
Abstract
Replication stress refers to slowing or stalling of replication fork progression during DNA synthesis that disrupts faithful copying of the genome. While long considered a nexus for DNA damage, the role of replication stress in aging is under-appreciated. The consequential role of replication stress in promotion of organismal aging phenotypes is evidenced by an extensive list of hereditary accelerated aging disorders marked by molecular defects in factors that promote replication fork progression and operate uniquely in the replication stress response. Additionally, recent studies have revealed cellular pathways and phenotypes elicited by replication stress that align with designated hallmarks of aging. Here we review recent advances demonstrating the role of replication stress as an ultimate driver of cellular senescence and aging. We discuss clinical implications of the intriguing links between cellular senescence and aging including application of senotherapeutic approaches in the context of replication stress.
Similar content being viewed by others


Introduction
Replication stress can be caused by an endogenous or environmental condition that disrupts the faithful copying of the genome (Table 1)1. In addition to metabolites, drugs, and radiation that can introduce nuclear or mitochondrial genomic DNA damage and interfere with replication-associated processes, genetic deficiencies can perturb replicative DNA synthesis or fork stability. Replication fork stalling and demise contribute profoundly to genomic DNA damage and cellular senescence characterized by irreversible replication arrest2. Understanding the molecular-genetic defects and environmental causes of replication stress is paramount to characterizing the mechanism of aging, determining biomarkers for aging, and developing new treatment strategies and cures for age-related diseases. Cells of neoplastic tissues experiencing replication stress must be refreshed with nutrient macromolecules so they can perform vital functions. Alternatively, to suppress aging, senescent cells are cleared and replaced with newly divided and differentiated cells. Thus, replication stress as a driver of cellular senescence and aging is critically important to characterize.
The dire consequences of a poor response to replication stress are clinically apparent in genetically inherited premature aging disorders3. A common feature of these diseases is chromosomal instability, prompting an investigation of the cellular pathways responsible for a compromised response to replication stress. Endogenous or exogenously induced DNA damage derails normal replicative DNA synthesis and contributes to aging at the cellular and tissue-specific levels.
Although replication stress has long been considered a nexus for DNA damage, its role in aging is under-appreciated. We describe phenotypes and pathways evoked by replication stress that align with hallmarks of aging. Furthermore, many age-related diseases are characterized by compromised replicative DNA synthesis and chromosomal instability, sparking renewed interest in the search for biomarkers of aging and potential targets to ameliorate phenotypes associated with replication stress that might translate to clinical therapies (e.g., senotherapeutics4).
Replication stress contributes to the dysregulation of many hallmarks of aging, which are extensive5. Difficulty in replicating chromosome ends causes telomere attrition or telomeric DNA damage that perturbs binding of the protective shelterin complex, commonly observed during cellular senescence6. Depletion of the stem cell (SC) reservoir by replication stress promotes decline of tissues and organs in a cell lineage-dependent manner7. For example, replication stress drives functional loss in old hematopoietic SCs8. Inducible double-strand breaks (DSBs) caused mouse liver aging9; these highly toxic lesions are associated with cellular senescence and apoptosis10 and potentially trigger the senescence-associated secretory phenotype (SASP) that mediates intercellular signaling and is a representative feature of aging cells11. Moreover, even a low level of inducible DSBs in a transgenic mouse model was found to cause epigenetic changes and dysfunctional transcription networks associated with age-related phenotypes12. Genetic backgrounds or cellular conditions conducive to perturbed replicative DNA synthesis display abnormal epigenetic regulation which affects transcription and histone recycling13. Moreover, retrotransposable elements that alter chromatin state can lead to a sustained inflammatory response14,15.
Replication stress in mitochondria (mt) compromises their function16, which is interconnected with other aging hallmarks that underlie poor energy production and frailty17. Cytosolic nucleic acids including DNA fragments from mitochondria or nuclei are detected by specialized sensors which trigger proinflammatory cytokines that induce an innate immune response by the cGAS-STING pathway18, resulting in senescence and compromised SC function. Altogether, nearly all hallmarks of aging are elicited by a poor response to replication stress.
In this review, we will discuss these topics to frame the case that replication stress represents a driving force for cellular senescence and aging phenotypes.
Hereditary diseases characterized by cellular phenotypes characteristic of a defective response to replication stress
There continues to be growing interest in rare hereditary diseases characterized by an accumulation of genomic DNA damage and accelerated aging that provide insights into normal aging19. Many genetic disorders characterized by clinical symptoms of premature aging display cellular phenotypes that also indicate a condition of heightened replication stress (Table 2). These hereditary diseases provide a window of opportunity to better understand pathways of genome metabolism responsible for chromosomal stability and underlying mechanisms in which replication stress is driving not only cellular senescence but also the accelerated decline of bodily function, including (but not exclusively) replicative tissues characterized by persistent inflammatory processes. Some salient and notable points are discussed in this section.
RECQ genetic disorders caused by deficiencies in helicases with unique but also overlapping functions
Of the five conserved human RECQ helicase genes, mutations in four (WRN, BLM, RECQL4, RECQL1) are linked to distinct hereditary premature aging disorders all characterized by genomic instability and a cellular phenotype of compromised response to replication stress (Table 2)20,21. While the RECQ helicases all share a conserved ATPase/helicase core domain and some overlap in their catalytic functions, nucleic acid substrate specificity, and protein interactions22, the growing consensus is that each RECQ has unique roles in pathways of genome metabolism that presumably reflect the different clinical features of each genetic disease; however, these relationships are complex. For example, studies of human cells and mouse models have demonstrated important roles of both the WRN23,24,25 and BLM25,26,27,28 helicases in telomere metabolism, suggesting that these two RECQs are specially tailored during cellular DNA replication to resolve unusual nucleic acid structures prone to occur at chromosome ends, as well as more broadly in the genome (Table 2).
Consistent with their unique nature, there are distinct genetic interactions of the five mammalian RECQ helicases29, a topic of great interest in cancer biology given their up-regulation in various cancers20 and potential as anti-cancer drug targets30. For example, WRN has been implicated as an essential factor for survival and proliferative capacity of tumors characterized by microsatellite instability (for review, see ref. 31). The discovery of helicase inhibitors such as one in a clinical trial (Study Details | Study of HRO761 Alone or in Combination in Cancer Patients With Specific DNA Alterations Called Microsatellite Instability or Mismatch Repair Deficiency. | ClinicalTrials.gov), and their applicability in vitro with human cancer cell lines32,33,34 and in vivo with a BRCA2 xenograft mouse model35 suggest that continued pursuit of RECQ pharmacological modulation to combat cancer in preclinical models may translate to efficacious strategies in the clinic.
Bone marrow failure diseases Fanconi anemia and dyskeratosis congenita
Characterized as a bone marrow failure disorder, Fanconi Anemia (FA) stems from mutations in >20 genes that play an integral role in the cellular response to DNA damage and replication stress36 (Table 2). While originally thought to be strictly responsible for repair of DNA interstrand cross-links (ICLs), the FA pathway with direct involvement of BRCA genes is implicated in DSB repair by homologous recombination (HR) and break-induced replication at collapsed or reconstructed replication forks. For many years, it was suggested that lipid peroxidation was the primary source of endogenous ICLs susceptible to repair by the FA pathway37; however, more recent evidence implicates the accumulation of formaldehyde which reacts with macromolecules, including DNA, lipids, and proteins, to be a major culprit38. Precisely how ICLs affect replication fork-associated events is still a major area of investigation, but it is evident that formaldehyde-induced DNA damage causes replication stress and the FA/BRCA genes play a significant role in its alleviation39,40,41. As discussed below, replication stress drives the decline of hematopoietic SCs (HSCs), a characteristic feature of FA, and may more broadly and adversely affect SC compartments implicated in aging of tissues and organs beyond the hematopoietic lineage.
Like FA, Dyskeratosis Congenita (DC) and the related, more severe disease Hoyeraal-Hreidarsson Syndrome (HHS) are also genetically complex disorders characterized by bone marrow failure42. Telomere defects are implicated in DC/HHS, with evidence that telomeric DNA synthesis is compromised (Table 2). Thus, an emerging theme is that difficult-to-replicate telomeres pose a unique challenge to cells that require specialized genetically controlled pathways to maintain their stability.
Hutchinson-Gilford progeria syndrome caused by perturbed nuclear envelope
A genetic splicing variant of the lamin A/C gene (LMNA) encodes an aberrant lamin A protein (progerin) that results in defective structural filamentous protein incorporated into the nuclear skeleton43. Transient nuclear ruptures perturb replication and induce DNA damage via changes to chromatin structure and reduced nuclear replication protein recruitment44. This discoordination of replication factors causes replication stress and p53-activated senescence of Hutchinson-Gilford Progeria Syndrome (HGPS) cells. The consequential elevation in DNA damage signaling activates cell cycle checkpoints that mediate reduced cell cycle progression in HGPS cells, which is believed to drive accelerated aging. Many advances have been made in understanding the mechanistic basis for HGPS phenotypes that revolve around a poor response to replication stress and telomere defects (Table 2), deriving from mechanical stress that causes problems with DNA synthesis and genomic stability that in turn drive aging processes. The Gonzalo lab and collaborators determined that cellular aging induced by progerin expression caused replication fork stalling and degradation accompanied by activation of the interferon (IFN)-like response45. Moreover, these characteristics could be rescued pharmacologically by cellular reprogramming agents.
Ataxia Telangiectasia and Seckel Syndrome display defects in signaling DNA damage and replication stress
Mutations in the ATM and ATR genes which encode protein signaling kinases are linked to the premature aging disorders Ataxia Telangiectasia (AT)46 and Seckel Syndrome (SS)47, respectively. Classically, ATM and ATR were assigned checkpoint roles in response to DSBs and replication stress, respectively; however, emerging evidence suggests that they both play roles during DNA synthesis that influence chromosomal stability and cellular senescence (Table 2), which together underlie the accelerated aging features characteristic of the two disorders. AT is classified as a neurodegenerative disorder, whereas SS is a growth and developmental disease with intellectual disability apparent. It is believed that loss of checkpoint functions due to these genetic mutations causes DNA damage-induced dysfunction of somatic SCs and defective tissue maintenance that are both critical for development and normal aging.
Cockayne syndrome
Cockayne Syndrome (CS), originally identified to be a disease defective in the preferential nucleotide excision repair of transcriptionally active genes, arises due to bi-allelic mutations in the CSA or CSB genes48. CSA encodes a protein with a tryptophan-aspartic acid (WD) repeat, a moiety that often serves as a scaffold for protein or DNA interactions involved in transcriptional regulation or chromatin remodeling. CSA is thought to mediate key interactions of proteins with RNA polymerase II stalled at sites of bulky DNA damage49, whereas the CSB protein is an ATP-dependent DNA translocase thought to push the stalled RNA polymerase II forward to bypass the obstacle50. In addition to its transcription-coupled repair mode, experimental evidence from cell-based assays places CSB as an important player in the response to replication stress (Table 2). CSB biochemically acts to reverse stalled replication fork-like structures in a manner similar to the DNA translocases SMARCAL1, ZRANB3, and HLTF51. In so doing, CSB acts in the same pathway as the structure-specific nuclease MRE11 to restart stalled replication forks in human cells. It remains unclear if or how CSB’s activity is orchestrated with the other DNA translocases implicated in fork reversal to regulate fork progression throughout the genome. However, it was determined that co-deficiency of CSB and SMARCAL1 synergistically promotes recruitment of the structure-specific nuclease MUS81 to telomeres in cells that operate by the alternative lengthening of telomeres (ALT) pathway dependent on HR52. Moreover, ALT cells co-deficient in CSB and SMARCAL1 display elevated fragile chromosomes which are believed to be a consequence of fork stalling at telomeric regions.
