
Abstract
Subjecting copper to short anodic pulses during the electrocatalytic reduction of carbon dioxide (CO2RR) has been shown to improve the activity and selectivity towards hydrocarbons and alcohols. Nonetheless, the nature of the active sites is still unclear. Here the evolution of the morphology, chemical state and crystal structure of Cu(100) exposed to potential pulses during the CO2RR was resolved by a combination of spectroscopy, microscopy and diffraction methods applied concurrently. Under anodic potential pulses, (n10) facets were formed. Moreover, alternating anodic to cathodic potential pulses during the CO2RR also lead to the stabilization of copper oxide species located either at the surface or directly underneath ultrathin metallic copper layers, depending on the specific pulse potential treatment applied. Both findings are key for the enhanced ethylene and ethanol production reported during pulsed CO2RR.

Similar content being viewed by others
Main
Globally increasing industrialization and massive deforestation are counted among the leading causes for the worrying increase in CO2 emissions observed over the last decades—the primary promoter of climate change1. Reducing CO2 emissions and the reutilization of CO2 through conversion into energetically valuable products are current societal and industrial challenges. The development of affordable renewable energy sources facilitates the use of electric power to promote such conversion. Among many proposed catalysts, copper proved to be the best candidate for the electrocatalytic reduction reaction of CO2 (CO2RR) due to its ability to form C2 + products, such as ethylene or ethanol2,3,4.
Understanding of the local processes taking place at the electrode surface during the CO2RR has gradually increased in recent years through the development and optimization of quasi in situ and operando characterization techniques. In particular, information on the catalyst transformations taking place during the formation of the catalytically active surface sites5,6,7,8 has been gained, and the structural/chemical state effects determining the activity/selectivity towards desired products have been discussed6,7,9,10,11,12,13,14. Relatedly, the presence of oxygen species on the surface, either through preferential copper oxidation11,15,16,17, the adsorption of oxygen at the surface or the presence of subsurface oxygen under reaction conditions18,19,20,21, was reported to enhance the selectivity of copper catalysts for C2+ products. A possible approach to stabilize oxygenated species on the surface of copper electrodes under the CO2RR is the application of a pulsed treatment that alternates between oxidizing anodic and working CO2RR cathodic potential regimes. In this way, during the anodic pulse, Cu(I)/Cu(II) species are formed on the electrode surface depending on the chosen electrode potential and, depending on the length of the cathodic pulse17, may still be present during the CO2 reduction step. Recently, it was proven that applying this type of pulsed potential treatment clearly influences the product distribution, with the selectivity being highly dependent on the chosen anodic potential voltage and time for both copper single crystals11 and nanoparticles (NPs)17. For instance, Aran-Ais et al. found evidence that continuous in situ regeneration of Cu(I), combined with surface restructuring, drastically enhances the selectivity of Cu(100) electrodes towards C2+ products11. Similar findings were reported by Timoshenko et al. for Cu NPs17, although it was concluded here that bulk-like Cu2O species mainly favour C1 products, and that reconstructed surfaces with highly distorted mixed Cu(I)/Cu(II) species are needed for the generation of oxygenates such as ethanol.
However, the identity of the specific structural motifs formed during a pulsed CO2RR has remained unclear to date and constitutes the main scientific question of the present study. This experimental identification is key information needed by theorists calculating reaction pathways on model flat surfaces (equivalent to our single-crystal precatalyst) who are as yet unable to predict ab initio the structural transformations occurring under the simultaneous effect of an applied external electric field, in this case a periodically changing one, and the presence of an explicit complex electrolyte. Here we systematically investigate how the application of pulsed treatments affects the morphology and chemical state of Cu(100). We use a quasi in situ approach in an experimental set-up that combines an electrochemical cell attached to the ultrahigh-vacuum (UHV) system of a low-energy electron microscope/X-ray photoemission electron microscope (LEEM-XPEEM) operating at the BESSY II synchrotron in Berlin22. This spectro-microscope offers the possibility of locally collecting crystallographic structure and chemical information on identical areas of the sample without air exposure. These distinctive capabilities have enabled us to decipher a more detailed picture of the structural motifs responsible for the enhanced C2+ selectivity of copper surfaces under pulsed electrolysis. In particular, we have unveiled that short electrochemical anodic pulses followed by cathodic pulses in the CO2RR voltage regime not only generate Cu+ species, but also lead to an increase in the surface roughness and defect density, with preferential (n10) facet restructuring. Such defective and oxygen-rich surfaces are responsible for the enhanced ethylene and ethanol selectivities previously reported.
Results
Morphological changes after quasi in situ pulsed CO2RR
Here we investigate the reasons behind the increased catalytic selectivity of potential pulsed-treated copper catalysts towards C2+ products, which has been at least partially attributed to the presence of Cu(I) or oxygen-derived species6,11,21,23,24,25,26 or adsorbed oxygen during the CO2RR. Therefore, we selectively modify the surface of Cu(100) by subjecting it to either a cathodic static potential or to alternating short cathodic/anodic pulse potentials6,7,8,11,15,16,18,19,20,21,27,28. Although the electrochemical performance of such treatments was previously reported by our group11 (and those selectivities are also reproduced here in Supplementary Fig. 1), we focus herein on shedding light on the active structures and species formed during the dynamic operation of Cu(100) CO2RR catalysts based on laterally resolved structural and chemical state measurements.
After an initial characterization of the surface to assess the crystallographic quality and chemical purity, the sample was transferred quasi in situ in an argon atmosphere to a glass chamber with an electrochemical (EC) cell, where the CO2RR was performed (Methods) for 15 min, a time that has proved to be sufficient to ensure a similar selectivity as the 1-h reaction described in ref. 11 and Supplementary Fig. 1. After the reaction, the sample surface was rinsed with Milli-Q water and transferred back to the analysis chamber of the microscope without exposure to reactive gases. Figure 1 presents an overview of LEEM images on relatively homogeneous surface areas after various EC treatments, indicated above the corresponding microscopy images, and those of a clean pristine Cu(100). The datasets corresponding to the same anodic value differ by the ending pulse, ending either at the anodic (+0.6 VRHE or +0.8 VRHE) or cathodic working potential (−1.0 VRHE), as represented in the schematics shown above the images. The morphological changes as a function of the EC treatments can be traced in the LEEM images.

