
Abstract
Artificial intelligence (AI) is being used in oncological drug development to address the high costs, low success rates, and long timelines that characterize traditional drug development pipelines. The use of machine learning (ML) and deep learning (DL) models in computer-aided drug design is constantly growing owing to their capacity to analyze large, heterogeneous datasets, their ability to capture nonlinear biological trends, and their integration of various molecular and clinical characteristics. AI applications accelerate target discovery by predicting protein structures, ranking disease-relevant genes, and assessing target drugability. AI can be used to conduct rapid searches of multiplexed chemical libraries, predict drug-target interactions, and optimize the pharmacological and physicochemical properties of drugs in virtual screening. Advanced neural network designs also aid in de novo drug design, which involves developing new molecular structures with therapeutic properties of interest. This review outlines how AI has been used for target identification, virtual screening, de novo molecular design, and, specifically, in cancer applications. It further discusses the major issues in AI-based drug development, such as data quality, model interpretation, computational constraints, and ethical and regulatory considerations, which remain essential obstacles to broader clinical translation.
Similar content being viewed by others
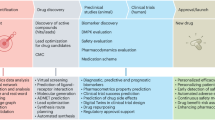

References
Gupta, R. et al. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol. Divers. 25, 1315–1360 (2021).
Tsou, L. K. et al. Comparative study between deep learning and QSAR classifications for TNBC inhibitors and novel GPCR agonist discovery. Sci. Rep. 10, https://doi.org/10.1038/s41598-020-73681-1 (2020).
Grabowski, H. & Long, G. Post-approval indications and clinical trials for cardiovascular drugs: some implications of the US Inflation Reduction Act. J. Med. Econ. 27, 463–472 (2024).
Barbolosi, D., André, N., Barlési, F., Lacarelle, B. & Ciccolini, J. Computational oncology–mathematical modelling of drug regimens for precision medicine. Nat. Rev. Clin. Oncol. 13, 242–254 (2015).
Bhange, M. & Telange, D. Convergence of nanotechnology and artificial intelligence in the fight against liver cancer: a comprehensive review. Discover Oncol. 16, https://doi.org/10.1007/s12672-025-01821-y (2025).
Biankin, A. V., Piantadosi, S. & Hollingsworth, S. J. Patient-centric trials for therapeutic development in precision oncology. Nature 526, 361–370 (2015).
Wang, K., Huang, Y., Wang, Y., You, Q. & Wang, L. Recent advances from computer-aided drug design to artificial intelligence drug design. RSC Med.Chem. 15, 3978–4000 (2024).
Rawat, S. et al. Drug repositioning using computer-aided drug design (CADD. Curr. Pharm. Biotechnol. 25, 301–312 (2024).
Afrose, N., Chakraborty, R., Bhowmick, M., Bhowmick, P. & Hazra, A. AI-driven drug discovery and development. Drug Discov Today. 26, 80–93 (2024).
Lee, J. W., Yoon, S., Maria-Solano, M. A., Choi, S. & Vu, T. N. L. Big data and artificial intelligence (AI) methodologies for computer-aided drug design (CADD. Biochem. Soc. Trans. 50, 241–252 (2022).
Udegbe, F., Ekesiobi, C., Ebulue, C. & Ebulue, O. Machine learning in drug discovery: a critical review of applications and challenges. Comput. Sci. IT Res. J. 5, 892–902 (2024).
Zhang, X. C. et al. MG-BERT: leveraging unsupervised atomic representation learning for molecular property prediction. Brief Bioinformatics 22, https://doi.org/10.1093/bib/bbab152 (2021).
Striuk, O. & Kondratenko, Y. Generative adversarial neural networks and deep learning: successful cases and advanced approaches. Int. J. Comput 20, 339–349 (2021).
Singh, A., Eising, C., Ven, P. & Denny, P. Dynamic filter application in graph convolutional networks for enhanced spectral feature analysis and class discrimination in medical imaging. IEEE Access 12, 113259–113274 (2024).
Cheung, M. & Moura, J. M. F. Graph Neural Networks for COVID-19 Drug Discovery. In Proc. IEEE International Conference on Big Data (Big Data) 5646–5648 (IEEE, 2020).
Hanassab, S. et al. The prospect of artificial intelligence to personalize assisted reproductive technology. npj Digit. Med. 7, 55 (2024).
Liu, X., Xue, Z., Luo, M., Ke, B. & Lv, J. Anesthetic drug discovery with computer-aided drug design and machine learning. Anesthesiol. Perioper. Sci. 2, https://doi.org/10.1007/s44254-023-00047-x (2024).
Wong, C. F. & McCammon, J. A. Protein flexibility and computer-aided drug design. Annu. Rev. Pharmacol. Toxicol. 43, 31–45 (2002).
Pasrija, P., Jha, P., Chopra, M., Upadhyaya, P. & Khan, M. S. Machine learning and artificial intelligence: a paradigm shift in big data-driven drug design and discovery. Curr. Top. Med. Chem. 22, 1692–1727 (2022).
