
Abstract
Covalent functionalisation can be a powerful lever to tune the properties and processability of graphene. After overcoming the low chemical reactivity of graphene, covalent functionalisation led to the generation of new hybrid materials, applicable in a broad variation of fields. Although the process of functionalising graphene is nowadays firmly established, fundamental aspects of the produced hybrid materials remain to be clarified. Especially the atomically resolved imaging is only scarcely explored. Here we show aberration corrected in situ high resolution TEM imaging of dodecyl functionalised monolayer graphene at atomic resolution after an effective mechanical filtering approach. The mechanical filtering allows to separate adsorbed contamination from the covalently bound functional molecules and thus opens the possibility for the observation of this hybrid material. The obtained data is validated by DFT calculations and by a novel image simulation approach based on molecular dynamics (MD) simulations at room temperature.
Similar content being viewed by others
Introduction
Chemical functionalisation is an excellent way to alter and improve graphene’s properties1 leading towards applications in optoelectronics2, sensing3, and energy conversion and storage4, to name a few. Especially covalent functionalisation has been proven tremendously useful for opening a bandgap5 or inducing magnetic behaviour in monolayer graphene6. Furthermore, using a wet-chemical approach to functionalise graphene also presents a straightforward way to scale up the process. This approach has been continuously improved to overcome challenges such as graphene’s low chemical reactivity7. While spectroscopic and chemical evidence of covalently functionalised graphene has been well established8, direct imaging of single molecules attached to graphene has only been achieved by localised in vacuo functionalisation9,10. High-resolution imaging of wet-chemically functionalised graphene is, however, necessary to understand key factors, such as spatial homogeneity of addends and the degree of functionalisation.
Here we show atomically resolved transmission electron microscopy (TEM) imaging of dodecyl-functionalised freestanding monolayer graphene. We use a mechanical cleaning approach11, that can also work as an effective filter to separate covalently from non-covalently bound moieties. The high-resolution imaging is backed up and compared to extensive molecular dynamics (MD) and image simulations. We find that the functionalised graphene hybrid can be studied in situ for up to several minutes and we can estimate an overall degree of functionalisation. The surprising stability is explained by the size of the synthesised macromolecule that allows to study beam induced rearrangements for prolonged periods of time. Finally, silicon impurities in graphene are discussed as possible alternative explanation for the observed contrasts.
Results and discussion
A deliberate tuning of the properties of graphene can be achieved by introducing functional molecules covalently anchored to the basal plane. The generation of isolated sp³ defects was shown to induce a band gap12, generate binding sites for protein probing13, and even leads to a tuneable generation of magnetic behaviour in graphene6,14.
While covalent functionalisation has been widely studied, the data on detailed atomically resolved descriptions of such hybrid materials are extremely scarce. The generation of functionalised graphene is either achieved by gas-phase reactions with reactive precursors or, as in this case, following a wet-chemical functionalisation approach employing solubilised reagents15,16. The wet-chemical route is easily upscaleable but a subsequent atomic resolved investigation of the produced material is conventionally hampered by extended surface contamination.
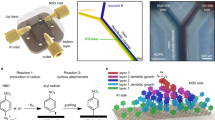
For a deeply fundamental understanding of functionalised graphene, we deliberately introduced dodecyl chains that are covalently bound to suspended monolayer CVD graphene and investigated the hybrid material in atomic resolution using high resolution TEM (HRTEM). Therefore, we directly functionalised free-standing graphene membranes, which can be cleaned and viewed in TEM directly after the synthesis (see Fig. 1a for the general approach). The functionalisation is achieved using a reductive alkylation of chemically charged graphene with dodecyliodide8 (see Fig. 1b). Chemical activation is performed by drop casting a Na/K-DME solution after which dodecyl iodide, as reactive agent was added (see Fig. 1c, d). The reagents were rinsed with DME after 15 min of reaction time (details of the functionalisation procedure can be found in the Methods Section). Unavoidably, residuals from the synthesis accumulate on the graphene surface, necessitating a cleaning method that leaves the graphene-hybrid intact while removing physisorbed contaminants. An in situ mechanical cleaning and filtering method (detailed below) is employed that can be directly followed up with imaging, without exposing the specimen to further sources of contamination.

a Graphical representation of the experimental procedure: graphene is covalently functionalised, mechanically filtered, and subsequently imaged using HRTEM. b Basic reaction pathway of covalent functionalisation using dodecyl iodide on graphene. c Free-standing graphene on TEM grids is used for the functionalisation procedure. d Eutectic Na/K alloy used in the synthesis. e Raman spectroscopy point measurements on the free-standing graphene before functionalisation show a negligible intensity of the defect-induced D band. f After covalent functionalisation the binding of dodecyl to graphene’s basal plane induces local re-hybridisations from sp² to sp³ and thus a remarkable increase of the D-band intensity. The appearance of D’ and D+D’ bands are a clear indication of the successful covalent addition of the dodecyl precursors by supratopic addition.
