
Abstract
Chorion trophoblasts (CTCs) and immune cell-enriched decidua (DECs) comprise the maternal-fetal membrane interface called the chorio-decidual interface (CDi) which constantly gets exposed to maternal stressors without leading to labor activation. This study explored how CTCs act as a barrier at CDi. The roles of human leukocyte antigen (HLA)-G and progesterone receptor membrane component 2 (PGRMC2) in mediating immune homeostasis were also investigated. The CDi was recreated in a two-chamber microfluidic device (CDi-on-chip) with an outer chamber of primary DECs and immune cell line-derived innate immune cells and an inner chamber of wild-type or PGRMC2 or HLA-G knockout immortalized CTCs. To mimic maternal insults, DECs were treated with lipopolysaccharide, poly(I:C), or oxidative stress inducer cigarette smoke extract. Expression levels of inflammation and immunity genes via targeted RNA sequencing, production of soluble mediators, and immune cell migration into CTCs were determined. In CDi-on-chip, decidua and immune cells became inflammatory in response to insults while CTCs were refractory, highlighting their barrier function. HLA-G and PGRMC2 are found to be vital to immune homeostasis at the CDi, with PGRMC2 serving as an upstream regulator of inflammation, HLA-G expression, and mesenchymal-epithelial transition, and HLA-G serving as a frontline immunomodulatory molecule, thus preventing fetal membrane compromise.
Similar content being viewed by others


Introduction
Labor is an inflammatory process at the maternal-fetal interface characterized by leukocyte activation, migration, and recruitment, and a shift to a pro-inflammatory microenvironment1, leading to childbirth. Premature activation of these processes can lead to spontaneous preterm birth, a major complication of pregnancy with no known treatment2. The activation signals pathologically come from maternal exposure to various risk factors such as diet, environment, and lifestyle activities, and interact at the two maternal-fetal interfaces: the thoroughly investigated placenta-decidual interface at the placental disc and the often-forgotten chorio-decidual interface (CDi) at the fetal membranes. The latter is less understood since fetal membranes have been considered as a mere appendage of the placental disc providing only mechanical support to the fetus, but recent studies point to endocrine3,4, metabolic5, transport6,7,8,9,10 anti-microbial11, and immune12,13 functions.
The CDi consists of fetally derived chorion trophoblasts (CTCs) and immune cell-enriched maternal decidua (Fig. 1A). Insults into this interface can come from the maternal side, whether because of systemic viral or ascending bacterial infection or accumulation of reactive oxygen species (oxidative stress) from maternal lifestyle factors. Despite its proximity to the decidua and the absence of an extracellular matrix between the two layers, the chorion does not experience immune cell infiltration except during inflammation of the fetal membranes or chorioamnionitis. We postulate that chorion prevents such infiltration by acting as a barrier, providing structural integrity, and producing various endocrine and immune mediators14,15,16,17 including progesterone18, human leukocyte antigen (HLA)-G19,20, and anti-inflammatory interleukin (IL)-1021. In addition, CTCs do not express nuclear progesterone receptors22,23,24. Instead, like other fetal membrane cells, CTCs express progesterone receptor membrane components (PGRMC) 1 and 2. PGRMC2 in particular, which has been implicated in maintaining amnion integrity by promoting mesenchymal-epithelial transition (MET)25, is downregulated in fetal membranes during onset of labor at term and in preterm premature rupture of membranes18. Further investigation is required to understand the factors providing barrier functions at this maternal-fetal interface and how this structure is compromised.

A Conceptual framework of how the chorio-decidual interface responds to maternal insults. We hypothesize that the decidua and resident innate immune cells counter maternal insults by promoting inflammation, while the chorion produces immunoregulatory molecules such as HLA-G and PGRMC2. With intact barrier functions, the insult should resolve without causing fetal inflammatory response. When HLA-G or PGRMC2 is compromised, this should result in excessive inflammatory response that may lead to premature labor activation. B Schematic diagram of the chorio-decidual interface-on-chip and treatments. Both (A) and (B) are created with BioRender.com. C Immune cell subpopulations in the decidua parietalis at term prior to labor (n = 3) based on Mosebarger et al. 36. D Phase-contrast images of the CDi-on-chip. Yellow arrowheads point to cell line-derived immune cells. E Immunostaining of CD45 (red) and vimentin (green) expression of cells in the CDi-on-chip. DAPI (blue) was used for nuclear staining. Separation between two chambers is highlighted in yellow dashed circle. Inset images show migrating and migrated cell line-derived immune cells (yellow arrowheads). F Timelapse stills at a device microchannel with a cell line-derived immune cell (yellow arrowhead) migrating towards CTC. Scale bar for (D–F), 100 µm. ROS: reactive oxygen species, NK: natural killer, HLA-G: human leukocyte antigen G, PGRMC2: progesterone receptor membrane component 2, CTC: chorion trophoblast cell, DEC: decidual cell, CD45: cluster of differentiation 45, LPS: lipopolysaccharide, pIC: poly(I:C), mOS: maternal oxidative stress.
