
Abstract
Patients with high-grade serous ovarian carcinoma (HGSC) are usually diagnosed with advanced-stage disease, and the tumors often have immunosuppressive characteristics. Together, these factors are important for disease progression, drug resistance, and mortality. In this study, we used a combination of single-cell sequencing and spatial transcriptomics to identify the molecular mechanisms that lead to immunosuppression in HGSC. Primary tumors consistently showed a more active immune microenvironment than did omental tumors. In addition, we found that untreated primary tumors were mostly populated by dysfunctional CD4 and CD8 T cells in later stages of differentiation; this, in turn, was correlated with expression changes in the interferon α and γ pathways in epithelial cells, showing that cross-communication between the epithelial and immune compartments is important for immune suppression in HGSC. These findings could have implications for the design of clinical trials with immune-modulating drugs.
Similar content being viewed by others


Introduction
Ovarian cancer is a heterogeneous disease with multiple histological subtypes, the most common of which is high-grade serous ovarian carcinoma (HGSC)1. Patients are usually diagnosed with advanced stage disease2,3 and often develop resistance to conventional therapy with carboplatin and paclitaxel4,5, making it the deadliest gynecological malignancy in the United States and other developed countries6. Despite technological advances, ovarian cancer is commonly detected when the disease has already spread widely in the abdominal cavity. Ovarian cancer has a tropism for adipose-rich tissues such as the omentum7,8 and often causes large volumes of ascites9. Epithelial ovarian tumors have been shown to be rich in CD8+ lymphocytes, which infiltrate both primary and omental tumor sites; in recent years, several prominent studies have highlighted a positive association between the presence of these immune cells and favorable prognosis10,11. Nonetheless, clinical trials of immune checkpoint inhibitors have so far shown unsatisfactory results in ovarian cancer, with a clinical benefit reported in less than 15% of patients12,13,14,15,16,17. Thus, deciphering the complexity of the immunosuppressive tumor microenvironment (TME) in ovarian cancer represents an important unmet need.
The complexity of the HGSC TME, its high level of heterogeneity, and its immunosuppressive characteristics18,19,20 are important factors in disease evolution, disease progression, drug resistance, and mortality, thereby representing one of the biggest challenges in cancer therapy21. While some meaningful attempts to decipher the heterogeneity of HGSC have been made22,23,24,25,26,27,28,29,30, overcoming its aggressiveness requires a deeper understanding of its molecular features. Our aim was to identify the molecular mechanisms that could lead to immune suppression in primary and metastatic HGSC. To achieve this aim, we used in-depth single-cell RNA sequencing (scRNA-Seq) to determine the molecular characteristics of primary tumors and metastatic omental tumors and of treated and untreated tumors. We found notable differences in immune cell composition and activation status between both comparison groups.
Results
Landscape of tissue sample characteristics and single-cell transcriptome data
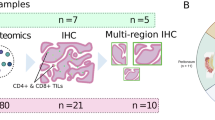
To determine the cellular heterogeneity and architecture of HGSC, 19 patients with advanced HGSC and 1 healthy control individual were enrolled in this study. In total, 28 tumor samples and 1 healthy ovary were obtained (Fig. 1A; Table 1). Six patients with HGSC were treated with carboplatin and paclitaxel and the samples were obtained during interval tumor reductive surgery (Supplementary Fig. 1A). The other patients had not been treated when the samples were acquired. Patients’ clinical and histopathological characteristics are summarized in Table 1. CD45+ and CD45- cells were collected from 9 patients with HGSC by fluorescence-activated cell sorting (FACS) to characterize immune features and dynamics. The transcriptomes of individual cells were obtained by scRNA-seq via a droplet-based 10× Genomics platform.

A Schematic workflow. A total of 19 freshly resected HGSC specimens were collected. Samples were processed using 10X genomic Chromium Single cell 3' v3. Created with BioRender.com using data contained elsewhere in the paper. B UMAP visualization of major cell types that were used for the subsequent analysis by cell type, tissue type, treatment, and tissue sample (patients). C UMAP visualization of the lineage canonical marker. D Cell count (up) and cell percentage (down) of the major cell types for each sample analyzed.
Cellular heterogeneity of primary and metastatic HGSC
After initial quality filtering, 100,480 cells passed quality control and were included in subsequent analyses. Batch effects were assessed and corrected using Batch Balance KNN (BBKNN)31. Six major cell types were identified (Fig. 1B, C; Supplementary Fig. 1B–E); each was observed across all patients (Fig. 1D). A multistep approach was then applied to distinguish malignant cells (see Methods). As expected, these epithelial tumor cells were clustered by patient, irrespective of tissue type, treatment, or tumor location (Fig. 2A and Supplementary Fig. 2A). This finding demonstrates that epithelial cells exhibit not only intrapatient but also interpatient heterogeneity; only a few patients showed more than 1 identifiable epithelial cell cluster. Next, unsupervised cell clustering was carried out on epithelial cancer cells. A total of 21,144 cells were divided into 20 unique clusters. Often, single patients presented a single dominant cluster of epithelial cells; in 3 cases, more than 1 cluster was recognizable (Fig. 2A).

