxidized graphene’ formation, stating that the energized water molecules get intercalated between the graphite layers during the mild sonication followed by insertion of \(\:{NO}_{3}^{-}\)/\(\:{Na}^{+}\) ions, which leads to swelling and exfoliation of graphite layers17,44,45,46,47,48,49,50,51,52. The ultrasonication process initially can create some edge defects via bubble pressure to weaken the van der Waals forces act between the graphite layers and the presence of Na+/NO₃⁻ ions can provide an additional driving force for the expansion process17,44,45,46,47,48,49,50,51,52. To facilitate the mild oxidation process and to diminish the van der Waals interaction between the layers, the addition of KMnO4 is followed, and thus the complete exfoliation of reduced graphene oxide-like sheets can be achieved. The as-prepared ‘partially oxidized graphene’ will be highly beneficial for the design and development of electrodes, carbon ink, conducting paste etc.
Experimental section
Synthesis of the ‘partially oxidized graphene’ material
The direct but controlled synthesis of the ‘partially oxidized graphene’ material from the graphite powder was performed in a water medium. As a first step, a solution of sodium nitrate (NaNO3) (0.625 g) in deionised (DI) water was prepared under constant magnetic stirring (800 rpm). This is followed by the addition of 1.25 g graphite powder (Sigma Aldrich, particle size < 50 μm) while sonicating the dispersion to expedite the intercalation process. Afterwards, to facilitate the exfoliation process, we introduced potassium permanganate (KMnO4) (6.25 g, S. D. Fine-Chem Limited) slowly to the sodium nitrate aqueous solution. In a standard procedure3, the required graphite to KMnO4 wt% ratio is 1:3 to ensure the completion of the reaction. However, typically an excess KMnO4 is taken53,54,55,56,57,58,59,60, since it has an optimum oxidizing capacity at higher concentrations61,62. In the next step, 50 ml of DI water was added drop-wise to the reaction solution under constant stirring (500 rpm) and the mixture was heated at a temperature of ~ 70 ± 5 oC for 50 min. Termination of the reaction was done with the addition of deionized water/hydrogen peroxide (Honeywell). The obtained solution was kept overnight at rest. Later, the supernatant was decanted and the obtained part was cleaned (8000 rpm) several times using deionised water. The final dark-brown dispersion which contains rGO-like nano-sheets was achieved by the ultra-sonication treatment of the cleaned sample.
For a clear comparison of the physicochemical properties of the obtained ‘partially oxidized graphene’ material with standard GO and rGO, we have also synthesized GO by the conventional modified Hummers method and rGO using a standard hydrazine hydrate reduction process39,53,54,55,56,57,58,59,60. GO was prepared via modified Hummers method using concentrated (99%) sulphuric acid (H2SO4), graphite powder, potassium permanganate, and hydrogen peroxide53,54,55,56,57,58,59,60. The reduction of the obtained GO was performed through chemical treatment using hydrazine hydrate at a temperature of 100 ± 5 o C for 24 h and dried the obtained clean product in an open air environment39. We named our ‘partially oxidized graphene’ material POG, reduced graphene oxide of the standard reduction method as rGO and graphene oxide of modified Hummers method as GO.
Characterization techniques
To analyse the absorbance and the bandgap of the exfoliated POG material, UV-visible spectroscopy measurements were performed on a PerkinElmer UV/VIS Spectrometer (Model Lambda 950). The phase and crystal structure of the acquired POG material were analysed by X-ray diffraction (XRD) experiments on PANalytical (Empyrean, Netherlands) X-ray diffractometer using Cu Kα radiation (λ ~ 1.5404 Å). Surface morphology and thickness of the as-synthesized POG material was investigated using Atomic Force Microscopy (AFM, Multimode Nanoscope-IV) in a tapping mode, Field Emission Gun Transmission Electron Microscopy (FEG-TEM, JEOL, JEM-2100 F) and Field Emission Scanning Electron Microscopy (FESEM, Zeiss/Ultra 55). Raman spectroscopic measurements were performed (HR800-UV confocal micro-Raman spectrometer) to quantify the defect density and sp2 domains in the material with an excitation wavelength of 532 nm. The specific surface area (BET: Brunauer-Emmett-Teller) and pore size distribution (BJH: Barrett-Joyner-Halenda) measurements of the POG sample were analyzed using the surface area analyzer: Quantachrome Autosorb AS-1Version-1.55. The surface elemental compositions and chemical states of the POG, rGO, and GO samples were investigated via X-ray Photoelectron Spectroscopy (XPS - Kratos Analytical, AXIS Supra) operated using Al, Kα (hν = 1486.6 eV) radiation. The stability of the as-prepared POG water dispersion was analysed via zeta potential and dynamic light scattering (DLS, Brookhaven Instruments) measurements. The presence of various oxygen functional groups in the POG material was studied using Fourier Transform Infrared Spectroscopy (FTIR); 3000 Hyperion Microscopic spectrometer model from Bruker, Germany. Two probe current-voltage measurements were recorded for the as-synthesized POG material, rGO, and GO samples using a dual-probe KEITHLEY, 2602-A source meter. The error values included in the experimental data each performed at least three repeated times are expressed as mean ± standard deviation.
