
Abstract
Building accurate prediction models and identifying predictive biomarkers for treatment response in Muscle-Invasive Bladder Cancer (MIBC) are essential for improving patient survival but remain challenging due to tumor heterogeneity, despite numerous related studies. To address this unmet need, we developed an interpretable Graph-based Multimodal Late Fusion (GMLF) deep learning framework. Integrating histopathology and cell type data from standard H&E images with gene expression profiles derived from RNA sequencing from the SWOG S1314-COXEN clinical trial (ClinicalTrials.gov NCT02177695 2014-06-25), GMLF uncovered new histopathological, cellular, and molecular determinants of response to neoadjuvant chemotherapy. Specifically, we identified key gene signatures that drive the predictive power of our model, including alterations in TP63, CCL5, and DCN. Our discovery can optimize treatment strategies for patients with MIBC, e.g., improving clinical outcomes, avoiding unnecessary treatment, and ultimately, bladder preservation. Additionally, our approach could be used to uncover predictors for other cancers.
Similar content being viewed by others


Introduction
Every year, approximately 81,000 new bladder cancer cases are diagnosed in the United States, resulting in 17,000 annual deaths1. Muscle-invasive bladder cancer (MIBC) is a high-grade type of bladder cancer characterized by tumors invading the detrusor muscle of the bladder2. Neoadjuvant chemotherapy (NAC) followed by radical cystectomy (RC) has been considered the gold standard treatment for MIBC3. However, RC results in high mortality rates (0.3–5.7%)4 and significant surgical morbidity, with 64% of patients experiencing postoperative complications within 90 days of RC5. About 35% of MIBC patients achieve complete pathologic response (pCR) with no residual tumor after treatment with NAC6. Achieving pCR to NAC is a well-established prognostic predictor of overall survival in patients with MIBC7,8. In our analysis of long-term outcomes of patients enrolled in the SWOG S1314 trial, we found that pCR is strongly correlated with survival with a 5-year overall survival rate of 90%8. Given the current paradigm for NAC of “one size fits all,” which carries the burden of acute and chronic toxicities, there is significant interest in a precision medicine approach to predicting complete response to NAC. Accurately predicting response to NAC will allow for the selective use of NAC in patients who are more likely to benefit from treatment while minimizing treatment-related toxicity and delayed access to surgery in patients who are less likely to respond to NAC9.
Tumor heterogeneity in MIBC has been demonstrated at both the molecular and histologic levels10, posing challenges to building accurate prediction models as well as identifying predictive biomarkers for treatment response10,11. Previous studies have investigated different predictors for treatment response in MIBC, including germline biomarkers for cisplatin sensitivity12, immunohistochemical subtyping13,14, defects in DNA repair genes15, radiomics16, gene expression17, and molecular subtypes18. However, no study has established a robust and accurate method for predicting response to NAC for MIBC patients19.
Computational pathology has emerged as a promising tool for analyzing histology images from whole-slide images (WSIs) beyond the routine manual examination of cancer slides20. Deep learning approaches incorporating WSIs and multi-omics data have demonstrated outstanding potential for predicting clinical outcomes21. Previous studies have shown that deep learning models based on WSI can accurately predict bladder cancer molecular subtypes22,23, cancer recurrence24, and sensitivity to chemotherapy25. Moreover, these models can also serve as effective tools for extracting features from the tumor and predicting biological interactions underpinning tumor behavior26.
WSIs datasets of hematoxylin and eosin (H&E) stained tissue images have unraveled the potential of deep learning in linking complex associations of histology data with patient outcomes27. However, predicting treatment outcomes directly from WSIs faces limitations due to the need for large datasets with matched imaging and response data. Previous studies have found an improvement in the predictive performance of deep learning models when integrating multiple data types28,29. By integrating multimodal data, including histopathology images and gene expression profiles, deep learning models can provide more reliable results and identify relevant biological pathways30.
In this study, we aim to leverage data from WSIs, and gene expression profiles prospectively collected from patients enrolled in the SWOG S1314 clinical trial (NCT02177695) to predict NAC response using deep learning. We hypothesize that multimodal integration of accessible H&E images and molecular data using deep learning can accurately stratify MIBC patients based on their response to NAC independently of clinical features such as age and stage. By using different interpretation approaches, including Shapley Additive Explanation (SHAP)31, we can identify molecular and histologic biomarkers associated with clinical outcomes that can serve as predictors of NAC response in patients with MIBC.
Results
Study cohort
Our study included prospectively collected data from patients enrolled in the SWOG S1314 clinical trial (NCT02177695). S1314 is a randomized phase II trial to study co-expression extrapolation (COXEN), a gene expression model, as a predictive biomarker for response to NAC in MIBC. Cisplatin-eligible 237 patients with cT2-T4a N0 M0 urothelial cancer were randomized to receive either dose-dense Methotrexate-Vinblastine-Adriamycin/doxorubicin-Cisplatin (ddMVAC) every 14 days for 4 cycles or Gemcitabine-Cisplatin (GC) every 21 days for 4 cycles8,17,18. Among 167 evaluable patients, 42% and 36% achieved pCR in ddMVAC and GC groups, respectively8.
Our study analyzed 182 gigapixel WSIs and microarray gene expression data from 180 patients enrolled in S1314. Of 237 patients enrolled in the S1314 trial, we included 180 with available WSIs and gene expression data. The clinical characteristics of included patients are summarized in Supplementary Table 1. Our dataset included 56 (30.8%) WSIs of patients who achieved pCR (pT0 after RC) and 126 (69.2%) WSIs for those who had a partial response (PR, ≤pT1 but not pT0 at RC) or no response (NR, >pT1 at RC). To convert the prediction task into a clinically relevant binary classification problem, patients who achieved complete pathologic response were labeled as responders, and patients who had partial response or no response were labeled as non-responders, as only complete pathologic response would potentially enable future bladder preservation. Each WSI was coupled to a 1,071-dimensional microarray gene expression (GEX) vector of the same patient, forming a multimodal input data structure for our model (Methods).
Determining the most effective model architecture for handling whole slide images
To maximize the overall performance of our model, we sought to identify the best model to handle the H&E-stained histology imaging data. Analyzing WSIs is particularly challenging due to the complex tissue patterns, intricate cellular details, hyper-resolution, immense size, and computational demands. We tested three recently-developed weakly-supervised WSI-analysis approaches including 1. Patch-based model32, 2. CLAM33, and 3. SlideGraph+34 using deep learning-derived features. The results of these three approaches are shown in Supplementary Table 2. We found that SlideGraph+, a graph neural network, outperformed the other two approaches in predicting response to NAC measured by Area Under the Receiver Operating Characteristic Curve (AUROC). Specifically, SlideGraph+ achieved an AUROC of 0.67, followed by CLAM with an AUROC of 0.60. SlideGraph+ focuses on the spatial correlation between local features of patches, allowing for capturing contextual information and complex interactions in a holistic model instead of analyzing local features. Therefore, we selected Slidegraph+ architecture as the backbone of the histology data analysis branches in our Graph-based Multimodal Late Fusion (GMLF) model.
GMLF: multimodal integration of histology WSIs and gene expression for predicting response to NAC
We used GMLF to integrate the histologic and transcriptomic information to predict response to NAC. The model used SlideGraph+ to analyze the tumor spatial information at both tissue and cellular levels from WSIs and a multilayer perceptron for analyzing gene expression data (Fig. 1). For evaluating the model performance in predicting response to NAC, we used two strategies: 5-fold-cross-validation (5-fold CV) and 80/20 training testing split (Fig. 2). In 5-fold CV, the GMLF model achieved performance in predicting response to NAC with a mean AUC of 0.74 ( ± 0.1). In an 80/20 split, the model achieved an AUC of 0.72 in the testing set (Fig. 3).