Nayarisseri, A. et al. Artificial Intelligence. Big data and machine learning approaches in precision medicine & drug discovery. Curr. Drug Targets 22, 631–655 (2021).
Cesaro, A., Bagheri, M., Torres, M., Wan, F. & Fuente-Nunez, C. Deep learning tools to accelerate antibiotic discovery. Expert Opin Drug Discov. 18, 1245–1257 (2023).
Nguyen, A. T. N. et al. The application of artificial intelligence to accelerate G protein-coupled receptor drug discovery. Br. J. Pharmacol. 181, 2371–2384 (2023).
Mouchlis, V. D. et al. Advances in de novo drug design: from conventional to machine learning methods. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22041676 (2021).
Lin, E., Lin, C. H. & Lane, H. Y. De novo peptide and protein design using generative adversarial networks: an update. J. Chem. Inf. Model. 62, 761–774 (2022).
Fu, C. & Chen, Q. The future of pharmaceuticals: artificial intelligence in drug discovery and development. J. Pharm. Anal. 15, 101248. https://doi.org/10.1016/j.jpha.2025.101248 (2025).
Kant, S., Deepika, D. & Roy, S. Artificial intelligence in drug discovery and development: transforming challenges into opportunities. Discov. Pharm. Sci. 1, https://doi.org/10.1007/s44395-025-00007-3 (2025).
Ocana, A. et al. Integrating artificial intelligence in drug discovery and early drug development: a transformative approach. Biomarker Res. 13, https://doi.org/10.1186/s40364-025-00758-2 (2025).
Pammolli, F. et al. The endless frontier? The recent increase of R&D productivity in pharmaceuticals. J. Translat. Med. 18, https://doi.org/10.1186/s12967-020-02313-z (2020).
Dhudum, R., Pawar, A. & Ganeshpurkar, A. Revolutionizing drug discovery: a comprehensive review of AI applications. Drugs Drug Candidates 3, 148–171 (2024).
Shaki, F., Jahani, D., Amilrkhanloo, M. & Chahrdori, M. Artificial intelligence in pharmaceuticals: exploring applications and legal challenges. Pharm. Biomed. Res. 10, 1–10 (2024).
Mukaidaisi, M., Grantham, K., Vu, A., Li, Y. & Tchagang, A. Multi-objective drug design based on graph-fragment molecular representation and deep evolutionary learning. Front. Pharmacol. 13, https://doi.org/10.3389/fphar.2022.920747 (2022).
Khan, M. K. et al. The recent advances in the approach of artificial intelligence (AI) towards drug discovery. Front. Chem. 12, https://doi.org/10.3389/fchem.2024.1408740 (2024).
Li, K., Du, Y., Li, L. & Wei, D. Q. Bioinformatics approaches for anti-cancer drug discovery. Curr. Drug Targets 21, 3–17 (2019).
Doherty, G. J. et al. Cancer treatment in the genomic era. Annu. Rev. Biochem. 88, 247–280 (2019).
Kumar, B., Singh, S., Kumar, V. & Skvortsova, I. Promising targets in anti-cancer drug development: recent updates. Curr. Med. Chem. 24, https://doi.org/10.2174/0929867324666170331123648 (2018).
Garg, P. et al. Artificial intelligence–driven computational approaches in the development of anticancer drugs. Cancers 16, 3884 (2024).
Hachem, S. et al. Contemporary update on clinical and experimental prostate cancer biomarkers: a multi-omics-focused approach to detection and risk stratification. Biology 13, https://doi.org/10.3390/biology13100762 (2024).
Arya, K. R. et al. Multitarget-directed multiple ligands in anti-VEGF resistant glioblastoma therapeutics. https://doi.org/10.47852/bonviewMEDIN52023816 (2025).
Akbani, R. et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 5, https://doi.org/10.1038/ncomms4887 (2014).
Mottaghi-Dastjerdi, N. & Soltany-Rezaee-Rad, M. Advancements and applications of artificial intelligence in pharmaceutical sciences: a comprehensive review. Iran. J. Pharm. Res. IJPR 23, https://doi.org/10.5812/ijpr-150510 (2024).
Kamya, P. et al. PandaOmics: an AI-driven platform for therapeutic target and biomarker discovery. J. Chem. Inf. Model. 64, 3961–3969 (2024).
Raufaste-Cazavieille, V., Droit, A. & Santiago, R. Multi-omics analysis: paving the path toward achieving precision medicine in cancer treatment and immuno-oncology. Front. Mol. Biosci. 9, https://doi.org/10.3389/fmolb.2022.962743 (2022).
Kamya, P. et al. PandaOmics: an AI-driven platform for therapeutictarget and biomarker discovery. J. Chem. Inf. Model. 64, 3961–3969 (2024).
Ivanenkov, Y. A. et al. Chemistry42: an AI-driven platform for molecular design and optimization. J. Chem. Inf. Model. 63, 695–701 (2023).
Patel, M. N., Halling-Brown, M. D., Al-Lazikani, B., Tym, J. E. & Workman, P. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discov. 12, 35–50 (2012).