Raman spectroscopy measurements performed directly on the TEM grid before (Fig. 1e) and after (Fig. 1f) the functionalisation reveal a drastic increase in D band intensity at approximately 1350 cm−1, as well as the appearance of a distinct D’ signal at 1622 cm−1 and the occurrence of D + D’ at 2930 cm−1. This clearly indicates a structural alteration of the graphene substrate, induced by the reactive dodecyl precursor. The local re-hybridisations from an all-sp² system to isolated sp³ hybridised C−C bonds is induced by the covalent attachment of dodecyl chains to the graphene basal plane17. The C−C bonds are established between one of graphene’s carbon atoms and the terminal positions of the employed dodecyl precursors8,18, where the iodine leaving groups were formerly located.
As the addition proceeds via a radical-mediated reaction, involving one electron from graphene’s π-system19, an unpaired electron is remaining after the addition of the dodecyl moiety. For this unpaired electron, the following scenarios can be considered: persistence of a stabilized radical, transformation to an ion, or an additional functionalisation. To investigate the likelihood of these scenarios, we performed DFT calculations on the cleavage energy of the established C−C bond (see Supplementary Fig. 1 and Supplementary Table 1 for an overview). A radical or ionic stabilisation results only in a very weak C−C bond between graphene and the dodecyl moiety (0.01 to 0.3 eV), in contrast to a second functionalisation, which strongly increases the bond strength. The strongest bond considered here is created through a pairwise addition of dodecyl and hydrogen on neighbouring (1,2)-positions in an anti configuration, with a C−C bond strength of 1.55 eV. Other binding motifs, for instance, saturation with a second dodecyl, also result in a reasonable bond strength. Due to the cleaning and filtering method (detailed below) we can rule out weakly bound binding motifs with an energy that is in the range of or only slightly above that of van der Waals forces. We therefore only consider the case of double functionalisation in the following, with a focus on hydrogen saturation.
Mechanical cleaning and filtering
One inherent consequence of producing materials through wet-chemical processes is the inadvertent occurrence of contamination, e.g. hydrocarbons contained in solvents. The existence of those contaminants is mainly accounted to background noise in established analytical procedures, while in atomically resolved solid state investigations (TEM, AFM, STM), these residues render the in-depth study of wet-chemically synthesised materials extremely challenging. In our synthesised graphene-dodecyl hybrid, the contamination remains ubiquitous, even though the reactions were carried out under strict inert conditions in a glove box, using highly purified solvents and reagents. The contamination can be seen as a thin amorphous film on both sides of the freestanding graphene-dodecyl membranes (see Fig. 2b and Supplementary Fig. 2). This coverage obviously obstructs the detailed microscopic investigation of the local structure of molecular addends. To alleviate this problem, the contamination needs to be removed while keeping the graphene-dodecyl hybrid intact. Common approaches to remove physisorbed contamination from a freestanding graphene (or any other 2D material) surface are based on thermal annealing20,21, plasma cleaning22, local laser irradiation23 or chemical activation24. All of these methods, however, can introduce a sufficient amount of energy leading to a cleavage of the covalent bonds8,18.

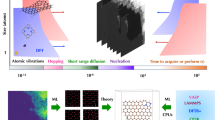
a Schematic representation of the cleaning approach. A tip is pressed onto the graphene surface and removes physisorbed contaminations while leaving covalent bonds intact. b TEM image acquired during the in situ cleaning process. c The same window after the process, showing no traces of residual contamination. d HRTEM image of the cleaned area showing several contrast features. e Magnified views from d showing molecular attachment sites (black, blue, purple) and the pure graphene lattice (red).
Instead of these methods, here we employ a purely mechanical in situ cleaning approach in the TEM11 which functions also as a filtering device (schematic representation in Fig. 2a, setup and tip preparation in Supplementary Fig. 3). Concretely, the removal of surface adsorbents is accomplished by bringing a fine gold tip in physical contact with the graphene surface and moving it in a sweeping manner. This procedure is performed on both sides of the graphene membrane. In Fig. 2b remaining amorphous contamination during the cleaning process is visible, while after the complete removal in Fig. 2c, the membrane, suspended over the hole of a Quantifoil substrate, appears completely featureless and thus atomically clean on an area of ca. 5 µm² (in situ cleaning shown in Supplementary Movies 1 and 2). The process is unique regarding the selectivity in removing physisorbed contamination: the typical energy of cohesive bonds in graphitic systems is in the range of 40 meV per atom25, which is much lower than the ~1 eV of the covalent bond between a dodecyl chain and graphene (see Supplementary Table 1). This energy difference enables a selectivity of the mechanical cleaning method, transforming it effectively into a filtering method as well. To ensure the stability of the covalently bound moieties during the mechanical filtering process, we conducted Raman spectroscopic measurements after the mechanical removal of the surface contamination and can clearly state that no bond cleavage is generated by our presented approach (see Supplementary Figs. 9 and 10).