Ruijs-Aalfs syndrome
The SPRTN gene, mutated in the premature aging disorder Ruijs-Aalfs Syndrome, encodes a PCNA-interacting protein that operates in DNA damage tolerance53. Phenotypic deficiency of SPRTN was first observed in transgenic mice which displayed chromosomal instability, cellular senescence, and premature aging features including cataracts, lordokyphosis (curvature of the spine), and cachexia (wasting) early in life54. Biochemical studies revealed that SPRTN encodes a metalloprotease that digests covalent DNA-protein complexes (DPCs) and that the clinical mutations reduced its substrate cleavage activity compared to recombinant wild-type SPRTN55. It remains to be fully understood how defective DPC processing gives rise to the characteristic premature aging features and predisposition to liver cancer. However, it was determined that SPRTN-deficient cells display reduced replication fork speed following subjection to formaldehyde that induces DPCs56 (Table 2), suggesting a role of SPRTN in facilitating replication via its DPC processing function. Subsequent research demonstrated that SPRTN proteolytically attacks the checkpoint kinase CHK1, activating CHK1’s phosphorylation of SPRTN which enhances its recruitment to chromatin to allow smooth replication and DPC repair57.
Warsaw Breakage Syndrome
Warsaw Breakage Syndrome (WABS) is classified as a cohesinopathy disorder characterized by developmental abnormalities58. Although WABS does not fully resemble a classic premature aging disorder, the pre- and post-natal growth inhibition is accompanied by chromosomal instability, a hallmark of many more traditional hereditary accelerated aging diseases. The mutated DDX11 gene encodes a DNA helicase that interacts with proteins involved in replication fork protection and stability59 (Table 2). Cells from WABS patients display reduced replication fork progression59. Moreover, cancer cell lines depleted of DDX11 by RNA interference or in which DDX11 was deleted by CRISPR were found to be hypersensitive to chemotherapy drugs that induce replication stress60. DDX11 is believed to resolve G-quadruplex (G4) DNA to enable smooth replication61,62,63. However, the precise relationship of G4 DNA metabolism to aging and the mechanistic function(s) of DDX11 in G4-induced replication stress have not yet been fully elucidated.
Fork instability is a common feature of replicative stress
At the molecular level, accumulation of replicatively senescent cells characterizes biological aging. Replication stress induced by various exogenous and endogenous sources triggers DNA damage response (DDR) pathways that act as barriers to prevent neoplastic transformation of cells harboring damaged DNA. However, chronic DDR activation may also lead to premature replicative senescence and accumulation of senescent cells over time that contribute to human aging. Therefore, replication stress is thought to directly contribute to biological aging processes64,65. Replication stress is defined as stalling or slowing of replication fork progression which may lead to replication collapse and DNA damage. Stalled forks need to be protected and recovered to resume DNA synthesis and prevent genomic instability, a hallmark of aging. A direct link between compromised stalled fork recovery and aging has been established by characterizing the molecular phenotypes of cells isolated from individuals with Werner Syndrome (WS), an autosomal recessive premature aging disease resulting from loss-of-function mutations in the WRN gene (Table 2). Over the years, experimental evidence has demonstrated critical functions of the RECQ helicase WRN in stability and recovery of stalled replication forks under conditions of replication stress. Consistent with the roles of WRN in processing and stabilizing stalled forks, WS fibroblasts show reduced DNA replication capacity, fork asymmetry, heightened genomic instability, and premature replicative senescence. These phenotypes are likely attributed to failure to resolve complex replication intermediates resulting from stalled replication forks upon functional loss of WRN.
In addition to WS, hereditary diseases linked to other RECQ helicases implicated in stalled fork processing (BLM, RECQL1) exhibit some molecular phenotypes related to premature aging (Table 2). Thus, replication fork instability resulting from the loss of critical fork maintenance proteins potentially drives the onset of replicative senescence which in turn leads to accelerated decline of function at the cellular, tissue, and organ levels. Below, we discuss how coordinated functions of various proteins implicated in DNA processing ensure replication fork stability and DNA synthesis resumption following fork stalling (Fig. 1).

Replication stalling or fork collapse upon exposure to various exogeneous and endogenous stressors activates DNA damage response pathways critical for stalled fork stabilization or restoration and replication restart. a A replication fork encountering an ssDNA break/gap can be recovered by generating a DSB, thereby allowing HR-dependent fork restoration. Nucleases involved in DNA end resection such as MRE11 or EXO1 process the break and generate a 3’ ssDNA overhang necessary to initiate HR at the broken fork. ssDNA-bound RPA is replaced by RAD51 recombinase which in turn allows for strand invasion into the sister chromatid, thereby forming a displacement (D)-loop structure to initiate break-induced replication using the sister chromatid as template. The D-loop can be cleaved by structure-specific endonucleases such as MUS81 to restore the replication fork and continue DNA synthesis. b A dysfunctional fork such as one stalled due to nucleotide depletion is acted upon by the coordinated actions of fork remodeling enzymes and RAD51 to generate a reversed fork intermediate. Fork protecting factors such as BRCA1, BRCA2, RAD51, WRN, and RECQL1 protect reversed forks from unscheduled pathological degradation by nucleases. However, helicase-assisted nucleolytic processing (e.g., WRN-DNA2) of stalled forks (1) in a controlled manner promotes replication restart through HR. Alternatively, a reversed fork can be restored via the branch-migration activity of RECQL1 helicase (2). The reversed fork can also be cleaved by structure-specific endonucleases (e.g., MUS81) to generate a single-ended double strand break (seDSB) at the fork that undergoes homology-directed fork restoration (3), as described in a. Cells genetically deficient in fork-stabilizing proteins (e.g., WRN helicase mutated in WS) accumulate DSBs at forks and experience gross chromosomal instability which may lead to replicative senescence and accelerated aging phenotypes.
DNA helicases are key players in replication fork integrity
Cells have distinct but interconnected mechanisms that ensure protection and accurate restart of stalled replication forks. A battery of helicases and nucleases act on stalled or damaged replication forks and catalytically process replication fork intermediates. State-of-the-art cell-based techniques such as Isolation of Proteins on Nascent DNA (iPOND)66 or In-Situ Protein Interactions with Nascent DNA Replication Forks (SIRF)67 have provided direct evidence for enrichment of these enzymes specifically at forks in a replication stress-dependent manner. The RECQ class of ATP-dependent DNA helicases (RECQL1, WRN, BLM, RECQL4, and RECQL5) are fork-associated proteins that play vital roles in resolving complex replication intermediates generated during replication, recombination, and repair processes. Fork restoration activity of RECQL1, mutated in the genomic instability disorder RECON syndrome21,68, converts a stalled replication fork from its regressed state to an active fork, thereby promoting replication restart69. In addition, RECQL1 can protect stalled replication forks from DNA2 nuclease-mediated degradation70. Accordingly, clinically relevant RECQL1 mutations impair its fork restoration activity in vitro as well as reduce fork restart ability and increase fork degradation as demonstrated by cell-based DNA fiber experiments68.
The increased spontaneous and drug-induced chromosomal aberrations (chromatin breaks and gaps) in RECON syndrome patient-derived cells underscore the importance of RECQL1-mediated fork integrity maintenance to suppress chromosomal instability. BLM and WRN are two helicases vital to rescue stalled replication forks and maintain genomic stability. BLM helicase, mutated in Bloom’s Syndrome (BS), mitigates replication stress by promoting efficient fork restart and suppressing new replication origin firing in a manner that depends on its helicase activity and genetic interaction with the FA pathway protein FANCD271,72. BS patients exhibit characteristics of premature aging, and human BLM expressed in yeast has been shown to suppress premature aging phenotypes of sgs1Δ mutants73, suggesting that persistent replication stress serves as a driver of aging. Like BLM, WRN helicase also promotes fork restart by assisting nucleolytic processing of stalled forks by DNA2 exonuclease, thereby preventing replication fork collapse and DSB generation at forks70. In addition, WRN exonuclease activity protects stalled forks from unscheduled nucleolytic degradation by MRE11 nuclease, thus promoting genome stability74. In cooperation with RAD51, WRN also plays a structural role in counteracting unscheduled degradation of stalled forks by MRE11 nuclease75. Moreover, WRN helicase has been reported to promote stabilization and recovery of deprotected stalled forks in BRCA2-deficient cells35,76. Given the essential role of WRN in fork recovery, it follows that WS cells exhibit spontaneous fork stalling, increased chromosomal instability, and early senescence features.
Another important aspect of WRN-mediated genome stability maintenance is its well-characterized role in supporting replication of telomeric DNA rich in repeat sequences. This is attributed to its functions in resolving DNA topological structures such as G4s or T-loops prevalent in telomeres. Importantly, consistent with its telomere localization and role in telomere replication24,77, WRN deficiency results in telomeric defects coupled with premature aging phenotypes in late-generation and telomerase-deficient mice harboring ‘pre-shortened’ telomeres23,25. RECQL4, mutated in Rothmund–Thomson syndrome (RTS), controls replication initiation by assisting with loading of the replicative helicase78,79. RTS and the RECQL4-related genetic diseases Baller-Gerold Syndrome and RAPADILINO are heterogeneous disorders with a spectrum of diverse clinical features, including poikiloderma, skeletal abnormalities, and craniosynostosis80. Although mutation of RECQL5 has not been linked to any genetic disorders to date, it has emerged as an important player in stabilizing stalled forks during replication stress81,82. RECQL5 also promotes DNA repair synthesis at common fragile sites during mitosis by facilitating MUS81-EME1 endonuclease-mediated cleavage of late replication intermediates83. Importantly, RECQL5 suppresses replication stress arising from replication-transcription conflicts by resolving DNA replication intermediates84.
In addition to the RECQ helicases, FANCJ and DDX11, belonging to the family of Iron-Sulfur (Fe-S) cluster helicases22, play crucial roles in the replication stress response. The BRCA1-interacting FANCJ helicase implicated in the BRCA-FA pathway protects replication forks from MRE11-mediated degradation by controlling HLTF-mediated remodeling of stalled forks, thereby counteracting replication stress85. FANCJ is also crucial for G4 resolution that can perturb fork progression86. DDX11 helicase, mutated in the chromosomal instability disorder WABS, mitigates replication stress by promoting stalled fork recovery in cooperation with Timeless, a component of the replication fork protection complex59, and by promoting DNA end resection at damaged replication forks60. Importantly, experimental evidence demonstrates that FANCJ86 and DDX1161,62,63 helicases facilitate replication and DNA damage repair through difficult-to-replicate G-rich genome sequences. FANCM, a DNA-dependent ATPase and translocase implicated in the FA pathway, also plays a vital role in accurate repair of stalled forks. Using its conserved ATPase activity, FANCM promotes stalled fork reversal and error-free HR-mediated stalled fork repair in conjunction with BLM helicase87,88,89,90.
Structure-specific nucleases and fork protection factors suppress replication stress by their action at stalled or regressed forks
When fork movement is challenged by replication stress, the forks undergo reversal to generate “four-way” reversed fork structures. This is a global response to replication stress-induced uncoupling of replicative polymerase and helicase and is primarily catalyzed by the HR factor RAD51 and vital DNA translocase enzymes such as SMARCAL1, HLTF, and ZRANB391. Fork reversal is critical to stabilizing stalled forks, allowing DNA synthesis past the stall-inducing lesion by a template-switching mechanism and repair of damaged DNA by excision repair or recombination-mediated repair. Reversed fork structures are preferred substrates for nucleolytic processing by nucleases implicated in stalled fork recovery and genome maintenance. One such nuclease is DNA2, which processes reversed fork ends following replication stalling in a WRN helicase-dependent manner and creates partially resected reversed fork intermediates amenable to restart by branch migration or HR mechanisms70. MRE11, CtIP, and EXO1 nucleases have also been reported to perform end processing of stalled fork intermediates to promote HR-mediated fork restart92,93,94; however, the underlying mechanisms are distinct from DNA2-mediated stalled fork processing and dependent on specific genetic backgrounds95. An important backup mechanism of stalled fork recovery is mediated by the structure-specific endonuclease MUS81, which becomes activated as a last resort when other fork recovery mechanisms become unavailable. MUS81 cleaves stalled fork intermediates to generate DSBs at forks, thereby favoring HR-mediated replication restart96,97.
Although nucleolytic processing of stalled forks is necessary for replication restart, uncontrolled nucleolytic degradation of stalled replication intermediates can lead to fork collapse and instability. Fortunately, stalled forks are protected from unscheduled nucleolytic degradation by the crucial FA/HR pathway proteins BRCA1, BRCA2, FANCD2, and RAD5195,98,99. In genetic backgrounds deficient in major fork-protecting factors, stalled forks undergo MRE11 and EXO1 nuclease-mediated pathogenic degradation, leading to genomic instability95,100. Importantly, experimental evidence suggests that BRCA1/2-mediated stalled fork protection is also critical for timely restart as BRCA1/2-deficient cells exhibit delayed restart following replication stalling35,101. Aside from BRCA1 and BRCA2, other members of the BRCA1-A complex (RAP80, ABRAXAS, MERIT40, BRCC36, and BRCC45) have been implicated in stalled fork protection and recovery following replication stress102,103,104. Thus, principal components of HR repair play vital roles in rescuing stalled replication forks, thereby maintaining genome integrity under conditions of replication stress.