a–f, LEEM images depicting variations in the morphology, crystallinity and chemical state of a Cu(100) single crystal acquired after different CO2RR treatments performed in 0.1 M KHCO3 electrolyte consisting of either different potentiostatic (−1.0 VRHE) or different pulsed electrochemical treatments (1 s alternating −1.0 VRHE and +0.6/+0.8 VRHE, stopping either at the cathodic or anodic potentials): as-prepared catalyst (a), working catalyst after performing standard CO2RR by applying constant −1 VRHE potential (b), working catalyst after performing pulsed CO2RR between −1 VRHE and +0.6 VRHE, stopping the protocol at the cathodic pulse (c), activated catalyst after performing pulsed CO2RR between −1 VRHE and +0.6 VRHE, stopping the protocol at the anodic pulse (d), working catalyst after performing pulsed CO2RR between −1 VRHE and +0.8 VRHE, stopping the protocol at the cathodic pulse (e) and activated catalyst after performing pulsed CO2RR between −1 VRHE and +0.8 VRHE, stopping the protocol at the cathodic pulse (f). The total reaction time for all images was 15 min. Electron energy, 2.2 eV. Scale bars, 2 µm. Inset, a: LEED pattern of pristine crystal (42 eV), with an inverted linear intensity scale. The red arrows indicate the square-like objects observed after the EC treatments. Above each image there is displayed a graphical representation of the experimental tratement, if applied. U, applied potential; t, time. Cathodic/anodic ending indicates the type of last applied pulse.
By monitoring the state of the copper surface after different electrochemical protocols ending in either anodic or cathodic pulses, we were able to reveal differences between the as-prepared catalyst (pristine surface), the activated catalyst (after the anodic ending pulse) and the working catalyst (after the cathodic ending pulse). In particular, we showed that the (n10) facets formed during the pulsing procedure, and stabilized on the anodic pulse, are metastable and lose their well-defined shape during the cathodic pulse. Although the CO2RR takes place under cathodic conditions, where the surface restructures further, what leads to the surface-enhanced performance in terms of increasing the product selectivity is the mixed symbiotic state of a working dynamic catalyst being continuously subjected to regeneration and structure tuning induced by the repeated anodic pulses.
Whereas the pristine Cu(100) surface shows flat terraces separated by step bunches and step edges visible as dark lines29 (Fig. 1a), the initial state is generally preserved after applying a constant negative potential (Fig. 1b). However, one can identify various morphological features after the surface is subjected to a pulsed EC treatment (Fig. 1c–f). First, enhanced but homogeneous roughness can be observed through the presence of granular features (Fig. 1c–f). The average size and distribution of these granular features can vary as a function of the anodic/cathodic potential, duration of the individual pulses or total duration of the potentiostatic treatment. In contrast, after 15 min of a constant potential treatment (−1 VRHE), step bunches can still be observed on the surface together with point-like defects (Fig. 1b), even though the step contrast is reduced due to local roughening. Additionally, we observe the formation of square-like objects of various sizes after the pulsed EC treatments (red arrows). Several such objects are represented in Supplementary Fig. 4. In previous studies, the surface roughness of UHV-prepared Cu(100) and Cu(111) single crystals was estimated by means of the electrochemical active surface area and it was shown that despite the distinct microscopically observed surface roughness, no drastic changes are observed in the electrochemical active surface area, which was in fact similar to the geometric surface area13,14. Thus, it is evident that the changes in the selectivity observed cannot be explained purely based on roughness effects, and that more detail must be known about the atomistic transformations taking place during the CO2RR, including the formation of specific defects as featured here.
A more thorough investigation of the morphological features reveals that the anodic ending pulses (+0.6 or 0.8 VRHE) exhibit well-defined square-like features, some even presenting an inner structure. Considering that it is generally not possible to extract absolute height information from LEEM data directly, one can qualitatively identify them as pyramid-like, given also the focusing effects. However, if the pulse ends at the cathodic potential, the surface displays only smaller square islands or faded contours of square/round islands that only resemble the ones observed in the case of anodic pulses. These observations can be understood on the basis of dissolution/redeposition cycles:
with n = 1 or 2. For instance, during the anodic pulse (equation (1)) at +0.6 or +0.8 VRHE, the copper surface oxidizes, forming Cu(I) or Cu(II) species under +0.6 VRHE or +0.8 VRHE pulse potential, respectively, some of which can then get dissolved into the electrolyte. Due to the short duration of the pulse, we anticipate that hydrated Cu(n) species remain in the vicinity of the electrode surface, within the region of the electrical double layer. Consequently, during the cathodic pulse (equation (2)), Cun+ ions in solution are reduced and redeposited onto the electrode surface. Ion dissolution was previously discussed in the framework of another study via inductively coupled plasma mass spectrometry30 and observed to take place even under potentiostatic conditions for extended operation times. Here the short duration of the pluses applied makes it possible to control the dissolution/redeposition process. Dissolution and redeposition effects of copper were proven to influence not only the stability of the electrode during the electrochemical treatments, but also its restructuring, both under cathodic and anodic conditions30,31,32,33,34,35. In support for such an interpretation, we address the growth of copper nanocubes directly from dissolved ions under electrochemical conditions characterized by similar pulse treatments. Grosse et al. showed that pulsing between specific oxidative/reduction potentials can promote the dissolution of non-cubic particles and the promotion of the growth of those with preferential {100} facets33, in line with a previous observation that non-cubic particles dissolve faster than cubic ones when subjected to anodic conditons33,35. In the current case of a stable (100) surface, local surface defects may act as starting points for copper dissolution, forming etch pits with the shape of inverted pyramids with (n10) side facets being favoured at the Cu(I) oxidation state in the applied potential window. It is important to note that both dissolution and redeposition are processes whose kinetics strongly depend on the structure of the substrate, thus allowing the formation of structures along preferential directions of the Cu(001) surface (Supplementary Fig. 4). See also Supplementary Fig. 2 for a graphical representation of the surface transformation during cathodic/anodic pulsing11 and Supplementary Fig. 3 for a schematic of the atomic processes taking place, as described previously. Similarly, during a pulsed CO2RR with Cu–N–C electrocatalysts, Timoshenko et al. showed the role played by copper–substrate interactions in the dissolution of cationic copper atoms under anodic conditions and subsequent redeposition and agglomeration during the cathodic pulse, and revealed that the formation of small copper clusters was associated with a higher CH4 selectivity, whereas C2H4 was only obtained when larger NPs (2.5 nm) were present35.
