
Abstract
Ultra-high-resolution mass spectrometry was used to investigate the characterization of aerosol organonitrates (ONs). ONs featured high-molecular-weight (HMW), high unsaturation, and high functionalization. Under high air pollution events, the number of N1Ox class ONs composed of aliphatic and aromatic compounds increased with high levels of oxidation. In the daytime, highly unsaturated aliphatic-ONs were continuously produced. In the nighttime, the atmospheric oxidation of NO3 radicals promoted ONs. N2Ox class ONs were mainly comprised of multi-generation oxidated aliphatic compounds with high unsaturation. Most of the N2Ox-ONs were from cooking. Biomass burning also played an indelible role in the formation of ONs. In the daytime, atmospheric photodegradation led to the removal of the HMW ONs, especially aliphatic ONs. During nighttime, the NO3 oxidation radicals inhibited the generation of ONs, especially anthropogenic ONs. This study improves the understanding of the source, formation, and evolution of HMW ONs under the demand for continuous PM mitigation.
Similar content being viewed by others

Introduction
Organonitrates (ONs) refer to the general term for esters and their derivatives containing nitric acid functional group (–ONO2) and peroxynitric acid (–O2NO2)1. ONs are important classes of active nitrogen oxides in the atmosphere as temporary storage of nitrogen oxides (NOx), affecting the atmospheric nitrogen cycle in various ways2,3.
ONs originate from direct emissions and atmospheric oxidation processes. Some short-chain alkyl nitrates, such as methyl nitrate, ethyl nitrate, and propyl nitrate, are emitted from the ocean and industry as the primary sources. These primary ONs majorly distributed in the gas phase and trivially in the particle phase4,5. ONs are mainly formed via atmospheric oxidation reactions between nitrogen oxides and volatile organic compounds (VOCs) with oxidative radicals such as OH, O3, and NO32. During the daytime, the oxidation reactions between anthropogenic precursor and OH and O3 are the main pathways for the formation of ONs6. These precursor compounds include typical polycyclic aromatic hydrocarbons (PAHs), toluene, xylene and acenaphthylene emitted from biomass burning vehicle exhaust and solid fuel combustion7. ONs are widely used in tracking anthropogenic SOA8,9. In heavily populated urban areas, anthropogenic VOCs may be more dominant precursors of particulate ONs than biogenic VOCs such as isoprene and pinene.
Additionally, some biogenic VOCs, e.g., pinene, as a typical natural source alkene, undergo photochemical reactions with OH radicals during the day, forming ONs through the RO2 + NO pathway10,11. At night, NO3 radicals can react with isoprene, pinene, limonene, and oleic acid to produce a significant amount of gas-phase and particulate-phase ONs12,13. ONs can subsequently partition into the particulate phase through gas-particle partitioning or heterogeneous reactions.
The formation of ONs is influenced not only by precursor and oxidant concentrations but also by aerosol physical-chemical properties, human activities, and atmospheric conditions. Changes in atmospheric conditions, such as temperature, precipitation, humidity, and meteorological factors, are crucial for ONs regional transport and impact the global nitrogen cycle14. Under light wind conditions, local circulations like sea-land breezes or valley winds can dominate pollutant transport. Studies have shown that valley winds significantly affect ONs formation8,15. Sadanaga et al. found that during cold seasons, the atmospheric lifetime of ONs is extended, and their concentration is influenced by regional air mass transport16.
Particle-phase ONs are often highly functionalized, which means containing nitric ester hydroxyl (–OH), carboxyl (–COO), carbonyl (–C = O), or multiple –ONO2 groups17. Due to the hydrophilicity of –OH, –COO, ONs can be somehow water-soluble and favorable for being removed by wet deposition6,18,19. These groups can reduce the saturation vapor pressure of ONs, leading to the partitioning into the particle phase20. By influencing cloud condensation nuclei, hygroscopicity of particles, etc., ONs directly impact the global climate21,22. Furthermore, they have an impact on human health and other organisms on the earth23.
Since the 1980s, particle-phase ONs have been detected and analyzed. Over time, ONs in particles have drawn attention as a class of secondary organic aerosol24. The contribution of ONs to the organic matter (OM) in PM2.5 ranges from ~ 5% to 40%25. ONs are significant organic compounds with diverse atmospheric sources. ONs have diversified sources26, including anthropogenic ONs27,28,29,30 represented by alkane, olefin, and biogenic ONs generated from atmospheric oxidation of isoprene and pinene12,31,32. Fisher et al. found that the contribution of anthropogenic ONs was as high as 40% and reached a peak in the early morning in forest areas3. The studies implied that NO3 oxidation could play an important role in particulate ONs formation at night17,25,33,34. Day et al. also observed a high abundance of ONs in the urban areas of Los Angeles, suggesting the contribution of human activities to ONs20.
Huang et al. identified ONs in nighttime particulates with carbon numbers ranging from five to fifteen, oxygen numbers from four to twelve, and a weighted average mass of 191.135. Xu et al. in their source apportionment of ONs, focused on m/z < 60 to accurately separate and quantify ONs in OA, discovering that winter atmospheric ONs were mainly influenced by biomass burning, while in the other three seasons, they primarily originated from secondary transformation processes36. In their study on the atmospheric cycling of nitrogen oxides, Wu et al. demonstrated the contribution of particulate ONs to OA mass. They explored the atmospheric transformation and migration processes of ONs with m/z below 10037. We found that most studies focused on particle ONs with small molecular weights based on the literatures. The knowledge of high molecular weight (HMW, m.w. > 500 Da) ONs, including their sources, formation, and evolution, was still limited. In particular, atmospheric formation and removal of HMW ONs need to be further investigated.
In this study, the chemical composition of ONs in a typical medium-sized city in the Yangtze River Delta (YRD) was analyzed using electrospray ionization (ESI) coupled with Fourier Transform Ion Cyclotron Resonance mass spectrometry (FT-ICR MS) to explore the atmospheric ONs behaviors based on four sampling covered whole 24 h. The sources, formation, and evolution of HMW ONs with varying nitrogen contents in different levels of pollution were discussed. This study aims to enhance the comprehension of ONs’ behaviors throughout the diurnal cycle and offers insights into the HMW components of SOA. This study can improve our knowledge of the behavior of high-molecular-weight organic compounds and their secondary surrogates.