Migliozzi, S. et al. Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy. Nat. Cancer 4, 181–202 (2023).
Tang, X. et al. A survey of generative AI for de novo drug design: new frontiers in molecule and protein generation. Briefings Bioinformatics 25, https://doi.org/10.1093/bib/bbae338 (2024).
Bilal, H. et al. An intelligent approach for early and accurate predication of cardiac disease using hybrid artificial intelligence techniques. https://doi.org/10.3390/bioengineering11121290 (2024).
Karimi, M., Wang, Z., Shen, Y. & Wu, D. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35, 3329–3338 (2019).
Lilhore, U. K. et al. ProtienCNN-BLSTM: an efficient deep neural network with amino acid embedding-based model of protein sequence classification and biological analysis. Comput. Intelligence 40, https://doi.org/10.1111/coin.12696 (2024).
Bull, S. C. & Doig, A. J. Properties of protein drug target classes. PLoS ONE 10, https://doi.org/10.1371/journal.pone.0117955 (2015).
Haider, S., Ghosh, S., Pal, R. & Rahman, R. A copula based approach for design of multivariate random forests for drug sensitivity prediction. PLOS ONE 10, 0144490 (2015).
Akter, S. AI-driven precision medicine: transforming personalized cancer treatment. J. AI Power. Med. Innov. 2, 10–21 (2024).
Masso, M. & Vaisman, I. I. AUTO-MUTE 2.0: a portable framework with enhanced capabilities for predicting protein functional consequences upon mutation. Adv. Bioinformatics 2014, 1–7 (2014).
Zhai, D. et al. Small-molecule targeting AMPA-mediated excitotoxicity has therapeutic effects in mouse models for multiple sclerosis. Sci. Adv. 9, https://doi.org/10.1126/sciadv.adj6187 (2023).
Song, K. et al. AlloDriver: a method for the identification and analysis of cancer driver targets. Nucleic Acids Res. 47, 315–321 (2019).
Larochelle, J. R. et al. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 9, https://doi.org/10.1038/s41467-018-06823-9 (2018).
Deng, X., Das, S., Valdez, K., Camphausen, K. & Shankavaram, U. SL-BioDP: Multi-cancer interactive tool for prediction of synthetic lethality and response to cancer treatment. Cancers 11, https://doi.org/10.3390/cancers11111682 (2019).
Das, S., Shankavaram, U., Deng, X. & Camphausen, K. DiscoverSL: an R package for multi-omic data driven prediction of synthetic lethality in cancers. Bioinformatics 35, 701–702 (2018).
Benfatto, S. et al. Uncovering cancer vulnerabilities by machine learning prediction of synthetic lethality. Mol. Cancer 20, https://doi.org/10.1186/s12943-021-01405-8 (2021).
Huang, J., Lu, F., Wu, M., Ou-Yang, L. & Zhu, Z. Predicting synthetic lethal interactions in human cancers using graph regularized self-representative matrix factorization. BMC Bioinformatics 20, https://doi.org/10.1186/s12859-019-3197-3 (2019).
Wang, Z. et al. BatmanNet: bi-branch masked graph transformer autoencoder for molecular representation. Brief. Bioinformatics 25, https://doi.org/10.1093/bib/bbad400 (2023).
Savage, S. R. et al. Pan-cancer proteogenomics expands the landscape of therapeutic targets. Cell 187, 4389–4407 4315 (2024).
Kokudeva, M., Vichev, M., Naseva, E., Miteva, D. G. & Velikova, T. Artificial intelligence as a tool in drug discovery and development. World J. Exp. Med. 14, https://doi.org/10.5493/wjem.v14.i3.96042 (2024).
Nelen, J. et al. ESSENCE-Dock: a consensus-based approach to enhance virtual screening enrichment in drug discovery. J. Chem. Inf. Model. 64, 1605–1614 (2024).
Bielska, E. et al. REVIEW PAPER: Virtual screening strategies in drug design – methods and applications. BioTechnologia 92, 249–264 (2011).
Polgar, T. & Keseru, M. Integration of virtual and high throughput screening in lead discovery settings. Combinat. Chem. High. Throughput Screen. 14, 889–897 (2011). & G.
Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 1, 882–894 (2002).
Grotsch, K. et al. Virtual screening of a chemically diverse “Superscaffold. Library Enables Ligand Discovery for a Key GPCR Target. ACS Chem. Biol. 19, 866–874 (2024).
Yang, Y. et al. D3AI-CoV: a deep learning platform for predicting drug targets and for virtual screening against COVID-19. Brief. Bioinformatics 23, https://doi.org/10.1093/bib/bbac147 (2022).
Cournia, Z. et al. Rigorous free energy simulations in virtual screening. J. Chem. Inf. Model. 60, 4153–4169 (2020).
Nascimento, L. D. et al. Applications of virtual screening in bioprospecting: facts, shifts, and perspectives to explore the chemo-structural diversity of natural products. Front. Chem. 9, https://doi.org/10.3389/fchem.2021.662688 (2021).