Unveiling restructuring and crystal orientation
The orientation of the observed structures was detailed by comparing the LEEM morphology (Fig. 1 and Supplementary Fig. 4) with the crystal orientation described by low-energy electron diffraction (LEED) (Supplementary Fig. 5). These correlations were further complemented with additional shadowing effects observed in PEEM due to the grazing incidence of the ultraviolet/X-ray radiation36. A first remark is that the LEED (1 × 1) spots characteristic of the Cu(100) surface are preserved after all EC treatments, and after subsequent water rinsing, denoting that the crystallinity of the topmost layers of the crystal is preserved, despite the roughening observed in LEEM. Moreover, no additional reconstruction is observed, indicating no long-range order of adsorbates or even restructuring of the surface.
A comparison of the Cu(100) LEED pattern (detailed in Supplementary Fig. 5) with the morphological features discussed in Fig. 1 and Supplementary Fig. 4 reveals the general orientation of the square objects along the [001] and [010] directions, which proves that the newly formed structures adopt the substrate crystal orientation. Figure 2 displays two LEEM selections presented in comparison with their PEEM counterparts. Additionally, PEEM image simulations are provided, in which the X-ray illumination angle (20°) is considered in relation to the structure geometry. One can observe in Fig. 2, which shows representative treatments with anodic ending pulses (+0.6 VRHE, +0.8 VRHE), that the features present an inner structure that resembles the shape of a pyramid or truncated pyramid. More details on how the combined information from XPEEM and LEED can be used to extract more information about the geometry of the features are provided in Supplementary Fig. 6. For example, the shadowing effect evidenced in Fig. 2b–e and Supplementary Fig. 6 indicates the geometry represented in Fig. 2h as an inner pyramid type. Nevertheless, since the pyramid edges are oriented along the Cu(100) crystal directions, the possible configuration of pyramid facets is constrained to a variety of (n10) directions, where n is an integer number, for example, (110), (310), (510). Such facets are composed from steps with a (110) character, which might be formed by preferential dissolution during the anodic pulses.

a, LEEM image (electron energy E0 = 48 eV) after EC pulsing during the CO2RR between −1 VRHE and +0.6 VRHE, 1-s cycle ending at +0.6 VRHE. The images were obtained after experimental protocols where the ending pulse is indicated in red. Scale bar, 1 μm (applies to all images). b, XPEEM image (hν = 933 eV, E0 = 2 eV) on the same area as a displaying shadowing effects. c, Schematic representation of the observed shadowing effect for the truncated pyramid shape, based on the X-ray incidence angle and sample orientation, as observed in (b). d, LEEM image (electron energy E0 = 2.4 eV) for a sample exposed to EC pulses during the CO2RR between −1 VRHE and +0.8 VRHE, ending at +0.8 VRHE. e, XPEEM image (hν = 933 eV, E0 = 2 eV) on the same area as d displaying shadowing effects. f, Schematic representation of the observed shadowing effect for the pyramid shape, based on the X-ray incidence angle and sample orientation, as observed in (e). g, Atomistic representation of copper planes forming (n10) facets, where different atom colours represent atoms in different atomic planes. h, Graphic representation of height–angle effects in PEEM. Some areas appear brighter or darker as a consequence of the facet inclination in relation to the X-ray illumination. i, Overview cartoon of the square structures in g formed during pulsed CO2RR on the (001) surface.
Previous studies proposed that low amounts of oxygen exposure are enough to induce important morphological changes on copper surfaces and described the stability of various facets under CO2 reduction conditions32. Interestingly, in our case the (n10) facets can only be observed when we end the pulse treatments at the oxidizing anodic potential, while ending at the cathodic potential (Fig. 1c,e) resulted only in the presence of smaller square objects or diffuse-shaped features on large flat areas. The dissolution at the anodic pulse corresponds roughly to thermal deposition in UHV. Furthermore, the weaker-bound copper atoms, such as single atoms on the plane, step edges or outer kink sites, will be dissolved first and the more strongly bound atoms will remain. Therefore, this selective dissolution leads to a faceting where only the most stable facets survive. At the cathodic pulse, all or a part of the dissolved copper will be redeposited onto the surface, whereas the copper does not necessarily adsorb at the surface site from which it was just desorbed. Instead, it lands somewhere on the plane, from which it diffuses on the surface. At the end of the diffusion process it can either be incorporated in an existing substrate step edge, leading to a step flow growth, or agglomerate with a second or more diffusing adatoms, forming a stable cluster, which can subsequently grow into a two-dimensional island. However, the limited diffusion across the step edges (due to the Ehrlich–Schwoebel barrier) is expected to lead to a mound-like growth (three-dimensional). All these growth modes, well known in UHV, are also observed by X-ray diffraction (XRD) in the liquid phase37,38. Additionally, due to the applied electric field, the surface morphology itself might influence the adsorption process; for example, a tip on the surface might enhance the local electric field, leading to a preferred growth of the tip, which would also explain the irregular growth observed in ref. 34. Thus, the redeposition of copper during the cathodic pulse occurs quasi-homogeneously, and due to the limited diffusivity at room temperature the copper atoms appear to redeposit at mounds, partially refilling the pyramidal voids generated during the anodic pulse. Nevertheless, providing a definitive explanation of the observed effect would require an in-depth theoretical exploration of the restructuring pathways, taking into consideration not only the surface structure, but also the electrolyte, the potential pulses, and considering also the potassium cations. The calculations would need to consider possible cathodic corrosion effects and would also need to incorporate the electric fields that we are changing by applying the potential pulses. Although such calculations could provide a notable contribution to the understanding of the observation, they are subjected to computational and time limitations and are outside the scope of this experimental investigation. Noteworthy in this regard is the operando electrochemical scanning tunnelling microscopy work of Kim et al., in which surface restructuring was demonstrated on Cu(100), Cu(111) and a polycrystalline (pc) copper electrode, both at constant potential −0.9 VSHE (standard hydrogen electrode (SHE)) in 0.1 M KOH, but also as a consequence of oxidation–reduction cycles39. The study points out that Cu(pc)–[Cu(111)] and Cu(pc)–[Cu(100)] facets can develop upon applying a constant potential and that Cu(pc)–[Cu(100)] can further restructure to Cu(pc)–[Cu(511)] during oxidation–reduction cycles. The former surfaces were shown to produce ethanol by differential electrochemical mass spectrometry.
Importantly, as previously discussed, even for samples only exposed to potentiostatic conditions40, the faceting that we have now intentionally generated under pulsed electrocatalytic conditions probably results in enhanced C2+ product generation11. Timoshenko et al. showed various possibilities for Cu nanocube restructuring under different pulsing conditions, and correlated the different structures achieved with distinct product selectivities. Moreover, such restructuring was observed even after longer constant cathodic potentials, indicating the possibility that cathodic corrosion occurred17. Although we do not yet have direct proof that such effects would manifest also for our system in a more practical environment, for example, in a gas diffusion electrode configuration, previous operando XAS experiments on a different nanocrystalline-copper-based system revealed behaviour similar to that observed in an H-cell, with drastic surface restructuring accompanied by the regeneration of Cu(I) and Cu(II) species. Nonetheless, accelerated changes in the material were observed under the higher current density operation conditions and distinct concentrations of the different oxide species41.
