
Abstract
The rapid development of deep learning has revolutionized medical image processing, including analyzing whole slide images (WSIs). Despite the demonstrated potential for characterizing gene mutations directly from WSIs in certain cancers, challenges remain due to image resolution and reliance on manual annotations for acute myeloid leukemia (AML). We, therefore, propose a deep learning model based on multiple instance learning (MIL) with ensemble techniques to predict gene mutations from AML WSIs. Our model predicts NPM1 mutations and FLT3-ITD without requiring patch-level or cell-level annotations. Using a dataset of 572 WSIs, the largest database with both WSI and genetic mutation information, our model achieved an AUC of 0.90 ± 0.08 for NPM1 and 0.80 ± 0.10 for FLT3-ITD in the testing cohort. Additionally, we found that blasts are pivotal indicators for gene mutation predictions, with their proportions varying between mutated and standard WSIs, highlighting the clinical potential of AML WSI analysis.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML), as an aggressive hematologic malignancy, exhibits significant biological and clinical heterogeneity, characterized by uncontrolled proliferation and impaired differentiation of hematopoietic precursors1. Precise risk stratification is the cornerstone for optimizing treatment efficacy and minimizing treatment-related complications. Currently, a range of cytogenetic changes and gene mutations have been integrated into the risk stratification, shaping the treatment landscape2. Nucleophosmin 1 (NPM1) and FMS-like tyrosine kinase-3 internal tandem duplication (FLT3-ITD) are the most prevalent recurrent gene mutations in patients with AML3,4. The NPM1 mutation occurs in 15-20% of cases, FLT3-ITD in 10-20%, and both mutations together in 10.5%5,6. Considerable efforts have been devoted to developing targeted therapies against these mutations7,8, highlighting their crucial significance in clinical practice. Performing molecular testing for these mutations presents significant challenges. While some institutions can provide results for FLT3-ITD and NPM1 mutations within days using straightforward PCR methods, disparities in resources and laboratory infrastructure across facilities often limit accessibility and prolong test turnaround times.
Whole slide images (WSIs) employ digital imaging technology to transform pathological specimens into high-resolution digital images detailing cellular and histological structures9. While deep learning has shown promise in tasks such as binary morphological classification and histological grading using WSIs10,11, challenges persist in analyzing bone marrow aspirates due to their complex cytological nature. Aspirates typically feature small, cluttered regions with various cell types and non-cellular debris. Identifying regions of interest (ROIs) and distinguishing individual cells or objects from the background require multi-step preprocessing, including segmentation and denoising12,13. Despite advances in deep learning for object detection, manual annotation of segmented cells by experts remains labor-intensive and time-consuming14. Developing a more efficient and accurate method for analyzing bone marrow aspirate WSIs is essential.
Contemporary histopathology research often follows a two-stage workflow, focusing on patch-level and slide-level training15. Initially, a Convolutional Neural Network (CNN) is trained on patches extracted from WSIs with patch-level annotations, learning complex patterns. In the second stage, features learned at the patch level are utilized to train a slide-level model, which is necessary for diagnosing WSIs, capitalizing on insights from patch-level analysis. Widely used for cancer identification16,17,18, classification19,20, and metastasis detection16, these approaches require substantial manual annotations. Multiple Instance Learning (MIL) has been employed to use slide-level labels directly21,22,23, reducing the annotation burden by classifying slides based on the highest-scoring patch.
Apart from patch-level MIL, numerous related studies have utilized individual cells as inputs for MIL training24,25, underscoring the existence of disease-associated cytomorphologies in cellular images26,27,28. Therefore, we hypothesized that deep learning could predict gene mutations based on cellular morphology. Here, we present an end-to-end artificial intelligence framework for bone marrow cytology, uniquely trained using WSIs with slide-level annotations. By leveraging annotation-free WSIs for gene mutation predictions, we demonstrate the capability of deep learning in predicting gene mutations. Our results highlight that models trained at the cell level outperform those trained at the patch level. Additionally, we illustrate how techniques, such as upsampling and ensemble learning, can enhance the predictive performance of the model, especially in scenarios with limited training data.