Imaging and image simulations
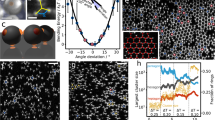
After cleaning and filtering, HRTEM imaging reveals several localised contrast features within the graphene lattice as shown in Fig. 2d, e. We take this as a first microscopic hint to identify these features as covalently bound molecules, based on a simulation assisted analysis. At first glance, the contrasts in the images may not appear to fit to an alkyl chain as they are spot-like rather than chain-like. However, it has to be taken into account that the measurements were taken at room temperature, which enables thermally activated rapid motion of the attached molecules. This motion cannot be directly included into conventional HRTEM image simulations, which only account for lattice vibrations in crystalline solids using, e.g., the frozen phonon approximation26. Instead, here we use a unique combination of molecular dynamics (MD) and HRTEM image simulations to include thermally driven molecular motions. Classical force-field MD simulations performed at 300 K show that dodecyl chains attached to graphene pivot around their site of attachment (see Fig. 3a and Supplementary Movie 3) within picoseconds. During the time frame of HRTEM image acquisition (0.5 to several seconds), the molecules can therefore ‘rotate’ around the attachment site millions of times, which means that the images capture an average of many different states of the molecule. Due to the proposed saturating H atom, the orientation of the dodecyl chain is biased to one side (Fig. 3b), however, overall the dodecyl carbon atoms are distributed fairly evenly (see Supplementary Fig. 4 for other binding motifs). To be able to account for the molecular motion, we used up to 30,000 individual frames of each 50 ns MD trajectory as input for HRTEM image simulations (see Supplementary Movie 4 and Supplementary Fig. 5). For each selected frame an image is simulated with different defocus values (see Fig. 3c). Averaging all images with the same defocus parameter leads to a final simulated image that includes the rapid motion of the molecules (see Supplementary Movie 5 for the averaged image in relation to the defocus). In this image the remaining contribution of the dodecyl molecule to the HRTEM image contrast is mainly due to the “static” part of the chain, which consists of the first carbon atoms in the dodecyl chain where the covalent bond to the graphene substrate is established. The rest of the chain essentially “disappears” due to its fast motion. As a side benefit of this simulation approach, thermal vibrations of the graphene lattice are also directly included in the image simulations. With this robust simulation setup, it is now possible to compare experimental and simulated images. Across different defocus values (see Fig. 3d–f), we obtain a fairly good agreement between image and simulation. Considering the different possible binding motifs, we find that a single dodecyl (with and without saturating hydrogen) gives the best fit between image and simulation (see Supplementary Fig. 6 for a comparison). The saturating hydrogen does have a small effect on the overall contrast, however, due to its invisibility and instability in TEM we cannot draw a definite conclusions about its long term presence.

a Graphical representation of the setup used in the MD simulations. A dodecyl chain is attached to a graphene sheet and can move due to thermal vibrations. b 2D histogram of the probability distribution of the dodecyl carbon atoms above the graphene sheet, extracted from a 50 ns MD simulation. c Two MD snapshots after 0 and 10 ns and corresponding image simulations at −5 nm and −17 nm defocus. Averaging over the entire trajectory leads to a much smaller footprint of the molecule (bottom row). d–f Experimental images and corresponding simulated images for different defocus values show a good agreement.
Dynamics of functionalised molecules during imaging
By the presented combination of ex situ chemical functionalisation, in situ mechanical filtering, high-resolution electron microscopy and simulation, we are able to postulate the presence of individual alkylic molecules, covalently bound to graphene. Moreover, based on overview HRTEM data (e.g. Figure 2d), we evaluated the area coverage of dodecyl groups to 0.06%, while the degree of functionalisation for comparable systems was shown to be ca. 0.1%27,28. In accordance with the presented Raman experiments, the mean “defect” distance should be ca. 7 nm for an ID/IG ratio of 2.3, as reflected in the data29.