The presence of residual Cu(I) species under these conditions might also play a role, as will be discussed below. It should be noted that recent reports have identified the (100)–(110) interface as responsible for the higher selectivity for C2+ products, in particular, ethanol production42,43. Moreover, Zhu et al. reported that not only is the crystallographic orientation of the terrace–step interface important, but also the step density. Thus, the pyramid-like structures composed of (n10) facets presented in Fig. 2 might be the most active areas of the electrode surface for CO2 to hydrocarbon and alcohol conversion43. In fact, previous results from Scholten et al. and Nguyen et al. showed that in the case of copper the orientation of the pristine electrode is not necessarily responsible for the catalyst performance, but that one needs to consider the possible pathways of defect formation and restructuring that manifest in the presence of the electrolyte, during the reaction and after several CO2RR cycles. Both these previous studies revealed that the pristine surfaces produce mostly H2, regardless of the (100) or (111) orientation of the precatalyst13,14. Interestingly, after a Cu(111) surface was subjected to several CO2RR cycles at a constant cathodic potential, features in the cyclic voltammetry (CV) curves of the then hydrocarbon-selective surface could be related to the presence of newly formed (110) facets14. Such results show that the initial state of the copper single-crystal surface might not play an important role in the selectivity trends towards C2+ products, but that one should consider the new surface structures that are formed during the CO2RR. In fact, our present data suggest that roughness is not the key parameter to consider because for initially flat single-crystal surfaces characterized by low relative roughness differences, very distinct product selectivities can be obtained, depending on the specific defects that are formed during operation. A high defect density has been proven to be important to produce hydrocarbons in detectable amounts13,14, but even more critical are the specific characteristics of such defects, such as the step orientations, which might evolve during extended CO2RRs. Roughness factors were previously estimated through capacitance measurements, but only small differences were found between the treatments with pulsing to anodic Ea = +0.6 VRHE (roughness factor, 1.0), in comparison to pulsing to Ea = +0.8 VRHE (roughness factor, 1.1), which indicated that there is no statistically notable difference in roughness between different treatments11. It should be mentioned that the average surface roughness alone is an overly simplistic descriptor for the characterization of the active species in copper surfaces during the CO2RR, and that specific facets or steps that must be either available in the precatalyst surface and stabilized during the CO2RR, or formed during the CO2RR, are key for an enhanced carbon–carbon coupling. Here, the application of potential pulses leads to the regeneration of the active morphological features. Nonetheless, it is not only the density of steps that matters, but also the step orientation on the surface. Additional complexity is added by the fact that both of these can also change during the CO2RR even under potentiostatic reaction conditions14.
Spectroscopic analysis of the final state of CO2RR pulsed treatments
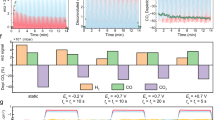
The chemical state of the surface was investigated, with spatial resolution, by near-edge X-ray absorption spectroscopy (NEXAFS) at the Cu L3 edge. Figure 3a–c displays NEXAFS spectra produced by intensity integration of a series of XPEEM images recorded with the signal of the secondary electrons (E0 ≈ 2 eV) scanned over the given photon energy range. Although this spectro-microscope makes it possible to distinguish areas manifesting different NEXAFS fingerprints with a spatial resolution down to 18 nm, we focus here on the spectra obtained from the intensity over the entire field of view (that is, 20 µm) to describe the chemical state of the entire surface. We note that this was done because no clear NEXAFS variations were observed when the square structures were investigated separately, indicating they were in a similar chemical state to the surrounding surface. Because the spectra are extracted from XPEEM images that are subjected to an energy gradient44, an uncertainty of ∼0.2 eV in the energy position occurs due to X-ray beam position shift. The difference between the spectra measured after various experimental stages was quantified by principal component analysis (PCA) combined with a multivariate curve resolution (MCR) approach as implemented in the PyFitIt code45. With this method we were able to identify two main species contributing to our experimental NEXAFS spectra (Fig. 3d). The first component corresponds to the spectrum of the pristine state, and has been assigned to the Cu(0) metal signal, characteristic of the pristine state of the crystal. The second spectral component was attributed to a cationic copper in a Cu2O-like phase, namely Cu(I) species. The contribution of this species depends strongly on the nature of the pulsing EC treatment. Supplementary Table 1 presents the concentrations of Cu(0) and Cu(I) extracted from the PCA model.

a–c, NEXAFS spectra (Cu L3 edge) recorded after different stages of the sample preparation, potentiostatic and pulsed CO2RR: as-prepared and working catalyst (a), activated catalyst (b) and working catalyst (c). d, The NEXAFS components corresponding to two different chemical environments extracted from the PCA analysis. e, Composition of the two components, metallic copper and Cu2O, extracted from the PCA depending on the different EC treatments. f, Schematics of the concentration profiles describing the experimental composition in e. The numerical values for the estimated thickness were obtained based on the model described in Supplementary Note 1.
We note here that the PCA/MCR approach does not suggest the presence of Cu(II) species in substantial amounts in any of our samples. Although the presence of Cu(II) was observed in nanoparticulate samples during operando XAS measurements17, we note that the concentration of those species was always very small in comparison with that of copper metal or Cu(I) species.
Focusing on the evolution of the copper oxidation state under EC treatments, Fig. 3e shows the changes in the concentration of the oxide phase. It can be seen that even in the chronoamperometry experiment, which involves a constant reducing potential, a small contribution of ~3% Cu2O is detected. The small difference between the pristine and the constant −1 VRHE experiment could arise from various adsorbates present on the surface or even introduced by the water rinsing, a step performed before the sample transfer in UHV to eliminate electrolyte residues from the surface. This 3% Cu2O is considered the maximum error of our experiments, and clearly smaller than the values obtained under the cathodic pulse potential after the anodic pulse oxidation.