Probable mechanism of POG material formation
Firstly, an aqueous solution of sodium nitrate is prepared under constant magnetic stirring. At the time of sodium nitrate addition, the pH of the solution mixture doesn’t change significantly; this is because when it dissolved in water the Na+/NO3− ions do not react with water molecules and the solution remains neutral with no change in pH (pH ~ 7.3–7.5). This aqueous solution is kept in bath sonication (300 W) while adding graphite in it (Fig. 1 step A). Typically, ionic solutions have surface energies (approx. 40–70 mN m− 1) similar to that of graphene and hence a suitable candidate to assist in sonication-based exfoliation63,64,65. We believed that a comparable surface energy of sodium nitrate solution to that of graphene (approx. 35–45 mN m− 1) may provide an energetically favourable ambience to assist in the first stage of exfoliation. Thermodynamically, a fine balance between the surface energies of the ionic solvent (ES) with graphene (EG) will only yield a minimal enthalpy of sonication that is,
Where V is the volume of the solution mixture, T is the thickness of the graphene sheets, and ФG is the volume fraction of the dispersed graphene. In earlier literature, researchers have used ionic liquids, surfactants, and polymers to exfoliate graphite to graphene but such processes render a poor yield (less than 1%) and are inefficient/laborious66,67. In this work, we are relying on the assistance of \(\:{NO}_{3}^{-}\)/\(\:{Na}^{+}\:\)ions and water molecules to intercalate through edges (via the assistance of mild sonication) and probably a mild oxidation via \(\:{NO}_{3}^{-}\) ions/water molecules17,44,45,46,47,48,49,50,51,52. The polyatomic structure of nitrate ions in sodium nitrate aqueous solution may increase the activity coefficient (via rate of increase in vibration) to normalize the ES to the level of EG. In addition to that, the cavitation induced via mechanical forces during bath sonication (with mild energy only) facilitates easy swelling of the graphitic layers (Fig. 1 step A)17,44,45,46,47,48,49,50,51,52. During this, to attain stable dispersion of the liquid phase, the energized water molecules (with the help of its bubble pressure created by mechanical cavitation events supported by the consistent acoustic flows) and the ions (\(\:{NO}_{3}^{-}\)/\(\:{Na}^{+}\)) will enter between the graphite layers, which lead to a significant reduction of the interlayer binding strength17,44,45,46,47,48,49. However, the force applied by the water molecules/ions (via sonication) is not strong enough to overcome the van der Waals interaction completely. Therefore, we take a mild solution of KMnO4 to minimally oxidize the layers which separates the graphene layers in a short time to obtain partially oxidized graphene material (Fig. 1 step B). In this step, the oxidation reaction is prompted and slightly increases the pH (pH ~ 8.40–8.80) of the solution. Towards the end, we add DI water/H2O2 to stop the oxidation reaction and hence remove metal ions and KMnO4 impurities (Fig. 1 step C). At this step, the reaction is exothermic and causes the pH to increase slightly (pH ~ 10.34–10.66). In the final stage, the solution is washed multiple times with the help of deionized water to remove unreacted residues and impurities. During each wash, the pH decreases gradually as the unreacted residues and impurities are diluted and removed. The process continues until the pH of the solution becomes neutral (pH ~ 7.0-7.5), ensuring the product is free from unwanted ions.

Schematic step-by-step view of the process for the exfoliation of POG material. NaNO3 aqueous solution is prepared under constant magnetic stirring, step A—addition of graphite under ultrasonication, step B—addition of KMnO4, step C—addition of DI water, step D—providing some heat of 70 °C, step E—supernatant collection, step F—centrifugation.
Results and discussion
The dispersibility and solubility of the as-prepared POG material have been studied with respect to GO, and rGO obtained from the standard method. Figure 2 shows the gram scale suspension behavior and the solution stability measurements in terms of absolute zeta potential of the as-synthesized POG material, rGO of the standard reduction method, and GO of the modified Hummers method. During the synthesis process, we start with 1.25 g of graphite powder (particle size < 50 μm, Sigma Aldrich) and end up with ~ 0.96 g of partially oxidized graphene, and the yield is found to be ~ 77%. Yield values of graphene exfoliated through different acid-less processes are shown in supplementary Table 1 (SI file). The GO dispersion is light-brown in colour, while the POG displays dark-brown colour (Figs. 2a; and S1a, b, Supporting Information). This kind of appearance is because of the fewer π–π conjugations in GO than the POG and rGO, i.e. presence of a very small number of oxygen functional groups in the POG as compared to the GO18,19,68,69.

(a) Digital photograph of POG material, rGO (standard reduction method), and GO (modified Hummers method) water dispersion; all pictures were taken on the same day. (b) zeta potential values for the fresh samples and after 3 months of storage time-duration. The POG material shows a good dispersion behavior, similar to GO in water and exhibits long-term stability than rGO and GO. Few additional digital photographs of the three suspensions are added in SI as Figure S1 (a, b).
Stability measurements (Fig. 2b) demonstrate the absolute value of the zeta potential of 39.38 mV, 42.63 mV, and 42.67 mV for the as-synthesized GO, rGO, and POG aqueous dispersions, respectively69,70,71. Usually, suspensions with absolute zeta potential values higher than 30 mV are physically stable, while suspensions below 20 mV have limited stability and suspensions below 5 mV face a rapid aggregation49,69. It is interesting to note that POG and rGO aqueous solutions have a higher initial absolute zeta potential value and even after 3 months of storage in normal ambience these POG and rGO solutions maintained a value of 41.62 mV and 38.76 mV, respectively34,68,69,71,72. These values of the zeta potential are also in good agreement with the reported zeta potential values of GO and rGO34,68,69,71. This clearly indicates that the POG aqueous dispersion has better stability than the aqueous solutions of both rGO and GO.
Figure 3a–c shows the UV-visible spectra of the as-synthesized POG, rGO, and GO. For UV-visible spectroscopy measurements, the samples were prepared by using the aqueous dispersion of POG flakes and quartz cuvettes to minimize interference from the container. It is evident that, the absorption spectrum of the POG is red-shifted as compared to the spectrum of rGO. The absorption maxima for the POG, rGO and GO are seen at ca. 282 nm, 272 nm, and 230 nm, respectively. The shift in spectra reveals higher π–π* conjugation in the POG and rGO which is attributed to the better retention of graphitic cyclic alkene (C=C) rings in the basal planes18,19,73,74,75. The n–π* electronic transition mode occurs when additional oxygen atoms are attached to the graphitic basal plane of GO18,19,73,74,75. The blue-shifted absorption band in GO (230 nm) because of the n–π* transition signifies the presence of a larger number of hydroxyl and epoxy groups in GO than in rGO and POG. The absorption shoulder corresponding to n–π* transition of C= O bonds is observed at ca. 295 nm for GO sample, while it is absent in both POG and rGO. When compared to the corresponding peak for GO (Fig. 3c), the UV-vis absorption spectrum of rGO exhibits a broad peak centred at 272 nm with steadily decreasing intensity up to 800 nm, which is consistent with the restoration of the electronic conjugation in the graphene sheets upon reduction76,77. However, for POG the peak is much broader (Fig. 3a) than rGO, which reveals the existence of a higher conjugated electronic structure in the POG.