Results
Overall composition characteristics of ONs
Generally, ONs refer to nitrogenous organic compounds (CHON) in which the ratio of the number of oxygen atoms to the number of nitrogen atoms is greater than or equal to 3 (O/N ≥ 3)25,38. CHON detected in negative ESI mode (-ESI) is more likely to be compounds of acidic-ONs containing –ONO or –ONO2 groups39. Previous studies suggested that ester compounds containing nitryl in CHONS were more likely to exist in the form of sulfate40,41. Therefore, only CHON compounds and ONs detected in -ESI mode were analyzed in this study.
Based on the time variation of PM2.5 mass concentration (Supplementary Fig. 1), we selected samples with a daily average PM2.5 concentration exceeding the air quality standard limit (75 μg/m³) for pollution day analysis. The pollution day was identified as April 29, 2021, with a 24 h average PM2.5 concentration of 90.6 μg/m³ from four consecutive sampling periods. Conversely, the clean day was May 1, 2021, with a 24-hour average PM2.5 concentration of 36.2 μg/m³ from the same periods, allowing us to study changes in the molecular composition of SOA in the ambient environment. A total of 8 samples were collected and analyzed. In the following text, to differentiate samples from different periods on polluted and clean days, we will add the prefix H and C, which stands for the hazy and clean days, respectively. For example, H-MEM represents the sample from the midnight and early morning (MEM) period on polluted days.
Evolution of ONs
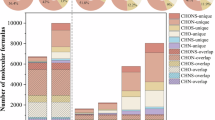
A number of 1,855 CHON molecules were detected, with a relative abundance of 28.24% (Supplementary Table 1) during the haze days. ONs molecules satisfying O/N ≥ 3 in CHON accounted for 80.1% of CHON (n = 1486) (Table 1). When atmospheric pollution occurred in northern China (Beijing), the relative abundance of CHON was 23%–25%42, with ONs constituting 80% of CHON43, similar to that in the YRD.
In addition, there were a total of 970 CHON molecules in the clean days with a relative abundance of 33.5%, with an abundance of ONs molecules for 74.7% (n = 725). ONs detected on haze days were almost 2 times that on clean days, consistent with our previous organosulfate study44. With the deterioration of air quality, the occurrence of ONs increased significantly.
The average molecular weight of ONs was 594.93 Da, and the carbon chain length was 32 on haze days, and 627.46 Da of molecular weight and 34 on the carbon chain length on clean days. It should be noted that ONs in this research area have higher molecular weights and longer carbon chains on both clean and haze days (Table 1). The molecular weight of ONs in this study was notably higher than those reported in studies by Huang et al. in rural southwestern Germany (300–450 Da)35 and Lee et al. in the southeastern United States (250–450 Da)38. It is indicated that the contribution of HMW compounds to ONs increased in the YRD region.
Figure 1 shows the 3D distribution of DBE, carbon number, and O/N ratio of ONs compounds in haze and clean days. ONs containing up to 57 carbon atoms were found with oxygen atoms of 5–15, indicating the high functionalization of ONs35.

3D diagram of DBE, C number, and O/N ratio of ONs compounds in haze and clean days.
Only 202 ONs (green) coexisted in both haze and clean days, 1278 ONs molecules (red) only in haze days, and 632 ONs compounds (blue) exist only in clean days. Higher DBE values (8.11 vs. 7.79) and a greater aromaticity index (1.72 vs. 1.59) were shown in clean days than those in haze days. On clean days, highly unsaturated ONs (DBE ≥ 5) were primarily compounds with 5–7 oxygen atoms and contain only one nitrogen atom, mainly appearing during the midnight and early morning (MEM) (Supplementary Fig. 2). This was likely generated from the nighttime oxidation reactions of NO3 radicals with biogenic alkenes (such as isoprene and pinene)12,13. The peak intensities of highly unsaturated ONs on polluted days consistently showed a significantly higher trend than clean days. The precursors were photo-oxidized and formed a substantial amount of ONs. The semi-volatile long-chain alkanes and polycyclic aromatic compounds could be recognized as a priority in control strategies. In addition, ONs found in polluted days had a wider distribution in carbon number than in clean days, suggesting the breaking-up of long-chain alkanes and PAHs was possible23.
Formation and removal of ONs
Figure 2 is a schematic diagram of ONs’ formation and removal based on the KMD plot. When atmospheric pollution occurred, the formation of ONs occurred along with their removal. It should be noted that enormous HMW ONs were generated in the afternoon compared to other periods, with a relative abundance accounting for about 86.5% of all newly formed ONs (Fig. 2b). Among these, HMW ONs with 8–9 oxygen atoms were particularly prominent, with a relative abundance of about 8.0%, which was roughly twice that of other ONs with different oxygen contents (Supplementary Fig. 3). This indicated that photooxidation promoted the generation of these HMW ONs.

(a–c) show the KMD diagrams for –CH2. (d–f) show the KMD diagrams for –COO.
While HMW ONs were mainly eliminated in the nighttime. In the afternoon, approximately 49.3% of HMW ONs were removed from the atmosphere by nightfall (Fig. 2c). These HMW ONs predominantly had oxygen atom numbers ranging from six to nine (Supplementary Fig. 3), showing little variation in their relative intensity. However, the nighttime atmospheric removal of HMW ONs primarily focused on compounds with higher DBE values (DBE ≥ 5, RI: 36.38%). Nearly a quarter of these compounds were polycyclic aromatic ONs with DBE values of twelve and fifteen (Supplementary Fig. 4), containing two or more benzene rings. Smog chamber studies had demonstrated that the heterogeneous oxidation of nitro-polycyclic aromatic hydrocarbons by O3 was a significant degradation pathway for these ONs at night45,46. Additionally, these HMW ONs were likely produced from multi-stage oxidation reactions during the day, so the formation of polyols through hydrolysis of HMW ONs at night may also be a primary reason for their atmospheric removal47,48.
The atmospheric formation and removal of low molecular weight (LMW) ONs exhibited a trend opposite to that of HMW ONs. As sunlight intensity increased during the day, the removal of LMW ONs was enhanced. In the morning, only 9.9% of LMW ONs were removed, but this figure nearly doubles by the afternoon, with ONs containing 7–9 oxygen atoms accounting for 10.5%. Daytime removal primarily targeted ONs from the N1O9 and N2O7 classes. In the morning, ONs of N1O9 class undergoing removal represented 34.0% of total LMW ONs (Supplementary Fig. 3). Unsaturated aliphatic compounds with multiple oxygen-containing groups (e.g., –COO, –OH, –C = O), such as C21H29N1O9, were cleared through double bond cleavage under intense photochemical oxidation (e.g., –OH, O3)49,50.