Ebalunode, J. O., Liang, J., Ouyang, Z. & Zheng, W. Novel approach to structure-based pharmacophore search using computational geometry and shape matching techniques. J. Chem. Inf. Model. 48, 889–901 (2008).
Niinivehmas, S. P., Manivannan, E., Rauhamäki, S., Huuskonen, J. & Pentikäinen, O. T. Identification of estrogen receptor α ligands with virtual screening techniques. J. Mol. Graph. Model. 64, 30–39 (2016).
Rudrapal, M. & Chetia, D. Virtual screening, molecular docking and QSAR studies in drug discovery and development programme. J. Drug Deliv. Ther. 10, 225–233 (2020).
Potlitz, F., Link, A. & Schulig, L. Advances in the discovery of new chemotypes through ultra-large library docking. Expert Opin. Drug Discov. 18, 303–313 (2023).
Jiang, P. et al. Molecular persistent spectral image (Mol-PSI) representation for machine learning models in drug design. Brief. Bioinformatics 23, https://doi.org/10.1093/bib/bbab527 (2021).
Kuan, J., Cherkasov, A., Avenido, A., Gentile, F. & Radaeva, M. Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening. WIREs Comput. Mol. Sci. 13, https://doi.org/10.1002/wcms.1678 (2023).
Wang, S. et al. MSGNN-DTA: multi-scale topological feature fusion based on graph neural networks for drug-target binding affinity prediction. Int. J. Mol. Sci. 24, 8326 (2023).
Isert, C., Atz, K., Riniker, S. & Schneider, G. Exploring protein-ligand binding affinity prediction with electron density-based geometric deep learning. RSC Adv. 14, 4492–4502 (2024).
Zhou, G. et al. An artificial intelligence accelerated virtual screening platform for drug discovery. Nat. Commun. 15, https://doi.org/10.1038/s41467-024-52061-7 (2024).
Meli, R., Morris, G. M. & Biggin, P. C. Scoring functions for protein-ligand binding affinity prediction using structure-based deep learning. Rev. Front. Bioinformatics 2, https://doi.org/10.3389/fbinf.2022.885983 (2022).
Bekker, G. J., Kamiya, N., Fukuda, I., Fukunishi, Y. & Higo, J. Cryptic-site binding mechanism of medium-sized Bcl-xL inhibiting compounds elucidated by McMD-based dynamic docking simulations. Sci. Rep. 11, https://doi.org/10.1038/s41598-021-84488-z (2021).
Vidal-Limon, A., Aguilar-Toalá, J. E. & Liceaga, A. M. Integration of molecular docking analysis and molecular dynamics simulations for studying food proteins and bioactive peptides. J. Agric. Food Chem. 70, 934–943 (2022).
Beglov, D. et al. Exploring the structural origins of cryptic sites on proteins. Proc. Natl. Acad. Sci. USA 115, https://doi.org/10.1073/pnas.1711490115 (2018).
Naqvi, A. A. T., Hassan, M. I., Mohammad, T. & Hasan, G. M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 18, 1755–1768 (2018).
Gu, S. et al. Can molecular dynamics simulations improve predictions of protein-ligand binding affinity with machine learning? Brief. Bioinformatics 24, https://doi.org/10.1093/bib/bbad008 (2023).
Bentham Science Publisher, B. S. P. Docking and scoring - theoretically easy, practically impossible? Curr. Med.Chem. 13, 2995–3003 (2006).
Díaz-Holguín, A. et al. AlphaFold accelerated discovery of psychotropic agonists targeting the trace amine-associated receptor 1. Sci. Adv. 10, https://doi.org/10.1126/sciadv.adn1524 (2024).
Glukhov, E. et al. Phospho-Tune: Enhanced Structural Modeling of Phosphorylated Protein Interactions (Cold Spring Harbour Laboratory, 2024).
Liu, J., Neupane, P. & Cheng, J. in and AlphaFold3-Based Protein Complex Structure Prediction With MULTICOM4 in CASP16. Proteins (2025).
Krokidis, M. G. et al. AlphaFold3: an overview of applications and performance insights. Int. J. Mol. Sci. 26, 3671 (2025).
Gheidari, D., Mehrdad, M. & Bayat, M. Synthesis, docking, MD simulation, ADMET, drug likeness, and DFT studies of novel furo[2,3-b]indol-3a-ol as promising Cyclin-dependent kinase 2 inhibitors. Sci. Rep. 14, https://doi.org/10.1038/s41598-024-53514-1 (2024).
Kaveh, S., Mani-Varnosfaderani, A. & Neiband, M. S. Deriving general structure–activity/selectivity relationship patterns for different subfamilies of cyclin-dependent kinase inhibitors using machine learning methods. Sci. Rep. 14, https://doi.org/10.1038/s41598-024-66173-z (2024).
Sheik Amamuddy, O. et al. Integrated computational approaches and tools forallosteric drug discovery. Int. J. Mol. Sci. 21, 847 (2020).
Aderinwale, T. et al. Real-time structure search and structure classification for AlphaFold protein models. Commun. Biol. 5, https://doi.org/10.1038/s42003-022-03261-8 (2022).