Using such an approach, short potential pulses of ∆t = 1 s are supposed to regenerate Cu(I) ions which, in conjunction with metallic copper regions, have been suggested to enhance the C2+ product selectivity6,8,9,11,24,46. Thus, sequential pulsing between reducing and oxidizing potentials changes both the morphology of the surface through roughening11,12 and local faceting, and the chemical state, resulting in the build-up of a gradient of chemical species, for example, Cu(I)–Ox, in the topmost layers of the crystal. It should be noted that we do not observe local inhomogeneities regarding preferential localization of oxygen-rich areas in either of the experiments. The square objects do not show a different NEXAFS spectrum than the surrounding areas, and no other chemically inhomogeneous regions could be identified. This indicates that there is a gradient in the oxygen concentration subsurface, below the metallic surface, and that any such oxygen reservoir created during the oxidative anodic pulse decreases in concentration but does not disappear during the subsequent short cathodic pulse. Thus, our data reveal that one must consider the modifications of the properties of the copper surface by such subsurface oxygen species. Nevertheless, the ending applied pulse, that is, a reducing or oxidizing potential, determines whether notable oxidation takes place and partially remains even after the short (1 s) cathodic pulse is applied. The spectral decomposition procedure, introduced in the case of pulsed EC treatments, illustrates this aspect: while the pulsed sequences finishing at the cathodic potential (−1.0 VRHE) show a Cu2O contribution of 10.5% ± 0.5%, irrespective on the value of the oxidizing pulse, which is higher than that under constant applied −1 VRHE (maximum 3.3% Cu2O, possibly assigned to reoxidation from water rinsing in the quasi in situ approach), with treatments ending at the oxidizing pulse, a higher value is obtained, namely, 23% Cu2O for +0.6 VRHE pulses and 34% Cu2O for 0.8 VRHE pulses. Similar values were reported by Aran-Ais et al., as detected by X-ray photoelectron spectroscopy (XPS)11, namely, 16% Cu2O after pulsing to 0.6 VRHE, and 20% Cu2O + 4% CuO after EC pulsing at +0.8 VRHE. A maximum error of 5% from the calculated percentage is considered as an uncertainty. These variations of the NEXAFS fingerprints do not necessarily represent a degree of surface oxidation, but may also be an indication of possible subsurface oxygen dispersed under the first layers of the surface27,47. It is commonly accepted that the NEXAFS technique is more bulk sensitive than conventional XPS48, in particular in the case when the measurement is performed in total electron yield (TEY) or secondary electron yield (SEY), as in the current dataset. Although the electron yield increases the surface sensitivity, due to the limitation induced by the electron inelastic mean free path (λ, IMFP), one can estimate values of λ = 3 nm (±20%) for the electrons with a kinetic energy of ∼2–3 eV selected with an energy analyser at the maximum of the secondary electron onset28. Therefore, after the anodic pulses, one can estimate a range of at least 3 × 3 nm ≈ 9 nm (3 × IMFP at a certain KE) below the surface as being enriched with oxygen-derived copper26 based on the data of Fig. 3. More importantly, Fig. 3e also revealed that ending subsequently (after a pulse treatment) at the cathodic pulse (1 s) does not result in a completely metallic copper surface. The different IMFP values for the different techniques could also explain the small variation in the estimated concentration between the previous report11 and the current results.
The selectivity results11 (Supplementary Fig. 1) suggest that the surface is more selective towards C2+ products during pulsing treatments between −1 VRHE and +0.6 VRHE, and it is worth mentioning that we observe a similar evolution of the surface structure with both anodic pulses (+0.6 VRHE and +0.8 VRHE), meaning that both treatments produce similar (n10) facets. In addition, both treatments result in an increase in the Cu(I) concentration, with this content being even larger in the case of Ea = +0.8 VRHE. However, the later anodic pulses also generate simultaneously Cu(II) species, which are less stable and more prone to dissolution in the electrolyte used, as demonstrated by ICP-MS30. Based on the data reported by Speck et al., one can estimate a value of 0.5 ng s−1 cm−2 in the case of a pulse at +0.6 VRHE in a pH 6.8 electrolyte, like CO2-saturated KHCO3. Moreover, not only is the specific content of Cu(I)/Cu(II) species in the catalyst important, but also the degree of atomic order of such species, which strongly depends on the reaction conditions used, including the selected anodic and cathodic potentials and the duration of the applied potential pulses17. In fact, it has been shown that the formation of bulk-like ordered oxides during pulsed CO2RR does not lead to C2 but to C1 products17. Thus, it is plausible that the better C2 selectivity of our Cu(100) surface following treatment at the lower anodic potential (Ea = +0.6 VRHE) might be due to the more disordered structure of the copper oxides formed.
Finally, one can use the PCA/MCR-derived concentration variation to estimate a quantitative thickness of the oxide species after different EC treatments. Based on the escape depth of the electrons during the photoemission process49, and assuming that the oxidation/reduction always starts from the topmost layer of the surface and processes towards the bulk, we can build a layer model displayed in Fig. 3f, to which we apply a numerical estimate of the thickness (see Supplementary Note 1 for further details). It is worth mentioning that we can exclude the possibility of laterally coexisting Cu(0) and Cu(I) domains because we observed microscopically a homogeneously oxidized surface. Thus, we conclude that we have here subsurface oxygen-rich layers, with a thickness and oxygen gradient concentration that strongly depend on the specific pulse parameters used during the CO2RR. A multistacking of Cu(0) and Cu(I) layers was reported by Grosse et al.33 during the electrochemical growth of Cu2O cubes from a 5 mM CuSO4 solution via pulsed electrodeposition. In our case we do not have a net growth of the copper surface due to periodic dissolution and redeposition phenomena. As indicated already by the PCA analysis, the constant −1 VRHE treatment and the two pulsing treatments ending at the anodic potential leave behind a surface enriched with an oxide-like layer, the thickness of which depends on the nature of the treatment. While applying consecutive pulses, one possibility is that the strength of the oxidation and reduction is not the same; that is, reduction might occur without reducing completely the oxide layer formed within the previous pulse. Therefore, in the case of treatments ending with cathodic pulses, the signal originating from the oxide component would be dampened by a metallic layer formed on top. An estimate of the layer thickness developed in these cases can be obtained based on the concentrations given by the PCA analysis; this procedure is detailed in Supplementary Note 1 and presented in Supplementary Table 2. With the −1 VRHE treatment, we determine a thickness of 0.1 nm corresponding to the 3% Cu2O given by the PCA analysis; this value must represent a dispersed amount of species, most probably generated during the rinsing stage after the EC treatment. Nevertheless, the 0.6 VRHE and 0.8 VRHE anodic 1-s pulses produce Cu2O layers with thicknesses of 1.0 nm and 1.3 nm, respectively, which are only partially reduced if they end at the cathodic potential. From the analysis shown in Fig. 3f,e, it also appears that after the two different pulse treatments at +0.6 VRHE and +0.8 VRHE, and subsequent reduction (1 s) at −1.0 VRHE, a sublayer of Cu2O-like species 0.6–0.8 nm thick stills remains, probably covered by a 0.4-nm to 0.5-nm layer of metallic copper. Such a situation occurs in the case of cathodic pulses, when a reduction occurs from the outer layers of a previously formed oxide during the anodic pulses.