UV–visible spectra (a–c) and their corresponding Tauc plots (d–f) for the POG, rGO, and GO samples, respectively. The as-synthesized POG has a lower band gap (1.42 ± 0.01 eV) than the rGO (1.66 ± 0.03 eV). The GO has an optical band gap of 2.91 ± 0.02 eV.
The optical band gaps calculated from the corresponding Tauc plots (Fig. 3d–f) of the POG, rGO and GO are 1.42 ± 0.01 eV, 1.66 ± 0.03 eV and 2.91 ± 0.02 eV, respectively. The band gap value of the POG material is also found to be lower than the reported band gap values of rGO25,26,27,28,29,30,31,32,33,34,76,77,78,79. The lower band gap (0.24 eV and 1.49 eV than rGO and GO respectively) of the POG in comparison to the rGO and GO suggests better π–π* interaction and absence of insulating functional groups in POG73,74,75,76,77.
Primarily, X-ray diffraction analysis is performed to identify the structural alterations in the graphite flakes. As can be seen in Fig. 4, an intense sharp peak corresponding to (002) diffraction in the graphite powder is observed at 2θ = 26.33° (lattice spacing, d = 3.4 Å). The conserved and organised stacking along the c-axis is responsible for the strong and identifiable peak in the graphite XRD pattern80,81. For the GO sample, the peak shifts to a lower value of 2θ = 9.07°, suggesting the successful oxidation of graphene sheets with a wider d-spacing of 9.8 Å. The increase in lattice plane separation occurs due to the intercalation with water molecules and the presence of epoxide and hydroxyl oxygen functional groups on the basal plane of the graphene layers18,19,80,81,82. After hydrazine reduction (towards rGO from GO), the peak shifts to a higher 2θ value (21.02°) with an interlayer spacing of 4.2 Å. It is observed from the POG XRD pattern in Fig. 4, the partial collapse of the long-range ordered graphene layers in the graphite powder and simultaneous introduction of mild oxidation leads to a decrease in the peak intensity (2θ = 21.79°) and peak broadening (FWHM = 7.23 ± 0.03°) for the POG sample. The interlayer separation of the POG sample is estimated to be ~ 4.0 Å. This value is slightly higher than the natural graphite (3.4 Å), but substantially lower than the GO precursor (~ 9.8 Å).

XRD patterns of graphite powder, POG, rGO, and GO. A minimal lower 2θ shift (from 26.33 to 21.79°) suggests the mild oxidation of the POG material.
The decrease in the interlayer spacing of POG, with respect to GO as well as rGO, indicates the presence of higher sp2 domains and less oxygenated functional groups in the POG material80,81,83,84. The lower percentage of functional groups in the POG than that in GO and rGO samples causes lesser stretching between the crystallites, which explains the smaller crystallite size and larger peak broadening in the XRD spectrum of POG80,81,82. The POG XRD spectrum also matches well with several XRD spectra of good quality rGO as has been demonstrated in the existing literature18,19,73,74,75,80,81,82,83,84.
Figure 5a,b shows the atomic force microscopic images of the POG material at a scan size of 3 μm and 1 μm, respectively. For sample preparation, we used a Si/SiO2 substrate cleaned with acetone-IPA-DI water, followed by ten minutes of sonication in each case. Then, the POG sheets from an aqueous suspension were spin-coated on the cleaned Si/SiO2 substrates by a spin-coating technique and dried in a normal ambience. The POG sheet-coated Si/SiO2 substrates were used for AFM analysis. The calculated thickness of the flakes from the section profile analysis (Fig. 5c, d) (Figure S4, SI file) was found to be in the range of ca. 1.59 ± 0.14 nm to ~ 5.0 nm confirming the presence of bi-few layer graphene sheets in the as-prepared POG samples. This value of the flake thickness is consistent with the typical two-few layer thickness reported for rGO20,85,86,87. However, the edge-to-edge attraction generated by the hydrogen bonding between the very few oxygen functionalities might result in the crumpling and aggregation of POG sheets (Figure S4a–d)80,81.

AFM images of the POG sample at a scan size of (a) 3 μm and (b) 1 μm. (c) and (d) are sectional or height profile analysis at different sites of the same bi/multilayered POG. The as-synthesized POG material contains bi to few-layer graphene sheets.
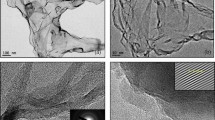
Further, Fig. 6a–d shows the FEG-TEM images of POG material. The layer-like POG sheets are seen with wrinkles and folds with high transparency indicating single/bi- to few-layer graphene (Figure S5c, d)88,89,90. The SAED pattern (inset: Fig. 6d) consists of concentric rings which indicate the crystalline character of the POG material. The HR-TEM image in Fig. 6d shows the well-defined lattice fringes. The (002) plane of the POG material corresponds to the fringe width with an interlayer spacing of 0.4 nm (inset: Fig. 6d).

TEM images (a–d) and SAED pattern (inset of d) of the POG material, illustrate proper exfoliation of graphite to single-few layers graphene with good crystallinity behavior.
Figure 7 displays the Raman spectra of the POG, rGO, and GO samples, respectively. In the case of GO, the well-ordered sp2 hybridized carbon structure is responsible for the G-band at 1586 cm− 1, and the structural defects in the hexagonal graphitic structure are responsible for the D-band at 1345 cm− 1. The intensity ratio (ID/IG) of these bands is found to be ~ 1.0 ± 0.03, which confirms the presence of numerous surface defects in the as-synthesized GO sample91. For the rGO sample, the removal of several structural defects causes the ID/IG ratio to reduce to 0.92 ± 0.05. A much smaller ID/IG value (0.80 ± 0.06) for the POG sample clearly indicates the existence of lesser number of structural defects than the rGO and GO samples (Supplementary Table 4)92,93,94,95,96,97. The 2D band observed at 2693 cm− 1 for the POG (Fig. 7), further supports the existence of graphene lattice with minimal defect sites66,92,93,94,95,96. Moreover, the scatter-plot data (Figure S9, SI file) confirm a clear trend between defect density and Γ2D (FWHM—full width at half maximum), suggests the presence of less disordered sp2 conjugation in the as-prepared POG, consistent with previous literature75. This result demonstrates that as-prepared POG material is of higher quality than that of rGO and GO.