Our model uses two paired data types from bladder cancer samples: gigapixel whole-slide images from routine Hematoxylin and Eosin (H&E) stained slides, and gene expression data from tissue microarrays. Our GMLF model consists of three branches: (1) WSI Neural Embeddings Branch: a GNN-based branch processing attributed graphs with nodal features as neural embeddings extracted by ResNet50 from WSIs, (2) WSI Cell-type and Morphological Branch: another GNN-based branch for graphs with nodal features comprising cell type and morphological features extracted by HoVer-Net from WSIs, and (3) Gene Expression Branch: a multilayer perceptron that processes the gene expression vector. Each branch i of the model yields a scalar score Si. We employ a multimodal late fusion strategy, aggregating these branch-level scores through summation, followed by Platt scaling to generate a prediction value. This value represents a probability between 0 and 1, where 1 indicates a complete response (pCR) to NAC.

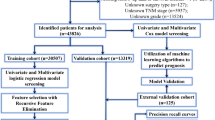
The dataset is initially split into an 80% discovery subset and a 20% hold-out test set, utilizing stratified random sampling at the patient level to ensure consistent data distribution among the different splits. Within the discovery subset, stratified 5-fold cross-validation is applied for model development and optimal parameter selection. The hold-out test set is then used to conduct an unbiased evaluation of the final model, assessing its performance on previously unseen data.

a Our comprehensive ablation study assesses the three-branch multimodal GMLF against different unimodal and bimodal baseline models formed based on the three distinct feature modalities. Specifically, Neural Embeddings refers to the GNN branch using ResNet50 for patch-level feature extraction, Cell Type and Morphology to another GNN branch using HoVer-Net for patch-level feature extraction, and Gene Expression to the branch analyzing patient-level gene expression data from tissue microarrays. b The AUROC (Area Under the Receiver Operating Characteristic) performance across different modality compositions is evaluated during the 5-fold cross-validation and tested on 20% internal validation data, with models trained on the 80% discovery dataset, for predicting response to neoadjuvant chemotherapy (NAC).
We hypothesized that integrating different data modalities, including gene expression and data extracted from WSIs, could improve the model performance compared to using a single data modality. To test our hypothesis, we conducted ablation studies in which we evaluated the performance of each modality (unimodal) or combined two modalities (bimodal) in predicting response to NAC compared to our multimodal GMLF model (Fig. 3a). Our multimodal model, which incorporates all three branches, outperformed unimodal and bimodal models (Fig. 3b). The second-best models were the unimodal SlideGraph+ branch for cell type and morphology with an AUC of 0.72 (± 0.14) and the gene expression branch with an AUC of 0.71 in 5-fold CV and 80/20 split, respectively (Fig. 3b).
Comparing the receiver operating characteristic (ROC) curves in specificity test35,36 showed that our GMLF model outperformed the second-best model in sensitivity with a P-value = 0.07 at 0.95 specificity.
Histopathological and molecular biomarker discovery through multimodal interpretation
As we demonstrated that a multimodal model is necessary for improving NAC response prediction performance, we sought to determine the features influencing model prediction. By leveraging model-agnostic Shapley Additive Explanation (SHAP)31,37, we were able to develop interpretation frameworks to analyze our trained GMLF model (Methods). Specifically, we used kernel SHAP31,37 together with our proxy model approach and graph-based visualization tools in our multimodal and multilevel interpretation framework.
In this SHAP interpretation analysis, we used the hold-out test set for our GMLF trained on the model development set38.
Inter-modality-level model interpretation
To explain how our multimodal model makes predictions, we quantified the contribution of each branch to the final model. This was achieved by applying the SHAP to the final layer of the GMLF for late fusion and prediction. This layer comprises a linear transformation, which takes each modality’s prediction score as input to compute a univariate raw score, followed by Platt scaling39, which converts the raw score into a prediction probability for a binary classification task (Methods). The contribution of each branch is shown in the SHAP summary plot (Fig. 4a) (Supplementary Data 1). Interestingly, we found that the GEX branch yielded SHAP values with the largest magnitude, indicating that it contributed more to the GMLF model than the two GNN branches. Moreover, we were able to quantify the contribution of each branch for each individual patient (i.e., a WSI paired with its corresponding gene expression vector) included in the hold-out test set, as shown in Fig. 4b (Supplementary Data 5). To evaluate the predictive power of each unimodality branch and the overall performance of our multimodal GMLF, we stratified patients in the hold-out test set by response status (pCR or non-pCR). For clarity, when comparing different branches within the overall GMLF framework, we refer to the output of each unimodality branch before it is combined with others in the final fully connected layer and adjusted by Platt scaling (see Methods, Fig. 1), as the prediction score of that branch. The final output of our GMLF framework is referred to as the overall prediction score. We then compared the prediction scores of each unimodality branch and the overall prediction score, respectively, between these two subgroups using the Mann-Whitney U test. The prediction scores from individual unimodal branches did not show statistical significance (GEX: P = 0.1834, CM: P = 0.6553, NE: P = 0.0741). In contrast, the overall prediction score was significantly different between the pCR and non-pCR subgroups (P = 0.0362 < 0.05), indicating that our multimodal prediction model can distinguish between response subgroups, whereas single unimodality branches cannot achieve this binary classification (Fig. 4c).

a Modality-level importance attributions across all patients in the hold-out test dataset are analyzed using a SHAP-based interpretation approach on a modality-level proxy model. b SHAP-based modality-level importance attribution for a representative patient (SAEAMD-0BS5RI-A1). c Comparison of prediction scores between responder and non-responder groups for the three individual unimodal branches of our multimodal framework GMLF: Neural Embeddings (NE), Cell-type and Morphology (CM), and Gene Expression (GE), and the overall prediction score from GMLF for predicting response to NAC. P-values in the boxplot subfigures were computed using the Mann-Whitney U test, with “*” indicating P-values < 0.05. d Gene (per alias) importance attributions across all patients in the hold-out test dataset are determined by applying SHAP to a proxy model that inputs the gene expression feature vector alongside predictions from the two GNN branches. The top 20 are presented. e Gene set enrichment analysis of the selected top 111 genes selected according to their SHAP-based gene importance attributions. Statistical significance is assessed by the hypergeometric test, using the overall investigated gene list as a background. f Visualization of node importance for the cell type and morphology branch overlaid on the original H&E slide for slide SADREE-0BGNRK-1A, correctly predicted as complete response (pCR). g Representative patches around the top 10th quantile of nodal importance associated with non-pCR (top row) and pCR (bottom row), annotated with HoVer-Net-estimated cell types for the same slide as (f). h Analysis of cell-type specific distributions based on the most contributive patches - i.e., the top 25% extremes of patch importance per slide. Boxplots for the average patch-level cell counts or tumor-stromal ratios for no pCR (red) or pCR (blue) predictive patches normalized by the average patch-level cell-type specific attribute of the entire WSI, with each point representing a distinct slide. The dotted line represents the average patch-level attribute (cell count or tumor-stromal ratio) for a given slide, indicating no enrichment for a particular cell type.