In the afternoon, the removal of LMW ONs of the N2O7 class primarily involved newly formed alkyl nitrates from the morning, with alkanes, alkenes, and other compounds likely serving as the main atmospheric sources of these alkyl nitrates through the “carbonyl-imine” conversion pathway51. Consistent with findings by O’Brien et al.52, ONs of N2Ox class predominantly appeared during midnight and morning periods, influenced by daytime anthropogenic activities such as biomass burning53,54. Intense photodegradation may cleave chemical bonds in complex ONs containing two nitrogen atoms. Furthermore, the dynamic equilibrium between particulate nitrate, gaseous nitric acid, and ammonia can lead to ammonium nitrate evaporation during high afternoon temperatures55, thereby reducing the abundance of particulate nitrate.
The formation and removal of atmospheric ONs in clean days were the same as those in haze days (Supplementary Fig. 3). However, ONs were only present in trace amounts compared to polluted days, owing to the limited availability of precursors (Supplementary Fig. 2). Furthermore, the generated new ONs were almost completely eliminated in the next period, nearly 98.34% of ONs undergo atmospheric removal on average during each time period. This result implies that a strong cleansing process44. Our results also suggested that those ONs were highly likely to be removed in the aerosol phase due to their high molecular weight and low volatility, improving our understanding of the atmospheric fates of HMW compounds in the aerosol phase.
Composition characteristics of ONs of N1Ox Class
Chemical composition
Based on the identified composition of ONs, they can be categorized into two classes: N1O4–N1O15 (N1Ox) with one nitrogen atom, and N2O7–N2O15 (N2Ox) contained two nitrogen atoms.
Figure 3 shows the distribution of DBE and C number of ONs of N1Ox class, indicating that all ONs molecules have other oxygen-containing functional groups53. ONs of N1O4 class containing only one –ONO2 group were mainly distributed in the range of DBE 10–13, and the C numbers of ONs molecular with the highest abundance were 44. ONs with abundance concentrated in the range with higher C numbers (C > 20). Additionally, as the oxygen content increased, the high abundance ONs progressively shifted towards the range of low DBE values. This is particularly evident for ONs with an oxygen count between 13 and 15, where ONs with high abundance were concentrated in the DBE range of 1 to 2 and primarily consist of long-chain aliphatic compounds. The compound of ONs of N1O14 class with the highest abundance was C12H22O14N1 with lower C numbers and a DBE value of 2. It is speculated that its structure may be aliphatic ONs.

The sizes of the circle in the figure indicate the relative abundance (100%) of the compound.
Comparing polluted days with clean days (Supplementary Fig. 6), N1O6–N1O9 compounds were the main ONs compound in haze days. The relative abundance of N1O8–N1O9 with high oxygen numbers decreased in clean days, and the contribution of ONs of N1O5–N1O7 classes increased significantly. Previous studies have confirmed that particle-phase ONs following multi-generation oxidation can contain 6 to 8, or even more oxygen atoms38. The highly oxidized macromolecular ONs make a significant contribution to SOA. The differences indicates that a large part of aerosol underwent secondary oxidation along with atmospheric pollution, leading to the appearance of aging ONs with high oxygen numbers.
By considering DBE, we found that ONs in polluted days were predominantly composed of highly unsaturated ONs with DBE ≥ 5. Similar to the CHON1 compounds found in aged aerosols by Brege et al. 56. The atmospheric oxidation of polycyclic aromatic hydrocarbon (PAH) precursors was likely the main cause, biomass burning was likely one of the inevitable major sources of ONs57. When PM2.5 concentration increased, the contribution of ONs with small DBE increased gradually, especially ONs with DBE between 2 and 5, showing a relative abundance increase of approximately 6 times compared to clean days (Supplementary Fig. 4). Under high NOx conditions, the photooxidation and photodegradation of VOCs were important formation pathways of these ONs58,59.
Diurnal variation of N1Ox
A Van Krevelen (VK) diagram of CHON was utilized using H/C ratio vs. O/C ratio. The aromaticity of CHON1 and ONs was characterized using AImod. We found that CHON1 was mainly distributed in the range of O/C ratio of 0–0.2 (mean 0.13) in haze days in Fig. 4a–d. ONs exhibited a higher oxidation state (O/Cw: 0.30). Combined with the previous description, it was further demonstrated that ONs of N1Ox class contained other oxygen-containing groups besides to a –ONO2 group.

(a–d) indicate MEM, M, A, and N periods in haze days respectively. (e–h) indicate those in clean days. The compounds were denoted as CHON1 compound (circle) and ONs (triangle) respectively.
CHON1 compounds exhibited a significantly higher oxidation state in haze days compared with that in clean days (Fig. 4e–h). The oxidation state of ONs was higher than that of other nitrogen-containing compounds in both haze and clean days, especially from the afternoon to night in haze days. The presence of many ONs with a high oxidation state indicated an obvious atmospheric aging process caused by high oxidants concentration in unfavorable air-stagnant conditions. For example, the concentrations of hydroxyl radicals (OH) were 7.25E–0.3 ppb and 2.13E–0.3 ppb, and the concentrations of nitrate radicals (NO3) were 1.01E–02 ppb and 4.70E–04 ppb, respectively, in Guangzhou during the atmospheric pollution period and non-pollution period60.
Because ONs had three or above more oxygen atoms than ordinary nitrogenous compounds, (O–2 N)/C was used to further truly reflect the oxidation state of ONs (Supplementary Fig. 7). The nitrogen-containing compounds were mostly aliphatic or highly unsaturated/phenolic compounds which may be aromatic-ONs formed by atmospheric oxidation of certain PAHs precursors61,62,63 (Fig. 4). Aliphatic and peptide compounds were prenominated during the midnight of the haze days instead of aromatic-ONs (Fig. 4d). These ONs also had a high O/N ratio, indicating that these compounds contained enormous unsaturated or saturated oxygen-containing groups, such as –COO, –C = O, –OH, etc. The atmospheric oxidation of oxygenated precursors was likely the main reason for their atmospheric formation3,20.