Replication stress induces changes in chromatin, and experimental evidence points toward important roles of stress-induced chromatin modifications in stalled fork protection and rescue. For example, methylation of histone H3 Lys4 (H3K4) by SETD1A methyl transferase enhances FANCD2-dependent chromatin remodeling, thereby stabilizing RAD51 nucleofilaments to prevent nucleolytic degradation of stalled forks105. In another study, Thakar et al. showed that ubiquitylation of PCNA protects stalled forks from extensive DNA2-mediated degradation by ensuring Okazaki fragment maturation and efficient nucleosome reassembly106. Consistent with the important functions of histone acetylation in chromatin accessibility and diverse cellular processes including transcription, replication, and DNA repair, PCAF-mediated acetylation of histone H4 Lys8 (H4K8ac) has been shown to engage MRE11 and EXO1 nucleases at stalled forks, resulting in enhanced fork degradation in BRCA1/2-deficient genetic backgrounds107. Thus, in response to replication stress, epigenetic modifications of fork histones determine stalled fork stability, and future studies would provide a better understanding of the epigenetic mechanisms of replication fork integrity.
Replication stress leads to genome instability if not resolved in a timely manner by the mechanisms described above. Unresolved replication fork intermediates result in irreversible fork collapse or DSB formation at forks that drive senescence and aging108. Other than telomere attrition, replication stress is considered one of the main sources of DNA breaks, which are common features of replicative senescence, oncogene-induced senescence, and aging108. Therefore, maintenance of replication fork integrity in response to replication stress is central to preventing genomic instability associated with senescence and accelerated aging.
Hallmarks of aging driven by replication stress
Lopez-Otin et al. described nine cardinal hallmarks of aging proposed to be common in multicellular organisms, with a special emphasis placed on aging processes in mammals5. The scheme has proven to be highly useful for drawing inferences about the mechanisms underlying aging and framing experimental studies to address their relative importance and how they are interconnected. The field of aging research is not static, and new hallmarks (expanded to 12) emerged with an even greater emphasis placed on robust response to stress being paramount to healthy aging109. In the following sections, we discuss selected hallmarks of aging in which replication stress is implicated. Because of their interconnected nature109, all hallmarks of aging would experience the impact of replication stress (Fig. 2).

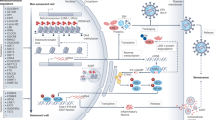
Evidence from the literature suggests that many of the previously described hallmarks of aging109 are driven by replication stress. Representative examples are cited below as well as in the text. Chronic Inflammation. Innate immune response activation by LINE-1 element derepression and cytosolic ssDNA incurred by replication stress promotes senescence induction and contributes to sterile inflammation112,257. Genomic Instability. Replicative stress incurs DNA damage in the form of chromosomal aberrations and DSBs that attenuate replicative lifespan of cells and induce aging-associated tissue dysfunction9,121,123. Telomere Attrition. Telomeres are fragile sites with DNA sequences prone to form G4 and T-loop secondary structures that break under conditions of replication stress, inducing telomere shortening that limits replicative potential133,134,139. Stem Cell Exhaustion. Replicative stress in SCs impairs the ability to produce SC progeny. Specific to HSCs, this leads to a variety of tissue-based and organismal dysfunctions including clonal drift and SC attrition147,151,258. Epigenetic Alterations. Replicative stress recruits a variety of proteins to the stalled replication fork that are involved in remodeling the DNA modification and histone landscapes around the stress. These changes can have long-term effects for cellular senescence12,259. Mitochondrial Dysfunction. Replication stress in the mitochondria leading to mtDNA mutations causes a deprivation of resources, culminating in premature aging in a pol γ proofreading mutant mouse model158. In addition, mtDNA replication errors have consequences for nuclear genome replication and stability, which contribute to aging phenotypes16. BioRender was used to create the figure.
Chronic inflammation
Loss of replicative potential is an underlying cause of cellular senescence, a hallmark of aging defined as a state of virtually irreversible cell cycle arrest characterized by macromolecular damage coupled with distinct secretory and metabolic profiles2,109. LINE-1 retrotransposable elements become transcriptionally derepressed during cellular senescence, reflecting a loss of heterochromatin and leading to activation of the type-I interferon (IFN-1) response characteristic of sterile inflammation110. The IFN-1 response induced by cGAS-STING recognition of LINE-1 cDNA contributes to the SASP, a state of inflammatory cytokine, proteinase, and growth modulator secretion that reinforces and promotes senescence induction in proximal cells2,110. Chronic inflammation contributes to the age-associated condition of frailty, which has been alleviated in aged mice following treatment with anti-inflammatory agents such as JAK1/2 inhibitors111. Inflammatory signaling perpetuated by senescent cell accrual thus contributes to both symptoms and propagation of cellular aging.
Replication stress-induced cytosolic self-DNA accumulation has been demonstrated to effect a senescence-promoting IFN-1 response initiated by cGAS-STING112. Cytosolic chromatin fragments in the form of micronuclei trigger the SASP and astrocyte senescence as well as premature neurodegeneration in human AT pluripotent SC-derived brain organoids112, exemplifying the role of replication stress in promoting cGAS-STING-mediated senescence induction which contributes to accelerated aging phenotypes.
Depletion of the fork protective factors Abro1 and FANCD2 promotes release of ssDNA fragments from stalled replication forks which leak into the cytosol and activate the cGAS-STING pathway113. cGAS-STING-mediated IFN-1 activation is similarly induced by impaired degradation of nascent DNA from stalled replication forks resulting from depletion of the dNTPase SAMHD1, mutated in the inflammatory disorder Aicardi-Goutières Syndrome (AGS)114. Moreover, knockdown of the fork protective factors Replication Protein A (RPA) and RAD51 exacerbates IFN-1 signaling in AGS patient fibroblasts that are deficient in TREX1 exonuclease, which functions in degradation of cytosolic single-stranded DNA (ssDNA)115. This suggests that replication stress resulting from RPA and RAD51 exhaustion contributes to autoinflammation that promotes senescence phenotypes observed in these cells, including upregulation of interferon-stimulated genes and β-galactosidase positivity115.
Cellular senescence and associated aging phenotypes promoted by replication stress are also induced by DNA damage or replicative exhaustion stemming from endogenous or genotoxic sources2. Senescence is promoted by a myriad of replication stress-induced processes, including genomic instability, telomere attrition, epigenetic modifications, mitochondrial dysfunction, and SC exhaustion2 (discussed below), and the processes of cellular senescence and aging are thus complementary and heavily interrelated in the context of replication stress.
Genomic instability
Replication stress is a primary source of genomic instability in the form of both chromosomal aberrations and DSBs, which have long been shown to play a causative role in aging109. Targeted DSB induction effectuates premature aging phenotypes in murine liver9, and systemic mtDNA DSBs induce progeroid phenotypes in rapidly proliferating murine tissues such as thymus and testes116. Persistent DSBs have been shown to cause segmental and whole-chromosome aneuploidies in human embryos resulting from chromosomal mis-segregation in mitosis117. Heightened chromosome mis-segregation in old-aged human dermal fibroblasts due to an increase in stable kinetochore-microtubule attachments enhances biomarkers of cellular senescence118, evidencing the role of chromosomal aberrations in promotion of aging and associated cellular phenotypes.
Nuclear deformation has recently been found to induce replication fork stalling, revealing replication stress as a source of elevated DSB formation observed in cells undergoing mechanical stress119. DSB formation is also incurred by replication fork blockage imposed by transcription-replication conflicts, and this DNA damage induction is enhanced in the presence of co-occurring three-stranded DNA-RNA hybrids with the associated non-template ssDNA (R-loops) that exacerbate replication fork stalling120. R-loop persistence in mitosis resulting from depletion of the R-loop resolving helicase senataxin (SETX) has also been shown to induce chromosomal fragility and DNA under-replication121. Deficiency in the SETX-interacting factor senataxin-associated exonuclease 1 (San1) induces early onset murine cardiomyopathy associated with elevated cardiomyocyte R-loop and DSB levels, evidencing the role of replication stress-induced DNA damage accrual in premature induction of age-related pathologies122.
S-phase replication fork stalling, low fork speed, and incomplete DNA replication have been identified as sources of DSB formation and subsequent chromosome breakage that result in segmental aneuploidies in human cleavage-stage embryos123. Replication stress incurred by the replicative B-family DNA polymerase inhibitor aphidicolin has been shown to directly stabilize spindle microtubules and induce premature centriole disengagement, causing lagging chromosomes and micronuclei formation124. Mitotic segregation defects are also effected by depletion of the structure-specific nucleases ARTEMIS and XPF-ERCC1, and resultant replication fork stalling has been found to increase the incidence of lagging chromosomes and DNA bridges125. Deletion of ERCC1 in hematopoietic cells induces immune cell senescence and premature age-related immune dysfunction in mice126. Subsequent age-associated tissue damage and senescence marker expression in peripheral organs accompanied by shortened lifespan implicates replication stress-induced DNA damage in promotion of systemic organismal aging.
Telomere attrition
Telomere instability is an established effector of aging and senescence-inducing replicative exhaustion109. Rich in G4 and T-loop secondary structures that impede replication fork progression, telomeres are fragile sites that pose a challenge to replication machinery and are prone to breakage under conditions of endogenous and genotoxin-induced replication stress127,128. Telomeric replication fork arrest effectuated by oxidative stress causes aging-associated telomere loss and dysfunction as well as premature cellular senescence129,130.
Persistent telomeric secondary structures resulting from RTEL1 helicase deficiency have been shown to cause accrual of reversed replication forks127. Reversed forks are aberrantly bound by telomerase, inhibiting replication fork restart and inducing telomere shortening and catastrophe which contribute to pathogenesis of telomere maintenance disorders. Telomere fragility and subsequent senescence induction resulting from replication fork stalling are also effectuated by disruption of RTEL1 helicase binding to the replisome131, evidencing the importance of effective replication machinery recruitment to chromosome ends in maintenance of telomere integrity.
Replication and fork protective factors are often localized to telomeres by their association with the shelterin complex, which promotes fork stability and efficient telomeric DNA replication in addition to its general role in telomere end protection132. TRF1, the core subunit of the shelterin complex, has been established as a primary facilitator of fork progression by recruitment of the secondary structure-resolving helicases RTEL1, BLM, and WRN, which promote lagging-strand telomere synthesis, as well as topoisomerase IIα (TopoIIα), which is essential for replication intermediate resolution128,133,134. Perturbation of TRF1 binding to WRN helicase during S-phase disrupts WRN association with telomeric chromatin, obstructing fork progression and inducing telomere fragility134. Depletion of TRF1 similarly destabilizes telomeres by reducing TopoIIα telomere association, impairing resolution of replication-incurred topological stress, and inducing mitotic defects in the form of ultrafine anaphase bridge (UFB) formation133. Shelterin subunits TRF1 and TRF2 also associate with the fork protection complex (FPC) subunit Timeless, which preserves telomere length by stabilizing stalled forks and regulating replisome progression through barriers such as the repetitive DNA sequences that constitute telomeres135. The FPC subunit Claspin is recruited to telomeres by a specific interaction with TRF2136, and overexpression of Claspin and Timeless has been shown to increase cellular replication stress tolerance137.
Recent studies have revealed the importance of TRF2 in promoting telomere stability by recruiting origin recognition complex subunit 2 (ORC2), thus promoting formation of telomeric replication origins which rescue compromised replication138,139. TRF2 suppresses fork stalling and telomere fragility resulting from transcription-replication conflict by binding the transcription repressor treacle ribosome biogenesis factor 1 (TCOF1) in an S-phase specific interaction140. TRF2 has recently been found to promote telomeric fork progression and stalled replication fork restart through S-phase recruitment of Microcephalin 1 (MCPH1), which is implicated in DNA damage signaling and repair and expressed during fetal brain development141. Fragile telomeres induced by MCPH1 depletion suggest that telomeric replication stress may contribute to primary microcephaly onset that presents as a clinical symptom of telomere disorder syndromes.