Methods
Dataset
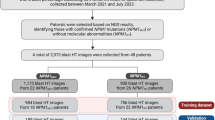
Between 1994 and 2015, 572 patients diagnosed with de novo AML at NTUH were enrolled in this study. Bone marrow smears and peripheral blood smears were scanned as WSIs after being stained with a modified Romanowsky stain (Fig. 2a). We utilized a Hamamatsu NanoZoomer Digital Slide Scanner equipped with a 40x NA 0.75 objective lens, achieving a resolution of approximately 0.23 μm/pixel. Gene mutation status was determined using the TruSight myeloid panel on the HiSeq platform (Illumina, San Diego, CA)6, and mutations were annotated as 1 (indicating the presence of pathogenic or likely pathogenic mutations) or 0 (indicating the absence of such mutations). In our cohort, a total of 34 genes were frequently found to be mutated, and in this study, we listed the mutation frequencies of more than 10% and selected the top two highest frequent mutations as the targets (Table 1). This retrospective study was approved by the NTUH Research Ethics Committee, and written informed consent was obtained from all participants in accordance with the Declaration of Helsinki (Approval number: 201802021RINC).
Cell image generation
The bone marrow smears of WSIs underwent a three-step filtering process to identify the cells for subsequent model training. First, we used the PyHIST tool to generate patches29. This tool was applied to filter out background regions and non-smear areas in the WSIs (Fig. 1a). Patches (512 × 512 pixels) were extracted at the highest resolution (×40 magnification), using a graph-based segmentation method30 as the parameter “generation method" in PyHIST, with a content threshold of 0.05. The parameters ’tilecross-downsample’ and ’mask-downsample’ adopted the default values, and the parameter ’output-downsample’ was set to 1 in order to obtain patches at the original resolution.

a Generation of patch images from bone marrow smears by PyHIST. b Selection of ROI patches using the model from a previous study31. c Detection of leukocyte cells using the model from a previous study31. d Preparation of bags and upsampling mutated WSIs to multiple bags. e MIL and ensemble learning. The MIL training procedure included a full inference pass through the dataset to rank the cells according to their probability of being mutated and learning about the top K cells per bag. The ensemble learning optimized the combination of base models by ss-based weights.
Secondly, the patches generated from the PyHIST tool were further classified into ROI and non-ROI patches. Due to the absence of manually annotated ROI and non-ROI data, we utilized an ROI detection model with training weights obtained from a previous study31. The model, based on DenseNet12132, was pre-trained and fine-tuned on labeled patches (ROI/non-ROI) in ref. 31. In this step (Fig. 1b), patches containing densely packed leukocytes or areas without any leukocytes were removed, significantly reducing the number of input data for the subsequent step: cell detection modeling.
After ROI selection, the ROI patches in each WSI would be used to get the cells in the third step. We applied the cell detection model using the training weights from the same study31. This model, based on YOLOv433, was trained in ref. 31 to predict the bounding boxes of leukocyte objects. In this step, all leukocytes in each ROI patch in bone marrow aspirates were detected (Fig. 1c).
Data upsampling
The WSI cohort exhibited a significant class imbalance, with relatively few mutated samples compared to a large number of standard samples. This imbalance could have influenced training outcomes, as the model might become biased towards predicting “standard” for all WSIs and consequently overlooking cases with mutations. To tackle this imbalance, we introduced an upsampling strategy by dividing the cells of a mutated WSI into multiple sets (bags), each containing a fixed number of cells (Fig. 1d). A previous study demonstrated that 500-cell sets are sufficient to capture the details of bone marrow34. In this study, we increased this number fourfold to 2000 cells to enhance the representativeness of each WSI. These bags were treated as separate samples during MIL training, effectively increasing the mutated class in the training data and resulting in a more balanced dataset.
In addition to addressing data imbalance, we faced the challenge of working with standard WSIs, which were not upsampled and each contained tens of thousands of cells. This large volume of input data could significantly slow down model training. To address this, we randomly selected a fixed number (2000) of cells per standard WSI as a bag, thereby expediting the training process by reducing input quantity.
Multiple instance learning
Fully supervised approaches for histopathology image analysis require detailed manual annotations, which are time-consuming and intrinsically ambiguous, even for well-trained experts. Standard unsupervised approaches usually fail due to their complicated patterns. MIL, a weakly supervised learning approach, addresses this challenge by utilizing labeled groups of instances, known as bags, instead of requiring individual instance labels. It works well for the current study because it takes advantage of supervised and unsupervised approaches. The main idea of MIL is to learn local patterns using global annotations. In MIL, each bag contains multiple instances, but only the bag is labeled with a class label, and the instances within the bag are unlabeled. Previous research transformed binary classification tasks into MIL problems by dividing WSIs into multiple instances. Here, the instances could be patches21 or cells24,25,26,27,28. Positive WSIs contain at least one positive instance, while negative WSIs do not contain any positive instances.