Raman spectra of POG, rGO, and GO samples. The lower ID/IG value of ~ 0.80 suggests that the POG material has lesser defects in the graphene lattice.
Field emission scanning electron microscopic observations were performed on the POG sample (Fig. 8) (Figure S5 a, b). It is observed that the exfoliated few layer POG flakes are homogeneously distributed on the Si/SiO2 surface and may be covered by many other flakes, and are also folded at many places. Flake size distribution of the as-prepared partially oxidized graphene shows that the average size of the as-prepared POG flakes is ~ 1.003 ± 0.272 μm (Figure S8, SI file). It is believed that because of the less number of oxygen functionalities, the stretching between graphene flakes decreases and the edge-to-edge attraction generated by the hydrogen bonding between the few oxygen functionalities might result in the crumpling and aggregation of POG as well as rGO sheets80,81,98,99. Furthermore, the overlapping of graphene or graphene oxide nanoplatelets gives rise to the large number of folds in the samples80,81,99,100. This result indicates POG has a similar behavior as rGO, wherein, the agglomeration of graphene or graphene oxide nanoplatelets is highly prominent98,99,100.

FESEM images of the POG sample at different magnifications; illustrate the homogeneous wide area distribution with aggregation and crumpling character of the few layer POG flakes.
Brunauer-Emmett-Teller (BET) surface analysis is a helpful measurement for the surface area of the prepared material. Figure S6a shows the adsorption and desorption BET isotherms of the as-prepared POG sample under nitrogen. The specific surface area of the POG sample is found to be 107.23 m2/g, which is in the range of many reported specific surface area values of standard rGO101,102,103. This could be due to the agglomeration of the graphenic sheets and overlap of the particularly smaller ones, reducing the surface area of the material101,102,103. Nonetheless, there are still a lot of exposed surface regions due to the wrinkled three-dimensional structure of the material. However, the BET isotherm curve suggests that the POG sample is porous in nature with hysteresis phenomenon at the elevated partial pressures. Moreover, to assess the average pore volume and average pore diameter of the fabricated POG sample, the BJH numerical integration method (Barrett-Joyner-Halenda) is being utilised (Fig. S6b). The calculated average pore diameter and average pore volume of the POG sample are 25 Å and 0.205 cc/g, respectively101,102,103. These investigations of BET and BJH indicate the formation of partially oxidized graphene.
X-ray photoelectron spectroscopy studies are conducted to comprehend the surface composition, quantitative functional group investigation, and chemical states of the POG, rGO, and GO samples. Figure 9a,b shows the high-resolution XPS spectra of the POG sample and Figure S2 explains the comparative survey spectra of the as-obtained POG, GO, and rGO samples. The POG sheets from an aqueous suspension were drop cast on the cleaned glass substrate and dried in a normal ambience. The thick POG material-coated substrates were used for XPS analysis to ensure that the signal collected was free of other oxygen signals coming from the substrate104. However, the relative sensitivity factors were used in the determination of carbon-to-oxygen (℅) ratios from the survey spectra105,106,107. The Gaussian-Lorentzian function is utilised to deconvolute the C 1s and O 1s spectra into several peak components. From the deconvoluted C 1s spectra of the POG sample (Fig. 10a), the sp2 hybridized carbon (C=C) has a binding energy of 284.80 eV, whereas, the peak components at binding energies of 285.58 eV, 286.95 eV, 289.37 eV, 283.00 eV are associated with the sp3 hybridized carbon (C–C), epoxy/hydroxyl group (C–O, C–OH), carboxyl group (O–C= O), and hydrocarbon group (C–H) respectively83,84,108,109. The broad C =C peak could arise from the heterogeneity of the sp2 domains in POG, that is the fabrication process could result in a mixture of larger and smaller sp2 domains110,111. The C-H peak, while less commonly highlighted, corresponds to edge hydrogenation or residual functional groups, as has been documented in reduced graphene oxide studies112,113. In the O 1s spectra of the POG sample (Fig. 9b), the peak components at 529.70 eV, 531.80 eV, 532.60 eV, 533.88 eV are related with carboxyl group (O–C =O), carbonyl group (C= O), hydroxyl/epoxy group (C–OH, C–O) and epoxide (C–O–C) group respectively108,109. The XP spectra of the as-prepared GO and rGO samples are analyzed in Figure S3 (a-d) (Supporting Information). The carbon-to-oxygen (C/O) ratio of the obtained POG sample is found to be 3.67, which is higher than that of GO (0.87) as well as rGO (2.42) (Supplementary Table 2). The higher carbon-to-oxygen (C/O) ratio suggests the formation of partially oxidized graphene with a lesser number of defects.

High-resolution X-ray photoelectron spectra (a) C 1s, (b) O 1s of the POG sample. The sample exhibits higher carbon-to-oxygen (C/O) ratio (3.67), which indicates the generation of partially oxidized graphene.
The FTIR spectroscopic measurements shown in Fig. 10, provide qualitative information about the vibrational properties of functional groups present in the graphite powder, POG, and GO samples. For FTIR spectroscopy measurements, the powder samples were mixed finely with potassium bromide and pressed into pellets. The broad absorption band located at ca. 3454 cm− 1 is attributed to the stretching of hydroxyl (–OH) groups present in the samples114. This band is similar in shape and becomes sharp or less intense for the POG material than the GO, however, the band shape changes and becomes much broader than that of the graphite powder. The band located at 2357 cm− 1 arises due to the stretching of CO2 groups115, and is similar in character for both GO and POG, but different for graphite. This indicates the presence of oxygen functional groups attached in POG basal planes. Moreover, the absorption bands located at 1728 cm− 1, 1628 cm− 1, and 1048–1260 cm− 1 are due to the stretching or bending of the ketonic group C=O, bending of hydroxyl group –OH, and C–O bonds, respectively114. The bands for the oxygenated functional groups are found to be less intense for the POG material than the GO sample, while these bands are completely suppressed in the spectrum of the graphite powder. The FTIR spectrum of the POG material is also in good agreement with the reported FTIR spectra of rGO25,26,27,28,29,30,31,32,33,34,78,79,115,116,117. This result gives a clear indication that the POG material is mildly oxidised with higher graphenic domains.