Intra-modality-level model interpretation
Within the gene expression branch, we tried to identify genes that played a more substantial role in predicting response to NAC. We built a proxy model that takes the GEX vector and the output prediction scores of the two GNN-based branches as inputs and the prediction scores of the full GMLF model as outputs (Methods). SHAP is then performed on this proxy model to quantify the contribution from individual genes to the model prediction.
A summary plot of the top 20 genes with the highest average SHAP value magnitude is shown in Fig. 4d, which shows that the model was able to pick up biologically relevant genes, including TP63, CCL5, and DCN, that have been previously found to be associated with response to NAC40,41,42,43,44,45. To further identify biological pathways predictive of response to NAC, we performed gene set enrichment analysis (GSEA) (see Methods). We conducted an exhaustive analysis of the top k gene aliases, sorted by their average SHAP value magnitude in descending order, with k ranging from 1 to the complete list of gene aliases. This also served as a sensitivity analysis and demonstrated stability in identifying highly enriched gene sets among the 15 in our study, particularly for k values between 50 and 300 (Supplementary Fig. 1, P < 0.05 for significant enrichment, P < 0.001 for highly significant enrichment). By associating the selected genes with the known biological processes and gene sets of interest using the combined P-value of the 15 gene sets computed based on GSEA, we identified a subset of the top 111 ranked genes as the key gene subset (see Methods). Gene set enrichment analysis of this top-111-gene subset revealed that basal differentiation and myofibroblasts are the most significant pathways predicting response to NAC, with FDR-adjusted P-values < 0.001 (Fig. 4e).
Within the GNN branch for cell type and morphological features, we sought to identify unique histopathological features influencing model prediction. In our framework, each node in the graph, derived from a WSI and used as input to a GNN branch, represents a specific patch or region of the WSI. The GNN branch assigns an importance value to each node, known as the node value. A lower node value suggests that the corresponding patch contributes towards predicting a complete response to NAC (Supplementary Data 2). These importance values are then pooled (i.e., summation over all nodes on a WSI) to get the output of this branch. Since lower values are associated with complete response, we sought to examine whether specific cell types or cell-type characteristics are linked to these nodes. To achieve this goal, we extracted the patches or regions from the WSI with the top 25% (lower node values) and bottom 25% (higher node values). We then quantified the cell-type specific characteristics on each patch using the cell counts of cancer cells, connective cells, immune cells, and necrotic cells, as well as the tumor-stromal ratio calculated by dividing the cancer cell count by the connective cell count.
For each cell type, we compared the average values of the top 25% of regions linked to complete response with the entire slide, and we did the same comparison for the bottom 25% of regions (Fig. 4h) (Supplementary Data 3).
In patches linked to complete response (low node values), we found an increase in cancer cell count and connective cell count but a decrease in necrosis cell count. We also found an increase in tumor-stromal ratio in these patches compared to patches with high node values (p-value < 0.0001).
To have a detailed analysis of the histologic features in individual WSIs, we overlaid the node values assigned by a GNN branch to the nodes (i.e., patches) on the original H&E-stained WSI, where each node’s importance value is mapped to its corresponding region or patch (Figs. 4f, 4g). We then compared cell type-specific cell counts between the responder-associated and the non-responder-associated regions (Supplementary Data 4). To identify potential histological markers, we focused on the patches that are enriched in a specific cell type (e.g., cancer cells) and associated with non-response (Fig. 4g Top) or response (Fig. 4g Bottom).
Evaluating the Influence of Intra-Tumor Heterogeneity on Model Performance
We quantified intra-tumor heterogeneity (ITH) using two approaches based on nuclei morphological features of cancer cells: the Median Diversity Rank (MDR)46 and the method based on the Shannon Diversity Index (SDI)47,48 (Methods). To evaluate the influence of the ITH degree on model performance, we compared ITH quantifications between the pCR and no pCR subgroups using the Mann-Whitney U test. No statistically significant differences were observed (P = 0.237 for MDR [Fig. 5a], P = 0.852 for SDI [Supplementary Fig. 2a]). These results indicate no clear association between ITH values and response status. Next, we stratified the instances into quantiles based on their ITH quantification values and evaluated the model performance within each subgroup using AUROC. The analysis was conducted across a varying number of quantiles. The MDR-based ITH quantification results revealed a general trend of improved model performance within the lower quantiles of ITH quantifications. In contrast, the lowest quantile did not consistently achieve the best AUROC (Fig. 5b). In comparison, SDI-based ITH showed no clear trend in its influence on model performance (Supplementary Fig. 2b).

ITH quantification was computed with the Median Deviation Ranking (MDR) approach in (a) and (b). a Boxplots of ITH metrics from the WSIs in pCR and no pCR subgroups. P-values computed by the Mann-Whitney U test. b Model performance evaluated by AUROC in different quantile subgroups stratified by ITH quantification. The x-axis indicates k, the number of quantiles, which ranges from 2 to the largest number before the first appearance of invalid quantile subgroups for computing AUROC.
Discussion
Relying on a single data modality to develop predictive models for complex diseases such as cancer may not offer adequate insights into disease heterogeneity. It is important to develop models that integrate multiple data modalities to capture complementary disease aspects, which can provide more precise insights for clinical decision-making. In this study, we developed a multimodal deep learning model, integrating tissue and cell information from WSIs with gene expression data to predict response to NAC in MIBC patients. Leveraging prospectively collected data from the SWOG S1314 clinical trial, our model integrated (1) tumor spatial details with cellular morphological features and cell type information from H&E-stained WSIs analyzed with GNNs and (2) tissue-level gene expressions analyzed with an MLP through a late fusion framework. Our model outperformed all unimodal and ablated models, highlighting the importance of integrating different data modalities in maximizing performance. The model was able to accurately predict response to NAC as well as identify prognostic biomarkers of response from WSIs and gene expression arrays only without including any clinical features, highlighting the strength of our model in extracting clinically relevant markers from images and molecular data.
Currently, there are no well-validated models for predicting the response to NAC in MIBC patients19. Font et al. have found that patients with basal/squamous tumors are more likely to achieve pCR13. On the other hand, Jütte et al. reported that tumors with high expression of luminal differentiation markers have a higher probability of achieving pCR49. Mi et al. proposed a machine-learning framework that integrated cellular, nuclear, and tissue architectural features from WSIs and immunohistochemistry staining with basic clinical features to predict response to NAC in MIBC patients. This framework was able to achieve 65%–73% accuracy25.
The limited performance of unimodal analysis, whether from gene expression or H&E-stained WSIs, drove our development of a multimodal framework. Although unimodal frameworks have their limitations, certain deep learning architectures have shown promise in other contexts32,33,34 by effectively extracting features and generating prediction scores from WSIs. We systematically compared various representative techniques to select the most suitable architecture for building the WSI-analysis branches that are integral to our multimodal framework.
In our experiments to identify the best model architecture for analyzing the gigapixel H&E-stained histology imaging data, SlideGraph+ demonstrated superior predictive power compared to approaches that did not consider spatial information. Spatial intratumoral heterogeneity is an important hallmark of cancer, which can drive therapy resistance and disease progression25,50. This is particularly important in bladder cancer, which presents with substantial heterogeneity and high mutational burden51. Our GMLF model was able to identify highly-attended patches associated with response to NAC, characterized by higher tumor cell content and altered immune and stromal profiles. Previous studies have found that integrating spatial information improves the performance of models predicting response to NAC25 and immune checkpoint blockade52. In MIBC, spatial organizations in tumor microenvironment have been linked to pCR with neoadjuvant chemoimmunotherapy53.