Under conditions of oncogene-induced telomeric DNA replication stress, human cells that lack telomerase activity experience telomere dysfunction-induced senescence (TDIS) characterized by telomere shortening and persistent DNA damage foci accompanied by stable proliferative arrest142. Preferential stalling of telomeric replication forks induced by oncogenic signals may suppress malignant cancer progression. Tumor suppression by this oncogene-induced senescence mechanism is mediated by a response to aberrant telomeres in telomerase-negative cells, emphasizing the biological and potential clinical relevance of telomeric replication stress (for a perspective, see ref. 143).
Stem cell exhaustion
The function of an SC is to renew both itself and differentiated progeny throughout an organism’s lifetime144. Thus, replication stress occurring within SCs is significantly detrimental to cell populations as well as organismal health. Replication stress in SCs, as such, is a multifaceted issue resulting in clonal drift or complete inability to produce progeny and increased DNA damage, both affecting progeny and DNA damage-associated checkpoints as well as decreasing functionality of specific organismal tissues145,146,147. Deletion of the key replication stress response checkpoint kinase regulator ATR in mice causes a diverse array of age-associated phenotypes, particularly in rapidly proliferating tissues148, suggesting that SC exhaustion has disease implications beyond the classic hematopoietic compartment.
Nonetheless, as highly replicative cells, HSCs are a prime example of the consequences of replicative stress. HSCs confronting replication stress and associated cellular damage experience high levels of attrition, contributing to the pathology of the bone marrow failure disorder FA147,149. The decline of HSCs accompanies global tissue degeneration and malignant transformation in cancer patients147. With a lack of blood cell production, tissues atrophy due to decreased oxygen supply, become susceptible to infection, and exhibit a reduced ability to protect against endogenous or exogenously induced DNA damage150,151.
Once DNA damage arises from dysfunctional replication, a series of checkpoints are activated to deal with the cellular stress that often detriment the function of SCs, notably in the hematopoietic compartment151. Upon checkpoint activation, a decline in SC proliferation occurs followed closely by a SASP-mediated inflammatory response and clonal drift. This often leads to selection of malignant clones and impaired tissue maintenance. Moreover, chronic inflammation compromises the immune system and others (e.g., cardiovascular)152. On a larger physiological basis and from the perspective of aging-related phenotypes, SC attrition can cause a myriad of different premature aging disorders and related symptoms. While replicative stress in HSCs is often considered in the context of FA, SC replicative stress in general is associated with muscle atrophy, impaired immune response, metabolic issues such as obesity and diabetes, inflammation, osteoporosis, WS-related cardiac decline, and other cardinal features of aging8,153.
While SC attrition and clonal drift induced by replicative stress are evidenced in many tissues, these defective processes may arise by a related mechanism147,151. For example, the same cellular issues that are responsible for HSC attrition and contribute to FA are also implicated in inflammatory responses and the SASP147,151. As such, SC replicative stress may be a good therapeutic target to mitigate aging-related pathologies, counteracting activated checkpoints, the inflammatory response, or negative selection of affected SCs. This would most likely be accomplished by inhibition of HSC-associated senescence but may lead to elevated cancer risk.
Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) has attracted interest in biomedical therapies. However, iPSCs are frequently characterized by genomic instability and elevated DNA damage, which may compromise their utility for replenishment treatments. Ironically, the rejuvenating Yamanaka factors (OCT4, SOX2, KLF4, cMYC) cause replication stress which in turn activates the DNA damage response154. Strategies such as CHK1 augmentation or nucleoside supplementation to limit replication stress during somatic cell reprogramming were shown to reduce genomic instability in iPSCs154, which may increase their efficacy for safe use in clinical therapies.
Epigenetic alterations
Replicative stress is not known to alter the epigenetic landscape in the vicinity of a stalled replication fork. Rather, the DNA damage response that is activated to alleviate replication stress can alter the epigenetic landscape and cause drastic long-term effects on the cellular and potentially organismal levels155. This occurs both as DNA-level modifications as well as changes to the histones in the vicinity of the stalled fork.
One of the initial steps in the replication stress response is recognition and binding of exposed ssDNA in the vicinity of the stalled fork by RPA. Subsequently, RPA interacts with the histone cell cycle regulator complex (HIRA) to deposit histone H3.3 and H4 proximal to promoters, enhancers, and other gene elements to modulate transcription of associated gene bodies155. When extensive fork stalling enables RPA to remain bound to ssDNA and form a HIRA complex for a prolonged period of time, histone placement and modification become varied, changing how those regions are normally transcribed155. In addition to RPA, other factors such as Atrx and Daxx associate with H3.3 at telomeres to repress telomeric RNA production, suggesting that both replication stress and factors associated with specific genomic loci play a role in H3.3 localization and resultant transcriptional regulation.
HIRA and another chromatin-regulating factor known as ASF1a (with which HIRA physically interacts) were found to play a role in the formation of senescence-associated heterochromatin foci (SAHF)156. During cellular senescence, ASF1a and HIRA act to promote assembly of proliferation-promoting genes into chromatin which discourages resident gene expression; moreover, HIRA-ASF1a complex-induced formation of SAHF provides the opportunity for cell cycle exit during senescence. SAHF are implicated in oncogene-induced replication stress and their formation is dependent on the DNA replication stress response checkpoint kinase ATR157. Thus, SAHF may serve as a valuable biomarker of oncogene-driven replication stress, signifying heterochromatin perturbation which in turn harnesses DNA damage response signaling. Epigenetic manipulation of the heterochromatin state in vivo to relax it with a histone deacetylase inhibitor enhanced the DNA damage response and triggered tumor regression157. This work supports efforts to pharmacologically modulate heterochromatin to treat tumors by targeting the DNA damage response.
Recently, DSBs have been evidenced to induce epigenetic modifications that are associated with aging-related phenotypes12. Specifically, histone H3 and H4 modification and rearrangement, causing differing levels of methylation and acetylation, decrease Waddington Valley differentiation and were shown to accelerate aging in mice12. It is likely that replication stress-induced DSB formation is largely responsible for this innate change in histone modifications. It is plausible that modification of histone H3.3 and H4 by the RPA-HIRA-histone H3.3 complex causes transcriptional changes associated with the same phenotypic changes evidenced in the study by ref. 12.
Interestingly, the organismal and phenotypic characteristics attributed to epigenetic changes are some of the most strongly correlated with aging and cellular degeneration. Yang et al. showed that DSB induction resulting in epigenetic rearrangement induced cellular and behavioral phenotypes of neural degeneration, including reduced memory12. Additionally, they demonstrated muscle atrophy, lack and discoloration of hair, skin abnormalities, and other phenotypes that closely mirror the pathology of advanced aging12. It is plausible that replicative stress that causes DNA breaks and histone rearrangements induces similar epigenetic rearrangements, thereby indirectly leading to an acceleration of the aging phenotype.
Mitochondrial dysfunction
A hugely consequential but often underappreciated effect of mitochondrial replicative stress on aging was evidenced by observations of mice genetically deficient in the proofreading subunit of the nuclear-encoded replicative DNA polymerase gamma (pol γ) that is responsible for replication of the mitochondrial genome158. The 3- to 5-fold elevated point mutations and increased deletions in the mitochondrial genome of pol γ mutant mice with a defective proofreading subunit cause reduced lifespan and accelerated aging phenotypes including reduced weight and subcutaneous fat, hair loss, spine curvature, osteoporosis, blood anemia, oversized heart, and compromised fertility. These symptoms first started to appear when the mice were ~25 weeks old, which is considered young adulthood. This was a major finding in gerontology research as it provided evidence to support the theory that mutation burden in the mitochondrial genome due to defective proofreading during replication may be a driving force of aging.
With this new advance, the field was intrigued by the possibility that mitochondrial mutation held rank over accumulation of mitochondrial reactive oxygen species (ROS) as the primary driver of aging. Indeed, Trifunovic et al. found that the level of ROS in the pol γ mutator mice was normal, and the mouse embryonic fibroblasts (MEFs) from these mice did not show a hypersensitivity to oxidative stress159. Collectively, the authors suggested that respiratory chain defects are not the driver of premature aging in the mtDNA mutator mice. Consistent with this conclusion, Vermulst et al. found that the pol γ mutator mice accumulated mtDNA deletions at an elevated rate with age in brain and heart tissue160. Although it is tempting to speculate that premature aging of the pol γ mutator mouse is attributed to error-prone replication, Martin and Loeb point out that it is dubious to discount the oxidative theory of aging as a driver of the mutator mouse phenotypes because the mutations in respiratory-chain proteins of the pol γ mutant mitochondria would result in elevated ROS and a leaky proton gradient161, representing a causative factor for the premature aging features.
Crosstalk between mitochondrial and nuclear genome metabolism is thought to play a role in aging, but the devil lies in the details. Using the pol γ proofreading mutant mouse model described above, Hamalainen et al. found that cells from the mutator mice displayed compromised nuclear DNA replication and accumulation of DSBs, especially in the SC compartment16. It was proposed that mitochondrial replication stress and mutation of the organelle’s genome deprive the nuclear genome of resources as the cell attempts to remedy the induced stress. Once mitochondrial replication stress occurs, replicative resources are sequestered in the mitochondria, causing an overall decrease in cellular nucleotide and deoxynucleoside triphosphate (dNTP) pools, therefore affecting nuclear genome stability and ability to replicate16. However, in a recent study, Sharma reported that genetic mtDNA polymerase γ proofreading deficiency in mouse embryos did not perturb total dNTP pools as demonstrated by a technique involving the HPLC/UV-detection approach, which is contended by the authors to produce highly reliable results162. Sharma et al. concluded that alterations in dNTP pools are likely not the underlying cause of premature aging in the pol γ proofreading mutant mouse model. Further studies are necessary. Despite this caveat, the central role of mitochondria in the cell suggests that when mitochondrial-encoded proteins are dysfunctional due to errors in mtDNA replication, other hallmarks of aging are likely to be affected. From depletion of cellular resources, increased ROS production, and decreased phosphate-rich energy production, mitochondrial replication stress likely directly contributes to age-related frailty; however, therapeutics targeting mitochondrial replication stress have not been widely developed to date.
Replicative stress as a biomarker or cause of cellular senescence and accelerated aging: evidence from translational models, genetic diseases, and clinical studies
There is much research supporting the theme that DNA damage universally causes organismal decline via its effect on multiple pillars of aging65. However, experimental evidence (as discussed above) demonstrates that replication stress causes genomic instability and endogenous DNA damage which also promotes aging and age-related diseases mediated in large part by advancing cellular senescence. Thus, aging researchers are very interested in genomic-based biomarkers for cellular senescence64,163. Many of these are based on DNA damage and the DNA damage response. Examples include γ-H2AX, MRE11, RAD50, NBS1, ATM, ATR, 53BP1, MDC1, RAD17, and telomere dysfunction-induced foci (TIF)164. In addition, a reduced level of DNA synthesis in cells or tissues detected by fluorescent staining of nucleoside analogs such as bromodeoxyuridine (BrdU) and ethynyl-2’-deoxyuridine (EdU) incorporated into genomic DNA serves as a reliable biomarker of replication stress in senescent cells. Cell cycle arrest-based markers align well with biomarkers of replication stress in senescent cells. Other biomarkers of cellular senescence such as the senescence-associated lysosomal enzyme β-galactosidase, epigenetic modifications, and the SASP are associated with replication stress. The distinction between biomarkers of the DNA damage response versus replication stress response may be in part blurred due to their overlapping nature. In fact, some of the classic DSB repair proteins such as MRE11 are implicated in stalled replication fork processing95. Many factors involved in replication fork reversal triggered by fork stalling are also involved in DSB repair165, making it difficult to strictly assign them as biomarkers of replication stress versus the DSB.
In the following sections, we will consider the experimental evidence and conceptual logic for replication stress as a driver of cellular senescence and aging and apply it to existing models and emerging topics that are relevant in translational and clinical realms. Characterizing how replication stress directly or indirectly drives phenotypes of accelerated aging will set the stage for developing a new understanding of the factors and conditions that might be modulated in replicative tissues to modulate debilitating mechanisms of cellular senescence and potentially ameliorate age-related disease pathways.