As the captured contribution of the functional moieties essentially only consists of the first static carbon atom, another plausible interpretation of the observed contrasts is the imaging of individual substitutional heteroatoms, such as silicon, commonly found in graphene. From only HRTEM data taken under the challenging conditions of the present in situ study (using a STM holder with limited mechanical stability) one cannot unambiguously distinguish between the two interpretations, as only subtle contrast differences are expectable that become almost indiscernible if the saturating functionalisation with hydrogen is lost (see Supplementary Fig. 8 for a direct comparison). However, here the potential of dedicated in situ microscopy proves itself useful, as it allows to record the structural changes presented in Fig. 4 on a fast enough timescale to practically exclude the imaging of heteroatomic impurities (see Supplementary Movie 7 for the unfiltered movie).

a–f Raw (top, with time and accumulated dose given as inset) and Gaussian filtered (bottom) experimental HRTEM images show the motion of a dodecyl group. During the migratory motion, the molecule repeatedly assumes similar states (compare a to d, b to e and c to f), while always staying centred on one hexagon. After a prolonged period of time (>45 s) the molecule detaches and leaves behind a perfect graphene lattice.
The long-term stability under the applied imaging conditions and the observed dynamic behaviour of the molecules therefore needs further analysis. For the local investigation of the functional sites, a dose rate of 0.1 C cm−2 s−1 is subjected to the illuminated area of the specimen. Generally, the hybrid material can withstand accumulated doses of up to 5 C cm−2 (50 s of high intensity illumination) before the contrast vanishes and a pristine graphene lattice remains. Interestingly, the defunctionalisation is also achievable by laser irradiation during Raman experiments contained in Supplementary Fig. 10. The high-energy electrons used in TEM are well known to have a detrimental effect on organic molecules, as they can knock-out hydrogen atoms and scissor/establish covalent bonds. We can therefore expect a profound effect of the electron beam on the functionalised molecules during continued imaging. Indeed, the critical doses for the disintegration of organic molecules is much lower than in our presented experiments, with a critical dose of around 0.5 C cm−2 for carbon loss in PMMA30. We address the higher stability of our hybrid material to the high conductivity of the graphene substrate and, more importantly, the size of the synthesised macromolecule. The covalent attachment of the dodecyl moieties at specific carbon atoms in the graphene framework classifies the resulting material as a de-facto macromolecule that allows stabilization of beam induced damage structures by mesomeric effects. It has been shown by Skowron et al.31, that graphene patches of minor sizes are tending to form edgeless fullerenes under electron illumination, depending on the dimensions. Contrarily, the here presented hybrid material is extremely large without defined edges. Also, the possibility of internal atomic reorganisations after partial dehydrogenation (Supplementary Fig. 7) of the functional moiety will substantially enhance the hybrids’ stability. For all molecules we find that exposure to the electron beam for extended periods of time results in the complete removal of the dodecyl chain, leaving a pristine graphene membrane behind. This very fact is also a further indication of the covalent addition of dodecyl chains to an otherwise unchanged lattice of graphene. Before final detachment, however, a plethora of dynamic movements, molecular migrations and state changes can be observed. Figure 4 demonstrates one such migration (see also Supplementary Movie 6 & 7).
One example of such concerted movement of mass on the graphene substrate can be followed over time and the temporal stability (as indicated by the progressing illumination time at an electron dose rate of 106 e/nm² s−1) clearly points to a covalent stabilisation of the molecule in each state, involving the breaking and the subsequent formation of a C-C bond between the molecule and the graphene substrate32,33. While the dodecyl moiety is bound at its initial site in Fig. 4a, the result of a jump after 2 s (0.2 C cm−2) of electron illumination towards the next site is observed. The change of bonding sites is accompanied by the alteration of the localised contrast produced by the addend. While the imaging of a Si impurity is another plausible way of interpreting the data, especially under the high electron doses, the asymmetric contrasts reproduced in Fig. 4 can hardly be matched by the symmetric image contrasts generated from a single atomic impurity (see Gaussian filtered images, Supplementary Fig. 8). The contrast alteration indicates structural changes to the molecule, which are most probably an effect of beam-induced dehydrogenation of the dodecyl chain9,34. Interestingly, the generated contrasts at each binding site are highly comparable (Fig. 4a, d, b, e, c, f), indicating similar structural conformations at each respective binding site. In the presented example, we observed 5 molecular jumps in a seemingly clockwise fashion, with the individual conformations being stable for several seconds. After more than 45 s of electron beam illumination (total dose 4.5 C cm−2), desorption of the remaining addend leads to a re-establishment of pristine and defect-free graphene, where the retro-functionalisation does not cause the generation of point or extended defects. Even though non-destructive retro-functionalisation has been also reported for impurity atoms35,36,37, we consider our interpretation of capturing the rearrangement and retro-functionalisation of the molecular moieties more plausible as the functionalization has been realized via a well-established wet-chemical route and our microscopic observations are backed up by the MD analysis of possible stabilisation pathways after the electron-beam induced dehydrogenation of the hybrid material (see below and Supplementary Fig. 7 and Supplementary Table 2).