Wu, N., Strömich, L. & Yaliraki, S. N. Prediction of allosteric sites and signaling: insights from benchmarking datasets. Patterns 3, 100408 (2021).
Ruiz-Carmona, S. et al. Dynamic undocking and the quasi-bound state as tools for drug discovery. Nat. Chem. 9, 201–206 (2016).
Sun, H. & Scott, D. O. Structure-based drug metabolism predictions for drug design. Chem. Biol. Drug Des. 75, 3–17 (2009).
Chen, L. et al. From laptop to benchtop to bedside: structure-based drug design on protein targets. Curr. Pharm. Des. 18, 1217–1239 (2012).
Ivanova, L. & Karelson, M. The impact of software used and the type of target protein on molecular docking accuracy. Molecules 27, 9041 (2022).
Pinzi, L. & Rastelli, G. Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci. 20, 4331 (2019).
Aboalroub, A. A. Virtual screening and molecular docking characterization of isoxazole-based small molecules as potential Hsp90 inhibitors: an in silico insight. Medinformatics 2, 154–166 (2025).
Luttens, A. et al. Rapid traversal of vast chemical space using machine learning-guided docking screens. Nat. Comput. Sci. 5, 301–312 (2025).
Pason, L. P. & Sotriffer, C. A. Empirical scoring functions for affinity prediction of protein-ligand complexes. Mol. Inform. 35, 541–548 (2016).
Morrone, J. A., Luo, H., Huynh, T., Weber, J. K. & Cornell, W. D. Combining docking pose rank and structure with deep learning improves protein-ligand binding mode prediction over a baseline docking approach. J. Chem. Inf. Model. 60, 4170–4179 (2020).
Lee, C. et al. GalaxyDock-DL: protein-ligand docking by global optimization and neural network energy. J. Chem. Theory Comput. https://doi.org/10.1021/acs.jctc.4c00385 (2024).
Buttenschoen, M., Morris, G. M. & Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130–3139 (2024).
Ghersi, D. & Sanchez, R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins 74, 417–424 (2008).
Arcon, J. P. et al. Molecular dynamics in mixed solvents reveals protein-ligand interactions, improves docking, and allows accurate binding free energy predictions. J. Chem. Inf. Model. 57, 846–863 (2017).
Kuzmanic, A., Bowman, G. R., Michel, J., Juarez-Jimenez, J. & Gervasio, F. L. Investigating cryptic binding sites by molecular dynamics simulations. Acc. Chem. Res. 53, 654–661 (2020).
Ge, Y., Pande, V., Seierstad, M. J. & Damm-Ganamet, K. L. Exploring the application of sitemap and site finder for focused cryptic pocket identification. J. Phys. Chem. B 128, 6233–6245 (2024).
Sabanés Zariquiey, F., Souza, J. V. & Bronowska, A. K. Cosolvent Analysis Toolkit (CAT): a robust hotspot identification platform for cosolvent simulations of proteins to expand the druggable proteome. Sci. Rep. 9, https://doi.org/10.1038/s41598-019-55394-2 (2019).
Kashyap, A., Singh, P. K. & Silakari, O. Counting on fragment based drug design approach for drug discovery. Curr. Top. Med. Chem. 18, 2284–2293 (2019).
Czub, N. et al. Artificial intelligence-based quantitative structure-property relationship model for predicting human intestinal absorption of compounds with serotonergic activity. Mol. Pharmaceutics 20, 2545–2555 (2023).
Qin, T., Zhu, Z., Wang, X. S., Xia, J. & Wu, S. Computational representations of protein–ligand interfaces for structure-based virtual screening. Expert Opin. Drug Discov. 16, 1175–1192 (2021).
Muhammed, M. T. & Aki-Yalcin, E. Pharmacophore modeling in drug discovery: methodology and current status. J. Turkish Chem. Soc. Sect. A Chem. 8, 749–762 (2021).
Akingbade, T. V. et al. In silico design of novel Artocarpus altilis-derived compounds targeting the c-Myc/Max heterodimer. Medinformatics 2, 181–194 (2025).
Fassio, A. V. et al. Prioritizing virtual screening with interpretable interaction fingerprints. J. Chem. Inf. Model. 62, 4300–4318 (2022).
Jiang, X., Tan, L. & Zou, Q. DGCL: dual-graph neural networks contrastive learning for molecular property prediction. Brief. Bioinformatics 25, https://doi.org/10.1093/bib/bbae474 (2024).
Menke, J. & Koch, O. Using domain-specific fingerprints generated through neural networks to enhance ligand-based virtual screening. J. Chem. Inf. Model. 61, 664–675 (2021).
Bathini, R., Fatima, S., Manga, V. & Sivan, S. K. Molecular docking, MM/GBSA and 3D-QSAR studies on EGFR inhibitors. J. Chem. Sci. 128, 1163–1173 (2016).
Pérez-Nueno, V. I., Ritchie, D. W., Pettersson, S., Borrell, J. I. & Teixidó, J. Discovery of novel HIV entry inhibitors for the CXCR4 receptor by prospective virtual screening. J. Chem. Inf. Model. 49, 810–823 (2009).