A UMAP visualization of epithelial cells by clusters and samples (patients). B CNV profiles using InferCNV to distinguish cancer cells from normal epithelial cells. The color-coded bars indicate sample, cluster, treatment, tissue type, and InferCNV prediction. C CNV profile of patient Pt_10 using InferCNV to distinguish subclusters of epithelial cells on the basis of their CNV characteristics. The color-coded bars indicate cluster (C1 and C2), treatment (untreated), and tissue type (primary). D UMAP visualization of C1 and C2 shows a clear distinction on the basis of CNV inference. E UMAP visualization of the CytoTrace score of patient Pt_10, showing C2 as a more differentiated cluster. F Pseudotime trajectory of the cancer cells from patient Pt_10. Pseudotime was colored in a gradient from dark to light blue. The start of pseudotime is indicated by dark blue and the end by light blue. ssGSEA of (G) omentum versus primary tumor tissues, H omentum versus primary treated tumor tissues, I primary untreated tumors versus primary NACT-treated tumors, and J NACT-treated versus untreated omentum tumors.
HGSC is well-known to have widespread copy number variations (CNVs). We used this feature to identify tumor epithelial cells. We inferred the CNV profiles using inferCNV (https://github.com/broadinstitute/inferCNV). All HGSC samples bore several genomic rearrangements (Fig. 2B). Primary untreated tumors bore major deletions on chromosomes 6 and 11, whereas primary tumors that had been treated with neoadjuvant chemotherapy (NACT) had major gains on chromosomes 6 and 11 (Supplementary Fig. 2B). Of note, chromosome 6 is known to contain human leukocyte antigen (HLA) genes32, whose expression could be affected by the presence of CNVs. To further investigate the impact of HLA gene expression, we performed a CNV analysis of specific HLA regions (Supplementary Fig. 3A). Multiple untreated primary tissues had deletions in several HLA genes. These rearrangements of the HLA regions in the epithelial cell compartment might have a role in the immune regulatory functions of the epithelial compartment of HGSC tumors.
A CNV analysis also revealed a high degree of inter-patient heterogeneity. Four patient tissue samples had multiple subpopulations of cancer cells with CNVs across chromosomes (Fig. 2B). In 1 patient (Pt_10; Fig. 2C), the CNV analysis showed 2 clusters of epithelial cells, 1 with a higher (C1) and 1 with a lower (C2) CNV score; this mirrored the transcriptomics analysis of the same sample, which identified 2 different populations of epithelial cells (Fig. 2D). To correlate the presence of CNVs with the degree of cell differentiation, we computed CytoTRACE33 scores of clusters C1 and C2. C2 exhibited a lower CNV score and higher CytoTRACE score, indicating that this population was more differentiated than C1 (Fig. 2E).
To further understand the differentiation trajectory, we performed a pseudotime analysis (Fig. 2F), which showed that C1 and C2 separated into divergent populations, forming 3 major branches of differentiation. We performed differential expression analysis to identify the differentially expressed genes (DEGs) between these clusters. In C1, there was marked upregulation of DAPL1 (death-associated protein like 1) (Supplementary Fig. 2C), upregulation of the inflammatory response pathway, and downregulation of the G2/M checkpoint pathway (Supplementary Fig. 2D). Interestingly, using The Cancer Genome Atlas database, we found that high expression of DAPL1 was associated with higher overall survival (p = 0.038) in patients with ovarian cancer.
Since the high-CNV score (C1) population also exhibited downregulated G2/M checkpoint pathway, we determined whether the expression of proliferation-associated genes was associated with the CNV pattern. Therefore, we measured the expression of MKI67, TOP2A, TYMS, STMN1, and TUBB, which are all involved in cell proliferation, and observed increased expression of these genes in the C2 population, indicative of a higher proliferative capacity (Supplementary Fig. 2E). Next, we applied the monocle pipeline34,35,36 to infer the differentiation trajectories of epithelial cells from single-cell data. For example, in patient Pt_10 described above, we can infer a trajectory in which the C2 populations differentiated into C1 populations, indicating that the C1 population was a subclone of C2 with accumulation of CNVs (Fig. 2E).
We next performed a pathway enrichment analysis (ssGSEA) to shed light on the differences between primary and metastatic tumors and between treated and untreated tumors (Fig. 2G–J). The major differences between primary and omentum tumors included enrichment of the epithelial-mesenchymal transition (EMT) and angiogenesis pathways and decrease in the interferon (IFN) α and IFNγ response pathways in omentum tumors (Fig. 2G). This finding was also validated using additional datasets (GSE180661 and GSE165897) (Supplementary Fig. 4A). The same trend of pathways was observed for treated primary tumors versus all omentum tumors as for primary versus omentum tumors (Fig. 2H). Moreover, IFNα and IFNγ response pathways and inflammatory response pathways were upregulated in NACT-treated primary tumors compared with the untreated primary tumors in which G2M checkpoint, TGF-β signaling, and EMT pathways were downregulated (Fig. 2I). Similarly, a comparison of untreated and NACT-treated omentum tissues revealed that the genes associated with EMT, TGF-β signaling, and WNT-β-catenin signaling pathways were enriched in NACT-treated tissues, and the reactive oxygen species pathway, oxidative phosphorylation, apoptosis pathways, and IFNγ response were downregulated (Fig. 2J). Together, these data indicate that the epithelial compartment of different tumor sites has marked differences in IFNγ and α, which could explain variable responses to specific treatments such as immune therapies. All of the enriched pathways are presented in Supplementary Tables 1–4.