The observed molecular motion is most likely related to an electron beam induced cleavage of C-H bonds but could also be an intrinsic property of functionalised graphene. Concerning the former case, the probability for a dehydrogenation of the alkyl chain is high, taking the elevated doses of electrons with a primary energy of 80 keV into account. Nevertheless, as observed in Fig. 4, the functional group is not instantly detached but undergoes several positional changes before a retro-functionalisation is effective. To further investigate this, we conducted an ab initio MD study of a hexyl-graphene hybrid, showing that, depending on the relative position of the dehydrogenation, a plethora of novel, stable conformations can appear (see Supplementary Fig. 7 and Supplementary Table 2). When removing two hydrogen atoms at random positions to simulate electron beam induced knock-off, the alkyl chain remains attached to the graphene in most cases (~96% probability) by forming additional bonds within the chain or with the graphene substrate. Among the novel conformations we encountered cyclisation of the alkyl chain towards graphene (~25% probability) that might be responsible for the contrasts shown in Fig. 4. The similar structural conformations at the different binding sites may also indicate that a migratory process could also be an intrinsic effect of functionalised graphene. Indeed, recent investigations of thermally induced retro-functionalisation of a dodecyl-graphene hybrid8 and theoretical studies of functionalised extended π-systems38 already point towards the tendency of a migration of molecular addends within the π-system to achieve a position of global minimum energy.
In conclusion, we presented an extensive electron microscopy study of covalently functionalised freestanding monolayer graphene, wherein we were able to resolve individual molecular addends at atomic resolution. To image the locally constrained molecules, we employed a dedicated mechanical in situ cleaning and filtering approach that allows to remove residuals from the wet-chemical synthesis. We verified our findings by the inclusion of thermally-induced motion of the alkyl chains for image simulation based on MD simulations. The obtained results indicate a dynamic rearrangement of the functional groups on the surface of graphene under electron beam irradiation. This study paves the way for further in-depth investigations of other 2D materials beyond graphene and will enable fundamental insights into the nature of covalent functionalisation in the 2D limit. Moreover, a thorough understanding of the dynamic behaviour down to the atomic level will help to control the physical properties, boosting novel device concepts based on 2D materials.
Methods
Graphene preparation
Freestanding monolayer graphene samples on Quantifoil grids were prepared from PMMA coated Trivial Transfer Graphene supplied by ACS Materials. The CVD graphene is delivered on a hydrophobic substrate that can be infiltrated by water to release the graphene with a PMMA transfer layer to float upon water. The film was picked up by a filter paper and cut in 2 × 2 mm2 rectangles, floated again on water and picked up with precleaned Quantifoil TEM grids with hole diameters of nominally 2 µm. The Quantifoil grids were cleaned by subsequent washing in chloroform, acetone and iso-propanol to dissolve mobile silicates from the amorphous Quantifoil film. After fishing the graphene/PMMA flakes from the water surface, the grids were dried on a hotplate at 100 °C for 10 min to enhance the contact between the sample grid and the graphene/PMMA flake. PMMA was dissolved in acetone by immersion in aceton followed by a washing step in aceton vapour under reflux for 2 h.
Graphene functionalisation
The TEM grid functionalisation was carried out in an argon filled LABmasterpro glove box (MBraun), equipped with a gas purifier and solvent vapour removal unit. The working conditions are <0.1 ppm for oxygen and <0.1 ppm for water. In the glove box, 150 mg Na/K alloy were dissolved in 5 mL absolute DME and stirred for 1 h. 30 µL of the deep blue Na/K solution were used to activate the CVD graphene. Afterwards, 30 µL of pure dodecyl iodide were added. The reaction was aborted after 15 min by removing the reactants with 50 µL absolute DME.
Raman spectroscopy
The Raman spectroscopic characterisation was carried out on a Horiba Jobin Yvon LabRAM Aramis. A laser with an excitation wavelength of 532 nm and a D2 filter (laser energy 0.17 mW) was used. The spectrometer was calibrated in frequency using crystalline graphite before the measurement. The TEM samples were measured with an Olympus LMPlan N 100x/0.90 objective, a 600 grating and 2 s acquisition time. The used software was LabSpec 6.