Various models are available to study physiological processes in pregnancy. Two-dimensional cultures of primary and immortalized cells have provided valuable insights into the molecular and cellular physiology of gestational cells. However, these models do not capture the architectural organization of cells and compartmentalization of various cell types seen with tissues26. While explants of fetal membranes preserve in vivo architecture27, conducting in vitro mechanistic studies on them is challenging. Mouse models permit pharmacologic and physiologic investigation into pregnancy and parturition. However, their relevance is limited by differences in anatomy and physiology during gestation—specifically, the chorion layer is not similar to that in humans28,29,30,31. Non-human primates can be used alternatively given the anatomic similarities of their fetal membranes to those of humans, but these models have long gestational periods and are costly to maintain, therefore preventing high-throughput studies32.
To develop an appropriate model to study the chorio-decidual innate immune interface and to investigate the role of PGRMC2 and HLA-G in mediating immune homeostasis at this interface, we recreated the CDi in a microfluidics-based microphysiological system (colloquially known as organ-on-chip) that allows for complex interactions between immortalized CTCs, primary decidual cells (DECs), and innate immune cells derived from immune cell lines. Using CRISPR-Cas9 gene editing technology, we explored the role of PGRMC2 or HLA-G in the intricacies of these intercellular interactions. We then profiled RNA expression of genes involved in inflammation and immunity and the production of cytokines and other soluble mediators in response to lipopolysaccharide (LPS), poly(I:C) (pIC), and cigarette smoke extract (CSE; a known oxidative stress inducer) (Fig. 1B). By incorporating PGRMC2 and HLA-G knockout CTCs in our microphysiological system, we report the utility of our model for mechanistic investigation.
Results
Design of chorio-decidual interface-on-chip (CDi-on-chip)
To model the chorio-decidual interface (Supplementary Fig. 1A), we repurposed a two-chamber microphysiological device previously used for amnion membrane-on-chip33 (Fig. 1B). This design reflects both the compartmentalization and interconnectedness of the fetal chorion and the maternal decidua parietalis that lines the chorion. We first seeded immortalized chorion trophoblast cells (Supplementary Fig. 1B) in the inner chamber (blue) and primary decidual stromal cells (Supplementary Fig. 1C) in the outer chamber (green) to allow cellular attachment. Immortalized CTCs were used due to their capacity to continually divide without prematurely becoming senescent, all while having comparable gene expression pattern to primary CTCs34. This was followed the next day by seeding innate immune cells and introducing treatments on the decidual side. For the innate immune cells, we used HL-60, THP-1, and NK-92 cell lines to differentiate into neutrophils, macrophages, and decidual NK cells, respectively (Supplementary Fig. 1D–G). This approach addressed sampling difficulties and inconsistencies, cell sorting, patient-to-patient variability, and short lifespan in vitro35. These differentiated immune cells were added in a 14:3:1 NK cell: macrophage: neutrophil ratio to reflect their population in the decidua in vivo (Fig. 1C)36. The cells maintained their cell morphology and cell-specific markers for at least 72 h, with immortalized CTCs appearing epithelioid, primary DECs appearing as vimentin-positive elongated stromal cells, and immune cells appearing as circular 10 µm CD45-positive cells (Fig. 1D). The two concentric elliptical chambers were linked by an array of microchannels that allowed intercellular interaction via exchange of paracrine mediators such as soluble molecules and extracellular vesicles37 between the two chambers as well as leukocyte trafficking and migration from the decidua into the chorion (Fig. 1E, F, Supplementary Movie 1).