In this study, “bags" refers to the bags created after upsampling, while “instances" denotes individual cells in bags. After the cell detection process described above, bags B = {Bi: i = 1, 2, …, n} are generated, where \({B}_{i}=\{{c}_{i,1},{c}_{i,2},\ldots ,{c}_{i,{m}_{i}}\}\) is a bag containing mi cells (Fig. 1e). For most bags, mi = 2000. However, a single bag from a mutated WSI might contain fewer than 2000 cell, as the total cell count of a WSI is typically not an exact multiple of 2000. We labeled bags as “mutated” or “standard” according to the labels of their originating WSIs. In a bag, all cells are classified using an embedding model, the training process for which is detailed in the following paragraph. The cells are then ranked based on their probability of being mutated. The MIL task involves learning a cell-level embedding that effectively identifies and distinguishes characteristic cells within mutated bags from standard cells.
During training (Fig. 1e), the MIL training process consists of two alternating phases: the inference phase and the learning phase. At first, the DenseNet121 model was employed and fine-tuned to serve as the embedding model. It was initialized with pre-trained weights provided by the PyTorch library, which were previously trained with the ImageNet dataset. The model is represented as a function fθ, where the current parameters θ map the input cells ci,j to probabilities of “mutated." In the cell inference phase of MIL training, a vector list \({P}_{i}=\{{p}_{i,1},{p}_{i,2},\ldots ,{p}_{i,{m}_{i}}\}\) is derived for the bag Bi, representing the probabilities of the “mutated” class for the cells ci,j (j = 1, 2, …, mi). The weights of the model function fθ are frozen during this phase, ensuring that the parameters remain fixed while producing the estimated probabilities.
During the learning phase of MIL training, we introduce a hyperparameter K to select the top K cells with the highest probabilities (pi,j) from each bag as training data for the embedding model. The true labels (yi,j) of these selected cells are determined based on the label of their respective bags: the top K cells in each mutated bag are labeled as mutated cells, while the top K cells in standard bags are labeled as standard cells. Then, the network’s output \({\widetilde{y}}_{i,j}={f}_{\theta }({c}_{i,j})={p}_{i,j}\) is compared to the label yi,j using the cross-entropy loss function l as in Eq. (1):
, where w1 is the number of mutated bags divided by the number of total bags and w0 = 1 − w1. Consequently, based on the loss function Eq. (1), the weights of fθ are updated to minimize the discrepancy between the predicted output and the actual label.
Ensemble Learning
Ensemble methods are a powerful approach that involves training and combining multiple models to tackle complex problems. The core idea is that a collection of individual “weak learners" can synergistically work to create a “strong learner." Each model contributes its perspective through voting, while the ensemble method combines these inputs to generate a final prediction. The overarching objective of ensembles is to mitigate bias and variance in predictions by harnessing the combined capabilities of multiple models instead of relying solely on one single model.
In this study, we employed an ensemble method that utilizes a loss-based weighting approach, where each model’s contribution within the ensemble is determined by its individual loss function \({l}_{{M}_{K}}\) as in Eq. (1). This weighting strategy assigns greater influence to models with lower loss values, thereby enhancing the impact of more accurate models in the ensemble’s final probability (Pensemble). Only the top three MIL models, determined by their loss values, were used as weak learners within the ensemble. This approach aimed to optimize the combination of base models \(\{{M}_{{K}_{1}},{M}_{{K}_{2}},{M}_{{K}_{3}}\}={\rm{argmin}}\,{M}_{K}\in \{{M}_{1},\ldots ,{M}_{30}\}\), where K1, K2, and K3 correspond to the three MIL models with the lowest loss values among the models in {M1, …, M30}. Each base model within the ensemble was assigned a weight Wi determined by its performance as in Eq. (2):
The ensemble model combined the probability (\(\max {P}_{i}\)) of these three models according to their respective weights (\({W}_{{M}_{K}}\)), as illustrated in Eq. (3):
Results
Automatic selection of ROI patches
In digital pathology, glass slides of bone marrow aspirate smears were scanned using a digital slide scanner to generate high-resolution WSIs for hematopathologist analysis (Fig. 2a). To initiate this process, a dataset was sampled from 572 bone marrow aspirate WSIs obtained at the National Taiwan University Hospital. To address the issue of detecting ROI patches, we developed a pipeline to select ROI patches in stages, automatically identifying areas within the bone marrow aspirate WSIs that are suitable for cytological analysis.