In order to accurately explain the characteristics of ONs’ aromaticity, the aromaticity index (Xc) was introduced. Figure 5 shows the number and relative abundance of aromatic and aliphatic-ONs detected in haze days. In Fig. 5a, b, the number of aliphatic compounds (n = 87–145, 45.4%) was slightly smaller than that of aromatic compounds (n = 114–189, 54.6%). As for relative abundance, the proportion of aliphatic-ONs in ONs was about 48.4%, slightly lower than aromatic compounds.

(a–c) indicate the number, number proportion, and relative abundance percentage of ONs in haze days. (d–f) indicate those in clean days.
Aliphatic-ONs showed a upward trend from the daytime (n = 194, 31.1%), to nighttime (n = 282, 38.6%), and reached a peak in the night (n = 145, RI: 39.9%). By contrast, aromatic compounds slightly decreased in the afternoon but increased during the nighttime. Compared with the clean period (Fig. 5d–f), the daily variations of aliphatic and aromatic ONs were consistent with those in haze days, indicating that the atmospheric photodegradation in the daytime mainly occurred on aromatic-ONs.
In addition, ONs increased in the night in both haze and clean days, indicating that the oxidation reaction led by NO3 radical in the nighttime had a positive effect on the generation of ONs of N1Ox class8,25.
During the haze days, aliphatic-ONs of N1Ox class mainly concentrated in the range of DBE 0–5 (Supplementary Fig. 8), with an average relative abundance (RI) of about 18.9%, which reached a peak in the MEM period (Fig. 5b) (RI: 21.4%). Low unsaturated ONs were most abundant with DBE equal to 4. According to their minimum carbon number, their structure may be as shown in Supplementary Fig. 9. Compared with clean period, ONs with DBE of 2–5 (Supplementary Fig. 6) and aliphatic-ONs with DBE of 4 showed the highest abundance when the oxygen content was equal to 8, then decreased with the improvement of air quality. The result suggests that the aliphatic-ONs could be more oxidized in haze days, showing higher oxygen atoms numbers. Some highly unsaturated compounds were also found in aliphatic-ONs in haze days (RI: 16.0%), and the relative abundance of these highly unsaturated aliphatic-ONs showed a small decreasing trend in clean days (RI: 11.0%).
Aromatic-ONs mainly existed in highly unsaturated (DBE > 7) compounds and reached a peak in the morning (RI: 38.5%), which was significantly affected by vehicle exhaust in the morning rush hour17. This indicates that the photooxidation of semi-volatile VOCs in the daytime contributed. Aromatic-ONs with high abundance appeared when DBE equaled 12 and oxygen content was between 6 and 8. The distribution was relatively uniform, and most were in the form of condensed aromatics. Combined with the above results, groups such as –COO and –C = O may be the main forms of oxygen-containing functional groups in aromatic-ONs.
Characteristics of ONs of N2Ox class
Chemical composition
Figure 6 shows the distribution of DBE and C number of ONs of N2O7–N2O15 classes. Previous studies revealed that ONs of N2Ox class could be generated from biomass burning, mainly heterocyclic nitric ester oxygen-containing derivatives53,54. Lignin and guaiacol were commonly used as tracers for biomass burning emissions64. Guaiacol can react with NO3 radicals in the atmosphere to form ONs of the N2Ox class (such as C12H7N2O4 and C12H10N2O7)65. We found ONs presented as C18H22N2O7 during the night on polluted days in this study, which may be homologs of nitroguaiacol. The result also suggested that biomass burning products were likely one of the main precursors of ONs in the YRD region.

The sizes of the dots in the figure indicate the relative abundance of the compound molecules.
As shown in Fig. 6, the DBE distribution of ONs of N2O7–N2O15 in the regions with of 15, 15, 5, 12, 0, and 7, and higher C numbers. Besides, the high abundance N2O11 compounds with DBE of 0 were a class of saturated long-chain aliphatic compounds, indicating that besides –ONO2 groups, and –OH was the main oxygen-containing functional group. The N2O11 compounds could be multi-generation hydroxyl nitrate oleic acids ester after the heterogeneous reaction of atmospheric NO3 radicals66,67,68. Therefore, the N2O11 class was likely to be from cooking, which is dominated by oleic acid or other homolog precursors.
In the clean days, the relative abundance of ONs of N2O8 class was the highest, about 10 times that of other oxygen-containing ONs, and the relative abundance decreased significantly when pollution appeared (Supplementary Fig. 10). ONs of this class were mainly composed of aliphatic compounds with DBE between 0 and 2 with plant sources as precursors (e.g. Limonene12). In the haze days, the overall relative abundance of ONs with oxygen content between 7 and 15 did not show significant differences. Meanwhile, the relative abundance contribution of ONs with higher DBE values increased in haze days (65.2% in haze days and 30.6% in clean days). The result was consistent with the presentation of ONs of N1Ox class due to atmospheric oxidation of PAHs precursors from anthropogenic sources.
Diurnal variations of N2Ox
CHON2 compounds in the haze days were mainly distributed within the O/C ratio of 0–0.2, while ONs showed high oxidation states (Fig. 7). As shown in Fig. 7e–h, the oxidation state of ONs was also significantly higher than that of other nitrogen-containing compounds in the clean days (Supplementary Fig. 11). The result suggested that, the N2Ox compounds had undergone significant atmospheric aging in both haze and clean days. CHON2 compounds were mostly aliphatic or highly unsaturated/phenolic compounds, consistent with the condition of containing only one nitrogen atom. The aromatic-N2Ox compounds showed a significant increase after atmospheric pollution events, and the dominating N2Ox had changed from highly aromatic compounds to aliphatic compounds, as shown in Fig. 7c, d, suggesting the accumulation of secondary products from cooking emissions.

(a–d) indicate MEM, M, A, and N periods in haze days respectively. (e–h) indicate those in clean days. The compounds were denoted as CHON1 compound (circle) and ONs (triangle) respectively.
Figure 8 shows the cumulative figure of the number and relative abundance of ONs of N2Ox class. Compared with Fig. 5, we found obvious differences in the number and relative abundance of ONs of N2Ox class. The variable environmental factors such as the concentration of atmospheric precursors and oxidants, were the main factors limiting two complex –ONO2 functional groups in the multi-generation oxidation products69,70. At the same time, the solubility of organic matter in different solvents was also a factor that cannot be ignored71.

(a–c) indicate the number, number proportion, and relative abundance percentage of ONs in haze days. (d–f) indicate those in clean days.