Liu, J. et al. Combined pharmacophore modeling, 3D-QSAR and docking studies to identify novel HDAC inhibitors using drug repurposing. J. Biomol. Struct. Dyn. 38, 533–547 (2019).
Keyvanpour, M. R. & Shirzad, M. B. An analysis of QSAR research based on machine learning concepts. Curr. Drug Discov. Technol. 18, 17–30 (2020).
Babajide Mustapha, I. & Saeed, F. Bioactive molecule prediction using extreme gradient boosting. Molecules 21, 983 (2016).
Ghaleb, A., Aouidate, A., Bouachrine, M., Lakhlifi, T. & Sbai, A. In silico exploration of Aryl halides analogues as checkpoint kinase 1 inhibitors by using 3D QSAR, molecular docking study, and ADMET screening. Adv. Pharm. Bull. 9, 84–92 (2019).
Halder, A. K. & Cordeiro, M. N. D. S. AKT inhibitors: the road ahead to computational modeling-guided discovery. Int. J. Mol. Sci. 22, 3944 (2021).
Xiong, G. et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49, 5–14 (2021).
Sá, A. G. C., Ascher, D. B. & Myung, Y. Deep-PK: deep learning for small molecule pharmacokinetic and toxicity prediction. Nucleic Acids Res. 52, 469–475 (2024).
Gangwal, A. et al. Generative artificial intelligence in drug discovery: basic framework, recent advances, challenges, and opportunities. Fron. Pharmacol. 15, https://doi.org/10.3389/fphar.2024.1331062 (2024).
Gupta, A. et al. Generative recurrent networks for de novo drug design. Mol. Inform. 37, 1700111 (2017).
Moret, M., Grisoni, F., Friedrich, L., Merk, D. & Schneider, G. Generative molecular design in low data regimes. Nat. Mach. Intell. 2, 171–180 (2020).
Loving, K., Alberts, I. & Sherman, W. Computational approaches for fragment-based and de novo design. Curr. Top. Med. Chem. 10, 14–32 (2010).
Gummesson Svensson, H., Tyrchan, C., Engkvist, O. & Haghir Chehreghani, M. Utilizing reinforcement learning for de novo drug design. Mach. Learn. 113, 4811–4843 (2024).
Sousa, T., Correia, J., Pereira, V. & Rocha, M. Generative deep learning for targeted compound design. J. Chem. Inf. Model. 61, 5343–5361 (2021).
Kotsias, P. C. et al. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2, 254–265 (2020).
Lavecchia, A. Navigating the frontier of drug-like chemical space with cutting-edge generative AI models. Drug Discovery Today 29, 104133 (2024).
Hong, S. H., Ryu, S., Lim, J. & Kim, W. Y. Molecular generative model based on an adversarially regularized autoencoder. J. Chem. Inf. Model. 60, 29–36 (2019).
Monti, M., Fiorentino, J., Gosti, G., Tartaglia, G. G. & Milanetti, E. Prediction of time series gene expression and structural analysis of gene regulatory networks using recurrent neural networks. Entropy 24, 141 (2022).
Mienye, I. D., Swart, T. G. & Obaido, G. Recurrent neural networks: a comprehensive review of architectures, variants, and applications. Information 15, 517 (2024).
Zheng, S. et al. Deep scaffold hopping with multimodal transformer neural networks. J. Cheminformatics 13, https://doi.org/10.1186/s13321-021-00565-5 (2021).
Bian, Y. & Xie, X. Q. Generative chemistry: drug discovery with deep learning generative models. J. Mol. Model. 27, https://doi.org/10.1007/s00894-021-04674-8 (2021).
Zhao, L., Wang, J., Pang, L., Liu, Y. & Zhang, J. GANsDTA: predicting drug-target binding affinity using GANs. Front. Genet. 10, https://doi.org/10.3389/fgene.2019.01243 (2020).
Baammi, S., El Allali, A. & Daoud, R. Potent VEGFR-2 inhibitors for resistant breast cancer: a comprehensive 3D-QSAR, ADMET, molecular docking and MMPBSA calculation on triazolopyrazine derivatives. Front. Mol. Biosci. 10, https://doi.org/10.3389/fmolb.2023.1288652 (2023).
Guo, J. et al. DockStream: a docking wrapper to enhance de novo molecular design. J. Cheminformatics 13, https://doi.org/10.1186/s13321-021-00563-7 (2021).
Abate, C., Cavalli, A. & Decherchi, S. Graph neural networks for conditional de novo drug design. WIREs Comput. Mol. Sci. 13, https://doi.org/10.1002/wcms.1651 (2023).
Born, J. et al in Designing Anticancer Drugs From Transcriptomic Data via Reinforcement Learning 231–233 (Springer, 2020).
Huang, D., Yang, C., Li, Z., Li, N. & Jiang, T. Artificial intelligence in lung cancer: current applications, future perspectives, and challenges. Front. Oncol. 14, https://doi.org/10.3389/fonc.2024.1486310 (2024).