FTIR spectra of graphite powder, POG material, and GO. The weak absorption bands in the POG spectrum illustrate mild oxidation character of the POG material.
The current-voltage measurements of the as-prepared air-dried samples at glass substrate are taken in the − 5 V to + 5 V range (Fig. 11). For the electrical conductivity measurements, films were fabricated on precleaned glass substrates (cleaning process:- 10 min sonication in each case: DI water⟶ acetone⟶ DI water⟶ IPA ⟶ DI water; then dried the substrates in a hot air oven at 70 °C for ~ 45 min) by drop cast as well as spin coat techniques using POG aqueous dispersion of 2 mg/ml concentration to create uniform thin films. The as-fabricated films were dried in an open-air environment for a day. The device geometry was 1 × 1 cm², with silver metal contact pads at the corners of the films. The metal contact pads have been used as positive and negative electrodes for the transport measurements, carried out using a KEITHLEY-2602 A source meter. The estimated film thicknesses were 150.4 ± 4.1 nm, 2.3 ± 0.2 μm, and 28.5 ± 1.0 μm for the POG, reduced graphene oxide (rGO), and graphene oxide (GO) films, respectively (Figure S7). The curves exhibit a linear current-voltage relation, which suggests an ohmic behavior with R2 values of 0.993, 0.994, and 0.971 for the POG, rGO, and GO, respectively. The calculated conductivities of the POG, rGO, and GO samples were found to be 9.22 ± 0.04 S/cm, 0.30 ± 0.03 S/cm, and \(\:(2\pm\:0.28)\times\:{10}^{-3}\:\)S/cm, respectively. The POG material demonstrates noticeably higher conductivity in comparison to both GO and rGO samples. The conductivity value of the POG material is higher or in the range of the reported conductivity values of rGO (Supplementary Table 3)25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,42,43.

Two-probe I-V measurements of POG, rGO, and GO drop casted thin films. The POG material has higher conductivity (9.22 ± 0.04 S/cm) than the rGO (0.30 ± 0.03 S/cm), while GO conductivity is found to be \(\:(2\pm\:0.28)\:\times\:\:{10}^{-3}\) S/cm.
Conclusions
Partially oxidized graphene is exfoliated at a gram scale directly from graphite powder without using any acids. The POG material phase formation (a peak at 2θ = 21.79°) is confirmed by the XRD analysis. From the UV-visible spectra, the lower band gap value (Eg = 1.42 ± 0.01 eV) suggests higher graphenic behavior of the POG material in comparison to the rGO (Eg = 1.66 ± 0.03 eV). FTIR signal (of POG) with diminished intensity reveals fewer oxygen functional groups on the surface of POG material. The bi-layer and few-layer characteristic (flake thickness ~ 1.5 nm to 3.6 nm) along with folds or aggregation tendency of the POG material are supported by the AFM, FEG-TEM, and FESEM analyses. The Raman spectra (ID/IG = 0.80 ± 0.06) suggest that the POG material is of higher quality with lesser number of defects. The XPS analysis (C/O = 3.67) and BET-surface study (107.23 m2/g) signify the exfoliated material is partially oxidized. Moreover, the conductivity of the POG material (9.22 ± 0.04 S/cm) is found to be higher than the rGO (0.30 ± 0.03 S/cm). The zeta potential analysis illustrates strong long-term stability (more than 3 months) of the POG water dispersion than that of GO and rGO dispersions. This acid-less novel route to obtain the ‘partially oxidized graphene’ directly from the graphite powder is a promising source of conducting electrodes for batteries or supercapacitors, carbon paste, etc.
Data availability
The data that support the findings of this study are available in the supplementary material of this article.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306(5696), 666–669 (2004).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6(3), 183–191 (2007).
Hummers, W. S. Jr & Offeman, R. E. Preparation of graphitic oxide. J. Am. Chem. Soc. 80(6), 1339–1339 (1958).
Pei, S. & Cheng, H. M. The reduction of graphene oxide. Carbon 50(9), 3210–3228 (2012).
Bacon, M., Bradley, S. J. & Nann, T. Graphene quantum dots. Part. Part. Syst. Charact. 31(4), 415–428 (2014).
Zhu, Y. et al. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 22(35), 3906–3924 (2010).
Qu, D. et al. Tailoring color emissions from N-doped graphene quantum dots for bioimaging applications. Light: Sci. Appl. 4(12), e364–e364 (2015).
Shen, Y. et al. Evolution of the band-gap and optical properties of graphene oxide with controllable reduction level. Carbon 62, 157–164 (2013).
Quhe, R. et al. Tunable band gap in few-layer graphene by surface adsorption. Sci. Rep. 3(1), 1794 (2013).
Nandee, R. et al. Band gap formation of 2D materialin graphene: Future prospect and challenges. Results Eng. 15, 100474 (2022).
Zhu, L. et al. Formation and electronic properties of hydrogenated few layer graphene. Nanotechnology 22(18), 185202 (2011).
Park, H. J. et al. Growth and properties of few-layer graphene prepared by chemical vapor deposition. Carbon 48(4), 1088–1094 (2010).
Wei, D. et al. Controllable chemical vapor deposition growth of few layer graphene for electronic devices. Acc. Chem. Res. 46(1), 106–115 (2013).
Pirzado, A. A. et al. Few-layer graphene from mechanical exfoliation of graphite-based materials: Structure-dependent characteristics. ChemEngineering 3(2), 37 (2019).
Lozano-Chico, M. et al. Water-based and tannin-assisted liquid-phase exfoliation for a sustainable production of graphene. Sustainable Mater. Technol. 40, e00956 (2024).
Ma, H. et al. Direct exfoliation of graphite in water with addition of ammonia solution. J. Colloid Interface Sci. 503, 68–75 (2017).