Differential transcription of inflammation and immunity genes at the chorio-decidual interface
We performed targeted RNA sequencing to determine the baseline differences in the expression of genes involved in inflammation and immunity between immortalized CTCs and primary DECs. Principal component analysis (PCA) of CTCs and DECs with or without cell line-derived immune cells is shown in Fig. 2A, revealing distinct clustering between CTCs and DECs, amounting to 45.18% of the explained variance. As compared to CTCs, clustering was less tight in DECs. Since the primary DECs were from a single placenta and thus no inter-individual variability, the observed heterogeneity was possibly due to the presence of immune cells, although this amounts to only 16.28% of the explained variance. Heatmap of inflammation and immunity genes showed differential expression of markers between CTCs and DECs (Fig. 2B). Immortalized CTCs expressed non-classical HLA-G, trophoblast differentiation markers such as PPARG and GATA3, and damage-associated molecular pattern HMGB1. On the other hand, primary DECs expressed classical HLAs such as HLA-A and HLA-B, and HIF1A. To mimic insults coming from the maternal side, the outer decidual chamber was treated with LPS, pIC, or CSE to represent bacterial infection, viral infection, or maternal oxidative stress (mOS), respectively (Fig. 1B). These stimulants can diffuse through microchannels as validated by diffusion studies33, immunofluorescence staining38, and metabolomics analysis39. There were 33 unique downregulated genes (Supplementary Fig. 2A) and 131 unique upregulated genes (Supplementary Fig. 2B) in DECs. Of the differentially expressed genes, one downregulated and 19 upregulated genes were shared across all treatments. In response to LPS, there were 100 differentially expressed genes in primary DECs: 21 downregulated and 79 upregulated (Fig. 2C). The top downregulated genes were EOMES, SIGIRR, GZMA, JAK2, and PPIL2, while the top upregulated genes were HLA-DPA1, IL18R1, IL10RA, CXCL13 and OAS1. In response to pIC, 5 downregulated and 61 upregulated genes were in DECs (Fig. 2D). The top downregulated genes were IL17RB, CXCL12, MMP3, LRP1 and NR4A1, while the top upregulated genes were IGSF6, CXCL5, CXCL3, CCR3 and CXCL11. Lastly, there were 10 downregulated and 64 upregulated DEC genes in response to mOS (Fig. 2E). The top downregulated genes were CXCL12, SLIT2, LRP1, EGR1, and FOS, while the top upregulated genes were BMP7, NOS2, ACKR2, CCR7 and TLR7. In contrast, only at most 3 differentially expressed genes were unique in immortalized CTCs in any treatment group. JUN, CCL4, and CCL3 were the only differentially expressed genes in immortalized CTCs in response to LPS, and JUN, FOS, and EGR1 in response to mOS. No differentially expressed genes were seen in immortalized CTCs in response to pIC.

A Principal component analysis (PCA) plots of cells based on transcriptomic profile. The first two principal components explained 61.46% of the variance. Colored symbols correspond to CTCs and DECs in both chambers of the CDi-on-chip. B Heatmap and hierarchical clustering of top 50 inflammation and immunity gene expression data for CTCs and DECs in CDi-on-chip. Upregulated genes are represented in red, and downregulated genes are represented in blue. Volcano plots of genes involved in inflammation and immunity in DEC and CTC in response to (C) LPS, (D) pIC, or (E) mOS. Significantly downregulated genes (–log10 adj. p-value > 1, log2 fold change < –1) are shown in blue dots, while significantly upregulated genes (–log10 adj. p value > 1, log2 fold change >1) are shown in red dots. Top differentially expressed genes are labeled. Measurement of (F) IL-6, (G) IL-8, (H) IL-10, (I) TNF-ɑ, (J) GM-CSF, (K) IL-1β, (L) sHLA-G and (M) progesterone production in CDi-on-chip. Multiple student’s (at least n = 4 devices from one placenta per treatment group). Data are presented as mean ± standard error of the mean (SEM). Statistics: Multiple student’s t tests (two-tailed). N Quantification of CD45+ cell migration from DEC into CTC (n = 5 devices from one placenta per treatment group). Statistics: Mann Whitney U test. p values are indicated. IL interleukin; TNF tumor necrosis factor; GM-CSF granulocyte-monocyte colony stimulating factor; sHLA-G soluble HLA-G; P4 progesterone.
