
Abstract
The DEFECTIVE KERNEL 1 (DEK1) protein plays essential functions throughout plant development. DEK1 is a multidomain 240 kDa protein with yet unsolved 3D structure. To facilitate structural and functional studies of DEK1, here we investigate its calpain protease core domain (CysPc) from Physcomitrium patens. Using integrated structural modelling we propose targeted mutagenesis of CysPc to enhance its solubility during recombinant protein production. We created a pipeline to predict the topology of the CysPc domain with improved precision, providing a robust framework for further exploration. We evaluated the native and mutant structures by MD simulations, concentrating on several solubility-related parameters. Following these features, we implemented specific single, double, and triple amino acid mutagenesis to select variants with improved solubility. Our method preserves overall structural integrity while reducing aggregation-prone traits. We advocate for the utilization of optimized data driven method that can effectively traverse the extensive combinatorial space and prioritize mutation sets with the greatest potential for enhancing solubility. This framework provides a logical, data-driven approach to improving protein solubility, particularly beneficial in situations lacking high-resolution structural data.
Similar content being viewed by others


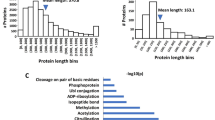
Data availability
The data generated and analysed during this study, including molecular dynamics simulation outputs, structural models, and solubility feature datasets and code resources will be made available from the corresponding authors upon reasonable request. Due to the size and computational nature of the datasets, they are not hosted in a public repository. Full mutagenesis dataset and related materials are provided in supplementary information.
References
Villaverde, A. Mar Carrió, M. Protein aggregation in Recombinant bacteria: biological role of inclusion bodies. Biotechnol. Lett. 25, 1385–1395 (2003).
Baneyx, F. & Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 22, 1399–1408 (2004).
Nemova, N. N., Lysenko, L. A. & Kantserova, N. P. Proteases of the Calpain family: structure and functions. Russ J. Dev. Biol. 41, 318–325 (2010).
Melloni, E., Salamino, F. & Sparatore, B. The calpain-calpastatin system in mammalian cells: properties and possible functions. Biochimie 74, 217–223 (1992).
Suzuki, K., Hata, S., Kawabata, Y. & Sorimachi, H. Structure, Activation, and biology of Calpain. Diabetes 53, S12–S18 (2004).
Zhao, S. et al. Massive expansion of the Calpain gene family in unicellular eukaryotes. BMC Evol. Biol. 12, 193 (2012).
Johansen, W. et al. The DEK1 calpain Linker functions in three-dimensional body patterning in Physcomitrella patens. Plant Physiol. 00925.https://doi.org/10.1104/pp.16.00925 (2016).
Johnson, K. L., Faulkner, C., Jeffree, C. E. & Ingram, G. C. The phytocalpain defective kernel 1 is a novel Arabidopsis growth regulator whose activity is regulated by proteolytic processing. Plant. Cell. 20, 2619–2630 (2008).
Lid, S. et al. (ed, E.) The defective kernel 1 (dek1) gene required for aleurone cell development in the endosperm of maize grains encodes a membrane protein of the Calpain gene superfamily. Proc. Natl. Acad. Sci. U S A 99 5460–5465 (2002).
Demko, V. et al. Regulation of developmental gatekeeping and cell fate transition by the Calpain protease DEK1 in physcomitrium patens. Commun. Biol. 7, 261 (2024).
Pantophlet, R., Wilson, I. A. & Burton, D. R. Improved design of an antigen with enhanced specificity for the broadly HIV-neutralizing antibody b12. Protein Eng. Des. Selection. 17, 749–758 (2004).
De Marco, A., Deuerling, E., Mogk, A., Tomoyasu, T. & Bukau, B. Chaperone-based procedure to increase yields of soluble Recombinant proteins produced in E. coli. BMC Biotechnol 7 (2007).
Gustafsson, C., Govindarajan, S. & Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 22, 346–353 (2004).
Chatterjee, D. K. & Esposito, D. Enhanced soluble protein expression using two new fusion tags. Protein Exp. Purif. 46, 122–129 (2006).
Esposito, D. & Chatterjee, D. K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 17, 353–358 (2006).
Sachdev, D. & Chirgwin, J. M. Properties of soluble fusions between mammalian aspartic proteinases and bacterial Maltose-Binding protein. J. Protein Chem. 18, 127–136 (1999).
Agostini, F., Cirillo, D., Bolognesi, B. & Tartaglia, G. G. X-inactivation: quantitative predictions of protein interactions in the Xist network. Nucleic Acids Res. 41, e31–e31 (2013).