In our analysis, we found an increase in cancer cell count and connective cell count and a decrease in necrosis cell count in WSIs of patients who achieved pCR. This suggests that our model can unravel the complex interactions between cancer cells and other cells in the tumor microenvironment. Interestingly, these patches showed a statistically significant increase in tumor-stromal ratio. This is consistent with studies that found tumor-stromal ratio an important predictor of response to NAC54, indicating that the model was able to autonomously identify clinically relevant predictors even without including clinical data as an input as in previously developed models25.
Through a SHAP-based analysis, we found that the gene expression branch contributed the most to the GMLF model compared to the two GNN branches for WSIs. Although the GNN branch of Neural Embeddings (NE) based on ResNet-5055 extracts embedding vectors that may not be biologically relevant, this branch was more important than the GNN branch of cell type and morphological features. This shows the inherent tradeoff between predictive power and the interpretability of the extracted features56. To interpret the transcriptomic data analysis part of the model, we performed GSEA on the selected top 111 genes ranked by their SHAP value magnitudes (see Methods for selecting the top 111 genes). This resulted in two significantly enriched pathways: myofibroblasts and basal differentiation. We have recently shown that the molecular subtypes of MIBC are a significant predictor of response to NAC18. This is consistent with our model interpretation with GSEA that our GMLF also recognized the significance of the basal differentiation. However, studies have reported conflicting results about whether the basal subtype is associated with increased13,40 or decreased57,58 response to NAC. This can be due to studies applying different methods to define molecular subtypes with molecular subtyping models are found to be inconsistent in their classification59,60. Given the unresolved dispute in the study of basal subtype, we studied the significance of enrichment of basal differentiation and other gene sets of interest by performing hypergeometric tests in GSEA. This approach utilizes the set sizes, not their expression levels, to avoid prematurely determining whether they are positively or negatively associated with the response to NAC.
SHAP-based interpretability analysis revealed several biologically established genes that the model considered prognostic for response to NAC, including TP63, CCL5, and DCN. TP63 has been shown to play a pivotal role in tumorigenesis, cancer progression, and resistance to chemotherapy61. TP63 expression has been identified as a biomarker for worse clinical outcomes in bladder cancer62. Moreover, dysregulated TP63 expression has been found to be associated with metastasis and higher stage63.
Interestingly, p53 plays an important role in controlling basal gene signature, and TP63 levels are found to be elevated in the basal subtype of MIBC40, which our GSEA found as a significantly enriched pathway.
SHAP-based analysis also identified important genes involved in DNA damage and repair as predictors of response to NAC, including PRRX1, RUNX3, PPARG, and ZEB2. PPRX1 regulates DNA repair pathways by cooperating with FOXM1, and PPRX1 downregulation was found to increase the sensitivity of osteosarcoma to cisplatin and doxorubicin64,65. RUNX proteins, including RUNX1 and RUNX3, regulate DNA damage response by facilitating the recruitment of FANCD2 to DNA repair foci66. Several studies have found that RUNX3 mediates resistance to cisplatin67, carboplatin68, and gemcitabine69 in different cancers. Li et al. have found that PPARG interacts with MRN complex (MRE11-RAD50-NBS1) to promote DNA repair70, and PPARG agonists were shown to enhance the efficacy of platinum-based compounds in several cancer types, including non-small cell lung cancer71, ovarian, and colon cancers72. ZEB2 can promote chemotherapy resistance by activating genes involved in nucleotide excision repair, including ERCC1 and ERCC473.
Our model also identified CCL5 as an important gene marker in predicting response to NAC, which has been reported to decrease chemotherapy activity in breast and prostate cancers74,75. This emphasizes the strength of our data-driven approach in identifying key molecular features crucial for predicting response to NAC in MIBC tumors.
Our study is not without limitations. Despite employing robust methods for training and testing, including 5-fold cross-validation and evaluating performance on a hold-out test set, the model was not externally validated using an external dataset other than SWOG S1314. Thus, further validation using an external dataset with larger sample sizes is needed to evaluate the model’s generalizability. In interpretability analysis, we assigned an importance score to each input gene instead of providing a specific subset of genes as molecular biomarkers. In gene enrichment analysis, we used an empirical cutoff of the top 111 important genes to be included. Our model employed a late fusion framework that aggregated univariate prediction scores from three different branches. Despite demonstrating superior prediction performance, it falls short in unraveling the intricate interactions between the valuable features learned from each different modality. Our model relied only on WSIs and gene expression. However, additional modalities could improve the model’s performance, such as digital spatial profiling and circulating tumor DNA. Previous studies have demonstrated that changes in ctDNA dynamics and digital spatial profiling are correlated with pathologic response53,76.
In summary, our study provides a novel framework for predicting response to NAC in MIBC patients from routinely collected H&E images and gene expression vectors. Predicting response to NAC in MIBC is crucial for personalizing treatment strategies, improving clinical outcomes, avoiding unnecessary treatment, and ultimately, bladder preservation77.
To the best of our knowledge, this is the first work to develop an interpretable model that integrates WSIs and gene expression for predicting response to NAC in MIBC.
Our findings suggest that the multimodal integration of tissue-level gene expression and tissue morphological and cell-type information extracted from histology WSIs can perform better than single unimodal models. An important strength of our model is being trained on prospectively collected data from the S1314 randomized controlled trial with rigorous validation methods. Our model used SlideGraph+ architecture for analyzing WSIs, which accounts for spatial information, allowing the model to capture the spatial intratumoral heterogeneity. We used robust interpretation methods to uncover the most important features that influenced the model’s predictions. Our model was able to autonomously reveal biologically relevant biomarkers and highly-attended patches from WSIs associated with response to NAC. Further research on larger datasets, as well as experimental validation, are needed to establish the identified molecular and histologic biomarkers for predicting response to NAC in MIBC. Given that H&E images and gene expression data are routinely collected, our study could potentially advance the stratification of patients with MIBC based on their response to NAC, allowing the integration of precision medicine in clinical decision-making.
Methods
Model evaluation strategies
We evaluated our model and competitive baseline methods through two different strategies (Fig. 2). The 180-patient dataset is split into two non-overlapping sets: one is the discovery set (80% of patients, 45 CR, 101 N/PR), and the other is the hold-out test set (20% of patients, 11 CR, 25 N/PR). In the first strategy, the models were trained and evaluated on the discovery set using 5-fold cross-validation (5-fold CV). In the second strategy, the models were trained using the discovery set divided into non-overlapping training and validation subsets and then tested using the hold-out test set. The second strategy is denoted 80/20 training-testing split according to the patient-level splitting ratio. We split data via stratified random sampling at the patient level for model training and testing to avoid data leakage bias, as some patients had multiple WSIs.
Our study used histopathology and cell type data from standard H&E images with gene expression profiles derived from RNA sequencing from the SWOG S1314-COXEN clinical trial (ClinicalTrials.gov NCT02177695 2014-06-25).
Baseline unimodal models
CLAM. The clustering-constrained-attention multiple-instance learning (CLAM)33 considers each WSI as a bag of non-overlapping patches and employs attention-based learning to identify patches of high diagnostic value to accurately classify whole slides and instance-level clustering over the identified representative patches to constrain and refine the feature space. Notably, CLAM operates without considering the spatial relationship between these subregions. Patches were extracted at 2048 x 2048 pixels at the highest resolution of the whole slide image, and features were extracted using the default modified ResNet-50 model. Default hyperparameters were used for the analysis.