a The WSI has an original resolution of approximately 200,000 × 100,000 pixels. b ROI patches cover areas with single or multiple leukocytes that are evenly distributed, clear, and not blurry. Non-ROI patches contain only red blood cells but no leukocytes, regions with cell overlaps making identification challenging, a considerable mix of heterogeneous tissues, areas devoid of blood cells within the designated range, or blurry patches. c The resolution of the patches is 512 × 512 pixels. The size of filtered cells is within the range of 51 × 51 pixels to 80 × 80 pixels and then resized to a uniform size of 64 × 64 pixels.
A single WSI might contain only a few areas suitable for cytology. These areas are sparsely distributed, with minimal cell overlap and staining artifacts, and exhibit subtle and intricate cytological features required for cell classification. To acquire such areas efficiently, the PyHIST tool29 was first used with the default graph method to remove the blank background regions, retaining only the stained portions. Subsequently, we employed a fine-tuned DenseNet121 architecture, utilizing pre-trained weights from previous research31 to classify individual patches as ROI patches or non-ROI patches (Fig. 2b).
We observed that the classification results aligned with expectation, i.e., a real-world scenario where usually only 10-20% of a WSI might be the ROI regions for cytology. The results obtained after applying DenseNet121 reveal that for most WSIs, the ROI patches were reduced to around 10-25% of their original counts after undergoing this selection process, indicating a substantial reduction in patch counts (Supplementary Fig. 1). This screening process significantly reduced the time required for subsequent cell detection processes and exhibited outstanding filtering effectiveness in removing areas with excessive cell overlap, excessive tissue artifacts, or patches devoid of blood cell presence within the designated range. These problematic patches were effectively eliminated, while, as shown in Supplementary Fig. 2, patches with a small number of cells were not erroneously dropped. This approach ensures that patches with few cells are retained, supporting data completeness and avoiding unnecessary loss of valuable information (Fig. 2b).
Leukocyte detection
After the ROI selection process, we employed a YOLOv4 model to automatically detect and classify cells and non-cellular objects within the selected ROI patches. In the absence of manually annotated cells, we used training weights that had been pre-trained and fine-tuned on labeled patches from the same study for selecting ROI patches31, applying them directly in this detection model. Using the ROI patches identified by the ROI detection model as input, this YOLOv4 model was designed to automatically detect and classify all cellular and non-cellular objects in bone marrow samples. We further set a confidence threshold of 0.5. Cells with confidence scores below this threshold were not captured, ensuring high-quality cell selection for subsequent MIL model training. In addition to managing confidence levels, we factored in cell size during the process. Under ultraviolet light with a 400 nm wavelength, the theoretical resolution is approximately 325.33 nm. We set the cell size range to 51 × 51 pixels to 80 × 80 pixels. This means that a cell size of 51 pixels represents approximately 16.6 μm, while 80 pixels corresponds to roughly 25.6 μm. This range was chosen based on the typical sizes of white blood cells (WBCs), which range from approximately 10–25 μm, and blasts, typically around 15-20 μm. By setting the selection threshold between 51 and 80 pixels, we aimed to capture the most representative cells that are complete and relevant for feature extraction. The chosen cells were directly resized to 64 × 64 pixels to standardize input size for MIL training, avoiding irrelevant features like neighboring cells or excessive background that could be introduced by alternative padding methods (Fig. 2c).
The number of cells left per WSI varied between 100 to 100,000, with the majority of cell counts being below 20,000 and an average of 11,273 (Fig. 3). This outcome effectively reduced the input quantity for the MIL model while maintaining the best quality of all input cells, accelerating training time without compromising accuracy. Therefore, we used leukocytes, including basophil, blast, eosinophil, lymphocyte, metamyelocyte, monocyte, myelocyte, neutrophil, and promyelocyte as inputs for MIL training, denoted as ’all cells’. Among these cells, MIL randomly selected 2000 cells as representative cells of a WSI, i.e., a bag. During the selection period, we applied the upsampling technique to the mutated WSIs to balance the data. By separating each mutated WSI into multiple bags, we can generate numerous mutated bags that constitute 2000 cells.

a Models trained with NPM1 mutations. b Models trained with FLT3-ITD. “Non-upsampling patch-level MIL" refers to patch-level MIL without upsampling and ensemble learning, “non-upsampling cell-level MIL" refers to cell-level MIL without upsampling and ensemble learning, “upsampling cell-level MIL" means cell-level MIL with upsampling bags but without ensemble learning and “ensemble + upsampling cell-level MIL" refers to cell-level MIL with both upsampling and ensemble learning.