Kulshreshtha, S., Chaudhary, V., Goswami, G. K. & Mathur, N. Computational approaches for predicting mutant protein stability. J. Comput. Aided Mol. Des. 30, 401–412 (2016).
Damborsky, J. & Brezovsky, J. Computational tools for designing and engineering enzymes. Curr. Opin. Chem. Biol. 19, 8–16 (2014).
Ebert, M. C. & Pelletier, J. N. Computational tools for enzyme improvement: why everyone can – and should – use them. Curr. Opin. Chem. Biol. 37, 89–96 (2017).
Broom, A., Jacobi, Z., Trainor, K. & Meiering, E. M. Computational tools help improve protein stability but with a solubility tradeoff. J. Biol. Chem. 292, 14349–14361 (2017).
Childers, M. C. & Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2, 9–33 (2017).
Rouhani, M., Khodabakhsh, F., Norouzian, D., Cohan, R. A. & Valizadeh, V. Molecular dynamics simulation for rational protein engineering: present and future prospectus. J. Mol. Graph. Model. 84, 43–53 (2018).
Pikkemaat, M. G., Linssen, A. B. M., Berendsen, H. J. C. & Janssen, D. B. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. Des. Selection. 15, 185–192 (2002).
Carballo-Amador, M. A., McKenzie, E. A., Dickson, A. J. & Warwicker, J. Surface patches on Recombinant erythropoietin predict protein solubility: engineering proteins to minimise aggregation. BMC Biotechnol 19 (2019).
Kumar, S., Kumar Bhardwaj, V., Singh, R. & Purohit, R. Explicit-solvent molecular dynamics simulations revealed conformational regain and aggregation Inhibition of I113T SOD1 by Himalayan bioactive molecules. J. Mol. Liq. 339, 116798 (2021).
Chennamsetty, N., Voynov, V., Kayser, V., Helk, B. & Trout, B. L. Prediction of aggregation prone regions of therapeutic proteins. J. Phys. Chem. B. 114, 6614–6624 (2010).
Agrawal, N. J. et al. Aggregation in Protein-Based biotherapeutics: computational studies and tools to identify Aggregation-Prone regions. J. Pharm. Sci. 100, 5081–5095 (2011).
Ako, A. E. et al. An intragenic mutagenesis strategy in physcomitrella patens to preserve intron splicing. Sci. Rep. 7, 5111 (2017).
Perroud, P. et al. Defective kernel 1 (DEK 1) is required for three-dimensional growth in P hyscomitrella patens. New Phytol. 203, 794–804 (2014).
Navarro, S. & Ventura, S. Computational re-design of protein structures to improve solubility. Expert Opin. Drug Discov. 14, 1077–1088 (2019).
Trainor, K., Broom, A. & Meiering, E. M. Exploring the relationships between protein sequence, structure and solubility. Curr. Opin. Struct. Biol. 42, 136–146 (2017).
Gupta, J., Nunes, C., Vyas, S. & Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B. 115, 2014–2023 (2011).
Ganugapati, J. & Akash, S. Multi-template homology based structure prediction and molecular Docking studies of protein ‘L’ of Zaire ebolavirus (EBOV). Inf. Med. Unlocked. 9, 68–75 (2017).
Lu, H., Cheng, Z., Hu, Y. & Tang, L. V. What can De Novo protein design bring to the treatment of hematological disorders? Biology 12, 166 (2023).
Roy, A., Yang, J. & Zhang, Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40, W471–W477 (2012).
Holm, L., Laiho, A., Törönen, P. & Salgado, M. DALI shines a light on remote homologs: one hundred discoveries. Protein Sci. 32, e4519 (2023).
Chan, P., Curtis, R. A. & Warwicker, J. Soluble expression of proteins correlates with a lack of positively-charged surface. Sci. Rep. 3, 3333 (2013).
Studer, G. et al. QMEANDisCo—distance constraints applied on model quality Estimation. Bioinformatics 36, 1765–1771 (2020).
Van Den Bedem, H. & Fraser, J. S. Integrative, dynamic structural biology at atomic resolution—it’s about time. Nat. Methods. 12, 307–318 (2015).
Heo, L. & Feig, M. High-accuracy protein structures by combining machine‐learning with physics‐based refinement. Proteins 88, 637–642 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Alden, K., Veretnik, S. & Bourne, P. E. dConsensus: a tool for displaying domain assignments by multiple structure-based algorithms and for construction of a consensus assignment. BMC Bioinform. 11, 310 (2010).