Simulations
The classical molecular dynamics simulations were done with the LAMMPS software package39 using the generalised Amber force field (GAFF)40. A single dodecyl chain was added to an orthorhombic (15 × 8√3) unit cell of graphene (480 carbon atoms). The simulations were performed in the canonical ensemble using a time step of 1 fs and a Nosé-Hoover thermostat enforcing a temperature of 300 K. Three sets of simulations were carried out: with an unsaturated single dodecyl chain, and with a chain saturated either by an additional H atom or a second dodecyl, both in the (1,2)-anti position with respect to the first addend. The systems were first equilibrated for 1 ns, followed by a production run of 50 ns. From the trajectories of the production runs 2D histograms of the probability distribution of the dodecyl carbon atom positions above the graphene plane were derived. The graphene plane was divided into quadratic bins with size of 0.1 Å and the number of trajectory frames with a dodecyl carbon atom above or below each bin was recorded (see Supplementary Fig. 4).
The DFT calculations were carried out with the plane-wave code PWscf of the Quantum Espresso software package41 using the Perdew-Burke-Ernzerhof PBE exchange-correlation functional42 together with Gimme’s semiempirical D3 dispersion correction with Beck-Johnson damping43,44. The interaction between electrons and ionic cores was described by Vanderbilt ultrasoft pseudopotentials45 with a plane-wave kinetic energy cutoff of 30 Ry for the expansion of the wave functions. Calculations were done for different addition patterns of alkyl chains and H atoms on neutral and charged graphene (see Supplementary Table 1). Graphene was represented by an orthorhombic (10 × 5√3) unit cell (200 carbon atoms), and the length of the alkyl chain was reduced to three (propyl). The strength of the C-C bond between the alkyl chain and the graphene sheet is characterized by its cleavage energy \(E_{{{{\mathrm{cleav}}}}}^{{{{\mathrm{C}}}} - {{{\mathrm{C}}}}}\), which was calculated as energy difference between the functionalized graphene \(E_{{{{\mathrm{graph}}}}}^{{{{\mathrm{funct}}}}}\) and the two fragments of the alkyl radical in the gas phase \(E_{{{{\mathrm{mol}}}}}^{{{{\mathrm{alkyl}}}}}\) and the remaining graphene \(E_{{{{\mathrm{graph}}}}}^{{{{\mathrm{rest}}}}}\) after the cleavage of the C-C bond:
In addition, we calculated the combined binding energy of all addends
In the case of the unsaturated functionalization with a single addend, Ebind is identical to the cleavage energy.
The Car-Parrinello ab initio molecular dynamics simulations were done with the new performance-optimized version of the CPMD software (www.cpmd.org)46, using the same functional, dispersion correction and pseudopotentials as in the geometry optimisations. The time step was 0.145 fs and the fictitious mass for the electron degrees of freedom was set to 700 a.u. A hexyl/hydrogen pair was added in a (1,2)-anti position to an orthorhombic (6 × 3√3) unit cell of graphene (72 carbon atoms). First a 72.6 ps simulation in the canonical ensemble at 300 K was performed by using Nosé-Hoover thermostats. Five configurations were chosen from the trajectory and from each of these snapshots randomly selected pairs of H atoms were removed to simulate electron beam induced knock-off of H atoms. Altogether, 105 configurations were created, for which the MD was continued for another 10.2 ps. A variety of chemical reactions occurred. Some examples of final configurations are shown in Supplementary Fig. 7 and a detailed analysis of the final reaction products is given in Supplementary Table 2.
Electron microscopy
Aberration corrected transmission electron microscopy was carried out using a FEI Titan Themis³ 300, operated at 80 kV to exclude knock-on damage to graphene. The spherical aberration coefficient was tuned to −10 µm and the Wien-type monochromator was excited to 1200 V to lower the chromatic aberration during high-resolution imaging, achieving an energy spread of ca. 200 meV. Image series were recorded with acquisition time of 500 ms on a FEI Ceta CMOS camera binned to 2k × 2k pixel and post-processed afterwards.
In situ cleaning
Amorphous contamination was removed from the surfaces of graphene by guiding an anodic etched gold double tip subsequently over the upper and the lower surface of the graphene film11. In situ cleaning was performed using a Nanofactory STM holder with the double tip mounted on the piezo-controllable spider hat. The TEM grid was cut in half using a razorblade, glued to a gold wire and fixed in the immobile STM mounting chip. The spider was controlled from the operator room while recording image series on the CMOS camera of the microscope.