Patch-based weakly-supervised Model. Patches were extracted at 1024 pixels x 1024 pixels at the highest resolution and down-sampled to 512 pixels x 512 pixels. Image patches were filtered out based on the percentage of tissue in the image (>40%), and blur detection was used to remove patches that were scanned out of focus78. Two different datasets were used. (1) All patches, 531,048, were used for the analysis, and (2) patches containing > 50% tumor purity as assessed by a trained HoVer-Net model (pre-trained on PanNuke79) to mimic the patch-based model32 that only used tumor regions for the analysis80.
A modified model and training protocol of the patch-based molecular subtype prediction model32 was used for this analysis. In short, each patch was given the same label for a given slide. Data augmentation was performed using a combination of PyTorch built-in functions (Resize: 256, random rotations: -90 to 270, Color Jitter: Brightness, contrast, saturation, and hue = 0.4 p = 0.8, RandomErasing, and mean/standard deviation normalization) and separate H&E slide specific transformations (HEDJitter – theta = 0.05)81. Batch size was set to 20, the learning rate was set to 1 x 10−4, weight decay was set to 1 x 10−3, and the Stochastic Gradient Descent (SGD) optimizer with momentum (0.9) was used. The model was EfficientNetV2_S with initial weight pre-trained on ImageNet. MixUp was used to train the model with BinaryCrossEntropywithLogits loss from PyTorch. All models were trained for five epochs.
Slidegraph + . SlideGraph+34 is a graph-based neural network model that can capture the overall organization and structure of the tissue. It does this by modeling the spatial relationships between cells in the tissue. The overall framework consists of four steps: (i) Feature Extraction: The WSI is preprocessed by masking out the background region and divided into non-overlapping patches of size 2048 x 2048 pixels at the highest resolution of the WSI. From each patch, a high-dimensional feature vector is extracted from a pre-trained deep-learning model. Depending on the context, we used ResNet-5055 to extract a 2048-dimensional embedding vector (namely, the neural embeddings) and HoVer-Net80 to extract 5 cell types and morphological features of nuclei from each cell type. (ii) Spatial Clustering: Similar patches are grouped together using an adaptive spatial agglomerative clustering, which relies on a patch-level similarity metric82. (iii) Graph Construction: A planar graph representation is built based on the clustered patches. In our work, each node of this graph representation consists of one patch. The graph edge set is built using Delauney triangulation based on the geometric coordinates of cluster centers with a maximum distance connectivity threshold of pixels83. This graph captures the spatial relationships and cellular organization of the tissue. (iv) Graph Neural Network Prediction: The constructed graph is fed into a graph neural network to predict the response to NAC at two levels: responders vs non-responders.
Graph-based Multimodal Late Fusion (GMLF) Framework
We built a Graph-based Multimodal Late Fusion (GMLF) model to integrate multimodal features from histology image data and gene expression data. Multiple branches are utilized to extract features from different modalities and generate a unimodal prediction score. We used the late fusion strategy to combine the unimodal prediction scores through a linear transformation into a univariate raw score, followed by the Platt scaling to this raw score into a prediction probability for the responder-vs-non-responder binary classification task. In this study, GMLF comprises three branches: two for histology imaging data (i.e., the WSIs) and one for gene expression data. The two WSI branches are based on SlideGraph+34 and differ in what features are extracted at the tile/patch level. Specifically, one used ResNet-5055 to extract 2048-dim features, namely the neural embeddings, as each individual feature has no specific biological interpretation. The other WSI branch used HoVer-Net80 to extract 155-dim features: 5-dim cell-type counts and \(5\times 30\)-dimensional feature vector, which contains the means and standard deviations of 15 different morphological properties34 of each cell type. We used a multilayer perceptron to generate a unimodal prediction score from gene expression.
Ablation study
We conducted an extensive ablation study to investigate the contribution of each feature modality. Besides the overall GMLF, we investigated three unimodal models that only used one of the three branches of GMLF and three bi-modal models that combined two of the three branches. Each bi-modal model also used the linear transformation to combine its two unimodal prediction scores. All these models used Platt scaling as the last step to convert the output into a probability of prediction.
Model evaluation
All models were chosen based on epoch with the lowest validation loss for Patch-based weakly supervised models and CLAM. AUROC was used to evaluate model performance across all experiments using scikit-learn.
Multimodal importance analysis
Proxy Models for Modality-level and Gene-level Feature Importance Analysis. We adapted SHapley Additive exPlanations (SHAP), which is a model-agnostic technique for interpreting complex machine learning models, to interpret our GMLF at different levels. The SHAP variants based on gradient-based feature attribution84,85 or backpropagation (e.g., DeepLIFT85,86) were not applied in our model interpretation framework. This is because their existing implementations are not directly applicable to our GMLF, which integrates both multilayer-perceptron and graph-neural-network components34, and they are reported to have limitations in interpreting graph-based deep models87. Instead, we leveraged model-agnostic SHAP31 by utilizing proxy models. A proxy model comprises part of the original trained model, redefines input data based on what is fed into this part, and generates the same final output as the original trained model for any test data. For the modality-level importance attribution, the proxy model comprises the fusion layer and the final prediction score. It redefines the input with the intermediate-output prediction score from each individual modality branch - i.e., a 3-dimensional vector. To obtain the molecular feature importance attribution, the proxy model comprises the MLP branch for gene features, the fusion layer, and the final prediction layer of GMLF. It redefines the input by appending the prediction scores of the two GNN-based branches (i.e., WSI Neural Embeddings and WSI Cell-type and Morphology) to the gene expression vector - i.e., an (n + 2)-dimensional vector where n is the length of input gene expression associated with a WSI.
Proxy models. For the modality-level importance attribution, we created a proxy model that can take the output prediction score from each individual modality branch as input and yield the same output as our trained GMLF model. This proxy-model-based technical approach is also applied to molecular feature importance attribution at the individual modality level. The input to this latter proxy model is created by appending the prediction scores of the two GNN-based branches (i.e., WSI Neural Embeddings and WSI Cell Type and Morphology) to the gene expression vector.
Gene Set Enrichment Analysis. A total of 15 different gene sets with a range of different sizes from the work on molecular classification of MIBC88 were used for interpreting gene expression. To interpret the gene sets most important for the prediction task, all gene aliases were sorted from the largest SHAP value magnitude to the lowest SHAP value magnitude. The input gene expression data in our study includes a total of 1071 gene aliases, corresponding to 818 unique gene symbols. We used gene symbols instead of gene aliases in GSEA. For genes with multiple aliases, each gene was counted only once, using the alias with the largest average SHAP value magnitude. To assess how sensitive the enrichment analysis is to different gene set sizes, a range of different subsets, from 1 to the length of gene aliases at intervals of 1, were used. A hypergeometric test was performed for each gene set at each subset size, and FDR correction was performed at each interval. We identified gene sets that were statistically significant at a P < 0.05 and highly significant at a P < 0.001 after correction for the top gene subset (cf. section Selection of the Top Gene Subset).