Batista, P. R. et al. Consensus modes, a robust description of protein collective motions from multiple-minima normal mode analysis—application to the HIV-1 protease. Phys. Chem. Chem. Phys. 12, 2850 (2010).
Lobanov, M. Y., Bogatyreva, N. S. & Galzitskaya, O. V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 42, 623–628 (2008).
Abouzied, A. S. et al. Structural and free energy landscape analysis for the discovery of antiviral compounds targeting the cap-binding domain of influenza polymerase PB2. Sci. Rep. 14, 25441 (2024).
Pace, C. N. et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 23, 652–661 (2014).
Jiang, L. & Lai, L. CH···O hydrogen bonds at Protein-Protein interfaces. J. Biol. Chem. 277, 37732–37740 (2002).
Tsumoto, K. et al. Role of arginine in protein Refolding, Solubilization, and purification. Biotechnol. Prog. 20, 1301–1308 (2004).
Strub, C. et al. Mutation of exposed hydrophobic amino acids to arginine to increase protein stability. BMC Biochem. 5, 9 (2004).
Warwicker, J., Charonis, S. & Curtis, R. A. Lysine and arginine content of proteins: computational analysis suggests a new tool for solubility design. Mol. Pharm. 11, 294–303 (2014).
Kramer, R. M., Shende, V. R., Motl, N., Pace, C. N. & Scholtz, J. M. Toward a molecular Understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys. J. 102, 1907–1915 (2012).
Mills, B. J. & Laurence Chadwick, J. S. Effects of localized interactions and surface properties on stability of protein-based therapeutics. J. Pharm. Pharmacol. 70, 609–624 (2018).
Trevino, S. R., Scholtz, J. M. & Pace, C. N. Measuring and increasing protein solubility. J. Pharm. Sci. 97, 4155–4166 (2008).
Kuhn, A. B. et al. Improved Solution-State properties of monoclonal antibodies by targeted mutations. J. Phys. Chem. B. 121, 10818–10827 (2017).
Ghahremanian, S., Rashidi, M. M., Raeisi, K. & Toghraie, D. Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: A structural review. J. Mol. Liq. 354, 118901 (2022).
Xiao, S. et al. Rational modification of protein stability by targeting surface sites leads to complicated results. Proc. Natl. Acad. Sci. U S A. 110, 11337–11342 (2013).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Mokmak, W., Chunsrivirot, S., Assawamakin, A., Choowongkomon, K. & Tongsima, S. Molecular dynamics simulations reveal structural instability of human trypsin inhibitor upon D50E and Y54H mutations. J. Mol. Model. 19, 521–528 (2013).
Zhou, H. X. & Pang, X. Electrostatic interactions in protein Structure, Folding, Binding, and condensation. Chem. Rev. 118, 1691–1741 (2018).
Gregory, K. P. et al. Understanding specific ion effects and the hofmeister series. Phys. Chem. Chem. Phys. 24, 12682–12718 (2022).
Hyde, A. M. et al. General principles and strategies for Salting-Out informed by the hofmeister series. Org. Process. Res. Dev. 21, 1355–1370 (2017).
Tadeo, X., López-Méndez, B., Castaño, D., Trigueros, T. & Millet, O. Protein stabilization and the hofmeister effect: the role of hydrophobic solvation. Biophys. J. 97, 2595–2603 (2009).
Tadeo, X., Pons, M. & Millet, O. Influence of the hofmeister anions on protein stability as studied by thermal denaturation and chemical shift perturbation. Biochemistry 46, 917–923 (2007).
Sammond, D. W. et al. Structure-based protocol for identifying mutations that enhance Protein–Protein binding affinities. J. Mol. Biol. 371, 1392–1404 (2007).
Xu, D., Tsai, C. J. & Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. Des. Selection. 10, 999–1012 (1997).
Goldenzweig, A. & Fleishman, S. J. Principles of protein stability and their application in computational design. Annu. Rev. Biochem. 87, 105–129 (2018).
Rosano, G. L. & Ceccarelli, E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5 (2014).
Zheng, W. et al. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell. Rep. Methods. 1, 100014 (2021).
Zhang, C., Freddolino, L. & Zhang, Y. COFACTOR: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 45, W291–W299 (2017).
Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of Spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Fiser, A., Šali, A. & Modeller Generation and refinement of Homology-Based protein structure models. in Methods in Enzymology vol. 374 461–491 (Elsevier, 2003).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods. 19, 679–682 (2022).
Waterhouse, A. M. et al. The structure assessment web server: for proteins, complexes and more. Nucleic Acids Res. 52, W318–W323 (2024).