Cellular heterogeneity of primary and metastatic HGSC: immune compartment
We next examined the immune microenvironment of HSGC. First, we used a graph-based clustering method to identify immune cell states in a total of 36 subclusters: B and plasma cells (6 subclusters), T cells and natural killer cells (19 subclusters), and myeloid cells (11 subclusters). Among the myeloid populations, we identified 4 subclusters of macrophages, 4 subclusters of dendritic cells, 1 subcluster of monocytes, and 2 subclusters of proliferative myeloid cells (Fig. 3A). The major cell types were identified on the basis of canonical lineage markers (Supplementary Fig. 1B–E).

A UMAP of immune cells from all patients, distributed by cell type: B cells, T cells, natural killer cells (lymphoid cells), and myeloid cells. B Immune cell distribution across all patients. Top: immune cell type by group; Bottom: tissue distribution of immune cells by Ro/e analysis in different groups. C Immune cell proportion of each group of patients showed statistically significantly different proportions between groups. D Correlation between immune cell proportion and overall survival of patients with HGSC. E Ecotypes (relative abundance) of immune cell types by cancer type and treatment.
We identified 6 distinct states of B cells, according to gene expression markers37. Plasma cells were identified according to their expression of CD79A, MZB1, and JCHAIN, and 2 subpopulations were subsequently defined, Plasma_C1 and Plasma_C2. The memory B cells were divided into non-switched and switched memory B cells. We also identified a population of naive B cells and a proliferative population characterized by MKI67 expression. The proportions of these cells varied across tissue type and treatment status (Fig. 3B). Plasma B cells were enriched in primary tumor tissues (p-value not significant). Plasma_C2 cells were enriched in untreated tissues while IL1RN-expressing macrophages were enriched in untreated primary tissues and NACT-treated omentum tissues (Fig. 3B). Interestingly, non-switched memory B cells were mainly enriched in omental tumors, while switched memory B cells were enriched in primary tumors. The most striking differences (p < 0.05) were observed between primary untreated tumor tissues and other groups. As shown in Fig. 3C, non-switched memory B cells were significantly enriched in omental tissues (p = 0.014). These cells are known to play an important role in lasting long-term immunity, but their precise role in the TME remains unclear. Some studies have shown that non-switched memory B cells have low PD-1 expression but high PD-L1 expression, making these cells a potential target for immune checkpoint blockade.
Next, we analyzed NK and T cells. The UMAP representation shows distinct clusters and subgroups in the different cell types (Fig. 3A). We identified two populations of NK cells, one characterized by the presence of GNLY and the other identified by CD16 expression. CD16 NK cells were more frequent in normal and omentum tissues, while GNLY NK cells were mainly observed in untreated omental tumor tissues (Fig. 3B).
We then focused on T cells, which were identified by canonical marker genes38. Within the CD4 T-cell cluster, we identified the following subclusters: naïve T cells, follicular helper T cells, exhausted T cells, active T cells, and regulatory T cells (Fig. 3A). Untreated primary tumors showed enrichment of CXCR13 exhausted T cells (p = 0.036) (Fig. 3C). Among CD8 T cells, we identified CD8 resident memory T cells, active T cells, effector T cells, effector memory T cells, exhausted T cells, and 2 populations of proliferative T cells. A statistical analysis showed that CD8 effector T cells were enriched in untreated omentum tumor tissues (p = 0.013) (Fig. 3C), while CD8 GZMK effector memory T cells were enriched in NACT-treated primary tissues (p = 0.03) (Fig. 3C). Interestingly, both populations of proliferative T cells (Prolif_STMN1 and Prolif_MKI67) were enriched in untreated primary tissues (p = 0.008 and p = 0.039, respectively) (Fig. 3C).
We identified 11 distinct clusters of myeloid cells: macrophages (Macro_C1QC, Macro_MACRO, Macro_IL1RN, and Macro_CTSK), dendritic cells (cDC2_CD1C, pDC_LILRA4, cDC3_LAMP3, and cDC1_CLEC9A), monocytes (mono_FCN1), and two populations of proliferative myeloid cells characterized by PCNA and MKI67 (Fig. 3A).
We examined the relative abundance of these immune cells and observed that our tissue samples were distributed in 3 ecotypes (the normal tissues were considered a separate ecotype), which exhibited clear tissue type preferences (Fig. 3E). The tissue enrichment for each ecotype was based on the ratio of observed to expected cell numbers in each cluster (Ro/e). One cluster, which contained only untreated primary tumors, was highly enriched in lymphoid cells, where active and exhausted T cells are observed, indicating a more active immune environment. The second cluster was composed of mixed primary and metastatic and treated and untreated tissues and was enriched in myeloid cells. The third cluster mainly consisted of NACT-treated tissues and was characterized by the enrichment of early-active lymphoid cells. The mixture of tumor sites and treatment statuses in the second cluster likely encompasses the heterogeneity of HGSC. These data clearly show distinct niches for immune cells and their correlation with specific locations or treatment conditions, where we observed enrichment of CD4 and CD8 exhausted T cells in untreated primary tumors, the same group where we observed changes in the IFNγ and IFNα pathways in the epithelial compartment. To validate these findings, we used 2 other datasets (GSE180661 and GSE165897), and we observed that primary untreated tissues showed an enrichment for active and exhausted T cells, demonstrating that the pattern observed in our initial analysis also is characteristic of other datasets (Supplementary Fig. 3B).