Tyurnina, A. V. et al. Ultrasonic exfoliation of graphene in water: A key parameter study. Carbon 168, 737–747 (2020).
Zhang, Q. et al. Roles of water in the formation and preparation of graphene oxide. RSC Adv. 11(26), 15808–15816 (2021).
De Silva, K. et al. Chemical reduction of graphene oxide using green reductants. Carbon 119, 190–199 (2017).
Fernández-Merino, M. J. et al. Vitamin C is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions. J. Phys. Chem. C. 114(14), 6426–6432 (2010).
Yun, Y. S. et al. Restoration of thermally reduced graphene oxide by atomic-level selenium doping. NPG Asia Mater. 8(12), e338–e338 (2016).
Li, J. et al. Selective oxidation of alcohols by graphene-like carbon with electrophilic oxygen and integrated pyridinic nitrogen active sites. Nanoscale 13(30), 12979–12990 (2021).
Kang, D. W. & Shin, H. S. Control of size and physical properties of graphene oxide by changing the oxidation temperature. Carbon Lett. 13(1), 39–43 (2012).
Zhang, L. et al. Size-controlled synthesis of graphene oxide sheets on a large scale using chemical exfoliation. Carbon 47(14), 3365–3368 (2009).
Fan, Y., Jiang, W. & Kawasaki, A. Highly conductive few-layer graphene/Al2O3 nanocomposites with tunable charge carrier type. Adv. Funct. Mater. 22(18), 3882–3889 (2012).
Eigler, S. A new parameter based on graphene for characterizing transparent, conductive materials. Carbon 47(12), 2936–2939 (2009).
Xu, Y. et al. Self-assembled graphene hydrogel via a one-step hydrothermal process. ACS Nano. 4(7), 4324–4330 (2010).
Bonavolontà, C. et al. Reduced graphene oxide on silicon-based structure as novel broadband photodetector. Sci. Rep. 11(1), 13015 (2021).
Park, H. et al. Electrical measurements of thermally reduced graphene oxide powders under pressure. Nanomaterials 9(10), 1387 (2019).
Zhu, Y. et al. Microwave assisted exfoliation and reduction of graphite oxide for ultracapacitors. Carbon 48(7), 2118–2122 (2010).
Stankovich, S. et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45(7), 1558–1565 (2007).
Moon, I. K. et al. Reduced graphene oxide by chemical graphitization. Nat. Commun. 1(1), 73 (2010).
Lotya, M. et al. Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions. J. Am. Chem. Soc. 131(10), 3611–3620 (2009).
Roy, I. et al. Physical and electrochemical characterization of reduced graphene oxide/silver nanocomposites synthesized by adopting a green approach. RSC Adv. 5(32), 25357–25364 (2015).
Eda, G., Fanchini, G. & Chhowalla, M. Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat. Nanotechnol. 3(5), 270–274 (2008).
Ludwig, T. et al. Boosting nitrogen-doping and controlling interlayer spacing in pre-reduced graphene oxides. Nano Energy. 78, 105286 (2020).
Sadek, R. et al. Tolerated Reduction Protocol of Graphene for High Performance Emi Shielding Materials. Charles and Tantawy, Hesham and Chaouki, Jamal, Tolerated Reduction Protocol of Graphene for High Performance Emi Shielding Materials.
Sieradzka, M. et al. Insight into the effect of graphite grain sizes on the morphology, structure and electrical properties of reduced graphene oxide. J. Mater. Res. Technol. 9(4), 7059–7067 (2020).
Xu, C., Yuan, R. & Wang, X. Selective reduction of graphene oxide. New Carbon Mater. 29(1), 61–66 (2014).
Shen, Y. et al. Chemical and thermal reduction of graphene oxide and its electrically conductive polylactic acid nanocomposites. Compos. Sci. Technol. 72(12), 1430–1435 (2012).
Sadhukhan, S. et al. Studies on synthesis of reduced graphene oxide (RGO) via green route and its electrical property. Mater. Res. Bull. 79, 41–51 (2016).
Lam, S. et al. On the rapid synthesis of reduced graphene oxide via ultrasound-promoted eco‐friendly reduction approach and its effects on physicochemical properties. ChemSusChem 17(24), e202400845 (2024).
Smith, A. T. et al. Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites. Nano Mater. Sci. 1(1), 31–47 (2019).
Chen, K. & Xue, D. Preparation of colloidal graphene in quantity by electrochemical exfoliation. J. Colloid Interface Sci. 436, 41–46 (2014).
Hamra, A. et al. Electro-exfoliating graphene from graphite for direct fabrication of supercapacitor. Appl. Surf. Sci. 360, 213–223 (2016).
Morton, J. A. et al. New insights into sono-exfoliation mechanisms of graphite: In situ high-speed imaging studies and acoustic measurements. Mater. Today 49, 10–22 (2021).
Parvez, K. et al. Exfoliation of graphite into graphene in aqueous solutions of inorganic salts. J. Am. Chem. Soc. 136(16), 6083–6091 (2014).
Tateishi, H. et al. Effect of the electrochemical oxidation/reduction cycle on the electrochemical capacitance of graphite oxide. Carbon 76, 40–45 (2014).
Wang, Q. et al. Chitosan and sodium alginate nanocarrier system: controlling the release of rapeseed-derived peptides and improving their therapeutic efficiency of anti-diabetes. Int. J. Biol. Macromol. 265, 130713 (2024).
Baig, Z. et al. Investigation of tip sonication effects on structural quality of graphene nanoplatelets (GNPs) for superior solvent dispersion. Ultrason. Sonochem. 45, 133–149 (2018).
Mombeshora, E. T., Ndungu, P. G. & Nyamori, V. O. Effect of graphite/sodium nitrate ratio and reaction time on the physicochemical properties of graphene oxide. New Carbon Mater. 32(2), 174–187 (2017).
Tyurnina, A. V. et al. Environment friendly dual-frequency ultrasonic exfoliation of few-layer graphene. Carbon 185, 536–545 (2021).
Marcano, D. C. et al. Improved synthesis of graphene oxide. ACS Nano. 4(8), 4806–4814 (2010).