Aliphatic ONs compounds were more abundant in both number and relative abundance (n = 65–81, 66.9%) than aromatic compounds (n = 32–40, 33.1%) (Fig. 8a, b). Regarding relative abundance, aliphatic-ONs constituted approximately 68.7% of the whole ONs, aligning with the trend observed in molecular numbers and significantly exceeding that of aromatic compounds.
The diurnal profiles of ONs showed a gradual downward trend from the morning in haze days (Fig. 8a). Aliphatic-ONs exhibited a trend of initially decreasing, then gradually increasing, and reached the minimum value in the afternoon (RI: 59.0%). Aromatic compounds peaked in the afternoon (RI: 41.0%), mainly composed of condensed aromatics (32.8%). There was no significant difference in the remaining periods, which were all around 28.0% (Fig. 8c). Unlike ONs of N1Ox class, this shows that photochemical oxidation in the daytime can promote the generation of aromatic-ONs with two nitrogen atoms.
Compared the haze days, ONs showed a trend of first decreasing and then increasing during the clean days (Fig. 8d–f). The minimum value was caused by strong atmospheric photodegradation in the afternoon, and the number gradually increased at night under the photooxidation of NO3 radicals. By comparing the diurnal variation of ONs between the haze and clean days, it is further indicated that the oxidation of NO3 radical at night has an inhibitory effect on the generation of ONs of N2Ox class mainly from anthropogenic sources.
In the haze days, ONs were distributed across DBE range of 0–11, with an average relative abundance of approximately 19.3% (Supplementary Fig. 12). Their peak occurrence was in the morning, where the RI reached 22.3% (Supplementary Fig. 12b). The average abundance of low unsaturated ONs (DBE: 0–4) was 9.7%, showing an obvious trend of a greater value in the daytime than in the nighttime. Compared to highly unsaturated aliphatic-ONs, they were most abundant when the DBE was 5 and the oxygen content was 9 (Supplementary Fig. 13). Groups such as –COO, –C = O, and –OH may be the main form of their existence. Because the molecule contains two nitrogen atoms, it is difficult to determine its specific structure.
Aromatic-ONs mainly existed in highly unsaturated (DBE: 8–18) compounds and reached a peak in the morning (RI: 14.1%). Aromatic-ONs with high abundance were evenly distributed when DBE equaled 15 and oxygen content was between 7 and 9. With a high carbon number, most of them were polymeric compounds with condensed aromatic hydrocarbons as the main component72.
Discussion
In this study, -ESI-FT-ICR MS was used to characterize the atmospheric environment of typical small and medium-sized urban areas in YRD, China. The composition, possible sources, and atmospheric evolution of ONs were discussed. High molecular weight, long carbon chain, and high unsaturation were the main molecular characteristics of ONs in the region.
The double bond equivalent (DBE) of ONs were generally consistent with those found in other cities in the YRD region, similar to those in Nanjing73 (3–10), the northern coastal city of Dalian74 (15.86 on polluted days and 10.94 on clean days) and the other cities like Beijing, Shanghai, and Guangzhou (DBE 3–25)75. We found that ONs in particulate matter in the YRD region mostly existed in the form of long carbon chain (C > 20) high molecular weight compounds. This result differed from past findings in the North China Plain, such as in rural areas of Beijing (m. w. 200–300 Da)76 and urban areas of Tianjin (around 300 Da)77.
Additionally, this study focused on the diurnal variation of ONs with different nitrogen atom numbers in the development of haze pollution. We have determined that the increase of ONs was mainly caused by the increase of highly unsaturated precursors. In the nighttime, the oxidation reaction dominated by NO3 promotes the generation of ONs of N1Ox class. This study determined the evolution of HMW aliphatic and aromatic compounds to form ONs in PM2.5.
However, in the face of variable atmospheric environments, such as emission sources, meteorological conditions, atmospheric chemical reactions, and regional transport, our understanding of the atmospheric generation driving factors and chemical mechanisms of ONs remains limited. In particular, the generation mechanism of ONs by nighttime NO3 radicals requires further in-depth study using techniques such as smog chambers. This will help us further understand the formation of ONs in the aerosol phase. At the same time, the technical improvement of FT-ICR MS in quantitative analyzing will also help to expand its application in aerosol chemistry.
In summary, the study nuanced understanding of ONs’ atmospheric behavior and the role of NO3 radicals provides crucial insights into the mechanisms driving organic aerosol evolution, and the importance of volatility of high molecular weight organic compounds in the secondary organic aerosol formation.
Methods
Sample collection
The sampling site was located on the rooftop of a building 45 m above ground level in a park in Huai’an City, Jiangsu Province, East China (119.0734°E, 33.6047°N). The site was surrounded by streets, national forest parks, and residential areas. Without industrial emissions, biomass, or the use of coal nearby, the site can be recognized as a typical urban environment. A full description is available in literature44.
Particles were collected using a high-flow particulate sampler (Thermo Inc, USA) with a flow rate of 1.13 m3/min. The sampling period was 23:00–04:00 (Midnight and Early Morning, labeled as MEM), 05:00–10:30 (Morning, labeled as M), 11:00–16:30 (Afternoon, labeled as A), and 17:00–22:30 (Night, labeled as N) from April 18 to May 11, 2021.
FT-ICR MS analysis
The organic chemical composition of aerosol was analyzed with 7.0 T Bruker Solarix 2XR FT-ICR MS equipped with the ESI ion source. Inorganic compounds in the sample could seriously interfere with the separation of organic matter under the ESI source, so pretreatment of the samples was necessary. 10 mL acetonitrile solution was used to soak a certain area of the filter membrane sample, and sonication was performed three times.
Concentrate the acetonitrile sample solution to 1 mL after sonication. Before FT-ICR MS, the mixture of acetonitrile extract was vaporized to dry using pure nitrogen. Then, the dry extract was reconstituted with water, and methanol was added proportionally to the water solution before FT-ICR MS analysis. The samples were dissolved in methanol at about 50 mg/L for the ESI analysis.
The treated sample solution was injected into the ESI source by a syringe pump with a flow rate of 120 µg/L. -ESI source operating conditions were set as follows: inlet capillary voltage of −4.0 kV and outlet voltage of −220 V. The ions were accumulated in the hexapole for 0.05 s and then transferred to the ICR pool with a time of flight of 0.7 ms. The ion conversion parameter was set to the mass-charge ratio (m/z) of 200 Da for the quadrupole (Q1), and the quality detection range was 150–1000 Da78.