Lastly, we correlated the cell types that were statistically significantly different between tumor sites with overall survival (Fig. 3D). The high proportions of non-switched memory B cells (p = 0.048), CD8 T effector cells (p = 0.0075), and active T CD8 cells (p = 0.039) were correlated with a decrease in overall survival, while high proportions of T exhausted CD4 cells (p = 0.0097) were correlated with better overall survival. These data could be explained by the presence of exhaustion markers, which are also indicative of the pre-existing functions of these cells, leading to better overall survival.
T-cell trajectory and function in HGSCs
The composition of primary untreated tumors is highly enriched in lymphoid cells in a late or exhausted stage of differentiation. This finding was corroborated by the results of our analysis of T-cell trajectories; both CD8 and CD4 T cells were present at different degrees of differentiation in primary untreated tumors (Fig. 4A, B). On the other hand, treated tumors (primary or omental) had an immune infiltrate that was marked by lymphoid cells in earlier stages of differentiation, such as naïve, central memory, and effector memory cells . Owing to the observation that primary untreated tumors are populated by a highly immunosuppressive TME, we inferred the degree of T-cell functionality using an analysis previously described by Li et al.39 Interestingly, we observed that T-cell functionality was clustered according to tissue location and treatment status. The cytotoxicity score, which was based on the expression of FGFBP2 and 30 correlated genes39, was lowest in the immune clusters of primary untreated tumors. In contrast, omental tumors (independent of treatment status) showed a higher cytotoxicity score (Fig. 4C).

Differentiation trajectory analysis of A CD8 T cells and B CD4 T cells projecting the expression of multiple cell types representing different stages of differentiation. Inference of T cell functionality based on gene expression modules for C T cell cytotoxicity, D T cell dysfunction, and E T-reg scores, showing clustering by tissue type and treatment.
Finally, we measured T-cell dysfunction (Fig. 4D) by analyzing the expression of a gene module that included LAG3 and 30 other genes, and measured the T-reg scores (Fig. 4E) by use of a gene module including IL2RA and 29 other genes that are correlated with it. Contrary to the cytotoxicity analysis, primary untreated tumors had the highest scores for both dysfunction and T-regs. Moreover, the omentum tissues all clustered together, independent of treatment status. NACT did not lead to a clear difference in cell states, as the tissue location did. The cytotoxicity, T-cell dysfunction, and T-reg scores were largely shared by all CD8 and CD4 cell subtypes, with few exceptions. Together, our findings on T-cell differentiation trajectories show that primary and omentum tissues are distinct niches for immune cells, with the primary site exhibiting considerably more immune-cell dysfunction than the omentum, according to what was demonstrated previously in the ecotype analysis. These findings could be useful for designing and planning immunomodulating treatments for HGSC.
Spatial analysis of the TME
To examine the crosstalk between distinct epithelial and immune compartments, we performed spatial transcriptomic analyses; the data were analyzed using the Visium platform on a subset of samples included in the single cell analysis. Specifically, we explored the relationship between the IFNα pathway in the epithelial compartment and sites of enriched CD8 cells using the TESLA pipeline. Table 2 shows the tissues used for Visium according to the original single cell data set.
As shown in Fig. 5, we examined the expression of CD8 T cells in tumor areas along with the IFNα and IFNγ response pathways. The IFNα pathway was largely found in areas of close contact between CD8 cells and epithelial cells, although not all areas with CD8 and epithelial cells necessarily demonstrated an IFNα response. The same trend was observed for IFNγ response but with a much larger expression of IFNγ response detected in areas that matched the epithelial areas. Similar patterns were observed in multiple tumors. The results of this analysis point to the importance of close contact between specific cells, in this case, CD8 and epithelial cells to spur increased expression of immune-related pathways, such as IFNα response. Due to this interesting correlation, we checked the correlation of IFN pathways and survival. The TCGA cohort showed a significant association between the Interferon gamma and Interferon alpha signature and overall survival in the ovarian cancer dataset (Supplementary Figs. 4B, C).

Representative tissue sections from three HGSC patients (Pt_12_OM, Pt_19_P, and Pt_19_OM), showing co-localization of CD8 T cells, tumor areas, and interferon signaling pathways (IFNα and IFNγ). Red areas indicate regions of high expression. Each row corresponds to a different patient sample, and each column displays the spatial distribution of one feature, demonstrating spatial relationships between immune infiltration, tumor regions, and interferon pathway activation.
Cellular heterogeneity of primary and metastatic HGSC: stromal compartment
In the stromal compartment, there were limited endothelial cells, cancer-associated fibroblasts (CAFs), and “normal” fibroblasts. The CAFs included 1 cluster of inflammatory CAFs, defined by α-SMAlow and IL-6high, and 2 populations of myofibroblastic CAFs (myCAFs), defined by α-SMAhigh and IL-6low (Supplementary Fig. 5A). The canonical markers used to define the populations are shown in Supplementary Fig. 5B. Statistically, there was no difference in the percentages of different CAF subgroups on the basis of the tumor location or treatment status (Supplementary Fig. 5C). A comparison between gene expression in inflammatory CAFs and myCAFs showed a marked enrichment of genes mapping to EMT, TNFα signaling and NFκβ pathways in inflammatory CAFs, while enrichment of metabolic pathways, such as fatty acid metabolism, reactive oxygen species metabolism, and oxidative phosphorylation was detected in myCAFs (Supplementary Fig. 5D).