Overrepresentation analysis (ORA) of upregulated genes in primary DECs revealed enrichment of inflammatory processes, with few differences observed across treatment groups. Specifically, cytokine-cytokine receptor interaction, TNF signaling pathway and Toll-like receptor signaling pathway were enriched in response to LPS (Supplementary Fig. 3A); chemokine signaling, Th1, Th2 and Th17 cell differentiation, and Toll-like receptor signaling pathway were enriched in response to pIC (Supplementary Fig. 3B); and chemokine signaling pathway, cytokine-cytokine receptor interaction, NF-kappa B signaling pathway and Toll-like receptor signaling pathway were enriched in response to mOS (Supplementary Fig. 3C). In summary, transcriptomic analysis captured the upregulation of pathways involved in innate immunity like TNF, TLR, and cytokine signaling pathways in addition to changes in adaptive immunity such as helper T cell differentiation. No transcriptomic changes were seen in immortalized CTCs regardless of treatment, which signifies their refractoriness to such insults. Consequently, the inflammatory response was localized only in the decidua without propagation towards the chorion, thus maintaining the barrier function at the CDi.
Production of soluble mediators and immune cell migration in response to maternal insults
We then investigated the functional response of the CDi by looking at the production of cytokines by both immortalized CTCs and primary DECs with cell line-derived immune cells, as well as soluble HLA-G and progesterone by CTC and immune cell migration to correlate with RNA sequencing results. In response to LPS, DECs and immune cells produced significantly increased levels of IL-6 (Fig. 2F) and GM-CSF (Fig. 2J), while CTCs produced substantially less IL-10 (Fig. 2H). An 11-fold decrease in IL-8 concentration in DEC and a 3-fold increase in CTC was observed, signifying increased IL-8 gradient towards the CTCs (Fig. 2G). Interestingly, pIC significantly decreased IL-10 and TNF-ɑ (Fig. 2I) levels in DEC, and decreased IL-8, IL-10, and GM-CSF levels in CTC. mOS significantly decreased TNF-ɑ level in DEC, and decreased IL-6, IL-8, and IL-10 levels in CTC. This observation may indicate the resolution phase of the inflammatory response in the presence of immune cells after 48 h of treatment, highlighting the relevance of cell line-derived immune cell inclusion in recapitulating the functions of this maternal-fetal interface. No changes in IL-1β levels were seen in both CTC and DEC chambers (Fig. 2K). Despite changes in cytokine levels, sHLA-G (Fig. 2L) and progesterone production (Fig. 2M) by immortalized CTCs remained constant, and no significant immune cell migration (Fig. 2N) was seen regardless of treatment. This implies that the immortalized CTCs produce constant levels of immunomodulatory molecules regardless of maternal insults, preventing immune cell infiltration into the CTCs.
PGRMC2 maintains the less inflammatory epithelioid phenotype of CTCs
PGRMC2 is a non-classical progesterone receptor expressed strongly in the cell membranes and perinuclear regions of amnion epithelial cells (AECs), CTCs, and DECs (Fig. 3A). To determine the functional effect of PGRMC2 at CDi, we knocked out this molecule in immortalized CTCs using CRISPR-Cas9 gene editing technology. This knockout neither impacted CTC growth nor produced any changes in overall cell morphology (Fig. 3B, C). PGRMC2 knockout resulted in 109 differentially expressed inflammation and immunity genes in CTCs (Fig. 3D): 32 downregulated and 77 upregulated compared to wild-type (WT) immortalized CTCs. The top downregulated genes were CXCL14, CXCR2, HLA-G, SYK, and ID2, while the top upregulated genes were TGFB2, IL23R, CD44, IL1A, and IL24. In response to PGRMC2 knockout, gene expression also changed in primary DECs, with 4 downregulated and 54 upregulated genes. This highlights the crosstalk between the two cell compartments our microphysiological system provides. Gene set enrichment analysis (GSEA) portrayed activation of cytokine-cytokine receptor signaling pathway and extracellular remodeling (proteoglycans in cancer), and suppression of pathways involved in physical barrier function (adherens junction and cell adhesion molecules), vesicle-based cellular pathways (autophagy and endocytosis), cAMP signaling, and other immune processes (B cell receptor signaling pathway and NK cell-mediated cytotoxicity) in PGRMC2 KO CTCs (Fig. 3E). On the other, primary DECs exhibited activation of immune rejection (graft-versus-host disease, allograft rejection) and T cell processes (Th1, Th2 and Th17 cell differentiation and T cell receptor signaling pathway) (Fig. 3F), suggesting a loss of immunotolerance at the maternal side.