Zheng, W. et al. Deep-learning-based single-domain and multidomain protein structure prediction with D-I-TASSER. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02654-4 (2025).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40, W294–W297 (2012).
Fiser, A. & Sali, A. ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 (2003).
Lüthy, R., Bowie, J. U. & Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 (1992).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods. 19, 1109–1115 (2022).
Xu, J. & Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26, 889–895 (2010).
Wiederstein, M. & Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 (2007).
Uziela, K., Menéndez Hurtado, D., Shu, N., Wallner, B. & Elofsson, A. ProQ3D: improved model quality assessments using deep learning. Bioinformatics 33, 1578–1580 (2017).
Ramachandran, G. N., Ramakrishnan, C. & Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99 (1963).
Lovell, S. C. et al. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 50, 437–450 (2003).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Brooks, B. R. et al. The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 14, 71–73 (2017).
MacKerell, A. D. et al. All-Atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102, 3586–3616 (1998).
Berendsen, H. J. C., Postma, J. P. M., Van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Allen, M. P. & Tildesley, D. J. Computer Simulation of Liquids (Oxford University PressOxford, 2017). https://doi.org/10.1093/oso/9780198803195.001.0001
Darden, T., York, D. & Pedersen, L. Particle mesh ewald: an N ⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Chialvo, A. A. & Debenedetti, P. G. On the use of the Verlet neighbor list in molecular dynamics. Comput. Phys. Commun. 60, 215–224 (1990).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Bauer, P., Hess, B. & Lindahl, E. GROMACS 2022 Source code. Zenodo https://doi.org/10.5281/ZENODO.6103835 (2022).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Galm, L., Amrhein, S. & Hubbuch, J. Predictive approach for protein aggregation: correlation of protein surface characteristics and conformational flexibility to protein aggregation propensity. Biotech. Bioeng. 114, 1170–1183 (2017).
Soares, C. M., Teixeira, V. H. & Baptista, A. M. Protein structure and dynamics in nonaqueous solvents: insights from molecular dynamics simulation studies. Biophys. J. 84, 1628–1641 (2003).
Friedman, R., Nachliel, E. & Gutman, M. Molecular dynamics of a protein surface: Ion-Residues interactions. Biophys. J. 89, 768–781 (2005).
Zhang, Y. & Cremer, P. Interactions between macromolecules and ions: the hofmeister series. Curr. Opin. Chem. Biol. 10, 658–663 (2006).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Arantes, P. R., Ligabue-Braun, R., Pedebos, C. & eRMSF: A python package for Ensemble-Based RMSF analysis of biomolecular systems. J Chem. Inf. Model. Acs Jcim. 5c02413 (2025).
Sormanni, P., Aprile, F. A. & Vendruscolo, M. The camsol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 427, 478–490 (2015).
Hebditch, M., Carballo-Amador, M. A., Charonis, S., Curtis, R. & Warwicker, J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 33, 3098–3100 (2017).
Meng, E. C., Pettersen, E. F., Couch, G. S., Huang, C. C. & Ferrin, T. E. Tools for integrated sequence-structure analysis with UCSF chimera. BMC Bioinform. 7, 339 (2006).
Abraham, M. J. et al. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2. GROMACS, 19–25 (2015).
Van Der Spoel, D., Van Maaren, P. J., Larsson, P. & Tîmneanu, N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B. 110, 4393–4398 (2006).
van der Bondi, A. Waals volumes and radii. J. Phys. Chem. 68, 441–451 (1964).
Schulman, J., Wolski, F., Dhariwal, P., Radford, A. & Klimov, O. Proximal Policy Optimization Algorithms. Preprint at (2017). https://doi.org/10.48550/ARXIV.1707.06347
Funding
This work is the result of implementation of Slovak Research and Development Agency grants APVV-21-0227, APVV-21-0215, APVV-22-0161, Comenius University grant UK/1088/2025 and by implementation of the project 101160008 “Fostering Excellence in Advanced Genomics and Proteomics Research at Comenius University in Bratislava – FORGENOM II” funded by the Horizon Europe program.
Author information
Authors and Affiliations
Contributions
MD and ZL: conceived and designed the study, MD: developed the structural prediction and mutagenesis workflow, and performed all molecular dynamics simulations and solubility analyses. Data processing, interpretation, and manuscript writing and editing were carried out by MD and ZL. VD and ES: Provided recommendations and scientific guidance of research. All aspects of the research were conducted under the academic supervision of JT, VD, VB and SS who provided critical feedback on the study design and manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep (2026). https://doi.org/10.1038/s41598-026-38805-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-026-38805-z