Discussion
In this study, we used an innovative combination of single-cell sequencing and spatial transcriptomics to identify the immune cell landscape in primary and omental HGSC patient tissue samples. We found several notable differences. Primary tumors consistently showed a more active immune microenvironment than did omental tumors. In addition, while several immune cell subtypes were present in all samples, their relative abundance differed by tumor type. Finally, we also identified differences in immune cell differentiation and functionality; primary tumors were mostly populated by dysfunctional CD4 and CD8 T cells in later stages of differentiation, while omental tumors were mostly populated by naïve immune cells.
In addition to the interpatient and intrapatient heterogeneity in CNV patterns, we showed how they differed by tumor site and treatment status. We compared epithelial cells in untreated primary tumors and omental metastases and found differences in IFNα and γ. Nonetheless, we identified distinct clustering patterns of immune cells in treated versus untreated tissues and in primary versus omental tissues. Our analysis also uncovered that primary untreated HGSCs were populated by exhausted T cells and epithelial cells, with activation of multiple immune-related pathways. In contrast, treated omental tumors were populated by T cells in earlier stages of differentiation, such as naïve T cells, central memory T cells, and effector memory T cells. The relationship between the epithelial and immune compartments suggests that immunotherapy could be effective against primary tumors in untreated patients.
An interesting finding in our analysis is the enrichment of genes related to the IFNα response pathway across tumor sites and treatment statuses in both epithelial and immune compartments. The IFN response pathways, when activated, mediate the expression of IFNs by immune cells. These pathways are an expression of the tight communication between multiple parts of the TME. The importance of the IFN signature for immune suppression has also been explored in other cancer types, such as melanoma, where the IFN-γ signature was shown to be important for determining the efficacy of immune checkpoint inhibitors. Other enriched signatures such as EMT, also have been correlated with immune suppression in ovarian cancer and melanoma. Interferon (IFN) signature is an important marker of increased IFN activity and is associated with various regulatory mechanisms across a range of diseases40,41. Type I IFNs trigger the expression of a wide array of interferon-stimulated genes (ISGs), which constitute the core of this signature42. In cancer, the IFN signature has a dual function, either supporting or hindering tumor progression, depending on the regulatory pathways involved and the tumor microenvironment. IFN signaling in tumors is complex and context dependent. While it can enhance immune surveillance and anti-tumor responses by increasing antigen presentation and stimulating cytotoxic T cells, prolonged IFN signaling can lead to immune suppression, enabling the tumor to evade immune detection.
While previous reports describing the immune microenvironment in ovarian cancer focused on the abundance of immune cells and their stromal or epithelial localization mainly in treatment naïve tissues43, our study provides a deeper understanding of the activity of these cells. To our knowledge, this is the first study to report a clear difference in the activation status of the immune cells populating untreated versus treated primary tumors, which was validated by using other datasets. Previous studies focusing on the immune microenvironment in HGSC identified the presence of exhausted immune cells as the cause of its poor sensitivity to immune-modulating agents; our data build on these findings by showing that it is specifically untreated primary HGSC that shows increased immune exhaustion, while NACT-treated tumors have immune cells in earlier stages of differentiation with higher immune-active potential. The differentiation stage of T cells in HGSC could significantly influence their cytotoxicity and dysfunction, impacting the effectiveness of immunomodulating treatments. In our study, T cells of untreated tumors were often more exhausted and less cytotoxic, which suggests that therapies such as checkpoint inhibitors targeting T cell exhaustion may be more effective in an upfront setting. Conversely, in treated tumors, T cells tend to be in earlier differentiation stages with higher immune activity, indicating that combining checkpoint inhibitors with other immunotherapies could further enhance their response. These differences highlight the potential for personalized immunotherapy strategies based on T cell differentiation status, optimizing treatment outcomes for patients with HGSC. These findings could have an impact on the timing and choice of immune-modulating drugs.
Our work underlines the importance of understanding the heterogeneity of cancer cells, whose behavior can differ even in the same tumor niche. Moreover, it suggests that external factors such as chemotherapy or surgery could affect the fundamental genetic programs in the cells composing the TME, which in turn modulates tumors’ sensitivity to additional treatments.
While our study demonstrates how single-cell gene expression profiling of clinical samples can uncover possible mechanisms of immune suppression and support the idea of site-specific immune suppression in ovarian cancer, additional analysis with larger number of samples per site may be needed. Further translational studies are needed to test the hypotheses generated here and to determine in a preclinical setting the mechanisms to guide the use of immune therapies in ovarian cancer models.
In conclusion, distinct intrinsic (e.g., tumor site) or extrinsic (e.g., treatment) factors determine the composition of specific immune niches in HGSC. We hope that additional studies like ours will dissect the heterogeneity of the ovarian cancer TME and guide treatments aimed at increasing overall survival and quality of life of patients affected by this malignancy.
Methods
Patient samples
HGSC surgical samples of primary and metastatic sites from 19 patients were collected under a protocol approved by the Institutional Review Board, and all patients provided written informed consent. We have complied with all ethical regulations, including the Declaration of Helsinki. The fresh tissues were obtained from the MD Anderson Gynecologic Tumor Bank, the Women’s Health Integrated Research Center for the Gynecologic Cancer Center of Excellence Program and the INOVA Health System, and the Stephenson Cancer Center at the University of Oklahoma.