Replicative senescence as a biomarker for aging
The cyclin-dependent kinase inhibitor and tumor suppressor p16INK4a has been useful in studying both neoplastic transformation during carcinogenesis and development of aging phenotypes in mouse models166. Expression of p16INK4a is significantly elevated during cellular senescence, prompting researchers to consider the signaling molecule as a potential biomarker for aging in humans as well (reviewed in ref. 167). Taking note that the transcription factor CUX1 regulates expression of p16INK4a and other tumor suppressor genes, Jiang et al. determined that endothelial cells in passage 10 compared to passage 5 displayed greater CUX1 and p16INK4a expression accompanied by elevated γ-H2AX and decreased telomere length, suggesting replicative senescence168. Conversely, depletion of CUX1 in late passage endothelial cells caused reduced p16INK4a expression, reduced β-galactosidase and γ-H2AX staining, restored BrdU incorporation indicative of more normal DNA replication, and ameliorated cell cycle arrest, all parameters pointing toward a role of CUX1 and p16INK4a in regulating replicative senescence. Similar findings were reported for human primary vascular smooth muscle cells, suggesting that the effects were not specific to endothelial cells.
Lifespan extension of ATR mutant mice by increased Rrm2 gene dosage
The central signaling axis for replication stress in mammals is mediated by the ATR kinase and its downstream target kinase CHK1. It follows that ATR or CHK1 may serve as a biomarker or even a therapeutic target for aging. Lopez-Contreras et al. explored the hypothesis that premature aging of transgenic ATR mutant mice could be suppressed by increased dosage of the Rrm2 gene, which encodes a regulatory subunit RRM2 of ribonucleotide reductase implicated in the production of dNTPs required for replicative DNA synthesis169, building on findings that replication stress is reduced by nucleoside addition170,171. Elevated Rrm2 gene dosage indeed prolonged survival of ATR mutant mice and depressed chromosomal breakage at fragile sites169. However, lifespan of wild-type laboratory mice was not extended by increased gene dosage of Rrm2 and/or Chk1, leading the authors to suggest that protection from replication stress using the transgenic mouse model system employed did not extend normal aging172.
Aging hematopoietic stem cells driven to the brink by replicative stress
A key advance fueling the theme that replicative stress drives aging was put forth by Flach et al. in 2014 with their Nature paper8 showing that cycling aged HSCs that display replication defects and reduced expression of the replicative DNA helicase MCM have compromised ribosomal (r) DNA gene status characterized by elevated γ-H2AX due to a mis-localized phosphatase, notably without the expected DNA damage response activation or a detectable level of DNA DSBs. The quiescent old HSCs display compromised self-renewal, a biased myeloid differentiation signal, transcriptional silencing, and reduced ribosome biogenesis, suggesting a mechanism of aging that is independent of DNA damage or DNA damage response activation and relies heavily upon replication stress as the driver of declined functional output. Epigenetic histone modifications, as suggested by the γ-H2AX enrichment, may drive changes in expression of replication machinery proteins (like the MCM helicase) as well as down-regulate rDNA transcription. Whether these changes can be remedied therapeutically in bone marrow failure syndromes remains to be seen. Replication stress may not be the only driver of aging for HSCs, as suggested by a recent study showing that genotoxic aldehyde which causes DNA damage drives aging in a p53-dependent manner41. Even these latest findings, however, do not discount the importance of replication stress in the aging pathway as metabolism-derived formaldehyde-induced DNA damage is likely to perturb fork progression as well, as suggested by studies that examined the role of the fork protection factors BRCA2 and FANCD2 in cells experiencing endogenous aldehyde toxicity173.
DNA damage tolerance (DDT) is an important pathway for dealing with stalled replication forks. Pilzecker et al. investigated the importance of DDT in HSCs and multipotent progenitors using a mouse PcnaK164R/K164R model engineered to be defective in transitioning from a replicative mode to a DNA damage-tolerant mode of replication174. By studying the bone marrow of DDT-deficient PcnaK164R/K164R mice, the researchers determined that the hematopoietic compartment displayed accelerated aging, indicative that the DDT response to replication stress in the murine model is critical for long-term HSC fitness and tissue homeostasis.
The role of replicative stress in aging of SCs other than those of the hematopoietic compartment remains to be fully characterized. However, disease models characterized by accelerated aging and elevated DNA damage in SCs have been described (for perspective, see ref. 175). As discussed by Goodell and Rando7, a myriad of factors revolving around genetic mutations, epigenetic changes, and environmental milieu play a role in SC functionality over the lifetime of an individual. It is reasonable to postulate that depletion of the SC reservoir by replication stress promotes decline of tissues and organs in a cell lineage-dependent manner. Although there was not a measurable level of DNA breaks detected in the aging HSCs described by ref. 8, in certain contexts inducible DSBs contribute to cellular senescence and aging (see below).
Double-strand breaks that may arise during replication stress cause accelerated aging
As detailed in previous sections, cellular senescence driven by replication defects is considered a hallmark of aging. This prompts one to consider replicative stress as a potentially useful biomarker for aging. Although cell metabolism markers such as β-galactosidase staining have been a popular marker for senescent cells176, markers of replication stress have not been as extensively studied. Rather, DNA damage emanating from replication stress or by other avenues (e.g., oxidative stress177) has been postulated as a key biomarker for cellular senescence and even organismal aging. One of the most prominent DNA lesions associated with changes to the genome that is implicated in (and perhaps a driving force of) aging is the DSB10, one of the most lethal forms of DNA damage and a source of great genomic instability due to its recombinogenic nature. A signature paper from the Vijg lab provided evidence that inducible DSBs cause accelerated aging of mouse liver9.
Recently, the Sinclair lab and collaborators developed an inducible DNA break mouse model that enabled them to investigate the importance of epigenetic changes induced by chromosome breaks for aging12. Alterations in epigenetic landscape in regions surrounding the DSBs were associated with aging phenotypes at the cellular and organismal levels. However, whether the aging phenotypes associated with epigenetic changes are reversible at the organismal level remains to be seen (for a perspective, see ref. 178). Nonetheless, the described model system will be useful for future work to study in vitro and in vivo aging. It remains to be determined if DSBs deriving from replication stress drive aging in replicative tissues by a mechanism that is different from the one described above, in which DSBs introduced frankly by the in vivo inducible restriction endonuclease system in both non-replicative and replicative tissues cause aging in a manner that is heavily dependent on epigenetic changes.
Although one could argue that DSBs represent only one of multiple DNA lesions to induce accelerated aging, the probability that they occur at the fork in replicative tissues in vivo is high. Replication fork stalling followed by blockage leads to single-stranded and ultimately DSBs, i.e., broken replication forks that cells must deal with using fork reconstruction pathways to preserve genomic stability (see Replication Fork Stalling Section). Typically, these repair mechanisms to heal DSBs at broken replication forks involve HR repair or the less faithful nonhomologous end-joining (NHEJ). Although stalled forks can be restarted by non-recombinogenic mechanisms, the transient ssDNA that arises is susceptible to breakage. Thus, it is difficult to tease out if a structural feature of the stalled or arrested replication fork, the fork-associated DSB itself, or both represent a key signaling event in cellular senescence and aging. Either way, in proliferating cells of rapidly turning over tissues, replication stress is a driving force for age-associated signaling pathways associated with delayed fork progression.
Chemotherapeutic senescence-causing drugs that interfere with replication fork progression
Therapy-induced senescence achieved by application of chemotherapeutic DNA damaging agents is a front-line strategy to combat cancer179. However, cancer survivors having undergone chemotherapy often display premature aging due to DNA damage by distinguishing underlying mechanisms, including the consequence of perturbing replication fork integrity180.
Common examples of chemotherapy drugs that not only induce caustic DNA damage but also deter replication fork progression include DNA cross-linking agents (cisplatin, methotrexate) and drugs that induce bulky DNA lesions (e.g., alkylating agents such as temozolomide, cyclophosphamide, etc.). However, even beyond such drugs that directly induce DNA damage are agents such as etoposide, mitoxantrone, camptothecin, irinotecan, topotecan, and derivatives that poison topoisomerases which relieve torsional stress at the fork to ensure smooth fork progression. Antimetabolites, including gemcitabine, hydroxyurea, and paclitaxel disturb the normal nucleotide pool, causing a reduction in replication fork rate. Even certain poly(ADP) ribose polymerase (PARP) inhibitors which were historically thought to solely exert their action by interfering with PARP’s role in the repair of DNA strand breaks are now also believed to exert cytotoxicity against cancer cells by trapping PARP on genomic DNA181, potentially interfering with cellular DNA replication progression and other nucleic acid metabolizing events.
Interestingly, certain topoisomerase inhibitors act by making toxic covalent DNA-topoisomerase protein complexes182, approximating the tight (but noncovalent) PARP-DNA complexes. In a broader sense, it is of growing interest to ascertain the role of DPCs in the mechanism of action for certain chemotherapy drugs and cellular proteases (e.g., SPRTN183) that act to degrade the DNA-interacting protein. Presumably, DPCs are a source of replication stress. As noted earlier, a hereditary premature aging disorder known as Ruijs-Aalfs syndrome is characterized by mutant alleles of the SPRTN protease implicated in DPC digestion53,54 (Table 2). While studies suggested SPRTN acts as a DPC protease specifically at replication forks184,185, a recent work provided evidence that SPRTN patient variants are defective in global genome DPC cleavage183. It will be of great interest to ascertain if SPRTN and other factors implicated in DPC repair (e.g., FANCJ helicase mutated in FA186) mediate functions in replication stress paramount to the suppression of aging phenotypes.
All of the scenarios mentioned above pose a challenge to cancer biologists to comprehend the dominant mechanism of action of senescence-causing drugs. What was once thought to be classic DNA damage therapy now entails replicative stress as a major component of not only the anti-cancer properties but also the potential age-associated symptoms experienced by many cancer patients undergoing chemotherapy treatments.
Senolytic drugs targeting replicative senescent cells to combat aging
As expertly summarized in recent reviews4,187, an opportunistic approach to extending health span is to target senescent cell viability with a new and emerging class of compounds known as senolytic drugs. The principle is simple: elimination of senescent cells by senolytic drugs would suppress the SASP, which is directly implicated in aging and age-related diseases. Alternatively, senomorphic drugs act to suppress cell-extrinsic effects of senescent cells (rather than eliminate senescent cells altogether); a good example is the SASP inhibitors. Senotherapeutics are being used to target adipose tissue, bone, skeletal muscle, brain, liver, and the cardiovascular system.
While DNA damage-induced senescent cells that elicit inflammatory pathways due to accumulation of cytosolic nucleic acids are frequently targeted by senotherapeutic drugs, the question arises: Can cells characterized by elevated replication stress be an optimal target of senolytic drugs? In other words, can senescence be prevented by targeting the heightened replication stress that may precede the DNA damage? Or does replication stress represent a biomarker for senolytic drugs? The fact that DNA damage drives cellular senescence prompts consideration of potential therapeutic strategies that exploit genetic and environmental factors to selectively eliminate cells that age prematurely (senescence) due to aberrant fork progression leading to DNA breaks, thereby keeping the level of SASP under control. It is plausible that characteristics of replicative stress might serve as novel biomarkers for senescence upstream of many of the cellular properties used to detect senescence. The activated signaling pathways (e.g., chemokines, interleukins, inflammatory factors, proteases, etc.) may ultimately derive from compromised replicative synthesis which may serve as a direct signal or be transmitted to the cellular machinery via DNA damage such as single-stranded and double-stranded DNA breaks.
Drug screening to identify classes of senolytic drugs that act to target cells that display compromised replicative DNA synthesis may prove to be informative and valuable, as replicative stress conceivably precedes genomic DNA damage in many cases. The targeted mechanism of action for drugs that target cells experiencing replication stress would well precede cell signaling pathways that may be less efficacious or pose undesired drug cytotoxicity toward normal cells.