Noorain, N., Parveen, R., Parveen, B. & Srivastava, V. Artificial intelligence in drug formulation and development: applications and future prospects. Curr. Drug Metab. 24, 622–634 (2023).
Hasselgren, C. & Oprea, T. I. Artificial intelligence for drug discovery: are we there yet? Annu. Rev. Pharmacol. Toxicol. 64, 527–550 (2023).
Larsson, P. et al. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 10, https://doi.org/10.1038/s41598-020-62848-5 (2020).
Schaduangrat, N. et al. Towards reproducible computational drug discovery. J. Cheminformatics 12, https://doi.org/10.1186/s13321-020-0408-x (2020).
Han, D., Huong, P. & Cheng, S. Enhancing semantic segmentation through reinforced active learning: combating dataset imbalances and bolstering annotation efficiency. J. Electron. Inf. Syst. 5, 45–60 (2023).
Karim, M. R. et al. Explainable AI for Bioinformatics: methods, tools and applications. Brief. Bioinformatics 24, https://doi.org/10.1093/bib/bbad236 (2023).
Singh, D. S., Pratap Singh, D. D. & Chandra, M. K. Enhancing transparency and interpretability in deep learning models: a comprehensive study on explainable AI techniques. Int. J. Sci. Res. Eng. Manag. 08, 1–13 (2024).
Alkhanbouli, R., Matar Abdulla Almadhaani, H., Alhosani, F. & Simsekler, M. C. E. The role of explainable artificial intelligence in disease prediction: a systematic literature review and future research directions. BMC Med. Inform. Decision Making 25, https://doi.org/10.1186/s12911-025-02944-6 (2025).
Başağaoğlu, H. et al. A review on interpretable and explainable artificial intelligence in hydroclimatic applications. Water 14, https://doi.org/10.3390/w14081230 (2022).
Korade, D. Unlocking machine learning model decisions: a comparative analysis of LIME and SHAP for enhanced interpretability. J. Electr. Syst. 20, 598–613 (2024).
Thalpage, N. Unlocking the black box: explainable artificial intelligence (XAI) for trust and transparency in AI systems. J. Digit. Art. Human. 4, 31–36 (2023).
Anang, A. et al. Explainable AI in financial technologies: balancing innovation with regulatory compliance. Int. J. Sci. Res. Arch. 13, 1793–1806 (2024).
Metta, C., Giannotti, F., Rinzivillo, S., Beretta, A. & Pellungrini, R. Towards transparent healthcare: advancing local explanation methods in explainable artificial intelligence. Bioengineering 11, 369 (2024).
Alizadehsani, R. et al. Explainable artificial intelligence for drug discovery and development: a comprehensive survey. IEEE Access 12, 35796–35812 (2024).
Kırboğa, K. K., Abbasi, S. & Küçüksille, E. U. Explainability and white box in drug discovery. Chem. Biol. Drug Des. 102, 217–233 (2023).
Jiang, Y. et al. Pharmacophoric-constrained heterogeneous graph transformer model for molecular property prediction. Commun. Chem. 6, https://doi.org/10.1038/s42004-023-00857-x (2023).
Yang, Z., Zhong, W., Zhao, L. & Yu-Chian Chen, C. MGraphDTA: deep multiscale graph neural network for explainable drug-target binding affinity prediction. Chem. Sci. 13, 816–833 (2022).
Li, S. et al. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity. 975–985 (2021).
Singhal, S. Data privacy, compliance, and security including AI ML. Healthcare 111–126 (2024).
Koo, T. H., Zakaria, A. D., Ng, J. K. & Leong, X. B. Systematic review of the application of artificial intelligence in healthcare and nursing care. Malays. J. Med. Sci. MJMS 31, 135–142 (2024).
Kalra, B., Khirasaria, R. & Batta, A. Trends in FDA drug approvals over last 2 decades: An observational study. J. Fam. Med. Prim. Care 9, 105 (2020).
Vora, L. K. et al. Artificial intelligence in pharmaceutical technology and drug delivery design. Pharmaceutics 15, https://doi.org/10.3390/pharmaceutics15071916 (2023).
O’Connor, L. M., O’Connor, B. A., Lim, S. B., Zeng, J. & Lo, C. H. Integrative multi-omics and systems bioinformatics in translational neuroscience: a data mining perspective. J. Pharm. Anal. 13, 836–850 (2023).
Alghushairy, O. et al. Machine learning-based model for accurate identification of druggable proteins using light extreme gradient boosting. J. Biomol. Struct. Dyn. 42, 12330–12341 (2023).
Haza, K. Z. et al. RAS-inhibiting biologics identify and probe druggable pockets, including an SII- u03b13 allosteric site. Nat. Commun. 12, https://doi.org/10.1038/s41467-021-24316-0 (2021).
Meng, L., Ye, Z., Yang, Y. & Zhao, H. DeepMCGCN: multi-channel deep graph neural networks. Int. J. Comput. Intell. Syst. 17, https://doi.org/10.1007/s44196-024-00432-9 (2024).