The data calibration method for FT-ICR MS followed the work of He et al. 78, using a reference list of known molecular formulas (12C0-100, 1H0-200, 14N0-10, 16O0-20, 32S0-2) to calibrate the complex mass spectrometry data, with accuracy under 1.0 ppm. The calculated formulas must adhere to specified element ratio limits79, as shown by Eqs. (1)–(3). Mass peaks with a signal-to-noise (S/N) ratio ≥ 4 were exported to a data sheet. Data analysis was performed using in-house software. The blank filters were processed similarly to sample filters.
FT-ICR MS data processing
In order to explore the molecular composition of ONs, the aromaticity index (AImod, Xc) and the number of the double bond equivalent (DBE) corresponding to each ONs molecule were calculated based on the research methods of Koch&Dittmar80 and Wang et al. 81, as shown in Eqs. (4)–(6).
Kendrick Mass Defect (KMD) was a commonly used data representation method in FT-ICR MS data analysis, providing an effective tool for visualizing complex organic mixtures82,83. The –CH2 and –COO groups were commonly used for the structural characterization of organic matter84,85,86. The KMD calculations for the two different groups were shown in Eqs. (7)–(8) below.
In addition, the ion with the highest peak intensity in the mass spectrum in each sample was defined as a relative abundance equivalent to 100%, and the peak intensity of all ions in the sample was measured using the normalization principle. The relative intensity after normalization was used to represent the relative abundance of each compound in the sample87. The percentage of each compound composed of different elements in the total number of sample compounds was referred to as the percentage of number. It was the compound molecular formulas detected in the blank filter that should be excluded, regardless of how high the response was in the sample compounds88,89.
Data availability
The datasets used and analysed during the current study available from the corresponding author on reasonable request.
References
Day, D. A. et al. On alkyl nitrates, O3, and the “missing NOy”. J. Geophys. Res. Atmos. 108, 4501 (2003).
Farmer, D. K. et al. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. P Natl Acad Sci. USA 107, 6670–6675 (2010).
Fisher, J. A. et al. Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US. Atmos. Chem. Phys. 16, 5969–5991 (2016).
Bertman, S. B. et al. Evolution of alkyl nitrates with air-mass age. J. Geophys. Res. Atmos 100, 22805–22813 (1995).
Perring, A. E. et al. Airborne observations of total RONO2: new constraints on the yield and lifetime of isoprene nitrates. Atmos. Chem. Phys. 9, 1451–1463 (2009).
Perring, A. E., Pusede, S. E. & Cohen, R. C. An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol. Chem. Rev. 113, 5848–5870, (2013).
Ravindra, K., Sokhi, R. & Van Grieken, R. Atmospheric polycyclic aromatic hydrocarbons: Source attribution, emission factors and regulation. Atmos. Environ. 42, 2895–2921 (2008).
Fry, J. L. et al. Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011. Atmos. Chem. Phys. 13, 8585–8605 (2013).
Fry, J. L. et al. Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model. Atmos. Chem. Phys. 9, 1431–1449 (2009).
Aschmann, S. M., Reisseil, A., Atkinson, R. & Arey, J. S. Products of the gas phase reactions of the OH radical with α‐ and β‐pinene in the presence of NO. J. Geophys. Res. Atmos. 103, 25553–25561 (1998).
Rindelaub, J. D. et al. The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact. Atmos. Chem. Phys. 16, 15425–15432 (2016).
Fry, J. L. et al. SOA from limonene: role of NO3 in its generation and degradation. Atmos. Chem. Phys. 11, 3879–3894 (2011).
Talbot, R. W. et al. Tropospheric reactive odd nitrogen over the South Pacific in austral springtime. J. Geophys. Res. Atmos 105, 6681–6694 (2000).
Tie, X. et al. A budget analysis of the formation of haze in Beijing. Atmos. Environ. 100, 25–36 (2015).
Salvador, C. M. et al. Measurements of submicron organonitrate particles: Implications for the impacts of NOx pollution in a subtropical forest. Atmos. Res. 245, 105080 (2020).
Sadanaga, Y. et al. Behavior of total peroxy and total organic nitrate concentrations at Suzu on the Noto Peninsula, Japan: Long-range transport and local photochemical production. Atmos. Environ. 196, 20–26 (2019).
Xu, L., Suresh, S., Guo, H., Weber, R. J. & Ng, N. L. Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates. Atmos. Chem. Phys. 15, 7307–7336 (2015).
Mazzoleni, L. R., Ehrmann, B. M., Shen, X., Marshall, A. G. & Collett, J. L. Jr Water-Soluble Atmospheric Organic Matter in Fog: Exact Masses and Chemical Formula Identification by Ultrahigh-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Environ. Sci. Technol. 44, 3690 (2010).
Neff, J. C., Holland, E. A., Dentener, F. J., McDowell, W. H. & Russell, K. M. The origin, composition and rates of organic nitrogen deposition: A missing piece of the nitrogen cycle? Biogeochemistry 57, 99–136 (2002).
Day, D. A., Liu, S., Russell, L. M. & Ziemann, P. J. Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California. Atmos. Environ. 44, 1970–1979 (2010).
Moise, T., Flores, J. M. & Rudich, Y. Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes. Chem. Rev. 115, 4400–4439, (2015).
Topping, D. O. & McFiggans, G. Tight coupling of particle size, number and composition in atmospheric cloud droplet activation. Atmos. Chem. Phys. 12, 3253–3260 (2012).
Ng, N. L. et al. Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 17, 2103–2162 (2017).
Hallquist, M. et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 3555–3762 (2009).
Rollins, A. W. et al. Evidence for NOx Control over Nighttime SOA Formation. Science 337, 1210 (2012).
Garnes, L. A. & Allen, D. T. Size Distributions of Organonitrates in Ambient Aerosol Collected in Houston, Texas. Aerosol Sci. Technol. 36, 983–992 (2002).
O’Brien, J. M. et al. Measurements of alkyl and multifunctional organic nitrates at a rural site in Ontario. J. Geophys. Res. Atmos. 100, 22795–22804 (1995).
Zhang, J. K. et al. Characterization of submicron particles during biomass burning and coal combustion periods in Beijing, China. Sci. Total Environ. 562, 812–821 (2016).
Lyu, X. P. et al. Re-examination of C1-C5 alkyl nitrates in Hong Kong using an observation-based model. Atmos. Environ. 120, 28–37 (2015).
Simpson, I. J. et al. Long-term atmospheric measurements of C1-C5 alkyl nitrates in the pearl river delta region of southeast China. Atmos. Environ. 40, 1619–1632 (2006).