Cell-Level Multiple Instance Learning
The total dataset of 572 WSIs was split into training, validation, and test sets at slide level, with a ratio of around 7:1:2 (400:56:116 WSIs). We used the DenseNet121 as the base embedding model, initialized with pre-trained weights provided by the PyTorch library based on training with the ImageNet dataset. The MIL model underwent training for 100 epochs with a learning rate set to 0.0001. The loss minimization is attained through stochastic gradient descent (SGD) utilizing the Adam optimizer. The batch size is determined by the number of cells within each bag, restricted to a maximum of 2000 instances (cells) per batch. Since the training data still exhibited minor class imbalance after upsampling, we set weights (w0, w1) to (0.51, 0.29) for NPM1 mutations and (0.37, 0.63) for FLT3-ITD (see Section 2.4.). No upsampling was applied to the validation or testing sets to emulate a real-world scenario. Each epoch was evaluated using an imbalance validation set of 56 WSIs, specifically, more standard than mutated WSIs. Early stopping was implemented to prevent overfitting. The separate testing set included 96 standard and 20 NPM1 mutations WSIs, along with 95 standard and 21 FLT3-ITD WSIs.
In addition to established MIL training, we leveraged ensemble learning by employing different values of K (from K = 1 to K = 30) in MIL to diversify the models’ perspectives on the data. Each K represents the number of positive instances considered within a bag during the MIL training process, leading to the creation of models with varying focuses on distinct data subsets. Based on Eq. (2) and Eq. (3), we calculated the respective weights Wi of the top 3 MIL models with the lowest loss values among the models and then estimated the final prediction. The models’ predictive performance on the training, validation, and test datasets is shown in Supplementary Fig. 4.
The integration of upsampling at the cell level improved model performance on the testing set for both gene mutations, achieving AUC values of 0.90 ± 0.08 for NPM1 mutations and 0.80 ± 0.10 for FLT3-ITD (Fig. 3). For NPM1 mutations, upsampling cell-level MIL achieved an AUC of 0.82 ± 0.11, which exceeded the non-upsampling approaches at both the patch and cell levels. The addition of ensemble learning further enhanced the AUC to 0.90 ± 0.08, comparable to results from models trained on cell-level annotations in prior studies28.
In the case of FLT3-ITD, upsampling cell-level MIL achieved an AUC of 0.77 ± 0.10, also higher than the non-upsampling patch-level and cell-level MIL models. With ensemble learning, the AUC further increased to 0.80 ± 0.10. This result demonstrates that upsampling at the cell level, combined with ensemble learning, addresses class imbalance and improves detection accuracy. The combination of these methods performs favorably compared to recent techniques based on cell-level annotation28 and exceeds previously reported results14. This approach provides a structured framework for identifying mutations in bone marrow samples, especially in cases with limited annotation resources.
Moreover, our ensemble cell-level MIL, when using sensitivity as the standard for the reliability analysis of clinical diagnosis, exhibited better performance than the traditional MIL methods. As the sensitivity was set to 0.75, our approach reduced the false positive rate from 0.36 to 0.09 for NPM1 mutations. For FLT3-ITD, there was also a notable reduction from 0.60 to 0.26 (Fig. 3). This outcome once again confirms that ensemble learning and upsampling can enhance the effectiveness of the MIL model.
To clarify the importance of cell features for prediction in the learning process of MIL, we compared the proportional representation of nine types of cells. Before applying the MIL model, all cell types in each WSI would be divided by the total number of cells of all types in that WSI to obtain the proportion of each type of cell. After prediction, we listed the top 100 images of cells with the highest probability of being predicted by the model as the highest correlated with mutations in each bag. Then, we calculated the proportion of all representative cells predicted by MIL in the original WSI based on their cell types (Fig. 4). This can reveal which cells are more meaningful for MIL predictions of gene mutations, even if we were only using slide-level annotations.

a boxplot of cell types with NPM1 mutations data. b boxplot of cell types with FLT3-ITD data. For each bag, the proportion of each cell type within the bag was first calculated prior to MIL prediction (referred to as 'all cells'). Bags were further categorized as mutated and standard, and these proportions were visualized using boxplots. After MIL prediction, the top 100 cells with the highest mutation probabilities in each bag were selected, their proportions were calculated, and their distributions by cell type were plotted. This highlights the cell types that might provide a stronger indication of mutated characteristics in the MIL model.