Conclusions
With our comprehensive approach combining microscopy, diffraction and spectroscopy techniques together with quasi in situ electrochemistry, we were able to fill the gap in understanding the reactivity of pulsed Cu(100) surfaces towards the formation of multicarbon products (C2+). Based on LEED and LEEM/PEEM measurements we demonstrated that the pulsing EC treatments induce a restructuring of the surface leading to the formation of truncated pyramids. We were able to identify the crystallographic orientation of the side facets of these pyramids as belonging to the (n10) family, previously predicted to be the most active facets for CO2RR conversion to ethylene and ethanol. Furthermore, our spectroscopic analysis unveiled the presence of oxygen/Cu(I) species near the surface of anodically pulsed Cu(100), even after a subsequent cathodic pulse. The resilience of such species against reduction is probably linked to the previous enhanced ethanol production reported for pulsed CO2RR under oxidative dynamic cycles. In summary, our work highlights that a combination of Cu(I)/Cu(0) species, together with specific (n10) crystallographic faceting of the surface are present under the pulse reaction conditions that have been shown optimum to facilitate carbon–carbon coupling, thereby resulting in an enhanced CO2RR selectivity for C2 products. Although EC pulsing protocols have previously been employed to improve the activity and selectivity of copper catalysts for C2+ products such as ethylene or ethanol, our results indicate that EC pulsing can also be used to selectively create and regenerate specific morphological features (that is, specific steps or facet orientations) that have been theoretically proposed as key for enhancing carbon–carbon coupling in the CO2RR. In this way, the working catalyst is trained during the reaction to generate specific active sites with an increased yield for specific C2+ products. Such insight can be extrapolated to other catalysts and electrochemical reactions, where specific facets are known to be more active or selective for a given desired product.
Methods
Sample preparation
Cu(100) single crystals (MaTeck) were cleaned in UHV through extensive sputtering and annealing cycles up to 600 °C, until flat terraces were observed in LEEM mode and a sharp background-free LEED was achieved. The sample was then transferred from UHV into a quasi in situ environment in an additional chamber that can be vented with argon (99.999%) and further transferred to a glass-cross chamber purged continuously with argon, where the EC cell is located. The transfer conditions have been extensively checked so that the transfer does not induce any exposure of the sample to air or any reactive gases.
EC treatment
The reactions were carried out in a customized tube-like EC cell made of Teflon using a conventional three-electrode set-up, where the copper crystal, as the working electrode, is softly pressed against a hole at the end of the EC tube. A platinum mesh and a leak-free Ag/AgCl electrode (LF-1, Innovative Instruments) were used as the counter-electrode and the reference electrode, respectively. The electrolyte used was 0.1 M KHCO3 (ACS reagent 99.7%, Sigma-Aldrich). Prior to usage, the electrolyte was treated with Chelex (Bio-Rad Laboratories) to remove trace metal impurities50. Before connecting the working electrode to the cell, the electrolyte was purged with CO2 gas (99.9995%, Air Liquide) for 30 min until a pH of 6.8 was reached. The reaction was investigated at different potentials either by chronoamperometry or by varying the applied potential between two values corresponding to oxidative or reductive conditions. After the EC treatment, the sample was quickly disconnected from the circuit and rinsed with water (Milli-Q, 18.2 MΩ) to remove the electrolyte from the surface. The water was purged for at least 30 min with argon before the rinsing procedure, to remove dissolved O2 molecules by saturation with argon. For more details and a scheme of the experimental design, see Supplementary Note 2 and Supplementary Fig. 8.
In this work, all potentials are referred to the reversible hydrogen electrode and labelled as VRHE. Here, use as-prepared to refer to the surface state of the pristine Cu(100) single crystal after sputter/annealing treatment, while the activated state corresponds to the same surface after a 15-min pulse CO2RR treatment ending at the anodic pulse (either +0.6 VRHE or +0.8 VRHE). The working catalyst label describes the copper surface after ending the CO2RR pulses at the cathodic pulse (−1 VRHE).
Characterization
The EC cell is mounted on a modified set-up attached to a preparation chamber of the LEEM/XPEEM (spectro-microscope with aberration correction for relevant techniques, SMART) microscope51, operated at the UE49PGM beamline at the BESSY II Synchrotron light source of the HZB in Berlin, Germany. This instrument enables rapid acquisition of LEEM, LEED, XPS and XPEEM, with a lateral resolution as good as 2.6 nm in LEEM and 18 nm in XPEEM mode and with an energy resolution of 150 meV. A number of other methods can also be implemented using the same instrument22. After the EC treatment is performed, the sample is transferred in an argon flow atmosphere to an intermediate UHV chamber, where it is pumped down to 10−6–10−7 mbar, and further transferred to UHV through a series of chambers until it finally gets located at the measuring position, in a low pressure of 10−9 mbar. Due to the ability to switch rapidly between different measurement modes in the SMART microscope, one can perform local characterization of a surface in a complementary manner by visualizing the real or the reciprocal space, and by capturing the spectroscopic fingerprint of the investigated areas. Microscopy can be achieved either by illuminating the sample surface with electrons from an electron gun and using the reflected electron to form an image, that is, LEEM mode, or by illuminating with X-rays and further detecting photoemitted electrons within a certain KE range, that is, XPEEM mode. Considering the low KE typically used in the SMART microscope (2–1,000 eV), the surface sensitivity in our experiments can be tuned to between 0.3 and 2 nm for XPS measurements. The secondary electrons usually involved in PEEM-NEXAFS measurements (SEY) are located at energies of 2–3 eV above the vacuum level (that is, about 5–7 eV above the Fermi level), a range that has been the subject of recent debates regarding the effective IMFP of very-low-energy electrons52,53. The photons reach the surface at a grazing angle of 20°. Most synchrotron-based NEXAFS measurements are performed in TEY mode, which is more sensitive to the bulk properties, and there has been little discussion of possible differences in the NEXAFS spectra when measured in distinctly different energy ranges because the secondary electrons should also be dominant in TEY mode.