Grossenbacher, J. W. et al. Measurements of isoprene nitrates above a forest canopy. J. Geophys. Res. Atmos 106, 24429–24438 (2001).
He, Q. F. et al. Organosulfates from Pinene and Isoprene over the Pearl River Delta, South China: Seasonal Variation and Implication in Formation Mechanisms. Environ. Sci. Technol. 48, 9236 (2014).
Rollins, A. W. et al. Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield. J. Geophys. Res. -Atmos. 118, 6651–6662 (2013).
Holzinger, R., Kasper-Giebl, A., Staudinger, M., Schauer, G. & Röckmann, T. Analysis of the chemical composition of organic aerosol at the Mt. Sonnblick observatory using a novel high mass resolution thermal-desorption proton-transfer-reaction mass-spectrometer (hr-TD-PTR-MS). Atmos. Chem. Phys. 10, 10111–10128 (2010).
Huang, W. et al. Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth. Environ. Sci. Technol. 53, 1165–1174 (2019).
Xu, W. et al. Seasonal Characterization of Organic Nitrogen in Atmospheric Aerosols Using High Resolution Aerosol Mass Spectrometry in Beijing, China. ACS Earth Space Chemistry 1, 673–682 (2017).
Wu, Y. et al. Enhanced formation of nitrogenous organic aerosols and brown carbon after aging in the planetary boundary layer. npj Clim. Atmos. Sci. 7, 179 (2024).
Lee, B. H. et al. Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets. Proc. Natl. Acad. Sci. USA 113, 1516–1521 (2016).
Jiang, H. X. et al. Factors Influencing the Molecular Compositions and Distributions of Atmospheric Nitrogen-Containing Compounds. J. Geophys. Res. Atmos. 127, 15 (2022).
Altieri, K. E., Turpin, B. J. & Seitzinger, S. P. Composition of Dissolved Organic Nitrogen in Continental Precipitation Investigated by Ultra-High Resolution FT-ICR Mass Spectrometry. Environ. Sci. Technol. 43, 6950–6955, (2009).
Wang, Y. J. et al. The formation of nitro-aromatic compounds under high NOx and anthropogenic VOC conditions in urban Beijing, China. Atmos. Chem. Phys. 19, 7649–7665 (2019).
Ning, C. et al. Urban particulate water-soluble organic matter in winter: Size-resolved molecular characterization, role of the S-containing compounds on haze formation. Sci. Total Environ. 875, 162657 (2023).
Kim, M. S. et al. Comparison of secondary organic aerosol (SOA)-associated molecular features at urban sites in China and Korea in winter and summer (2019). Atmos. Environ. 318, 120235 (2024).
Du, Y. et al. Characterization of Organosulfates (OSs) in typical urban areas in Eastern China: Source, process, and volatility. Atmos. Res. 301, 107258 (2024).
Yamasaki, H., Kuwata, K. & Miyamoto, H. Effects of ambient temperature on aspects of airborne polycyclic aromatic hydrocarbons. Environ. Sci. Technol. 16, 189–194 (1982).
Tokiwa, H. & Ohnishi, Y. Mutagenicity and carcinogenicity of nitroarenes and their sources in the environment. Crit. Rev. Toxicol. 17, 23–60 (1986).
Boyd, C. M. et al. Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate. Atmos. Chem. Phys. 15, 7497–7522 (2015).
Hu, K. S., Darer, A. I. & Elrod, M. J. Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates. Atmos. Chem. Phys. 11, 8307–8320 (2011).
Pavuluri, C. M., Kawamura, K., Mihalopoulos, N. & Swaminathan, T. Laboratory photochemical processing of aqueous aerosols: formation and degradation of dicarboxylic acids, oxocarboxylic acids and α-dicarbonyls. Atmos. Chem. Phys. 15, 7999–8012 (2015).
Hu, W. et al. Photochemical Degradation of Organic Matter in the Atmosphere. Adv. Sustain 5, 2100027 (2021).
Riva, M. et al. Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes. Atmos. Chem. Phys. 16, 11001–11018 (2016).
O’Brien, R. E. et al. Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass Spectrometry: CalNex 2010 field study. J. Geophys. Res. Atmos. 119, 12,706–712,720 (2014).
Song, J. et al. Molecular Characterization of Water-Soluble Humic like Substances in Smoke Particles Emitted from Combustion of Biomass Materials and Coal Using Ultrahigh-Resolution Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Environ. Sci. Technol. 52, 2575–2585 (2018).
Laskin, A., Smith, J. S. & Laskin, J. Molecular Characterization of Nitrogen-Containing Organic Compounds in Biomass Burning Aerosols Using High-Resolution Mass Spectrometry. Environ. Sci. Technol. 43, 3764–3771, (2009).
Wang, D. F. et al. Intense secondary aerosol formation due to strong atmospheric photochemical reactions in summer: observations at a rural site in eastern Yangtze River Delta of China. Sci. Total Environ. 571, 1454–1466 (2016).
Brege, M. et al. Molecular insights on aging and aqueous-phase processing from ambient biomass burning emissions-influenced Po Valley fog and aerosol. Atmos. Chem. Phys. 18, 13197–13214 (2018).
Atkinson, R., Tuazon, E. C., Bridier, I. & Arey, J. Reactions of NO3-naphthalene adducts with O2 and NO2. Int. J. Chem. Kinet. 26, 605–614 (1994).
Yuan, B. et al. Secondary formation of nitrated phenols: insights from observations during the Uintah Basin Winter Ozone Study (UBWOS) 2014. Atmos Chem. Phys. 16, 2139–2153 (2016).
Atkinson, R. & Arey, J. Atmospheric degradation of volatile organic compounds. Chem. Rev. 103, 4605–4638, (2003).
Li, W. Study on the sources and atmospheric processes of organonitrates in particulate matter in urban areas of the Pearl River Delta. Jinan University, (2022).
Jiang, H. et al. Molecular Dynamics and Light Absorption Properties of Atmospheric Dissolved Organic Matter. Environ. Sci. Technol. 55, 10268–10279 (2021).
Antony, R. et al. Origin and Sources of Dissolved Organic Matter in Snow on the East Antarctic Ice Sheet. Environ. Sci. Technol. 48, 6151–6159 (2014).
Medeiros, P. M. et al. Dissolved organic matter composition and photochemical transformations in the northern North Pacific Ocean. Geophys. Res. Lett. 42, 863–870 (2015).