From a more clinical viewpoint, collective evidence demonstrates that senescent cells that become enriched with age are found to be highly abundant at actual diseased sites (tissues, organs)188. The idea that senescence drives aging and age-related diseases in a broad spectrum of tissues including adipose, bone, skeletal muscle, brain, cardiovascular system, and liver is nicely summarized in a 2021 review by Robbins et al.4. A working model has arisen that reduction of senescent cell viability via senolytic drugs that kill senescent cells or senomorphic/senostatic drugs that suppress disease-causing phenotypes of senescent cells offers mechanistic approaches to extend health span. With many new and ongoing clinical trials for senotherapeutics underway, it is prudent to carefully study their mechanisms of action to ascertain if classes of drugs preferentially kill cells experiencing replication stress-induced senescence.
Summary
Replication stress has emerged as a central focus in eukaryotic chromosome biology and a driving force in hereditary diseases characterized by genomic instability and features of accelerated aging. Building upon these seminal human genetic studies, molecular biologists and nucleic acid biochemists have discovered many of the molecular mechanisms underlying replication stress that not only compromise the speed of DNA synthesis but also assault replication fork integrity. Both endogenously folded DNA structures such as G4s as well as exogenously induced DNA damage caused by environmental stress can induce replication stress; moreover, genetic deficiency of fork protection factors exacerbates the destabilization of replication forks. Combined, chemically induced and genetic synthetic lethality may result.
There is great interest in mechanisms whereby replication stress is suppressed or the outcomes induced by replication stress can be rescued (Fig. 3), as discussed in this review. A key DNA insult arising from replication stress is the DSB, which has been shown to cause epigenetic changes and drive cellular senescence and aging in elegant biological systems; resetting altered epigenetic landscapes caused by DNA damage to combat aging is gaining support. In addition to hereditary premature aging disorders, replication stress can also cause organ decline associated with normal aging.

The upper third of the hexagon depicts two mechanisms to avoid replication stress. The lower two-thirds of the hexagon depicts four strategies that alleviate the consequences of replication stress. See text for details. BioRender was used to create the figure.
As mentioned above, chemotherapeutic senescence-causing drugs or radiation exert negative cytotoxic side effects for normal cells and tissues, prompting new avenues of basic science and clinical research. For example, there is discussion of whether circadian clock genes can act to suppress tumors and if chronochemotherapy can be optimized189. Cancer cells have acquired mechanisms to withstand chemotherapy drug-induced replication stress, and transcriptional programs are proposed to serve as biomarkers and potential targets190. Combinations of chemotherapy drugs and immunotherapies have risen in the clinic191.
Replicative senescence has emerged as a biomarker for aging and age-related diseases. An ever-expanding body of evidence suggests that replication stress is the driving element in SC decline, particularly in the hematopoietic compartment. Thus, restoring the SC pool remains a viable option for some genetic diseases. As discussed on the Fanconi Anemia Research Fund (FARF) website, allogeneic HSC transplantation is the one durable cure for blood deficiencies in FA (Treatment | Fanconi Anemia Research Fund).
On the new frontier to promote healthy aging lie senolytic drugs that eliminate replicative senescent cells altogether. As the scientific and lay communities were poised to celebrate DNA repair in the 1990’s192 (Contents | Science 266, 5193), the 21st century brings robust interest in the replication stress response anticipated to be accompanied by new advances in medicine to improve cancer and aging outcomes.
References
Saxena, S. & Zou, L. Hallmarks of DNA replication stress. Mol. cell 82, 2298–2314 (2022).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Keijzers, G., Bakula, D. & Scheibye-Knudsen, M. Monogenic diseases of DNA repair. N. Engl. J. Med. 378, 491–492 (2018).
Robbins, P. D. et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev. Pharm. Toxicol. 61, 779–803 (2021).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Shay, J. W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 6, 584–593 (2016).
Goodell, M. A. & Rando, T. A. Stem cells and healthy aging. Science 350, 1199–1204 (2015).
Flach, J. et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014).
White, R. R. et al. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun. 6, 6790 (2015).
White, R. R. & Vijg, J. Do DNA double-strand breaks drive aging. Mol. Cell 63, 729–738 (2016).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-020-00314-w (2020).
Yang, J. H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326.e327 (2023).
Mognato, M., Burdak-Rothkamm, S. & Rothkamm, K. Interplay between DNA replication stress, chromatin dynamics and DNA-damage response for the maintenance of genome stability. Mutat. Res Rev. Mutat. Res. 787, 108346 (2021).
Saul, D. & Kosinsky, R. L. Epigenetics of aging and aging-associated diseases. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22010401 (2021).
Simon, M. et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 29, 871–885.e875 (2019).
Hämäläinen, R. H. et al. Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nat. Metab. 1, 958–965 (2019).
van der Rijt, S., Molenaars, M., McIntyre, R. L., Janssens, G. E. & Houtkooper, R. H. Integrating the hallmarks of aging throughout the Tree of Life: a focus on mitochondrial dysfunction. Front. Cell Dev. Biol. 8, 594416 https://doi.org/10.3389/fcell.2020.594416 (2020).
Ragu, S., Matos-Rodrigues, G. & Lopez, B. S. Replication Stress, DNA Damage, inflammatory cytokines and innate immune response. Genes 11. https://doi.org/10.3390/genes11040409 (2020).
Rieckher, M., Garinis, G. A. & Schumacher, B. Molecular pathology of rare progeroid diseases. Trends Mol. Med. 27, 907–922 (2021).
Brosh, R. M. Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 13, 542–558 (2013).
Datta, A. et al. Discovery of a new hereditary RECQ helicase disorder RECON syndrome positions the replication stress response and genome homeostasis as centrally important processes in aging and age-related disease. Ageing Res. Rev. 86, 101887 (2023).
Estep, K. N. & Brosh, R. M. Jr. RecQ and Fe-S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochem. Soc. Trans. 46, 77–95 (2018).
Chang, S. et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 36, 877–882 (2004).
Crabbe, L., Verdun, R. E., Haggblom, C. I. & Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953 (2004).
Du, X. et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol. Cell. Biol. 24, 8437–8446 (2004).
Drosopoulos, W. C., Kosiyatrakul, S. T. & Schildkraut, C. L. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J. Cell Biol. 210, 191–208 (2015).
Pan, X. et al. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl Acad. Sci. USA 114, E5940–e5949 (2017).
Zhang, J. M., Genois, M. M., Ouyang, J., Lan, L. & Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 81, 1027–1042.e1024 (2021).
Datta, A., Dhar, S., Awate, S. & Brosh, R. M. Jr. Synthetic lethal interactions of RECQ helicases. Trends Cancer 7, 146–161 (2021).
Datta, A. & Brosh, R. M. Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 5, 59 (2018).
Chan, E. M., Foster, K. J. & Bass, A. J. WRN Is a promising synthetic lethal target for cancers with microsatellite instability (MSI). Cancer Treat. Res. 186, 313–328 (2023).
Aggarwal, M., Banerjee, T., Sommers, J. A. & Brosh, R. M. Jr. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 12, 3329–3335 (2013).
Aggarwal, M. et al. Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional Fanconi Anemia pathway. Cancer Res. 73, 5497–5507 (2013).
Aggarwal, M., Sommers, J. A., Shoemaker, R. H. & Brosh, R. M. Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl Acad. Sci. USA 108, 1525–1530 (2011).
Datta, A. et al. WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nat. Commun. 12, 6561 (2021).
Peake, J. D. & Noguchi, E. Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum. Genet. 141, 1811–1836 (2022).
Pang, Q. & Andreassen, P. R. Fanconi anemia proteins and endogenous stresses. Mutat. Res. 668, 42–53 (2009).
Thada, V. & Greenberg, R. A. Unpaved roads: how the DNA damage response navigates endogenous genotoxins. DNA Repair 118, 103383 (2022).
Pontel, L. B. et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 60, 177–188 (2015).
Rosado, I. V., Langevin, F., Crossan, G. P., Takata, M. & Patel, K. J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 (2011).
Wang, M. et al. Genotoxic aldehyde stress prematurely ages hematopoietic stem cells in a p53-driven manner. Mol. Cell 83, 2417–2433.e2417 (2023).
Savage, S. A. Dyskeratosis congenita and telomere biology disorders. Hematol. Am. Soc. Hematol. Educ. Program 2022, 637–648 (2022).
Dreesen, O. Towards delineating the chain of events that cause premature senescence in the accelerated aging syndrome Hutchinson-Gilford progeria (HGPS). Biochem. Soc. Trans. 48, 981–991 (2020).
Wheaton, K. et al. Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford Progeria Syndrome. Mol. Cell. Biol. 37 https://doi.org/10.1128/mcb.00659-16 (2017).
Kreienkamp, R. et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by Progerin. Cell Rep. 22, 2006–2015 (2018).
Aguado, J. et al. The hallmarks of aging in Ataxia-Telangiectasia. Ageing Res. Rev. 79, 101653 (2022).
O’Driscoll, M., Gennery, A. R., Seidel, J., Concannon, P. & Jeggo, P. A. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair 3, 1227–1235 (2004).
Paccosi, E., Balajee, A. S. & Proietti-De-Santis, L. A matter of delicate balance: Loss and gain of Cockayne syndrome proteins in premature aging and cancer. Front. Aging 3, 960662 (2022).
van der Weegen, Y. et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 11, 2104 (2020).
Kokic, G., Wagner, F. R., Chernev, A., Urlaub, H. & Cramer, P. Structural basis of human transcription-DNA repair coupling. Nature 598, 368–372 (2021).
Batenburg, N. L. et al. Cockayne syndrome group B protein regulates fork restart, fork progression and MRE11-dependent fork degradation in BRCA1/2-deficient cells. Nucleic Acids Res. 49, 12836–12854 (2021).
Feng, E. et al. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 133 https://doi.org/10.1242/jcs.234914 (2020).
Lessel, D. et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 46, 1239–1244 (2014).
Maskey, R. S. et al. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 5, 5744 (2014).
Stingele, J. et al. Mechanism and regulation of DNA-Protein crosslink repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 64, 688–703 (2016).
Mórocz, M. et al. DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res. 45, 3172–3188 (2017).
Halder, S. et al. SPRTN protease and checkpoint kinase 1 cross-activation loop safeguards DNA replication. Nat. Commun. 10, 3142 (2019).
Alkhunaizi, E., Brosh, R. M. Jr., Alkuraya, F. S. & Chitayat, D. Warsaw Syndrome. In GeneReviews® [Internet] (eds Adam, M. P. et al.) (University of Washington, 1993–2024).
Cali, F., Bharti, S. K., Di Perna, R., Brosh, R. M. Jr. & Pisani, F. M. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res. 44, 705–717 (2016).
Jegadesan, N. K. & Branzei, D. DDX11 loss causes replication stress and pharmacologically exploitable DNA repair defects. Proc. Natl Acad. Sci. USA 118 https://doi.org/10.1073/pnas.2024258118 (2021).
Lerner, L. K. et al. Timeless couples G-quadruplex detection with processing by DDX11 helicase during DNA replication. EMBO J. 39, e104185 (2020).
van Schie, J. J. M. et al. Warsaw Breakage Syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat. Commun. 11, 4287 (2020).
Wu, Y., Sommers, J. A., Khan, I., de Winter, J. P. & Brosh, R. M. Jr. Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 287, 1007–1021 (2012).
Burhans, W. C. & Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 35, 7545–7556 (2007).
Yousefzadeh, M. et al. DNA damage-how and why we age? eLife 10 https://doi.org/10.7554/eLife.62852 (2021).
Sirbu, B. M., Couch, F. B. & Cortez, D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 7, 594–605 (2012).
Roy, S., Luzwick, J. W. & Schlacher, K. SIRF: quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 217, 1521–1536 (2018).
Abu-Libdeh, B. et al. RECON syndrome is a genome instability disorder caused by mutations in the DNA helicase RECQL1. J. Clin. Investiga. 132 https://doi.org/10.1172/jci147301 (2022).
Berti, M. et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354 (2013).
Thangavel, S. et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562 (2015).
Davies, S. L., North, P. S. & Hickson, I. D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 14, 677–679 (2007).
Pichierri, P., Franchitto, A. & Rosselli, F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23, 3154–3163 (2004).
Heo, S. J. et al. Bloom’s syndrome gene suppresses premature ageing caused by Sgs1 deficiency in yeast. Genes Cells 4, 619–625 (1999).
Iannascoli, C., Palermo, V., Murfuni, I., Franchitto, A. & Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 43, 9788–9803 (2015).
Su, F. et al. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 9, 1387–1401 (2014).