The spectra decomposition of the NEXAFS dataset, composed of six spectra shown in Fig. 3a–c, was realized by using the PyFitIt code45. The Malinowski IND factor54 was used to identify the number of principal components (PCs) sufficient to reproduce all the major intensity variations between different NEXAFS spectra. As can be seen from Supplementary Fig. 7, the latter shows a minimum in proximity to the second PC, indicating the presence of only two main chemical species responsible of the collected dataset.
The first two PCs extracted from the NEXAFS spectra were then linearly combined to form a new set of chemically/physically meaningful spectra corresponding to the spectra of pure species. For this purpose, we used the transformation matrix approach45,55, where we applied the following constraints56: the spectra for pure species should be non-negative and normalized. Furthermore, the weights (concentrations) of these two species in all experimentally acquired NEXAFS spectra must be non-negative and should sum to 1. Finally, we required that the spectrum for the first pure species (metallic copper) should match the experimental NEXAFS spectrum for our sample in its pristine state.
Although across the manuscript the concepts of oxidation state (Cu(0), Cu(I), Cu(II)) and chemical species (Cu, Cu2O, CuO) are both used, we note here that these refer to the same concept.
Data availability
The raw data consist of hierarchical data format (hdf) stacks, which require specialized software to process. The data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Chancel, L., Picketty, T. Carbon and Inequality: From Kyoto to Paris. Trends in the Global inequality of Carbon emissions (1998–2013) & Prospects for an Equitable Adaptation Fund (Paris School of Economics, 2015).
Gattrell, M., Gupta, N. & Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 594, 1–19 (2006).
Gao, D., Arán-Ais, R. M., Jeon, H. S. & Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2, 198–210 (2019).
Karapinar, D., Creissen, C. E., Rivera de la Cruz, J. G., Schreiber, M. W. & Fontecave, M. Electrochemical CO2 reduction to ethanol with copper-based catalysts. ACS Energy Lett. 6, 694–706 (2021).
Simon, G. H., Kley, C. S. & Roldan Cuenya, B. Potential-dependent morphology of copper catalysts during CO2 electroreduction revealed by in situ atomic force microscopy. Angew. Chem. Int. Ed. 60, 2561–2568 (2021).
Chou, T. C. et al. Controlling the oxidation state of the copper electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J. Am. Chem. Soc. 142, 2857–2867 (2020).
Velasco-Vélez, J.-J. et al. The role of the copper oxidation state in the electrocatalytic reduction of CO2 into valuable hydrocarbons. ACS Sustain. Chem. Eng. 7, 1485–1492 (2018).
Velasco-Velez, J. J. et al. Revealing the active phase of copper during the electroreduction of CO2 in aqueous electrolyte by correlating in situ X-ray spectroscopy and in situ electron microscopy. ACS Energy Lett. 5, 2106–2111 (2020).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016).
Moller, T. et al. Electrocatalytic CO2 reduction on CuOx nanocubes: tracking the evolution of chemical state, geometric structure, and catalytic selectivity using operando spectroscopy. Angew. Chem. Int. Ed. 59, 17974–17983 (2020).
Arán-Ais, R. M., Scholten, F., Kunze, S., Rizo, R. & Roldan Cuenya, B. The role of in situ generated morphological motifs and Cu(I) species in C2+ product selectivity during CO2 pulsed electroreduction. Nat. Energy 5, 317–325 (2020).
Gauthier, J. A., Stenlid, J. H., Abild-Pedersen, F., Head-Gordon, M. & Bell, A. T. The role of roughening to enhance selectivity to C2+ products during CO2 electroreduction on copper. ACS Energy Lett. https://doi.org/10.1021/acsenergylett.1c01485 (2021).
Scholten, F., Nguyen, K.-L. C., Bruce, J. P., Heyde, M. & Roldan Cuenya, B. Identifying structure–selectivity correlations in the electrochemical reduction of CO2: a comparison of well-ordered atomically clean and chemically etched copper single-crystal surfaces. Angew. Chem. Int. Ed. 60, 19169–19175 (2021).
Nguyen, K. C. et al. The influence of mesoscopic surface structure on the electrocatalytic selectivity of CO2 reduction with UHV-prepared Cu(111) single crystals. ACS Energy Lett. 9, 644–652 (2024).
Cheng, D. et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 12, 395 (2021).
Velasco-Velez, J. J. et al. On the activity/selectivity and phase stability of thermally grown copper oxides during the electrocatalytic reduction of CO2. ACS Catal. 10, 11510–11518 (2020).
Timoshenko, J. et al. Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses. Nat. Catal. 5, 259–267 (2022).
Cavalca, F. et al. Nature and distribution of stable subsurface oxygen in copper electrodes during electrochemical CO2 reduction. J. Phys. Chem. C 121, 25003–25009 (2017).
Fields, M., Hong, X., Nørskov, J. K. & Chan, K. Role of subsurface oxygen on Cu surfaces for CO2 electrochemical reduction. J. Phys. Chem. C 122, 16209–16215 (2018).
Garza, A. J., Bell, A. T. & Head-Gordon, M. Is subsurface oxygen necessary for the electrochemical reduction of CO2 on copper? J. Phys. Chem. Lett. 9, 601–606 (2018).
Favaro, M. et al. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6706–6711 (2017).
Schmidt, T., Sala, A., Marchetto, H., Umbach, E. & Freund, H. J. First experimental proof for aberration correction in XPEEM: resolution, transmission enhancement, and limitation by space charge effects. Ultramicroscopy 126, 23–32 (2013).
Dattila, F., Garcı́a-Muelas, R. & López, N. Active and selective ensembles in oxide-derived copper catalysts for CO2 reduction. ACS Energy Lett. 5, 3176–3184 (2020).
Zhou, Q., Zhang, W., Qiu, M. & Yu, Y. Role of oxygen in copper-based catalysts for carbon dioxide electrochemical reduction. Mater. Today Phys. 20, 100443 (2021).
He, M. et al. Oxygen induced promotion of electrochemical reduction of CO2 via co-electrolysis. Nat. Commun. 11, 3844 (2020).
Lian, Z., Dattila, F. & López, N. Stability and lifetime of diffusion-trapped oxygen in oxide-derived copper CO2 reduction electrocatalysts. Nat. Catal. 7, 401–411 (2024).
Eilert, A. et al. Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction. J. Phys. Chem. Lett. 8, 285–290 (2017).
Lee, S. H. et al. Oxidation state and surface reconstruction of Cu under CO2 reduction conditions from in situ X-ray characterization. J. Am. Chem. Soc. 143, 588–592 (2021).
Kunze, S. et al. Plasma-assisted oxidation of Cu(100) and Cu(111). Chem. Sci. 12, 14241–14253 (2021).