Deemer, E. M. et al. Consequence of oxidation method on graphene oxide produced with different size graphite precursors. Mater. Sci. Eng.: B 224, 150–157 (2017).
Kotsyubynsky, V. et al. Graphene oxide synthesis using modified tour method. Adv. Nat. Sci. NanoSci. NanoTechnol. 12(3), 035006 (2021).
Ranjan, P. et al. A low-cost non-explosive synthesis of graphene oxide for scalable applications. Sci. Rep. 8(1), 12007 (2018).
Alam, S. N., Sharma, N. & Kumar, L. Synthesis of graphene oxide (GO) by modified hummers method and its thermal reduction to obtain reduced graphene oxide (rGO). Graphene 6(1), 1–18 (2017).
Zaaba, N. et al. Synthesis of graphene oxide using modified hummers method: Solvent influence. Procedia Eng. 184, 469–477 (2017).
Benzait, Z., Chen, P. & Trabzon, L. Enhanced synthesis method of graphene oxide. Nanoscale Adv. 3(1), 223–230 (2021).
Habte, A. T. & Ayele, D. W. Synthesis and characterization of reduced graphene oxide (rGO) started from graphene oxide (GO) using the tour method with different parameters. Adv. Mater. Sci. Eng. 2019(1), 5058163 (2019).
Dimiev, A. et al. Pristine graphite oxide. J. Am. Chem. Soc. 134(5), 2815–2822 (2012).
Boehm, H. P. Graphene—how a laboratory curiosity suddenly became extremely interesting. Angew. Chem. Int. Ed. 49(49), 9332–9335 (2010).
Cai, M. et al. Methods of graphite exfoliation. J. Mater. Chem. 22(48), 24992–25002 (2012).
Shimizu, K. et al. Probing the surface tension of ionic liquids using the Langmuir principle. Langmuir 34(14), 4408–4416 (2018).
Ensminger, D. & Bond, L. J. Ultrasonics: Fundamentals, Technologies, and Applications (CRC, 2024).
Paton, K. R. et al. Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids. Nat. Mater. 13(6), 624–630 (2014).
Hernandez, Y. et al. High-yield production of graphene by liquid-phase exfoliation of graphite. Nat. Nanotechnol. 3(9), 563–568 (2008).
Abdolhosseinzadeh, S., Asgharzadeh, H. & Kim, H. S. Fast and fully-scalable synthesis of reduced graphene oxide. Sci. Rep. 5(1), 10160 (2015).
Cham sa-ard, W. et al. Synthesis, characterisation and thermo-physical properties of highly stable graphene oxide-based aqueous nanofluids for potential low-temperature direct absorption solar applications. Sci. Rep. 11(1), 16549 (2021).
Borode, A. O. et al. Effect of various surfactants on the viscosity, thermal and electrical conductivity of graphene nanoplatelets nanofluid. Int. J. Thermophys. 42(11), 158 (2021).
Wadzer, Y., Htwe, Y. Z. N. & Mamat, H. A review of ceramic nanofluids and their applications. J. Dispers. Sci. Technol. 1–24 (2024).
Cao, L. et al. Hydrophilic graphene preparation from gallic acid modified graphene oxide in magnesium self-propagating high temperature synthesis process. Sci. Rep. 6(1), 35184 (2016).
Yang, S. et al. A facile green strategy for rapid reduction of graphene oxide by metallic zinc. RSC Adv. 2 (23), 8827–8832 (2012).
Olumurewa, K. et al. Effect of hydrothermal and chemical treatment on the optical and electrical properties of reduced graphene oxide deposited on ITO glass. Mater. Res. Express. 7(10), 105606 (2020).
Cao, J. et al. Two-step electrochemical intercalation and oxidation of graphite for the mass production of graphene oxide. J. Am. Chem. Soc. 139(48), 17446–17456 (2017).
Rabchinskii, M. K. et al. Nanoscale perforation of graphene oxide during photoreduction process in the argon atmosphere. J. Phys. Chem. C 120(49), 28261–28269 (2016).
Wang, A. et al. Covalent functionalization of reduced graphene oxide with porphyrin by means of diazonium chemistry for nonlinear optical performance. Sci. Rep. 6(1), 23325 (2016).
Saxena, S. et al. Investigation of structural and electronic properties of graphene oxide. Appl. Phys. Lett. 99(1) (2011).
Eluyemi, M. et al. Synthesis and characterization of graphene oxide and reduced graphene oxide thin films deposited by spray pyrolysis method. Graphene 5(3), 143–154 (2016).
Huang, H. H. et al. Structural evolution of hydrothermally derived reduced graphene oxide. Sci. Rep. 8(1), 6849 (2018).
Loryuenyong, V. et al. Preparation and characterization of reduced graphene oxide sheets via water-based exfoliation and reduction methods. Adv. Mater. Sci. Eng. 2013 (1), 923403 (2013).
Thakur, S. & Karak, N. Green reduction of graphene oxide by aqueous phytoextracts. Carbon 50(14), 5331–5339 (2012).
Sharma, N. et al. A comparative study on gas-sensing behavior of reduced graphene oxide (rGO) synthesized by chemical and environment-friendly green method. Appl. Nanosci. 10, 517–528 (2020).
Siddiqui, M. S. & Aslam, M. Highly stable and reusable 3D graphene-quinizarin voltammetric pH sensor. J. Electrochem. Soc. 170(4), 047511 (2023).
Li, D. et al. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 3(2), 101–105 (2008).
Feng, H. et al. A low-temperature method to produce highly reduced graphene oxide. Nat. Commun. 4(1), 1539 (2013).
Paredes, J. et al. Atomic force and scanning tunneling microscopy imaging of graphene nanosheets derived from graphite oxide. Langmuir 25(10), 5957–5968 (2009).
Samson, V. A. F. et al. rGO Sheets/ZnFe2O4 nanocomposities as an efficient electro catalyst material for I3–/I – reaction for high performance DSSCs. J. Inorg. Organomet. Polym Mater. 32(3), 1183–1189 (2022).