Image processing
The image intensity equalisation was performed using a gamma correction function in combination with a brightness and contrast adjustment implemented in Python with the help of the OpenCV library. The image registration was done by calculating the discrete cross-correlation function between subsequent images. The peak of the correlation function was fitted using a two-dimensional Gaussian function to retrieve sub-pixel image shifts. Aligned frames were then averaged using a mean function.
Image simulation
Supercell HRTEM image simulations were performed using the simulation library provided by Kirkland26. For the dynamic simulations a wrapper programme was implemented in python. This programme automatically translates a molecular dynamics trajectory into single supercell states that are then used as a basis for image simulations using the Kirkland library. In the image simulations the following parameters are used: electron energy: 80 keV, Cs: −10 µm, opening angle: 25 mrad, Illumination semi-angle: 0.1 mrad, defocus spread: 2.5 nm. The defocus is varied during the simulation of each state. Finally, all images corresponding to one defocus from one MD trajectory are averaged using a mean function.
Data availability
All data is available from the corresponding author upon reasonable request.
References
Criado, A., Melchionna, M., Marchesan, S. & Prato, M. The covalent functionalization of graphene on substrates. Angew. Chem. Int. Ed. 54, 10734–10750 (2015).
Tetsuka, H., Nagoya, A., Fukusumi, T. & Matsui, T. Molecularly designed, nitrogen-functionalized graphene quantum dots for optoelectronic devices. Adv. Mater. 28, 4632–4638 (2016).
Tang, X., Mager, N., Vanhorenbeke, B., Hermans, S. & Raskin, J. P. Defect-free functionalized graphene sensor for formaldehyde detection. Nanotechnology 28, 055501 (2017).
Bonaccorso, F. et al. 2D materials. Graphene, related two-dimensional crystals, and hybrid systems for energy conversion and storage. Science 347, 1246501 (2015).
Jorgensen, J. H. et al. Symmetry-driven band gap engineering in hydrogen functionalized graphene. ACS Nano 10, 10798–10807 (2016).
Gonzalez-Herrero, H. et al. Atomic-scale control of graphene magnetism by using hydrogen atoms. Science 352, 437–441 (2016).
Sharma, R., Baik, J. H., Perera, C. J. & Strano, M. S. Anomalously large reactivity of single graphene layers and edges toward electron transfer chemistries. Nano Lett. 10, 398–405 (2010).
Abellan, G. et al. Unifying principles of the reductive covalent graphene functionalization. J. Am. Chem. Soc. 139, 5175–5182 (2017).
Markevich, A. et al. Electron beam controlled covalent attachment of small organic molecules to graphene. Nanoscale 8, 2711–2719 (2016).
He, Y. et al. Fusing tetrapyrroles to graphene edges by surface-assisted covalent coupling. Nat. Chem. 9, 33–38 (2017).
Schweizer, P. et al. Mechanical cleaning of graphene using in situ electron microscopy. Nat. Commun. 11, 1743 (2020).
Niyogi, S. et al. Spectroscopy of covalently functionalized graphene. Nano Lett. 10, 4061–4066 (2010).
Tyagi, A. et al. Modular functionalization of crystalline graphene by recombinant proteins: A nanoplatform for probing biomolecules. Nanoscale 10, 22572–22582 (2018).
Lado, J. L. & Fernández-Rossier, J. Unconventional Yu–Shiba–Rusinov states in hydrogenated graphene. 2D Mater. 3, 025001 (2016).
Yang, H. et al. Preferential and reversible fluorination of monolayer graphene. J. Phys. Chem. C. 115, 16844–16848 (2011).
Park, J. & Yan, M. Covalent functionalization of graphene with reactive intermediates. Acc. Chem. Res. 46, 181–189 (2013).
Eckmann, A. et al. Probing the nature of defects in graphene by Raman spectroscopy. Nano Lett. 12, 3925–3930 (2012).
Biswal, M. et al. Sodide and organic halides effect covalent functionalization of single-layer and bilayer graphene. J. Am. Chem. Soc. 139, 4202–4210 (2017).
Knirsch, K. C., Schafer, R. A., Hauke, F. & Hirsch, A. Mono- and ditopic bisfunctionalization of graphene. Angew. Chem. Int. Ed. 55, 5861–5864 (2016).
Lin, Y. C. et al. Graphene annealing: how clean can it be? Nano Lett. 12, 414–419 (2012).
Longchamp, J.-N., Escher, C. & Fink, H.-W. Ultraclean freestanding graphene by platinum-metal catalysis. J. Vac. Sci. Technol. B 31, 020605 (2013).
Cunge, G. et al. Dry efficient cleaning of poly-methyl-methacrylate residues from graphene with high-density H2 and H2-N2 plasmas. J. Appl. Phys. 118, 123302 (2015).