Datta, A. & Brosh, R. M. Jr. WRN rescues replication forks compromised by a BRCA2 deficiency: predictions for how inhibition of a helicase that suppresses premature aging tilts the balance to fork demise and chromosomal instability in cancer. BioEssays 44, e2200057 (2022).
Opresko, P. L. et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell 14, 763–774 (2004).
Im, J. S. et al. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl Acad. Sci. USA 106, 15628–15632 (2009).
Xu, X., Rochette, P. J., Feyissa, E. A., Su, T. V. & Liu, Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 28, 3005–3014 (2009).
Xu, X., Chang, C. W., Li, M., Liu, C. & Liu, Y. Molecular mechanisms of the RECQ4 pathogenic mutations. Front. Mol. Biosci. 8, 791194 (2021).
Chen, E. et al. RECQL5 suppresses oncogenic JAK2-induced replication stress and genomic instability. Cell Rep. 13, 2345–2352 (2015).
Kim, T. M. et al. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 43, 893–903 (2015).
Di Marco, S. et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at Common Fragile Sites during Mitosis. Mol. Cell 66, 658–671.e658 (2017).
Urban, V. et al. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 214, 401–415 (2016).
Peng, M. et al. Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep. 24, 3251–3261 (2018).
Brosh, R. M. Jr. & Wu, Y. An emerging picture of FANCJ’s role in G4 resolution to facilitate DNA replication. NAR Cancer 3, zcab034 (2021).
Gari, K., Decaillet, C., Delannoy, M., Wu, L. & Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl Acad. Sci. USA 105, 16107–16112 (2008).
Gari, K., Decaillet, C., Stasiak, A. Z., Stasiak, A. & Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 29, 141–148 (2008).
Panday, A. et al. FANCM regulates repair pathway choice at stalled replication forks. Mol. Cell 81, 2428–2444.e2426 (2021).
Schwab, R. A., Blackford, A. N. & Niedzwiedz, W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 29, 806–818 (2010).
Joseph, S. A. et al. Time for remodeling: SNF2-family DNA translocases in replication fork metabolism and human disease. DNA Repair 95, 102943 (2020).
Bryant, H. E. et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009).
Cotta-Ramusino, C. et al. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 17, 153–159 (2005).
Yeo, J. E., Lee, E. H., Hendrickson, E. A. & Sobeck, A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet. 23, 3695–3705 (2014).
Schlacher, K. et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011).
Franchitto, A. et al. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 183, 241–252 (2008).
Hanada, K. et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 14, 1096–1104 (2007).
Hashimoto, Y., Ray Chaudhuri, A., Lopes, M. & Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311 (2010).
Schlacher, K., Wu, H. & Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012).
Lemaçon, D. et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017).
Ray Chaudhuri, A. et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016).
Jiang, Q. et al. Autologous K63 deubiquitylation within the BRCA1-A complex licenses DNA damage recognition. J. Cell Biol. 221. https://doi.org/10.1083/jcb.202111050 (2022).
Jiang, Q. et al. MERIT40 cooperates with BRCA2 to resolve DNA interstrand cross-links. Genes Dev. 29, 1955–1968 (2015).
Jones, M. J. K. et al. Human DDK rescues stalled forks and counteracts checkpoint inhibition at unfired origins to complete DNA replication. Mol. Cell 81, 426–441.e428 (2021).
Higgs, M. R. et al. Histone methylation by SETD1A protects nascent DNA through the nucleosome chaperone activity of FANCD2. Mol. Cell 71, 25–41.e26 (2018).
Thakar, T. et al. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 11, 2147 (2020).
Kim, J. J. et al. PCAF-mediated Hhstone acetylation promotes replication fork degradation by MRE11 and EXO1 in BRCA-deficient cells. Mol. Cell 80, 327–344.e328 (2020).
Di Micco, R. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 (2006).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Aguado, J. et al. Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell 20, e13468 (2021).
Emam, A. et al. Stalled replication fork protection limits cGAS-STING and P-body-dependent innate immune signalling. Nat. Cell Biol. 24, 1154–1164 (2022).
Coquel, F. et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018).
Wolf, C. et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 7, 11752 (2016).
Pinto, M. et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 24, 288–299 (2017).
Zuccaro, M. V. et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell 183, 1650–1664.e1615 (2020).
Barroso-Vilares, M. et al. Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep. 21, e49248 (2020).
Shah, P. et al. Nuclear deformation causes DNA damage by increasing replication stress. Curr. Biol. 31, 753–765.e756 (2021).
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T. & Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786.e719 (2017).
Said, M. et al. FANCD2 promotes mitotic rescue from transcription-mediated replication stress in SETX-deficient cancer cells. Commun. Biol. 5, 1395 (2022).
Liu, Z. et al. San1 deficiency leads to cardiomyopathy due to excessive R-loop-associated DNA damage and cardiomyocyte hypoplasia. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166237 (2021).
Palmerola, K. L. et al. Replication stress impairs chromosome segregation and preimplantation development in human embryos. Cell 185, 2988–3007.e2920 (2022).
Wilhelm, T. et al. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 10, 3585 (2019).
Bétous, R. et al. DNA replication stress triggers rapid DNA replication fork breakage by Artemis and XPF. PLoS Genet. 14, e1007541 (2018).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Margalef, P. et al. Stabilization of reversed replication forks by telomerase drives telomere catastrophe. Cell 172, 439–453.e414 (2018).
Sfeir, A. et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009).
Coluzzi, E., Leone, S. & Sgura, A. Oxidative stress induces telomere dysfunction and senescence by replication fork arrest. Cells 8. https://doi.org/10.3390/cells8010019 (2019).
Fouquerel, E. et al. Targeted and persistent 8-Oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell 75, 117–130.e116 (2019).
Vannier, J. B. et al. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239–242 (2013).
Cicconi, A. & Chang, S. Shelterin and the replisome: at the intersection of telomere repair and replication. Curr. Opin. Genet. Dev. 60, 77–84 (2020).
d’Alcontres, M. S., Palacios, J. A., Mejias, D. & Blasco, M. A. TopoIIα prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle 13, 1463–1481 (2014).
Maresca, C. et al. PARP1 allows proper telomere replication through TRF1 poly (ADP-ribosyl)ation and helicase recruitment. Commun. Biol. 6, 234 (2023).
Leman, A. R. et al. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 11, 2337–2347 (2012).
Rai, R. et al. The replisome mediates A-NHEJ repair of telomeres lacking POT1-TPP1 independently of MRN function. Cell Rep. 29, 3708–3725.e3705 (2019).
Bianco, J. N. et al. Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun. 10, 910 (2019).
Drosopoulos, W. C. et al. TRF2 mediates replication initiation within human telomeres to prevent telomere dysfunction. Cell Rep. 33, 108379 (2020).
Higa, M. et al. TRF2-mediated ORC recruitment underlies telomere stability upon DNA replication stress. Nucleic Acids Res. 49, 12234–12251 (2021).
Nie, X. et al. TRF2 recruits nucleolar protein TCOF1 to coordinate telomere transcription and replication. Cell Death Differ. 28, 1062–1075 (2021).
Cicconi, A. et al. Microcephalin 1/BRIT1-TRF2 interaction promotes telomere replication and repair, linking telomere dysfunction to primary microcephaly. Nat. Commun. 11, 5861 (2020).
Suram, A. et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 31, 2839–2851 (2012).
Günes, C. & Rudolph, K. L. Telomere dysfunction puts the brakes on oncogene-induced cancers. EMBO J. 31, 2833–2834 (2012).
Li, L. & Xie, T. Stem cell niche: structure and function. Annu Rev. Cell Dev. Biol. 21, 605–631 (2005).
Rossi, D. J. et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 (2007).
Ruzankina, Y., Asare, A. & Brown, E. J. Replicative stress, stem cells and aging. Mech. Ageing Dev. 129, 460–466 (2008).
Walter, D. et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 (2015).
Ruzankina, Y. et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 (2007).
Datta, A. & Brosh, R. M., Jr. Holding All the Cards-How Fanconi Anemia proteins deal with replication stress and preserve genomic stability. Genes 10 https://doi.org/10.3390/genes10020170 (2019).
Brack, A. S., Bildsoe, H. & Hughes, S. M. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J. Cell Sci. 118, 4813–4821 (2005).
Lee, J., Yoon, S. R., Choi, I. & Jung, H. Causes and mechanisms of hematopoietic stem cell aging. International Journal of Molecular Sciences 20 https://doi.org/10.3390/ijms20061272 (2019).
Shevyrev, D., Tereshchenko, V., Berezina, T. N. & Rybtsov, S. Hematopoietic stem cells and the immune system in development and aging. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24065862 (2023).
Vijg, J. & Campisi, J. Puzzles, promises and a cure for ageing. Nature 454, 1065–1071 (2008).
Ruiz, S. et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 6, 8036 (2015).
Zhang, H. et al. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol. Cell 65, 272–284 (2017).
Zhang, R. et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8, 19–30 (2005).
Di Micco, R. et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 13, 292–302 (2011).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Trifunovic, A. et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl Acad. Sci. USA 102, 17993–17998 (2005).
Vermulst, M. et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 40, 392–394 (2008).
Martin, G. M. & Loeb, L. A. Ageing: mice and mitochondria. Nature 429, 357–359 (2004).
Sharma, S. et al. Proofreading deficiency in mitochondrial DNA polymerase does not affect total dNTP pools in mouse embryos. Nat. Metab. 2, 673–675 (2020).
Venkatachalam, G., Surana, U. & Clément, M. V. Replication stress-induced endogenous DNA damage drives cellular senescence induced by a sub-lethal oxidative stress. Nucleic Acids Res. 45, 10564–10582 (2017).
Kudlova, N., De Sanctis, J. B. & Hajduch, M. Cellular senescence: molecular targets, biomarkers, and senolytic drugs. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms23084168 (2022).
González-Acosta, D. & Lopes, M. DNA replication and replication stress response in the context of nuclear architecture. Chromosoma https://doi.org/10.1007/s00412-023-00813-7 (2023).
Burd, C. E. et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152, 340–351 (2013).
Safwan-Zaiter, H., Wagner, N. & Wagner, K. D. P16INK4A-more than a senescence marker. Life 12 https://doi.org/10.3390/life12091332 (2022).
Jiang, D. et al. Post-GWAS functional analysis identifies CUX1 as a regulator of p16(INK4a) and cellular senescence. Nat. Aging 2, 140–154 (2022).
Lopez-Contreras, A. J. et al. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 29, 690–695 (2015).
Bester, A. C. et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 (2011).
Danilova, N. et al. The role of the DNA damage response in zebrafish and cellular models of Diamond Blackfan anemia. Dis. Model Mech. 7, 895–905 (2014).
Albers, E., Avram, A., Sbroggio, M., Fernandez-Capetillo, O. & Lopez-Contreras, A. J. Supraphysiological protection from replication stress does not extend mammalian lifespan. Aging 12, 5612–5624 (2020).
Tacconi, E. M. et al. BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol. Med. 9, 1398–1414 (2017).
Pilzecker, B. et al. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl Acad. Sci. USA 114, E6875–e6883 (2017).
McNeely, T., Leone, M., Yanai, H. & Beerman, I. DNA damage in aging, the stem cell perspective. Hum. Genet. 139, 309–331 (2020).
Dimri, G. P. & Campisi, J. Molecular and cell biology of replicative senescence. Cold Spring Harb. Symp. Quant. Biol. 59, 67–73 (1994).
Maldonado, E., Morales-Pison, S., Urbina, F. & Solari, A. Aging hallmarks and the role of oxidative stress. Antioxidants 12 https://doi.org/10.3390/antiox12030651 (2023).
Schaffer, E. D., Beerman, I., de Cabo, R. & Brosh, R. M. Jr. Frontiers in aging special issue: DNA repair and interventions in aging perspective on “loss of epigenetic information as a cause of mammalian aging”. Front. Aging 4, 1199596 (2023).
Saleh, T. et al. Therapy-induced senescence: an “Old” friend becomes the enemy. Cancers 12 https://doi.org/10.3390/cancers12040822 (2020).
van den Boogaard, W. M. C., Komninos, D. S. J. & Vermeij, W. P. Chemotherapy side-effects: not all DNA damage Is equal. Cancers 14 https://doi.org/10.3390/cancers14030627 (2022).