Abid et al. Reduced graphene oxide (rGO) based wideband optical sensor and the role of temperature, defect States and quantum efficiency. Sci. Rep. 8(1), 3537 (2018).
Zhang, D. et al. Room-temperature macroscopic ferromagnetism in multilayered graphene oxide. Adv. Phys. Res. 3(5), 2300092 (2024).
Siddiqui, M. S. et al. Graphene oxide array for in-depth soil moisture sensing toward optimized irrigation. ACS Appl. Electron. Mater. 2(12), 4111–4121 (2020).
Scardaci, V. & Compagnini, G. Raman spectroscopy investigation of graphene oxide reduction by laser scribing. C 7(2), 48 (2021).
King, A. A. et al. A new Raman metric for the characterisation of graphene oxide and its derivatives. Sci. Rep. 6(1), 1–6 (2016).
Some, S. et al. High-quality reduced graphene oxide by a dual-function chemical reduction and healing process. Sci. Rep., 3(1), 1929 (2013).
Huynh, N. M. N. et al. Reduced graphene oxide as a water, carbon dioxide and oxygen barrier in plasticized poly (vinyl chloride) films. RSC Adv. 8(32), 17645–17655 (2018).
Ma, B. et al. Fabrication of WO 2.72/RGO nano-composites for enhanced photocatalysis. RSC Adv. 7(5), 2606–2614 (2017).
Das, P. et al. Facile Preparation of self-healable and recyclable multilayered graphene-based nanocomposites for electromagnetic interference shielding applications. Colloids Surf., A 676, 132244 (2023).
Mohammedture, M. et al. Impact of probe sonication and sulfuric acid pretreatment on graphene exfoliation in water. Sci. Rep. 13(1), 18523 (2023).
Mushfiq, S. W. & Afzalzadeh, R. Few-layered graphene production utilizing ultrasonic probe with simulations for optimization of geometrical parameters. Bull. Mater. Sci. 47(3), 196 (2024).
da Silva Gascho, J. L. et al. The effect of silver nanowires on the formation of aggregates of poly (3-hexylthiophene) in films deposited on reduced graphene oxide. J. Nanopart. Res. 23(7), 153 (2021).
Foschi, M. et al. Experimental design and response surface methodology applied to graphene oxide reduction for adsorption of triazine herbicides. ACS Omega 6(26), 16943–16954 (2021).
Paranthaman, V. et al. Investigation on the performance of reduced graphene oxide as counter electrode in dye sensitized solar cell applications. Phys. Status Solidi (a). 215 (18), 1800298 (2018).
Wang, H. et al. Preparation of RGO/TiO2/Ag aerogel and its photodegradation performance in gas phase formaldehyde. Sci. Rep. 9(1), 16314 (2019).
Mattevi, C. et al. Evolution of electrical, chemical, and structural properties of transparent and conducting chemically derived graphene thin films. Adv. Funct. Mater. 19(16), 2577–2583 (2009).
Ray, A. K. et al. Preparation and characterization of graphene and Ni-decorated graphene using flower petals as the precursor material. Carbon 50(11), 4123–4129 (2012).
Jankovský, O. et al. Use of deuterium labelling—evidence of graphene hydrogenation by reduction of graphite oxide using aluminium in sodium hydroxide. RSC Adv. 5(24), 18733–18739 (2015).
Poh, H. L. et al. Graphenes prepared by Staudenmaier, Hofmann and hummers methods with consequent thermal exfoliation exhibit very different electrochemical properties. Nanoscale 4(11), 3515–3522 (2012).
Gengler, R. Y. et al. Revealing the ultrafast process behind the photoreduction of graphene oxide. Nat. Commun. 4(1), 2560 (2013).
Al-Gaashani, R. et al. XPS and structural studies of high quality graphene oxide and reduced graphene oxide prepared by different chemical oxidation methods. Ceram. Int. 45(11), 14439–14448 (2019).
Kashyap, A., Barman, P. B. & Hazra, S. K. Response of rGO and Pd-decorated rGO to carbon monoxide gas. J. Electron. Mater. 52(3), 1999–2011 (2023).
Bueno-López, J. I. et al. Effects of graphene oxide and reduced graphene oxide on acetoclastic, hydrogenotrophic and methylotrophic methanogenesis. Biodegradation 31, 35–45 (2020).
Ricardo, K. B., Sendecki, A. & Liu, H. Surfactant-free exfoliation of graphite in aqueous solutions. Chem. Commun. 50(21), 2751–2754 (2014).
Taniguchi, T. et al. Reversible hydrogenation and irreversible epoxidation induced by graphene oxide electrolysis. Carbon 177, 26–34 (2021).
Gong, Y. et al. Influence of graphene microstructures on electrochemical performance for supercapacitors. Progr. Nat. Science: Mater. Int. 25(5), 379–385 (2015).
Faniyi, I. et al. The comparative analyses of reduced graphene oxide (RGO) prepared via green, mild and chemical approaches. SN Appl. Sci. 1, 1–7 (2019).
Kanta, U. et al. Preparations, characterizations, and a comparative study on photovoltaic performance of two different types of graphene/TiO2 nanocomposites photoelectrodes. J. Nanomater. 2017(1), 2758294 (2017).
Zhu, C. et al. Reducing sugar: New functional molecules for the green synthesis of graphene nanosheets. ACS Nano. 4(4), 2429–2437 (2010).
Acknowledgements
We would like to acknowledge SPM/AFM, ESCA facilities at Physics department, and Sophisticated Analytical Instrument Facility (SAIF), Indian Institute of Technology Bombay for providing the characterization support. J.A. Sarkar thanks IIT Bombay and MoE (Ministry of Education) for financial support.
Author information
Authors and Affiliations
Contributions
J.A.S.: Conceptualization, Methodology, Data curation, Formal analysis, Investigation, Software, Visualization, Writing—original draft, Writing-review & editing. M.S.S.: Writing—review & editing. M.A.: Supervision, Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sarkar, J.A., Siddiqui, M.S. & Aslam, M. Acid-less direct gram scale exfoliation of graphite to partially oxidized graphene. Sci Rep 15, 31505 (2025). https://doi.org/10.1038/s41598-025-99502-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99502-x