Selection of the Top Gene Subset. The top gene subset was derived from the gene alias list sorted by their average SHAP value magnitudes based on the association between the candidate gene subsets and the known biological pathways or gene sets of interest. Given the well-established use of GSEA for interpreting and justifying gene subset selection89, we developed an approach to identify the cutoff from the sorted gene alias list using GSEA. Specifically, for the subset size \(k\) ranging from 1 to the full length of the gene alias list, we selected the top \(k\) aliases and mapped them to their corresponding gene symbols as a candidate gene subset. We then measured the enrichment significance of each of the 15 gene sets of interest in each candidate gene subset. The combined p-value of all 15 gene sets was computed using Fisher’s method90. The \({k}^{* }\)-gene-alias subset yielding the highest \(-\mathrm{log}\,({combined\; p}-{value})\) was selected, and their corresponding gene symbols were used as the top gene subset of biological significance according to our input gene sets of interest.
Histological Feature Analysis. Cell type information was extracted for all patches, as mentioned previously, using a PanNuke pre-trained HoVer-Net model. To understand the cell types that were important for NAC response prediction, we identified the top 25% and bottom 25% of activations for the patches on the WSI cell type and morphological branch and compared them to all patches used for the analysis. We calculate the average patch-level cell feature for each slide. Tumor-stromal ratio was also assessed as a predictor of chemotherapy response as the per-patch cancer cell count was divided by the stromal cell count. We calculate the slide level average cell type feature and divide each subset (top 25% and bottom 25%) by the same metric for the entire slide to identify specific enrichment for the subset.
Intra-Tumor Heterogeneity (ITH) Quantification
We adapted two approaches for ITH quantification using the nuclei morphological features. We focused on cancer cells annotated by HoVer-Net80 and used the morphological features computed by the functions from the skimage.measure (label, regionprops, regionprops_table). (1) The Median Diversity Ranking (MDR) approach is adapted from a previous study on ITH with pan-cancer analysis46. An image-level diversity measure \({{d}_{f}}^{{WSI}}\) was first computed for each morphological feature using the Mean Absolute Deviation (MAD) across all cancer cell nuclei within this WSI - i.e., \({{d}_{f}}^{{WSI}}={MA}{D}_{{nuclei}}(f)\). Then, the nuclear diversity ranks \({{R}_{f}}^{{WSI}}\) were calculated for each morphological feature by sorting the WSIs according to the corresponding diversity measure. The final quantification of nuclear diversity D for each WSI was derived from the Median Diversity Rank (MDR) across all morphological features divided by the maximum MDR across all the WSIs - i.e., \({D}^{{WSI}}=\frac{{media}{n}_{f}({{R}_{f}}^{{WSI}})}{{ma}{x}_{{WSI}}({media}{n}_{f}({{R}_{f}}^{{WSI}}))}\). (2) The approach based on the Shannon Diversity Index91 is adapted from previous studies on heterogeneity in brain tumors and breast tumors47,48. The sampled cancer cell nuclei were firstly clustered into subgroups by hierarchical clustering Euclidean distance and Ward linkage, with the optimal number of clustering determined by the “silhouette” index. The Shannon Diversity Index91 (SDI) is computed over the cancer cell nuclei clusters for each WSI as its ITH quantification.
Data availability
The processed gene expression matrix and pathologic response data analyzed in this study are available in the Gene Expression Omnibus (GEO) under accession number GSE244266. The whole-slide images used in this study are maintained by SWOG and are available for qualified researchers upon reasonable request.
Code availability
The code repository for this work is available at https://github.com/ZB-WCM/GMLF_response_NAC_MIBC.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Park, J. C., Citrin, D. E., Agarwal, P. K. & Apolo, A. B. Multimodal management of muscle-invasive bladder cancer. Curr. Probl. Cancer 38, 80–108 (2014).
Zakaria, A. S. et al. Postoperative mortality and complications after radical cystectomy for bladder cancer in Quebec: A population-based analysis during the years 2000-2009. Can. Urol. Assoc. J. 8, 259–267 (2014).
Novara, G. et al. Complications and mortality after radical cystectomy for bladder transitional cell cancer. J. Urol. https://doi.org/10.1016/j.juro.2009.05.032 (2009).
Shabsigh, A. et al. Defining early morbidity of radical cystectomy for patients with bladder cancer using a standardized reporting methodology. Eur. Urol. 55, 164–174 (2009).
Chung, D. Y. et al. Comparison of oncologic outcomes of Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin (ddMVAC) with Gemcitabine and Cisplatin (GC) as neoadjuvant chemotherapy for muscle-invasive bladder cancer: systematic review and meta-analysis. Cancers 13, (2021).
Waingankar, N. et al. The impact of pathologic response to neoadjuvant chemotherapy on conditional survival among patients with muscle-invasive bladder cancer. Urol. Oncol. 37, 572.e21–572.e28 (2019).
Flaig, T. W. et al. Long-term outcomes from a Phase 2 study of neoadjuvant chemotherapy for muscle-invasive bladder cancer (SWOG S1314; NCT02177695). Eur. Urol. 84, 341–347 (2023).
Witjes, J. A. et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 79, 82–104 (2021).
Kang, H. W., Kim, W.-J., Choi, W. & Yun, S. J. Tumor heterogeneity in muscle-invasive bladder cancer. Transl. Androl. Urol. 9, 2866880–2862880 (2020).
Wezel, F., Vallo, S. & Roghmann, F. On behalf of the “Young Academic Urologi Carcinoma Group of the European Association of Urology.” Do we have biomarkers to predict response to neoadjuvant and adjuvant chemotherapy and immunotherapy in bladder cancer? Transl. Androl. Urol. 6, 1067080–1061080 (2017).
O’Donnell, P. H. et al. Clinical evaluation of Cisplatin sensitivity of Germline Polymorphisms in neoadjuvant chemotherapy for urothelial cancer. Clin. Genitourin. Cancer 14, 511–517 (2016).
Font, A. et al. Immunohistochemistry-based taxonomical classification of bladder cancer predicts response to neoadjuvant chemotherapy. Cancers 12, 1784 (2020).
Koskinen, I. et al. Prediction of neo-adjuvant chemotherapy response in bladder cancer: the impact of clinical parameters and routine biomarkers. Sjukskoterskan Scand. J. Urol. 55, 448–454 (2021).
Vale, C. L. Defects in DNA Repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur. Urol. 68, 959–967 (2015).
Choi, S. J. et al. Radiomics-based model for predicting pathological complete response to neoadjuvant chemotherapy in muscle-invasive bladder cancer. Clin. Radiol. 76, 627.e13–627.e21 (2021).
Flaig, T. W. et al. A randomized phase II study of coexpression extrapolation (COXEN) with neoadjuvant chemotherapy for bladder cancer (SWOG S1314; NCT02177695). Clin. Cancer Res. 27, 2435–2441 (2021).
Lerner, S. P. et al. Association of molecular subtypes with pathologic response, PFS, and OS in a phase II study of COXEN with neoadjuvant chemotherapy for muscle-invasive bladder cancer. Clin. Cancer Res. 30, 444–449 (2024).
Motterle, G., Andrews, J. R., Morlacco, A. & Karnes, R. J. Predicting response to neoadjuvant chemotherapy in bladder cancer. Eur. Urol. Focus 6, 642–649 (2020).
Vora, L. K. et al. Artificial Intelligence in pharmaceutical technology and drug delivery design. Pharmaceutics 15, (2023).
Cui, M. & Zhang, D. Y. Artificial intelligence and computational pathology. Lab. Invest. 101, 412–422 (2021).
Loeffler, C. M. L. et al. Artificial Intelligence-based detection of FGFR3 mutational status directly from routine histology in bladder cancer: a possible preselection for molecular testing? Eur. Urol. Focus 8, 472–479 (2022).