Yazdani, A. et al. Chemical evolution of primary and secondary biomass burning aerosols during daytime and nighttime. Atmos. Chem. Phys. 23, 7461–7477 (2023).
Lauraguais, A. et al. Atmospheric reactivity of hydroxyl radicals with guaiacol (2-methoxyphenol), a biomass burning emitted compound: Secondary organic aerosol formation and gas-phase oxidation products. Atmos. Environ. 86, 155–163 (2014).
Hung, H.-M., Katrib, Y. & Martin, S. T. Products and Mechanisms of the Reaction of Oleic Acid with Ozone and Nitrate Radical. J. Geophys. Res. Atmos. 109, 4517–4530 (2005).
Zhao, Z., Husainy, S., Stoudemayer, C. T. & Smith, G. D. Reactive uptake of NO3 radicals by unsaturated fatty acid particles. Phys. Chem. Chem. Phys. 13, 17809–17817, (2011).
Docherty, K. S. & Ziemann, P. J. Reaction of oleic acid particles with NO3 radicals: Products, mechanism, and implications for radical-initiated organic aerosol oxidation. J. Geophys. Res. Atmos. 110, 3567–3577 (2006).
Ng, N. L. et al. Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3). Atmos. Chem. Phys. 8, 4117–4140 (2008).
Kwan, A. J. et al. Peroxy radical chemistry and OH radical production during the NO3 initiated oxidation of isoprene. Atmos. Chem. Phys. 12, 7499–7515 (2012).
Jiang, B. et al. Molecular composition of urban organic aerosols on clear and hazy days in Beijing: a comparative study using FT-ICR MS. Environ. Chem. 13, 888–901 (2016).
Claflin, M. S. & Ziemann, P. J. Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms. J. Geophys. Res. Atmos. 122, 3640–3652 (2018).
Bao, M. et al. Impact of fossil and non-fossil fuel sources on the molecular compositions of water-soluble humic-like substances in PM2.5 at a suburban site of Yangtze River Delta, China. Atmos. Chem. Phys. 23, 8305–8324 (2023).
Ning, C. et al. Molecular characterization of dissolved organic matters in winter atmospheric fine particulate matters PM2.5 from a coastal city of northeast China. Sci. Total Environ. 689, 312–321 (2019).
Zhang, M. et al. Molecular characterization of atmospheric organic aerosols in typical megacities in China. npj Clim. Atmos. Sci. 7, 230 (2024).
Zhu, Q. et al. Characterization of Organic Aerosol at a Rural Site in the North China Plain Region: Sources, Volatility and Organonitrates. Adv. Atmos. Sci. 38, 1115–1127 (2021).
Zhong, S. et al. Impact of biogenic secondary organic aerosol (SOA) loading on the molecular composition of wintertime PM2.5 in urban Tianjin: an insight from Fourier transform ion cyclotron resonance mass spectrometry. Atmos. Chem. Phys. 23, 2061–2077 (2023).
He, C. et al. In-House Standard Method for Molecular Characterization of Dissolved Organic Matter by FT-ICR Mass Spectrometry. ACS omega 5, 11730–11736 (2020).
Kujawinski, E. B. & Behn, M. D. Automated analysis of electrospray ionization Fourier transform ion cyclotron resonance mass spectra of natural organic matter. Anal. Chem. 78, 4363–4373, (2006).
Koch, B. P. & Dittmar, T. From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 20, 926–932 (2006).
Wang, X. K. et al. Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River. Atmos. Chem. Phys. 16, 2285–2298 (2016).
Stenson, A. C., Marshall, A. G. & Cooper, W. T. Exact masses and chemical formulas of individual Suwannee River fulvic acids from ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectra. Anal. Chem. 75, 1275–1284, (2003).
Kendrick, E. A Mass Scale Based on CH2 = 14.0000 for High Resolution Mass Spectrometry of Organic Compounds. Anal Chem. 35, 2146–2154 (1962).
Qi, Y., Fu, P. & Volmer, D. A. Analysis of natural organic matter via fourier transform ion cyclotron resonance mass spectrometry: an overview of recent non-petroleum applications. Mass Spectrom. Rev. 41, 647–661 (2022).
Sleighter, R. L. & Hatcher, P. G. The application of electrospray ionization coupled to ultrahigh resolution mass spectrometry for the molecular characterization of natural organic matter. J. Mass Spectrom. 42, 559–574 (2007).
Zhang, B. et al. Transformation of dissolved organic matter during full-scale treatment of integrated chemical wastewater: Molecular composition correlated with spectral indexes and acute toxicity. Water Res. 157, 472–482 (2019).
Wu, Z. G., Rodgers, R. P. & Marshall, A. G. Characterization of vegetable oils: Detailed compositional fingerprints derived from electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Agric. Food. Chem. 52, 5322–5328 (2004).
Lin, P., Rincon, A. G., Kalberer, M. & Yu, J. Z. Elemental Composition of HULIS in the Pearl River Delta Region, China: Results Inferred from Positive and Negative Electrospray High Resolution Mass Spectrometric Data. Environ. Sci. Technol. 46, 7454–7462, (2012).
Lin, P., Yu, J. Z., Engling, G. & Kalberer, M. Organosulfates in Humic-like Substance Fraction Isolated from Aerosols at Seven Locations in East Asia: A Study by Ultra-High-Resolution Mass Spectrometry. Environ. Sci. Technol. 46, 13118–13127 (2012).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (grant no. 42107452, 42075109 and 42003059) and the Postdoctoral Science Foundation of Chongqing (grant no. CSTB2022 NSCQ-BHX0033).
Author information
Authors and Affiliations
Contributions
Y.D. conducted data analysis and wrote the original draft. H.X.C., J.W.Z., and Q.L. collected samples. Z.E.B., Y.L.L., Y.H., L.S.M, X.L., X.Q., X.J.Y, and S.M.Z. developed the concept and established the methodology. C.H., Q.S., M.H, and C.M.C. analyzed samples and provided technical support. Y.C. acquired funding, provided resources, and reviewed and edited the writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Du, Y., Che, H., Bao, Z. et al. Evolution of atmospheric high-molecular-weight Organonitrates (HMW ONs) in urban Yangtze River Delta (YRD), China. npj Clim Atmos Sci 8, 50 (2025). https://doi.org/10.1038/s41612-025-00928-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41612-025-00928-x