Tripathi, M. et al. Cleaning graphene: Comparing heat treatments in air and in vacuum. Phys. Status Solidi Rapid Res Lett. 11, 1700124 (2017).
Algara-Siller, G., Lehtinen, O., Turchanin, A. & Kaiser, U. Dry-cleaning of graphene. Appl. Phys. Lett. 104, 153115 (2014).
Koren, E., Lortscher, E., Rawlings, C., Knoll, A. W. & Duerig, U. Surface science. Adhesion and friction in mesoscopic graphite contacts. Science 348, 679–683 (2015).
Kirkland, E. J. Computation in electron microscopy. Acta Crystallogr. A 72, 1–27 (2016).
Englert, J. M. et al. Covalent bulk functionalization of graphene. Nat. Chem. 3, 279–286 (2011).
Schafer, R. A. et al. Substrate-modulated reductive graphene functionalization. Angew. Chem. Int. Ed. 55, 14858–14862 (2016).
Cancado, L. G. et al. Quantifying defects in graphene via Raman spectroscopy at different excitation energies. Nano Lett. 11, 3190–3196 (2011).
Egerton, R. F. Radiation damage to organic and inorganic specimens in the TEM. Micron 119, 72–87 (2019).
Skowron, S. T. et al. Chemical reactions of molecules promoted and simultaneously imaged by the electron beam in transmission electron microscopy. Acc. Chem. Res. 50, 1797–1807 (2017).
Schaffel, F., Wilson, M. & Warner, J. H. Motion of light adatoms and molecules on the surface of few-layer graphene. ACS Nano 5, 9428–9441 (2011).
Susi, T., Meyer, J. C. & Kotakoski, J. Manipulating low-dimensional materials down to the level of single atoms with electron irradiation. Ultramicroscopy 180, 163–172 (2017).
Mittelberger, A., Kramberger, C. & Meyer, J. C. Insights into radiation damage from atomic resolution scanning transmission electron microscopy imaging of mono-layer CuPcCl16 films on graphene. Sci. Rep. 8, 4813 (2018).
Dyck, O. et al. Building structures atom by atom via electron beam manipulation. Small 14, e1801771 (2018).
Mustonen, K. et al. Electron‐beam manipulation of silicon impurities in single‐walled carbon nanotubes. Adv. Funct. Mater. 29, 1901327 (2019).
Su, C. et al. Engineering single-atom dynamics with electron irradiation. Sci. Adv. 5, eaav2252 (2019).
Holzwarth, J. et al. Highly regioselective alkylation of hexabenzocoronenes: fundamental insights into the covalent chemistry of graphene. Angew. Chem. Int. Ed. 56, 12184–12190 (2017).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput Chem. 25, 1157–1174 (2004).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput Chem. 32, 1456–1465 (2011).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev., B Condens. Matter 41, 7892–7895 (1990).
Klöffel, T., Mathias, G. & Meyer, B. Integrating state of the art compute, communication, and autotuning strategies to multiply the performance of ab initio molecular dynamics on massively parallel multi-core supercomputers. Comput Phys. Commun. 260, 107745 (2021).
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support through the Sonderforschungsbereich SFB 953 “Synthetic carbon allotropes”, the Cluster of Excellence EXC 315 “Engineering of advanced materials” (EAM), and Research Training Group GRK 1896. The work has been further supported by the European Union (ERC-2018-StG 804110-2D-PnictoChem to G.A.). G.A. acknowledges support by the Generalitat Valenciana (CIDEGENT/2018/001), the Spanish MICINN (PID2019-111742GA-I00 and Excellence Unit María de Maeztu CEX2019-000919-M), and the DFG (FLAG-ERA AB694/2-1). Computational resources were provided by RRZE Erlangen and LRZ Garching (project pn98fa).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Functionalisation of graphene and Raman spectroscopy was carried out by D.D., and G.A., microscopy experiments were conceived, carried out and interpreted by C.D., P.S. and E.S.; S.G., R.M., and B.M. carried out the DFT calculations and MD simulations. E.S., B.M., G.A., F.H., and A.H. supervised the project. All authors discussed the obtained results. C.D., P.S., G.A., B.M., and E.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial or non-financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dolle, C., Schweizer, P., Dasler, D. et al. Atomically resolved TEM imaging of covalently functionalised graphene. npj 2D Mater Appl 6, 29 (2022). https://doi.org/10.1038/s41699-022-00304-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41699-022-00304-w
This article is cited by
-
High-power temperature resilient ionic liquid-driven graphene supercapacitor for an effective solar-powered energy backup system
Advanced Composites and Hybrid Materials (2025)