Velmahos, C. S., Badgeley, M. & Lo, Y.-C. Using deep learning to identify bladder cancers with FGFR-activating mutations from histology images. Cancer Med 10, 4805–4813 (2021).
Tokuyama, N. et al. Prediction of non-muscle invasive bladder cancer recurrence using machine learning of quantitative nuclear features. Mod. Pathol. 35, 533–538 (2022).
Mi, H. et al. Predictive models of response to neoadjuvant chemotherapy in muscle-invasive bladder cancer using nuclear morphology and tissue architecture. Cell Rep. Med 2, 100382 (2021).
Krishnamurthy, S. et al. Predicting response of triple-negative breast cancer to neoadjuvant chemotherapy using a deep convolutional neural network–based artificial intelligence tool. JCO Clin. Cancer Inform. https://doi.org/10.1200/CCI.22.00181 (2023).
Yamashita, R. et al. Deep learning model for the prediction of microsatellite instability in colorectal cancer: a diagnostic study. Lancet Oncol. 22, 132–141 (2021).
Kline, A. et al. Multimodal machine learning in precision health: A scoping review. npj Digital Med. 5, 1–14 (2022).
TTMFN: Two-stream transformer-based multimodal fusion network for survival prediction. ar5iv https://ar5iv.labs.arxiv.org/html/2311.07033.
Steyaert, S. et al. Multimodal deep learning to predict prognosis in adult and pediatric brain tumors. Commun. Med. 3, 1–15 (2023).
Scott M. Lundberg Paul G. Allen School of Computer Science, University of Washington, Seattle, WA & Su-In Lee Paul G. Allen School of Computer Science, Department of Genome Sciences, University of Washington, Seattle, WA. A unified approach to interpreting model predictions. https://doi.org/10.5555/3295222.3295230
Woerl, A.-C. et al. Deep learning predicts molecular subtype of muscle-invasive bladder cancer from conventional histopathological slides. Eur. Urol. 78, 256–264 (2020).
Lu, M. Y. et al. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat. Biomed. Eng. 5, 555–570 (2021).
Lu, W. et al. SlideGraph: Whole slide image level graphs to predict HER2 status in breast cancer. Med. Image Anal. 80, 102486 (2022).
Hanley, J. A. & McNeil, B. J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Pepe, M. S., Longton, G. & Janes, H. Estimation and comparison of receiver operating characteristic curves. Stata J. https://doi.org/10.1177/1536867X0900900101 (2009).
shap.KernelExplainer — SHAP latest documentation. https://shap-lrjball.readthedocs.io/en/latest/generated/shap.KernelExplainer.html.
Chen, R. J. et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell 40, 865–878.e6 (2022).
Platt, J. et al. Probabilistic Outputs for Support Vector Machines and Comparisons to Regularized Likelihood Methods. Adv. Large Margin Classif. 10, 61–74 (1999).
Choi, W. et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25, 152–165 (2014).
Robertson, A. G. et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 174, 1033 (2018).
Ascierto, M. L. et al. Abstract 2167: Immune-related biomarkers associated with pathologic complete response (pCR) to neoadjuvant cisplatin-based chemotherapy in patients with muscle-invasive bladder cancer undergoing cystectomy. Cancer Res 83, 2167–2167 (2023).
Sinn, B. V. et al. Immune-related gene expression predicts response to neoadjuvant chemotherapy but not additional benefit from PD-L1 inhibition in women with early triple-negative breast cancer. Clin. Cancer Res. 27, 2584–2591 (2021).
Li, P. et al. Identification of prognostic biomarkers associated with stromal cell infiltration in muscle-invasive bladder cancer by bioinformatics analyses. Cancer Med 9, 7253–7267 (2020).
Wang, J. et al. Integrating single-cell RNA-seq to identify fibroblast-based molecular subtypes for predicting prognosis and therapeutic response in bladder cancer. Aging 16, 11385–11408 (2024).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 22, 105–113 (2016).
Faust, K. et al. Unsupervised resolution of histomorphologic heterogeneity in renal cell carcinoma using a brain tumor-educated neural network. JCO Clin. Cancer Inform. 4, 811–821 (2020).
Danenberg, E. et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat. Genet. 54, 660–669 (2022).
Jütte, H. et al. KRT20, KRT5, ESR1 and ERBB2 expression can predict pathologic outcome in patients undergoing neoadjuvant chemotherapy and radical cystectomy for muscle-invasive bladder cancer. J. Pers. Med. 11, 473 (2021).
Marusyk, A., Janiszewska, M. & Polyak, K. Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Lavallee, E., Sfakianos, J. P. & Mulholland, D. J. Tumor heterogeneity and consequences for bladder cancer treatment. Cancers 13, 5297 (2011).
Nyman, J. et al. Spatially aware deep learning reveals tumor heterogeneity patterns that encode distinct kidney cancer states. Cron. Med. 4, 101189 (2023).
Beckabir, W. et al. Spatial relationships in the tumor microenvironment demonstrate association with pathologic response to neoadjuvant chemoimmunotherapy in muscle-invasive bladder cancer. Eur. Urol. 85, 242–253 (2024).
Li, F. et al. Combining the tumor-stroma ratio with tumor-infiltrating lymphocytes improves the prediction of pathological complete response in breast cancer patients. Breast Cancer Res. Treat. 202, 173–183 (2023).
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. https://ieeexplore.ieee.org/document/7780459.
Linardatos, P., Papastefanopoulos, V. & Kotsiantis, S. Explainable AI: A review of machine learning interpretability methods. Entropy 23, 18 (2020).
Taber, A. et al. Molecular correlates of cisplatin-based chemotherapy response in muscle invasive bladder cancer by integrated multi-omics analysis. Nat. Commun. 11, 4858 (2020).
Sjödahl, G. et al. Different responses to neoadjuvant chemotherapy in urothelial carcinoma molecular subtypes. Eur. Urol. 81, 523–532 (2022).
Sjödahl, G. et al. Molecular subtypes as a basis for stratified use of neoadjuvant chemotherapy for muscle-invasive bladder cancer-a narrative review. Cancers 14, 1692 (2022).
Seiler, R. et al. Divergent biological response to neoadjuvant chemotherapy in muscle-invasive bladder cancer. Clin. Cancer Res. 25, 5082–5093 (2019).
Patel, A. et al. Targeting p63 upregulation abrogates resistance to MAPK inhibitors in Melanoma. Cancer Res 80, 2676–2688 (2020).
Choi, W. et al. p63 EXPRESSION DEFINES A LETHAL SUBSET OF MUSCLE-INVASIVE BLADDER CANcers. PLoS One 7, e30206 (2012).
Koga, F. et al. Impaired p63 expression associates with poor prognosis and Uroplakin III Expression in invasive urothelial carcinoma of the bladder. Clin. Cancer Res. 9, 5501–5507 (2003).
Marchand, B. et al. PRRX1 isoforms cooperate with FOXM1 to regulate the DNA damage response in pancreatic cancer cells. Oncogene 38, 4325–4339 (2019).
Joko, R. et al. PRRX1 promotes malignant properties in human osteosarcoma. Transl. Oncol. 14, 100960 (2021).
Wang, C. Q. et al. Disruption of Runx1 and Runx3 leads to bone marrow failure and leukemia predisposition due to transcriptional and DNA repair defects. Cell Rep. 8, 767–782 (2014).