Sample collection and tissue dissociation
HGSC surgical samples were collected during surgery. The samples were kept in Dulbecco’s modified Eagle’s medium (DMEM) during transportation and prior to dissociation. Accumax cell detachment solution (StemCell Technologies, cat# 07921) was used for tissue dissociation. Briefly, tissues were rinsed with phosphate-buffered saline (PBS) and then minced. Next, the tissue was incubated in Accumax at 37 °C until partially dissociated, no longer than 15 min. Samples were passed through a 70-μm filter and centrifuged (1500 rpm for 5 min). After centrifugation, cells were frozen with a freezing medium containing 90% fetal bovine serum (FBS) and 10% DMSO and stored at −80 °C.
Cell preparation for single-cell profiling
The stored samples were retrieved right before processing. To maximize the recovery of viable cells, the complete medium (DMEM supplemented with 10% FBS) was warmed in a 37 °C incubator. Cells were then gently thawed in a water bath at 37 °C, transferred to 10 mL of medium, and centrifuged (at 450 × g for 5 min). After removal of the supernatant, the cell pellet was washed once with 10 mL PBS and then centrifuged (1500 rpm for 5 min).
To prepare cells for flow cytometry, cells were resuspended in FACS buffer (PBS supplemented with 5% FBS) and blocked using Human BD Fc Block (BD Bioscience, cat# 564219) for 30 min. To determine cell viability, cells were stained with LIVE/DEAD fixable aqua dead cell stain (ThermoFisher, cat# L34957). To sort CD45+ and CD45- cell populations, cells were incubated for 30 min with an APC anti-human CD45 antibody (BioLegend, cat# 368511). After incubation, cells were washed and resuspended with FACS buffer and filtered through a 35-μm strainer (BD Falcon 5 mL round-bottom tubes with cell strainer cap) to eliminate cell clumps. Cells were then sorted using a BD FACSAria Fusion flow cytometer.
Single-cell RNA sequencing
Sorted cells were washed with PBS and suspended in PBS containing 0.04% bovine serum albumin (BSA). Cells were double-checked for viability and cell number by using a Countess II FL automated cell counter (Thermo Fisher) and a microscope. All cells were diluted to a concentration of 500–1000 cells/μL in PBS/0.04% BSA before use.
Single cells were captured using the 10X Genomics controller according to the bead types and chip used for the experiments. The 10X Genomics Chromium Single Cell 3′ Next GEM library and gel bead kit v3 (cat# 1000075) and Chromium Chip B single cell kit (cat# 1000073) were used to capture cells on the controller. The cell recovery target was from 5000 to 10,000 cells. Captured cells were then subjected to gel-beads-in-emulsion–reverse transcription (GEM-RT), cDNA amplification, and purification according to the 10X Genomics protocol. Cleanup cDNA was checked via a TapeStation automated electrophoresis platform (Agilent 4200) using high-sensitivity D5000 reagents (cat# 5067-5593) for cDNA traces. The library was generated using 25% of the cDNA by using the Chromium i7 multiplex kit (cat #120262) to identify each sample.
The library was cleaned up using AMPure beads, and quality control was again conducted with TapeStation D1000 tapes (cat# 5067-5583). Estimated concentration of library was measured by a Tecan Qubit fluorometer.
Samples were then diluted to 10 nM. Quantitative PCR (qPCR) was performed for the concentration of the library. Ten libraries were pooled to give a final concentration of 10 nM. The pooled samples were again analyzed by qPCR at the final concentration before submission for sequencing, which was performed by the Advanced Technology Genomics Core facility at MD Anderson with a NovaSeq6000 S2 sequencer, using 28 cycles for read 1, 8 cycles for the i7 index, and 91 cycles for read 2. Sequence data were then analyzed using the 10X Genomics Cell Ranger 3.1 pipeline. QC and Fastq files were obtained and checked to verify the quality of the data. Fastq files were used for further analyses.
Single-cell RNA-Seq data processing
The Cell Ranger toolkit (version 3.1.0) from 10x Genomics was applied to aggregate raw data, filter low-quality reads, align reads to a human reference genome (GRCh38), assign cell barcodes, and generate the unique molecular identifier (UMI) matrix. A Python-based toolkit, Scanpy (version 2.3.4)44, was used for analyzing the scRNA-Seq data. Specifically, the raw UMI matrix was processed to filter out genes detected in fewer than 3 cells and cells with fewer than 200 genes. We further quantified the number of genes and the UMI count for each cell and kept high-quality cells with thresholds of more than 1000 UMIs, 500 to 5500 genes, and less than 15% mitochondrial genes to ensure that most of the heterogeneous cell types were included in downstream analyses. Scrublet45 was then applied to each sequencing library to remove potential doublets, with an expected doublet rate of 6%, and cells with a doubletScore above the 90% quantile were filtered out. The normalized expression matrix was calculated based on the raw UMI counts after normalizing total counts per cell (library size) and then scaled by 1e6 and logarithmically transformed.
Dimension reduction and unsupervised clustering for scRNA-Seq data
Dimension reduction and unsupervised clustering were performed according to the standard workflow in Scanpy44. Briefly, the top 3000 highly variable genes (HVGs) were detected using dispersion-based methods, where the normalized dispersion is obtained by scaling with the mean and standard deviation of the dispersions for genes falling into a given bin46. Unwanted sources of variation, including total gene counts, percentages of mitochondrial gene counts, and heat shock protein (HSP)-related gene counts, were further regressed out from the normalized expression matrices. principal component analysis (PCA) was performed on the variable gene matrix to reduce noise, and the top 50 components were used for downstream analyses. To correct the batch effects from different patients, we applied BBKNN to generate a batch-balanced k nearest-neighbor (KNN) graph, which identifies the top neighbors of each cell in each batch separately instead of the entire cell pool31. Then, The Leiden algorithm was applied to these nearest-neighbor graphs to detect communities and find cell clusters47. Of note, the same principal components were also used for nonlinear dimension reduction to generate the UMAP for visualization.