Bhalla, S. & Laganà, A. Artificial intelligence for precision oncology. Adv. Exp. Med. Biol. 1361, 249–268 (2022).
Eckhart, L., Lenhof, K., Rolli, L. M. & Lenhof, H. P. A comprehensive benchmarking of machine learning algorithms and dimensionality reduction methods for drug sensitivity prediction. Brief. Bioinformatics 25, https://doi.org/10.1093/bib/bbae242 (2024).
McDonagh, E. M. et al. Human genetics and genomics for drug target identification and prioritization: open targets’ perspective. Annu. Rev. Biomed. Data Sci. 7, 59–81 (2024).
Kim, M., Oh, I. & Ahn, J. An improved method for prediction of cancer prognosis by network learning. Genes 9, 478 (2018).
Dezső, Z. & Ceccarelli, M. Machine learning prediction of oncology drug targets based on protein and network properties. BMC Bioinformatics 21, https://doi.org/10.1186/s12859-020-3442-9 (2020).
Raies, A. et al. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 5, https://doi.org/10.1038/s42003-022-04245-4 (2022).
Nussinov, R., Jang, H. & Tsai, C. Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J. 34, 16–29 (2019).
Song, Q. et al. DeepAlloDriver: a deep learning-based strategy to predict cancer driver mutations. Nucleic Acids Res. 51, 129–133 (2023).
Park, N., Dong, X. L., Kan, A., Zhao, T. & Faloutsos, C. Estimating Node Importance In Knowledge Graphs Using Graph Neural Networks 596–606 (Association for Computing Machinery, 2019).
Li, P. et al. Improving drug response prediction via integrating gene relationships with deep learning. Brief. Bioinformatics 25, https://doi.org/10.1093/bib/bbae153 (2024).
Purwono, P., Wulandari, A. N. E., Salah, W. A. & Ma’Arif, A. Understanding Generative Adversarial Networks (GANs): A Review. Control Syst. Optim. Lett. 3, 36–45 (2025).
Ni, Y., Song, D., Liao, L., Zhang, X. & Wu, H. CAGAN: Consistent Adversarial Training Enhanced GANs https://doi.org/10.24963/ijcai.2018/359 (2018).
Chen, Y. et al. Molecular language models: RNNs or transformer? Brief. Funct. Genomics 22, 392–400 (2023).
Laurent, C., Zhang, Y., Pereyra, G., Brakel, P. & Bengio, Y. Batch normalized recurrent neural networks. In Proc. IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP) 2657-2661 (IEEE, 2016).
Bagal, V., Vinod, P. K., Priyakumar, U. D. & Aggarwal, R. MolGPT: molecular generation using a transformer-decoder model. J. Chem. Inf. Model. 62, 2064–2076 (2021).
Ross, J. et al. GP-MoLFormer: a foundation model for molecular generation. https://doi.org/10.48550/arxiv.2405.04912 (2024).
Miletic, M. & Sariyar, M. Challenges of using synthetic data generation methods for tabular microdata. Appl. Sci. 14, 5975 (2024).
Yu, M. S. et al. A variational autoencoder cascade generative adversarial network for scalable 3D object generation and reconstruction. https://doi.org/10.3390/s24030751 (2024).
Cai, L. et al. AEGNN-M:A 3D Graph-Spatial Co-Representation Model for Molecular Property Prediction. IEEE J. Biomed. Health Inform. 29, 1726–1734 (2025).
Cao, N. & Kipf, T. MolGAN: An Implicit Generative Model For Small Molecular Graphs (Cornell University, 2018).
Urbina, F., Lowden, C. T., Culberson, J. C. & Ekins, S. MegaSyn: integrating generative molecular design, automated analog designer, and synthetic viability prediction. ACS Omega 7, 18699–18713 (2022).
Xu, C., Zheng, P., Chen, H., Wang, H. & Wang, W. Geometric-facilitated denoising diffusion model for 3D molecule generation. Proc. AAAI Conf. Artif. Intell. 38, 338–346 (2024).
Regenwetter, L., Nobari, A. H. & Ahmed, F. Deep generative models in engineering design. A Review. J. Mech. Design 144, https://doi.org/10.1115/1.4053859 (2022).
Liu, X. et al. DrugEx v2: de novo design of drug molecules by Pareto-based multi-objective reinforcement learning in polypharmacology. J. Cheminformatics 13, https://doi.org/10.1186/s13321-021-00561-9 (2021).
Acknowledgements
This study was funded by the Liaoning Provincial Science and Technology Joint Program (Natural Science Foundation General Program, Grant No. 2024-MSLH-536).
Author information
Authors and Affiliations
Contributions
Da Li. Sanbao Shi.: Conceptualization, methodology, writing—original draft; Writing— review. Zhiyu Yu. Peng Xu.: Data curation, Formal analysis; Writing–review and editing. Cheng Zhang.: Supervision, writing–review and editing. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, D., Shi, S., Yu, Z. et al. AI accelerate the identification of druggable targets by 3D structures of proteins and compounds. npj Precis. Onc. (2026). https://doi.org/10.1038/s41698-026-01310-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-026-01310-7