It can be observed that the trend for blasts remains consistent regardless of whether it is the NPM1 or FLT3-ITD model. In bags with either genetic mutations, after applying MIL, there’s a notable increase in the proportion of blasts among the top 100 representative cells. On the other hand, in bags without genetic mutations, after applying MIL, there’s a decrease in the proportion of blasts among the top 100 representative cells. This trend indicates that the presence of blasts is an essential indicator for determining whether there are mutations in deep learning. Additionally, in bags with genetic mutations, the proportions of other cell types decrease, indicating that the majority of features used in machine learning to determine the presence of mutations are in the blast category. Therefore, in non-mutated bags, because these features cannot be found in blast cells, the proportions of other cell types increase while the proportion of blast significantly decreases.
Discussion
Analysis of AML cells often involves morphological examination24,25,26,27,28 and complete blood count-based approaches35, which have been applied in subtype classification and disease-associated cytomorphologies. Compared to previous studies, this work explores the potential of cell-level analysis using annotation-free data, demonstrating promising performance in predicting NPM1 mutations and FLT3-ITD. Furthermore, by applying MIL with upsampling and ensemble techniques, our research achieved AUC values of 0.90 ± 0.08 for NPM1 mutations and 0.80 ± 0.10 for FLT3-ITD. This underscores the strength of our approach in addressing the challenges of gene mutation prediction without the need for manual annotation of cell images.
We further explored the impact of gene interactions in our models. We observed that the presence of FLT3-ITD does not significantly influence the predictions of the NPM1 model (Supplementary Fig. 5.a). Conversely, for the FLT3-ITD model, the absence of concurrent NPM1 mutations leads to superior predictive performance compared to cases with concurrent NPM1 mutations (Supplementary Fig. 5.b). This suggests that predictions of FLT3-ITD in the presence of NPM1 mutations require greater scrutiny and caution, as the morphological impact of NPM1 mutations may significantly obscure or overshadow the features associated with FLT3-ITD. This overlap in morphological characteristics could potentially reduce the model’s ability to discern FLT3-related features accurately. We also investigated the concurrence of DNMT3A. As shown in Supplementary Fig. 6, the presence or absence of DNMT3A mutations does not appear to have a substantial impact on the performance of either model.
In addition to examining the impact of concurrent gene mutations on model performance, we also explored other factors. Age was found to have no significant effect on the models’ performance. For hyperleukocytosis, defined as WBCs ≥ 50,000/μL, the NPM1 model showed a decline in performance for subsets with hyperleukocytosis (Supplementary Fig. 7.a), while the FLT3-ITD model was relatively unaffected (Supplementary Fig. 7.b). Furthermore, we evaluated the impact of the NPM1 VAF cutoff using a threshold of 0.4436. The NPM1 model performed exceptionally well in the high-VAF subgroup, achieving a PPV of 100% (Supplementary Fig. 8.a), though the small sample size requires further validation. Interestingly, for the FLT3-ITD model, performance declined in the high-VAF subgroup (Supplementary Fig. 8.b), consistent with the observation that NPM1 mutations negatively affect FLT3-ITD predictions. These findings demonstrate the potential for our models to assist clinicians in re-evaluating mutation status, opening opportunities for targeted therapies. Importantly, this methodology enables the reduction of false positive rate for NPM1 mutations (from 0.36 to 0.09) and FLT3-ITD (from 0.60 to 0.26), highlighting its utility in enhancing diagnostic confidence in mutation identification.
Our model demonstrates significant promise for clinical application. It is well-established that FLT3-ITD is an unstable mutation that can undergo clonal evolution during treatment37. While the false positive rate of our model for predicting FLT3-ITD is not fully optimized, the results could encourage clinicians to perform confirmatory testing. This is particularly valuable given the availability of effective targeted therapies for patients with FLT3-ITD, even when the mutation is acquired during relapsed or refractory stages. Moreover, this study demonstrates the importance of different cell types in predicting gene mutations using MIL. Specifically, blasts were identified as critical indicators for both NPM1 mutations and FLT3-ITD prediction. Our model significantly enhanced the representation of blasts in mutated whole slide images (WSIs), underscoring their importance in mutation detection. Conversely, in standard WSIs, the increased proportion of other cell types alongside a reduced blast representation highlights a potential morphological distinction between mutated and standard cases.
Despite the advantages of our model, we acknowledge that a 26% false positive rate for FLT3-ITD mutations is not ideal for clinical application, particularly if this method were to be employed as the sole diagnostic tool during initial diagnosis. We currently recommend using a companion GENESCAN to confirm FLT3-ITD mutation status. Therefore, potential improvements of our model include embedding more detailed features that capture the relationships between additional myeloid-related gene mutations, which depend heavily on next-generation sequencing (NGS). Since NGS typically requires around three weeks to deliver results, incorporating these predictions into our model could dramatically shorten the time to actionable insights in clinical practice, enhancing its utility in time-sensitive scenarios. Additionally, enhancing the resolution of smear images would enable more precise visualization of morphological regions affected by mutations, providing clearer links between cellular morphology and gene mutations.