Kim, C., Chen, C. & Yu, Y. Avoid the trap: Targeting PARP1 beyond human malignancy. Cell Chem. Biol. 28, 456–462 (2021).
Sun, Y., Saha, L. K., Saha, S., Jo, U. & Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair 94, 102926 (2020).
Weickert, P. et al. SPRTN patient variants cause global-genome DNA-protein crosslink repair defects. Nat. Commun. 14, 352 (2023).
Larsen, N. B. et al. Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Mol. Cell 73, 574–588.e577 (2019).
Ruggiano, A. et al. The protease SPRTN and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. Cell Rep. 37, 110080 (2021).
Yaneva, D. et al. The FANCJ helicase unfolds DNA-protein crosslinks to promote their repair. Mol. Cell 83, 43–56.e10 (2023).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Naylor, R. M., Baker, D. J. & van Deursen, J. M. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin. Pharm. Ther. 93, 105–116 (2013).
Sancar, A. & Van Gelder, R. N. Clocks, cancer, and chronochemotherapy. Science 371 https://doi.org/10.1126/science.abb0738 (2021).
Segeren, H. A. & Westendorp, B. Mechanisms used by cancer cells to tolerate drug-induced replication stress. Cancer Lett. 544, 215804 (2022).
Barros, E. M., McIntosh, S. A. & Savage, K. I. The DNA damage induced immune response: implications for cancer therapy. DNA Repair 120, 103409 (2022).
Koshland, D. E. Jr. Molecule of the year: the DNA repair enzyme. Science 266, 1925 (1994).
Kumar, C. & Remus, D. Looping out of control: R-loops in transcription-replication conflict. Chromosoma https://doi.org/10.1007/s00412-023-00804-8. (2023).
Howell, R. M., Woodford, K. J., Weitzmann, M. N. & Usdin, K. The chicken beta-globin gene promoter forms a novel “cinched” tetrahelical structure. J. Biol. Chem. 271, 5208–5214 (1996).
Mellor, C., Perez, C. & Sale, J. E. Creation and resolution of non-B-DNA structural impediments during replication. Crit. Rev. Biochem. Mol. Biol. 57, 412–442 (2022).
Spiegel, J., Adhikari, S. & Balasubramanian, S. The Structure and function of DNA G-quadruplexes. Trends Chem. 2, 123–136 (2020).
Williams, S. L. et al. Replication-induced DNA secondary structures drive fork uncoupling and breakage. EMBO J. e114334 https://doi.org/10.15252/embj.2023114334 (2023).
Woodford, K. J., Howell, R. M. & Usdin, K. A novel K(+)-dependent DNA synthesis arrest site in a commonly occurring sequence motif in eukaryotes. J. Biol. Chem. 269, 27029–27035 (1994).
Zeraati, M. et al. I-motif DNA structures are formed in the nuclei of human cells. Nat. Chem. 10, 631–637 (2018).
Barnes, R. P., Thosar, S. A. & Opresko, P. L. Telomere fragility and MiDAS: managing the gaps at the end of the road. Genes 14 https://doi.org/10.3390/genes14020348 (2023).
Griffith, J. D. et al. Mammalian telomeres end in a large duplex loop. Cell 97, 503–514 (1999).
Andrs, M. et al. Excessive reactive oxygen species induce transcription-dependent replication stress. Nat. Commun. 14, 1791 (2023).
Burgos-Barragan, G. et al. Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548, 549–554 (2017).
Klages-Mundt, N. L. & Li, L. Formation and repair of DNA-protein crosslink damage. Sci. China Life Sci. 60, 1065–1076 (2017).
Sparks, J. L. et al. The CMG helicase bypasses DNA-protein cross-links to facilitate their repair. Cell 176, 167–181.e121 (2019).
Stingele, J., Bellelli, R. & Boulton, S. J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 18, 563–573 (2017).
Byun, T. S., Pacek, M., Yee, M. C., Walter, J. C. & Cimprich, K. A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19, 1040–1052 (2005).
Taylor, M. R. G. & Yeeles, J. T. P. The initial response of a eukaryotic replisome to DNA damage. Mol. Cell 70, 1067–1080.e1012 (2018).
Cordeiro-Stone, M., Boyer, J. C., Smith, B. A. & Kaufmann, W. K. Xeroderma pigmentosum variant and normal fibroblasts show the same response to the inhibition of DNA replication by benzo[a]pyrene-diol-epoxide-I. Carcinogenesis 7, 1783–1786 (1986).
Cordeiro-Stone, M., Boyer, J. C., Smith, B. A. & Kaufmann, W. K. Effect of benzo[a]pyrene-diol-epoxide-I on growth of nascent DNA in synchronized human fibroblasts. Carcinogenesis 7, 1775–1781 (1986).
Vare, D. et al. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA Repair 11, 976–985 (2012).
Drabløs, F. et al. Alkylation damage in DNA and RNA–repair mechanisms and medical significance. DNA Repair 3, 1389–1407 (2004).
Gu, L., Hickey, R. J. & Malkas, L. H. Therapeutic targeting of DNA replication stress in cancer. Genes 14 https://doi.org/10.3390/genes14071346 (2023).
Barlow, J. H. et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 (2013).
Musiałek, M. W. & Rybaczek, D. Hydroxyurea-The Good, the Bad and the Ugly. Genes 12 https://doi.org/10.3390/genes12071096 (2021).
Ray Chaudhuri, A. et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 19, 417–423 (2012).
Berdis, A. J. Inhibiting DNA polymerases as a therapeutic intervention against cancer. Front. Mol. Biosci. 4, 78 (2017).
Savitsky, K. et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753 (1995).
Aird, K. M. et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 11, 893–901 (2015).
Shiloh, Y. & Lederman, H. M. Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res. Rev. 33, 76–88 (2017).
Oh, J., Lee, S. J., Rothstein, R. & Symington, L. S. Xrs2 and Tel1 independently contribute to MR-mediated DNA tethering and replisome stability. Cell Rep. 25, 1681–1692.e1684 (2018).
Ellis, N. A. et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 (1995).
Hand, R. & German, J. A retarded rate of DNA chain growth in Bloom’s syndrome. Proc. Natl Acad. Sci. USA 72, 758–762 (1975).
Hand, R. & German, J. Bloom’s syndrome: DNA replication in cultured fibroblasts and lymphocytes. Hum. Genet. 38, 297–306 (1977).
Lönn, U., Lönn, S., Nylen, U., Winblad, G. & German, J. An abnormal profile of DNA replication intermediates in Bloom’s syndrome. Cancer Res. 50, 3141–3145 (1990).
Sidorova, J. M., Kehrli, K., Mao, F. & Monnat, R. Jr. Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair 12, 128–139 (2013).
Davies, S. L., North, P. S., Dart, A., Lakin, N. D. & Hickson, I. D. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell. Biol. 24, 1279–1291 (2004).
Mao, F. J., Sidorova, J. M., Lauper, J. M., Emond, M. J. & Monnat, R. J. The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res. 70, 6548–6555 (2010).
Barefield, C. & Karlseder, J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 40, 7358–7367 (2012).
Henning, K. A. et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82, 555–564 (1995).
Troelstra, C. et al. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 71, 939–953 (1992).
Walker, J. R. & Zhu, X. D. Role of Cockayne Syndrome Group B protein in replication stress: implications for cancer therapy. Int. J. Mol. Sci. 23. https://doi.org/10.3390/ijms231810212 (2022)
Cui, S., Walker, J. R., Batenburg, N. L. & Zhu, X. D. Cockayne syndrome group B protein uses its DNA translocase activity to promote mitotic DNA synthesis. DNA Repair 116, 103354 (2022).
Grill, S. & Nandakumar, J. Molecular mechanisms of telomere biology disorders. J. Biol. Chem. 296, 100064 (2021).
Savage, S. A. & Niewisch, M. R. Dyskeratosis Congenita and Related Telomere Biology Disorders. In GeneReviews® [Internet]. (eds Adam, M. P. et al.) (University of Washington, 1993–2023).
Björkman, A. et al. Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes Dev. 34, 1065–1074 (2020).
Takedachi, A. et al. SLX4 interacts with RTEL1 to prevent transcription-mediated DNA replication perturbations. Nat. Struct. Mol. Biol. 27, 438–449 (2020).
Audry, J. et al. RPA prevents G-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 34, 1942–1958 (2015).
Gu, P. et al. CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell 17, e12783 (2018).
Zimmermann, M., Kibe, T., Kabir, S. & de Lange, T. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 28, 2477–2491 (2014).
Xu, X. et al. Fanconi anemia proteins participate in a break-induced-replication-like pathway to counter replication stress. Nat. Struct. Mol. Biol. 28, 487–500 (2021).
Eriksson, M. et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003).
Gonzalez-Suarez, I. et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 28, 2414–2427 (2009).
Decker, M. L., Chavez, E., Vulto, I. & Lansdorp, P. M. Telomere length in Hutchinson-Gilford progeria syndrome. Mech. Ageing Dev. 130, 377–383 (2009).
Singh, M. et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol. Cell. Biol. 33, 1210–1222 (2013).
Vaz, B. et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Mol. Cell 64, 704–719 (2016).
O’Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A. & Goodship, J. A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501 (2003).
Murga, M. et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 41, 891–898 (2009).
Casper, A. M., Durkin, S. G., Arlt, M. F. & Glover, T. W. Chromosomal instability at common fragile sites in Seckel syndrome. Am. J. Hum. Genet. 75, 654–660 (2004).
van der Lelij, P. et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 86, 262–266 (2010).
Cortone, G. et al. Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork-protection factor Timeless promotes sister chromatid cohesion. PLoS Genet. 14, e1007622 (2018).
Yu, C. E. et al. Positional cloning of the Werner’s syndrome gene. Science 272, 258–262 (1996).
Hanaoka, F. et al. Autoradiographic studies of DNA replication in Werner’s syndrome cells. Adv. Exp. Med. Biol. 190, 439–457 (1985).
Takeuchi, F. et al. Altered frequency of initiation sites of DNA replication in Werner’s syndrome cells. Hum. Genet. 60, 365–368 (1982).
Takeuchi, F., Hanaoka, F., Goto, M., Yamada, M. & Miyamoto, T. Prolongation of S phase and whole cell cycle in Werner’s syndrome fibroblasts. Exp. Gerontol. 17, 473–480 (1982).
Sidorova, J. M., Li, N., Folch, A. & Monnat, R. J. Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7, 796–807 (2008).
De Cian, A., Guittat, L., Shin-ya, K., Riou, J. F. & Mergny, J. L. Affinity and selectivity of G4 ligands measured by FRET. Nucleic Acids Symp. Ser. 235–236 https://doi.org/10.1093/nass/49.1.235 (2005).
Ganuza, M. et al. The global clonal complexity of the murine blood system declines throughout life and after serial transplantation. Blood 133, 1927–1942 (2019).
Goldberg, A. D. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 (2010).
Acknowledgements
This work was supported in full by the Intramural Research Program, National Institute on Aging, NIH. Biorender was used to create Figs. 2 and 3. We wish to thank the many scientists whose seminal discoveries have advanced the study of replication stress and its role in cellular senescence and aging and apologize to those whose work we were not able to cite due to the expansive field and manuscript length restrictions.
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
L.M.H., E.D.S., K.F.F., A.D., and R.M.B. wrote and edited text and constructed tables and figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Dana Branzei and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Valeria Naim and Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herr, L.M., Schaffer, E.D., Fuchs, K.F. et al. Replication stress as a driver of cellular senescence and aging. Commun Biol 7, 616 (2024). https://doi.org/10.1038/s42003-024-06263-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06263-w
This article is cited by
-
Low-Intensity Electric Field Stimulation Modulates Proliferation, Stemness, and Chondrogenesis of Tonsil-Derived Mesenchymal Stem Cells
Tissue Engineering and Regenerative Medicine (2026)
-
Canonical and non-canonical functions of the non-coding RNA component (TERC) of telomerase complex
Cell & Bioscience (2025)
-
Molecular cartography of the human down syndrome and trisomic mouse brain
Nature Communications (2025)
-
Regulation of endothelial cell senescence via the miR-217/FOXO3 axis
Biogerontology (2025)
-
Putrescine functions as a metabolic checkpoint in replication stress-induced senescence
Cellular and Molecular Life Sciences (2025)