After the first round of unsupervised clustering, we annotated each cell cluster according to canonical immune cell markers and identified the major immune cell types, including T cells, NK cells, B/plasma cells, myeloid cells, and stromal cells37,38,46,48,49,50. To obtain the high-resolution map of each cell type, the second-round subclustering procedure was the same as the first-round clustering, but used the filtered UMI matrix from the first round and was conducted by using methods from the R package “Seurat.” We then identified HVGs (n = 2000), calculated the PCA matrix, corrected batch effects with ‘RunHarmony’ from the R package “Harmony,” detected cell clusters with the “FindClusters” algorithm, and performed dimensionality reduction for visualization by using “RunUMAP.” Marker genes were detected using the “FindAllMarkers” function with parameters of method “Wilcoxon.”
Tissue enrichment and time-point distribution analysis
To quantify the enrichment of immune cell types across different conditions of sample collection, we compared the observed and expected cell numbers for each cluster in each condition according to the following formula, as we previously described49,51: Ro/e = Observed/Expected, where the expected cell numbers of immune cell clusters in a given tissue or at a given time point were calculated by the chi-square test. We assumed that one cluster was enriched in a specific tissue or at a specific time point if Ro/e > 1.
Cell function and trajectory analyses
To identify differences in cell function in different areas, the single-sample gene set enrichment analysis (ssGSEA) algorithm in the gene set variation analysis (GSVA)52 package was used to evaluate the activity of different pathways (from the Molecular Signatures Database [MsigDB])53,54 in each cell, and the ssGSEA score was used to reflect the activation or inhibition of the pathway in each cell. The method “scVelo” was used to reconstruct the development and differentiation trajectories of immune cells55.
Survival analysis
The Cox proportional hazards model implemented in the R package “survival” was used to perform survival analyses. The mean percentage of each cell type among the patients was used as the threshold to classify patients into 2 groups. The multivariate survival analysis was performed by correcting for the effects of other clinical factors, including treatment status, tumor stage, and patient age.
Statistical methods
Differentially expressed genes (DEGs) were defined as genes with cutoffs of min.pct = 0.1, logfc.threshold >1 and adj.p < 0.05. The ssGSEA scores were compared between the 2 groups by using the R package “limma.” The pathways with adj.p < 0.05 were considered as significantly different between the 2 groups. Box plots, violin plots, and histograms in the “ggplot2” (ggplot2: Elegant Graphics for Data Analysis) package were used for visualization. All statistical analyses were performed using Python (version 3.8.5) and R (version 3.6.1).
Data availability
The single-cell RNA sequencing data generated in this study have been submitted to the European Genome-Phenome Archive (EGA) and will be publicly available at EGAD50000000057 upon publication of this manuscript.
References
Matulonis, U. A. et al. Ovarian cancer. Nat. Rev. Dis. Prim. 2, 16061 (2016).
Mullins, M. A. et al. Trends and racial disparities in aggressive end-of-life care for a national sample of women with ovarian. Cancer Cancer 127, 2229–2237 (2021).
Coalition Woc. The World Ovarian Cancer Coalition atlas. The Every Woman Study 2018. https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf) (2018).
Lheureux, S., Braunstein, M. & Oza, A. M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 69, 280–304 (2019).
Berchuck, A. et al. Patterns of gene expression that characterize long-term survival in advanced stage serous ovarian cancers. Clin. Cancer Res. 11, 3686–3696 (2005).
Society A. C. Cancer Facts & Figures 2020. (American Cancer Society, 2020).
Motohara, T. et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 38, 2885–2898 (2019).
Iwagoi, Y. et al. Omental metastasis as a predictive risk factor for unfavorable prognosis in patients with stage III-IV epithelial ovarian cancer. Int. J. Clin. Oncol. 26, 995–1004 (2021).
Etzerodt, A. et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 217, https://doi.org/10.1084/jem.20191869 (2020).
Zhang, L. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348, 203–213 (2003).
Biswas, S. et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 591, 464–470 (2021).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Disis, M. L. et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: phase 1b results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 5, 393–401 (2019).
Hamanishi, J. et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 33, 4015–4022 (2015).
Hodi, F. S. et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl Acad. Sci. USA 105, 3005–3010 (2008).
Hodi, F. S. et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl Acad. Sci. USA 100, 4712–4717 (2003).
Matulonis, U. A. et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann. Oncol. 30, 1080–1087 (2019).
De Nola, R. et al. The crowded crosstalk between cancer cells and stromal microenvironment in gynecological malignancies: biological pathways and therapeutic implication. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20102401 (2019).
Sun, B., Hyun, H., Li, L. T. & Wang, A. Z. Harnessing nanomedicine to overcome the immunosuppressive tumor microenvironment. Acta Pharm. Sin. 41, 970–985 (2020).
Cai, D. L. & Jin, L. P. Immune cell population in ovarian tumor microenvironment. J. Cancer 8, 2915–2923 (2017).
Stanta, G. & Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med.5, 85 (2018.
Lee, S. et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 31, 107502 (2020).
Patch, A. M. et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494 (2015).
Deng, Y. et al. Single-cell RNA-sequencing atlas reveals the tumor microenvironment of metastatic high-grade serous ovarian carcinoma. Front. Immunol. 13, 923194 (2022).