Li, D.-J., Shi, M. & Wang, Z. RUNX3 reverses cisplatin resistance in esophageal squamous cell carcinoma via suppression of the protein kinase B pathway. Thorac. Cancer 7, 570–580 (2016).
Barghout, S. H. et al. RUNX3 contributes to carboplatin resistance in epithelial ovarian cancer cells. Gynecol. Oncol. 138, 647–655 (2015).
Horiguchi, S. et al. Loss of runt-related transcription factor 3 induces gemcitabine resistance in pancreatic cancer. Mol. Oncol. 7, 840–849 (2013).
Li, C. G. et al. PPARγ interaction with UBR5/ATMIN promotes DNA repair to maintain endothelial homeostasis. Cell Rep. 26, 1333–1343.e7 (2019).
Reddy, R. C. et al. Chemotherapeutic drugs induce PPAR-γ expression and show sequence-specific synergy with PPAR-γ ligands in inhibition of non–small cell lung cancer. Neoplasia 10, 597–603 (2008).
Girnun, G. D. et al. Synergy between PPARγ ligands and platinum-based drugs in cancer. Cancer Cell 11, 395–406 (2007).
Sreekumar, R. et al. The ZEB2-dependent EMT transcriptional programme drives therapy resistance by activating nucleotide excision repair genes ERCC1 and ERCC4 in colorectal cancer. Mol. Oncol. 15, 2065–2083 (2021).
Xiang, P. et al. Infiltrating CD4+ T cells attenuate chemotherapy sensitivity in prostate cancer via CCL5 signaling. Prostate 79, 1018–1031 (2019).
Yaal-Hahoshen, N. et al. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clin. Cancer Res 12, 4474–4480 (2006).
Christensen, E. et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol. https://doi.org/10.1200/JCO.18.02052. (2019)
Rehme, C. et al. Clinical outcome and quality of life in octogenarian patients with muscle-invasive urothelial carcinoma of the bladder treated with radical cystectomy or transurethral resection of the bladder tumor: a retrospective analysis of 143 patients. Int. Urol. Nephrol. 54, 71–79 (2021).
Brendel, M. et al. Weakly-supervised tumor purity prediction from frozen H&E stained slides. EBioMedicine 80, 104067 (2022).
Gamper, J. et al. PanNuke Dataset Extension, Insights and Baselines. (2020).
Graham, S. et al. Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 58, 101563 (2019).
GitHub - gatsby2016/Augmentation-PyTorch-Transforms: Image data augmentation on-the-fly by add new class on transforms in PyTorch and torchvision. GitHub https://github.com/gatsby2016/Augmentation-PyTorch-Transforms.
Müllner, D. Modern hierarchical, agglomerative clustering algorithms. (2011).
Paul Chew, L. Constrained delaunay triangulations. Algorithmica 4, 97–108 (1989).
Sundararajan, M., Taly, A. & Yan, Q. Axiomatic Attribution for Deep Networks. in International Conference on Machine Learning 3319–3328 (PMLR, 2017).
Kokhlikyan, N. et al. Captum: A unified and generic model interpretability library for PyTorch. (2020).
Avanti Shrikumar Stanford University, Stanford, California, Peyton Greenside Stanford University, Stanford, California & Anshul Kundaje Stanford University, Stanford, California. Learning important features through propagating activation differences. https://doi.org/10.5555/3305890.3306006.
Yuan, H., Yu, H., Gui, S. & Ji, S. Explainability in graph neural networks: a taxonomic survey. IEEE Trans. Pattern Anal. Mach. Intell. 45, 5782–5799 (2023).
Kamoun, A. et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 77, (2020).
Abdolahi, F., Shahraki, A., Sheervalilou, R. & Mortazavi, S. S. Identification of differentially expressed genes associated with the pathogenesis of gastric cancer by bioinformatics analysis. BMC Med. Genomics 16, 311 (2023).
H., M & Fisher, R. A. Statistical methods for research workers. J. Am. Stat. Assoc. 67, 248 (1972).
Park, S. Y., Gönen, M., Kim, H. J., Michor, F. & Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Investig. 120, 636–644 (2010).
Acknowledgements
This work is supported by the National Institutes of Health (NIH) / National Cancer Institute (NCI) under Award Nos. U10CA180888 and U10CA180819. It is also supported by the National Science Foundation under Award No. 1750326.
Author information
Authors and Affiliations
Contributions
Z.B., M.B., B.M.F., and F.W. initiated and designed the study. C.M.T., T.W.F., I.M.T., M.P., M.S.L., D.T., D.G., S.D., J.J.M., W.C., C.P.N.D., S.P.L., and D.J.M. conducted subject enrollment and sample collection. Z.B., M.B., M.O., and O.E. conducted deep learning and statistical analysis. B.M.F. and F.W. supervised the research. Z.B., M.B., and M.O. wrote the first draft of the manuscript. All authors contributed to the writing and editing of the revised manuscript and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Bishoy M Faltas: Consulting or Advisory Role: QED therapeutics, Boston Gene, Astrin Biosciences Merck, Immunomedics/Gilead, QED therapeutics, Guardant, Janssen. Patent Royalties: Immunomedics/Gilead. Research support: Eli Lilly. Honoraria: Urotoday. Grants and research support: NIH, DoD-CDMRP, Starr Cancer Consortium, P-1000 Consortium. Olivier Elemento: Stock and Other Ownership Interests: Freenome, OneThree Biotech, Owkin, Volastra Therapeutics. Personal fees: Pionyr Immunotherapeutics, Champions Oncology. Seth P Lerner: Research support for Clinical trials - Aura Bioscience, FKD, JBL (SWOG), Genentech (SWOG), Merck (Alliance), QED Therapeutics, Surge Therapeutics, Vaxiion; Consultant/Advisory Board - Aura Bioscience, BMS, C2iGenomics, Immunity Bio, Incyte, Gilead, Pfizer/EMD Serono, Protara, Surge Therapeutics, UroGen, Vaxiion, Verity; Patent – TCGA classifier; Honoraria – Grand Rounds Urology, UroToday. Zilong Bai, Mohamed Osman, Matthew Brendel, Catherine M. Tangen, Thomas W. Flaig, Ian M. Thompson, Melissa Plets, M. Scott Lucia, Dan Theodorescu, Daniel Gustafson, Siamak Daneshmand, Joshua J. Meeks, Woonyoung Choi, Colin P. N. Dinney, David J. McConkey, and Fei Wang declare no competing interests.
Ethics approval
The study was reviewed and received approval by the National Cancer Institute (NCI) Central Institutional Review Board (CIRB), and patients provided written, informed consent; it was conducted according to the Declaration of Helsinki guidelines17.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, Z., Osman, M., Brendel, M. et al. Predicting response to neoadjuvant chemotherapy in muscle-invasive bladder cancer via interpretable multimodal deep learning. npj Digit. Med. 8, 174 (2025). https://doi.org/10.1038/s41746-025-01560-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41746-025-01560-y
This article is cited by
-
UroFusion-X: a unified multimodal deep learning framework for robust diagnosis, subtyping, and prognosis of urological cancers
npj Digital Medicine (2026)
-
Cytoskeletal protein KRT14 governs cisplatin resistance by modulating eIF4H-dependent ACOX2 translation and lipid metabolism in bladder cancer
Cell Death & Disease (2025)
-
Common inflammatory proteins linking frailty and area-level deprivation as key drivers of cardiovascular risk in women
Communications Medicine (2025)