Our findings underscore the immense potential of ensemble learning, upsampling techniques, and MIL in predicting gene mutations in AML patients. Importantly, we accomplished this using training data labeled exclusively at the slide level, eliminating the labor-intensive manual annotation required for cell images. This streamlined approach emphasizes the value of integrating advanced machine-learning techniques to address complex real-world challenges in medical image analysis, offering a robust foundation for further exploration of the correlation between genetic status and cellular morphology.
In conclusion, our research underscores the immense potential of ensemble learning, upsampling techniques, and MIL in predicting gene mutations in AML patients. Importantly, we accomplished this using training data labeled exclusively at the slide level, eliminating the labor-intensive manual annotation required for cell images. This approach streamlined the end-to-end training and prediction process, emphasizing the value of integrating advanced machine-learning approaches to address complex real-world challenges in medical image analysis.
Data availability
The gene mutation data used in this study was published in a previous work6. The WSIs contain sensitive patient information and are, therefore, not publicly available. However, the corresponding author can provide access to the WSIs upon request, subject to data-sharing agreements and privacy considerations.
Code availability
The code used for computation analysis in this study can be found at https://github.com/c4lab/AML-WSI.git. For any additional questions, please contact the corresponding author.
References
Döhner, H., Weisdorf, D. J. & Bloomfield, C. D. Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152 (2015).
Döhner, H. et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 140, 1345–1377 (2022).
Stone, R. M. et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a flt3 mutation. N. Engl. J. Med. 377, 454–464 (2017).
Erba, H. P. et al. Quizartinib plus chemotherapy in newly diagnosed patients with flt3-internal-tandem-duplication-positive acute myeloid leukaemia (quantum-first): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 401, 1571–1583 (2023).
Papaemmanuil, E. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016).
Tsai, X. C.-H. et al. Poor prognostic implications of myelodysplasia-related mutations in both older and younger patients with de novo aml. Blood Cancer J. 13, 4 (2023).
Perl, A. E. et al. Gilteritinib or chemotherapy for relapsed or refractory flt3-mutated aml. N. Engl. J. Med. 381, 1728–1740 (2019).
Issa, G. C. et al. The menin inhibitor revumenib in kmt2a-rearranged or npm1-mutant leukaemia. Nature 615, 920–924 (2023).
Bera, K., Schalper, K. A., Rimm, D. L., Velcheti, V. & Madabhushi, A. Artificial intelligence in digital pathology-new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 16, 703–715 (2019).
Sirinukunwattana, K. et al. Gland segmentation in colon histology images: the glas challenge contest. Med. Image Anal. 35, 489–502 (2017).
McKinney, S. M. et al. International evaluation of an AI system for breast cancer screening. Nature 577, 89–94 (2020).
Chandradevan, R. et al. Machine-based detection and classification for bone marrow aspirate differential counts: initial development focusing on nonneoplastic cells. Lab. Investig. 100, 98–109 (2020).
Reta, C. et al. Segmentation and classification of bone marrow cells images using contextual information for medical diagnosis of acute leukemias. PLoS ONE 10, e0130805 (2015).
Kockwelp, J. et al. Deep learning predicts therapy-relevant genetics in acute myeloid leukemia from pappenheim-stained bone marrow smears. Blood Adv. 8, 70–79 (2024).
Dimitriou, N., Arandjelović, O. & Caie, P. D. Deep learning for whole slide image analysis: an overview. Front. Med. 6, 264 (2019).
Bejnordi, B. E. et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 318, 2199–2210 (2017).
Burlutskiy, N., Gu, F., Wilen, L. K., Backman, M. & Micke, P. A deep learning framework for automatic diagnosis in lung cancer. Preprint at https://arxiv.org/abs/1807.10466 (2018).
Chuang, W.-Y. et al. Successful identification of nasopharyngeal carcinoma in nasopharyngeal biopsies using deep learning. Cancers 12, 507 (2020).
Coudray, N. et al. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 24, 1559–1567 (2018).
Santos, A. G., da Rocha, G. O. & de Andrade, J. B. Occurrence of the potent mutagens 2-nitrobenzanthrone and 3-nitrobenzanthrone in fine airborne particles. Sci. Rep. 9, 1 (2019).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25, 1301–1309 (2019).