Kan, T. et al. Single-cell RNA-seq recognized the initiator of epithelial ovarian cancer recurrence. Oncogene 41, 895–906 (2022).
Izar, B. et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 26, 1271–1279 (2020).
Olbrecht, S., et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 13, 111 (2021).
Carvalho, R. F.et al. Single-cell and bulk RNA sequencing reveal ligands and receptors associated with worse overall survival in serous ovarian cancer. Cell Commun. Signal 20, 176 (2022).
Xu, J. et al. Single-cell RNA sequencing reveals the tissue architecture in human high-grade serous ovarian cancer. Clin. Cancer Res. 28, 3590–3602 (2022).
Launonen, I. M. et al. Single-cell tumor-immune microenvironment of BRCA1/2 mutated high-grade serous ovarian cancer. Nat. Commun. 13, 835 (2022).
Polanski, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Shiina, T., Hosomichi, K., Inoko, H. & Kulski, J. K. The HLA genomic loci map: expression, interaction, diversity and disease. J. Hum. Genet. 54, 15–39 (2009).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Qiu, X. et al. Single-cell mRNA quantification and differential analysis with census. Nat. Methods 14, 309–315 (2017).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Hao, D. et al. The single-cell immunogenomic landscape of B and plasma cells in early-stage lung adenocarcinoma. Cancer Discov. 12, 2626–2645 (2022).
Chu, Y. et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat. Med. https://doi.org/10.1038/s41591-023-02371-y (2023).
Li, H., et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789.e18 (2019).
Carnero, E. et al. Type I interferon regulates the expression of long non-coding RNAs. Front Immunol. 5, 548 (2014).
Mostafavi, S. et al. Parsing the interferon transcriptional network and its disease associations. Cell 164, 564–578 (2016).
Padariya, M. et al. Functional interfaces, biological pathways, and regulations of interferon-related DNA damage resistance signature (IRDS) genes. Biomolecules 11, https://doi.org/10.3390/biom11050622 (2021).
Vazquez-Garcia, I. et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature 612, 778–786 (2022).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Zhang, Y., et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593.e8 (2021).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Cheng, S., et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e23 (2021).
Guo, X. et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985 (2018).
Zheng, C., et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017).
Zhang, L. et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 14, 7 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Acknowledgements
We thank A. Ninetto and A. Sutton (Research Medical Library, The University of Texas MD Anderson Cancer Center, Houston, Texas) for editorial assistance. This research was supported, in part, by the MD Anderson Ovarian Cancer SPORE (P50 CA281701), grants R35 CA209904 and U01 CA213759, American Cancer Society, Ovarian Cancer Research Alliance, the LeBert Suess Family Endowment for Ovarian Cancer, the Dunwoody Fund, the Blanton-Davis Ovarian Cancer Research Program, Judy’s Mission, the Cabes Family and Jane’s Joyride, and the Frank McGraw Memorial Chair in Cancer Research. E.S. is supported by the Ovarian Cancer Research Alliance (OCRA number FP00006137). S.C. is funded by the Foundation Amy Krouse Rosenthal Ovarian Cancer Early Detection Research Grant through the Foundation for Women’s Cancer 2020/2021 (AWD00005708). N.S. is a CPRIT Scholar in Cancer Research with funding from the Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant RR160021 and a recipient of an Early Career Award from the Ovarian Cancer Research Alliance (grant 649968) and Blanton-Davis Ovarian Cancer Research Program. Sample and data collection and distribution from the Women’s Health Integrated Research Center for the Gynecologic Cancer Center of Excellence Program were funded by awards from the Uniformed Services University of the Health Sciences from the Defense Health Program to the Henry M Jackson Foundation for the Advancement of Military Medicine Inc. including awards HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, HU0001-22-2-0016 to the Gynecologic Cancer Center of Excellence Program (PI: Chad A. Hamilton, Yovanni Casablanca, or Neil T. Phippen; Co-PI: G. Larry Maxwell). I.V.-G. was supported by a Mentored Investigator Award from the Ovarian Cancer Research Alliance [650687].
Author information
Authors and Affiliations
Contributions
E.S. and F.P. designed and conducted the experiments and analysis and wrote the manuscript; P.N.T. contributed to sample collection; E.B. contributed to sample collection and data interpretation; M.H., I.V.G and S.P.S assisted with the analysis; N.S. and S.C. contributed to data interpretation and paper writing; D.B., S.L., K.N.M., K.M.D., G.L.M., T.C., J.C., N.F. contributed to sample and patient data collection; N.S., N.W.B. and N.E.N. contributed to data analysis and interpretation; and L.W. and A.K.S. designed and supervised the study and provided guidance on manuscript development. All of the authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
A.K.S. is a consultant for Merck, GSK, Astra Zeneca, ImmunoGen, Iylon, Onxeo and DSMB for Advenchen and Mural Oncology. N.D.F. is a consultant for GlaxoSmithKline and Immunogen. S.P.S. reports research funding from AstraZeneca and Bristol Myers Squibb, outside the scope of this work; S.P.S. is a consultant and shareholder of Canexia Health Inc. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stur, E., Peng, F., Teng, Pn. et al. The dynamic immune behavior of primary and metastatic ovarian carcinoma. npj Precis. Onc. 9, 120 (2025). https://doi.org/10.1038/s41698-025-00818-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00818-8