Speck, F. D. & Cherevko, S. Electrochemical copper dissolution: a benchmark for stable CO2 reduction on copper electrocatalysts. Electrochem. Commun. 115, 106739 (2020).
Popovic, S., Bele, M. & Hodnik, N. Reconstruction of copper nanoparticles at electrochemical CO2 reduction reaction conditions occurs via two-step dissolution/redeposition mechanism. ChemElectroChem 8, 2634–2639 (2021).
Raaijman, S. J., Arulmozhi, N. & Koper, M. T. M. Morphological stability of copper surfaces under reducing conditions. ACS Appl. Mater. Interfaces 13, 48730–48744 (2021).
Amirbeigiarab, R. et al. Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions. Nat. Catal. 6, 837–846 (2023).
Grosse, P., Yoon, A., Rettenmaier, C., Chee, S. W. & Cuenya, B. R. Growth dynamics and processes governing the stability of electrodeposited size-controlled cubic Cu catalysts. J. Phys. Chem. C 124, 26908–26915 (2020).
Timoshenko, J. et al. Reversible metal cluster formation on nitrogen-doped carbon controlling electrocatalyst particle size with subnanometer accuracy. Nat. Commun. 15, 6111 (2024).
Schmidt, T. et al. Optical layout of a beamline for photoemission microscopy. J. Synchrotron Radiat. 6, 957–963 (1999).
Krug, K., Stettner, J. & Magnussen, O. M. In situ surface X-ray diffraction studies of homoepitaxial electrochemical growth on Au(100). Phys. Rev. Lett. 96, 246101 (2006).
Magnussen, O. M. et al. In situ and operando X-ray scattering methods in electrochemistry and electrocatalysis. Chem. Rev. 124, 629–721 (2024).
Kim, Y.-G. et al. Surface reconstruction of pure-Cu single-crystal electrodes under CO-reduction potentials in alkaline solutions: a study by seriatim ECSTM-DEMS. J. Electroanal. Chem. 780, 290–295 (2016).
Hori, Y., Takahashi, I., Koga, O. & Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A 199, 39–47 (2003).
Jeon, H. S. et al. Selectivity control of Cu nanocrystals in a gas-fed flow cell through CO2 pulsed electroreduction. J. Am. Chem. Soc. 143, 7578–7587 (2021).
Bagger, A., Ju, W., Varela, A. S., Strasser, P. & Rossmeisl, J. Electrochemical CO2 reduction: classifying Cu facets. ACS Catal. 9, 7894–7899 (2019).
Zhu, C. et al. Enhanced electrochemical CO2-to-C2+ conversion from synergistic interaction between terrace and step sites on monocrystalline high-index Cu facets. J. Energy Chem. 70, 382–387 (2022).
Günther, S. et al. Versatile procedure for the correction of non-isochromatism in XPEEM spectroscopic imaging. Ultramicroscopy 250, 113756 (2023).
Martini, A. et al. PyFitit: the software for quantitative analysis of XANES spectra using machine-learning algorithms. Comput. Phys. Commun. 250, 107064 (2020).
Hahn, C. et al. Engineering Cu surfaces for the electrocatalytic conversion of CO2: controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl Acad. Sci. USA 114, 5918–5923 (2017).
Wang, H.-Y. et al. Direct evidence of subsurface oxygen formation in oxide-derived Cu by X-ray photoelectron spectroscopy. Angew. Chem. Int. Ed. 61, e202111021 (2022).
Jiang, P. et al. Experimental and theoretical investigation of the electronic structure of Cu2O and CuO thin films on Cu(110) using X-ray photoelectron and absorption spectroscopy. J. Chem. Phys. 138, 024704 (2013).
Hüfner, S. Photoelectron Spectroscopy: Principles and Applications (Springer, 2013).
Hall, A. S., Yoon, Y., Wuttig, A. & Surendranath, Y. Mesostructure-induced selectivity in CO2 reduction catalysis. J. Am. Chem. Soc. 137, 14834–14837 (2015).
Schmidt, T. et al. Double aberration correction in a low-energy electron microscope. Ultramicroscopy 110, 1358–1361 (2010).
Bourke, J. D. & Chantler, C. T. Measurements of electron inelastic mean free paths in materials. Phys. Rev. Lett. 104, 206601 (2010).
Ridzel, O. Y., Astasauskas, V. & Werner, W. S. M. Low energy (1–100 eV) electron inelastic mean free path (IMFP) values determined from analysis of secondary electron yields (SEY) in the incident energy range of 0.1–10 keV. J. Electron Spectrosc. 241, 146824 (2020).
Malinowski, E. R. Theory of error for target factor analysis with applications to mass spectrometry and nuclear magnetic resonance spectrometry. Anal. Chim. Acta 103, 339–354 (1978).
Smolentsev, G., Guilera, G., Tromp, M., Pascarelli, S. & Soldatov, A. V. Local structure of reaction intermediates probed by time-resolved X-ray absorption near edge structure spectroscopy. J. Chem. Phys. 130, 174508 (2009).
Martini, A. & Borfecchia, E. Spectral decomposition of X-ray absorption spectroscopy datasets: methods and applications. Crystals 10, 664 (2020).
Acknowledgements
This work was funded by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) under grant number 03EW0015B (CatLab). L.d.S.C. is grateful for the funding of the DFG under Germany’s Excellence Strategy–EXC 2008–390540038–UniSysCat. We also thank our engineers M. Springer and S. Pohl for their support and the Helmholtz Center Berlin for Materials and Energy (HZB) for the allocation of synchrotron radiation beam time.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
All authors contributed to the development of the electrochemical cell set-up at the SMART microscope. L.C.T., M.J.P., L.d.C.S., A.T., F.S., P.G. and T.S. performed the sample preparation and electrochemical experiments. L.C.T., M.J.P., L.d.C.S., A.T. and T.S. analysed the results. A.M. and J.T. performed the PCA analysis of the NEXAFS data. L.C.T. wrote the manuscript with support from M.J.P., T.S. and B.R.C. B.R.C. co-designed the experiments and guided and supervised the project. All authors discussed the results, agreed with the content and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–8, Tables 1 and 2, and Notes 1 and 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tănase, L.C., Prieto, M.J., de Souza Caldas, L. et al. Morphological and chemical state effects in pulsed CO2 electroreduction on Cu(100) unveiled by correlated spectro-microscopy. Nat Catal 8, 881–890 (2025). https://doi.org/10.1038/s41929-025-01387-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41929-025-01387-6