Wang, X. et al. Weakly supervised deep learning for whole slide lung cancer image analysis. IEEE Trans. Cybern. 50, 3950–3962 (2019).
Pan, S. & Secrier, M. Histomil: a Python package for training multiple instance learning models on histopathology slides. Iscience 26, 108073 (2023).
Yudistira, N., Kavitha, M. S., Rajan, J. & Kurita, T. Attention-effective multiple instance learning on weakly stem cell colony segmentation. Intell. Syst. Appl. 17, 200187 (2023).
Sadafi, A. et al. Attention based multiple instance learning for classification of blood cell disorders. In: Medical Image Computing and Computer Assisted Intervention–MICCAI 2020: 23rd International Conference, Lima, Peru, October 4–8, 2020, Proceedings, Part V 23, (eds Martel, A. L., et al.) 246–256 (Springer, 2020).
Sidhom, J.-W. et al. Deep learning for diagnosis of acute promyelocytic leukemia via recognition of genomically imprinted morphologic features. NPJ Precis. Oncol. 5, 38 (2021).
de Almeida, J. G. et al. Computational analysis of peripheral blood smears detects disease-associated cytomorphologies. Nat. Commun. 14, 4378 (2023).
Eckardt, J.-N. et al. Deep learning detects acute myeloid leukemia and predicts npm1 mutation status from bone marrow smears. Leukemia 36, 111–118 (2022).
Muñoz-Aguirre, M., Ntasis, V. F., Rojas, S. & Guigó, R. Pyhist: a histological image segmentation tool. PLoS Comput. Biol. 16, e1008349 (2020).
Felzenszwalb, P. F. & Huttenlocher, D. P. Efficient graph-based image segmentation. Int. J. Comput. Vis. 59, 167–181 (2004).
Tayebi, R. M. et al. Automated bone marrow cytology using deep learning to generate a histogram of cell types. Commun. Med. 2, 45 (2022).
Huang, G., Liu, Z., Van Der Maaten, L. & Weinberger, K. Q. Densely connected convolutional networks. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition 4700–4708 (IEEE, 2017).
Bochkovskiy, A., Wang, C.-Y. & Liao, H.-Y. M. Yolov4: Optimal speed and accuracy of object detection. Preprint at https://arxiv.org/abs/2004.10934 (2020).
Abdulrahman, A. A. et al. Is a 500-cell count necessary for bone marrow differentials? a proposed analytical method for validating a lower cutoff. Am. J. Clin. Pathol. 150, 84–91 (2018).
Alcazer, V. et al. Evaluation of a machine-learning model based on laboratory parameters for the prediction of acute leukaemia subtypes: a multicentre model development and validation study in France. Lancet Digital Health 6, e323–e333 (2024).
Patel, S. S. et al. High npm1-mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood 131, 2816–2825 (2018).
Schmalbrock, L. K. et al. Clonal evolution of acute myeloid leukemia with flt3-itd mutation under treatment with midostaurin. Blood 137, 3093–3104 (2021).
Acknowledgements
This project was supported by the research grants from National Science and Technology Council in Taiwan (NSTC 112-2221-E-002-184-MY3 and NSTC 112-2314-B-002-116-MY3). The authors express our sincere gratitude to the laboratory department staff at NTUH for their exceptional management of the WSIs and to the National Center for High-performance Computing (NCHC) for providing computational and storage resources.
Author information
Authors and Affiliations
Contributions
Bo-Han Wei, Xavier Cheng-Hong Tsai, and Chien-Yu Chen developed the overall research idea; Bo-Han Wei and Xavier Cheng-Hong Tsai designed methods and procedures; Bo-Han Wei implemented analytic pipeline; Xavier Cheng-Hong Tsai, Min-Yen Lo, and Sheng Yu Hung collected image and clinical data; Bo-Han Wei and Kuo-Jui Sun contributed analyzed data; Bo-Han Wei, Xavier Cheng-Hong Tsai, Kuo-Jui Sun, and Chien-Yu Chen wrote the manuscript; Xavier Cheng-Hong Tsai, Wen-Chien Chou, Hwei-Fang Tien, Hsin-An Hou, and Chien-Yu Chen reviewed and edited the manuscript; and all authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, BH., Tsai, X.CH., Sun, KJ. et al. Annotation-free deep learning for predicting gene mutations from whole slide images of acute myeloid leukemia. npj Precis. Onc. 9, 35 (2025). https://doi.org/10.1038/s41698-025-00804-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00804-0