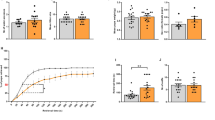
A Immunohistochemistry of human fetal membranes, with PGRMC2 staining brown. B Phase-contrast images of WT and PGRMC2 KO CTC. Scale bar, 100 µm. C Western blot analysis of PGRMC2 expression in WT and PGRMC2 KO CTCs. Beta-actin serves as control. D Volcano plots of genes involved in inflammation and immunity in PGRMC2 KO CTC and DEC compared to WT in control conditions. Significantly downregulated genes (–log10 adj. p value > 1, log2 fold change < –1) are shown in blue dots, while significantly upregulated genes (–log10 adj. p value > 1, log2 fold change > 1) are shown in red dots. GSEA dot plots of top significantly-enriched KEGG pathways in (E) PGRMC2 KO CTC and (F) corresponding DEC. Circle size denotes number of enriched genes in a pathway. Gene ratio denotes the quotient between number of enriched genes and total number of genes in the said pathway. Color denotes p value. G Venn diagram of common differentially expressed genes in PGRMC2 KO CTC compared to wild-type CTC and their corresponding DEC in various conditions. GSEA ridge plots of top significantly-enriched KEGG pathways in PGRMC2 KO CTC and their enrichment scores (x-axis) in response to (H) LPS, (I) pIC and (J) mOS. Peak corresponds to enrichment score; enrichment score below 0 denotes suppressed pathways, while enrichment score above zero denotes activated pathways. Color denotes p value. K Immunostaining of and vimentin (green) expression of WT and PGRMC2 KO CTCs in CDi-on-chip. DAPI (blue) was used for nuclear staining. Scale bar, 100 µm. Measurement of (L) chorionic IL-6 and TNF-ɑ, (M) decidual IL-6 and GM-CSF, (N) sHLA-G and (P) progesterone production in CDi-on-chip (at least n = 4 devices from one placenta per treatment group). Data are presented as mean ± SEM. Statistics: Multiple student’s t-tests (two-tailed). O Western blot analysis of HLA-G expression in wild-type and PGRMC1 or PGRMC2 KO CTCs. Beta-actin serves as control. Q Quantification of CD45+ cell migration from DEC into CTC (n = 5 devices from one placenta per treatment group). Data are presented as median (2nd quartile; solid line) and 1st and 3rd quartiles (dashed lines). Statistics: Multiple Mann Whitney U tests (two-tailed). AEC amnion epithelial cell, AMC amnion mesenchymal cell, CMC chorion mesenchymal cell, WT wild-type, KO knockout, GSEA Gene set enrichment analysis.
Figure 3G shows the differentially expressed genes in PGRMC2 KO CTCs and primary DECs across various treatments. In PGRMC2 KO CTCs, 41 downregulated genes were seen in treatments other than control, of which 8 were unique to LPS, 1 to pIC, and 17 to mOS. Similarly, 28 upregulated genes were observed in non-control groups, of which 2 were unique to LPS, 1 to pIC, and 6 to mOS. This signifies that in the absence of PGRMC2, the baseline response of immortalized CTCs drastically changed even in control conditions. Of the downregulated genes in primary DECs, 4 were unique to the control group and 24 to the mOS group, with no common downregulated genes between the two groups. For upregulated genes, 53 were unique to the control group, 9 to the pIC group, and 4 to the mOS group. This implies that the baseline primary DEC response changed as a result of the absence of PGRMC2 and in the presence of mOS and pIC.
Looking into stimulant-specific responses, 43 downregulated genes and 65 upregulated genes were seen in PGRMC2 KO CTCs compared to wild-type immortalized CTCs in response to LPS (Supplementary Fig. 4A). RUNX3, NFATC1, SYKK, NOS2 and IL10RA were the top downregulated genes, while TGFB2, MX1, OAS2, IFI27 and CD70 were the top upregulated genes. No differentially expressed genes were seen in primary DECs. Figure 3H shows the enriched KEGG pathways after GSEA. Common in all three treatments was the activation of TNF signaling pathway and inflammatory conditions (Fig. 3H–J). Of note in LPS was the activation of IL-17 signaling pathway and pathways involved in extracellular matrix remodeling (proteoglycans in cancer and ECM-receptor interaction) in PGRMC2 KO compared to wild-type immortalized CTCs.
In response to pIC, 24 downregulated genes and 69 upregulated genes were seen in PGRMC2 KO CTCs compared to wild-type immortalized CTCs (Supplementary Fig. 4B). SYK, NFATC1, ITGAM, NOS2 and CCL3 were the top downregulated genes, while MX1, MMP9, CD70, TGFB2 and IFI16 were the top upregulated genes. Ten upregulated genes were seen in primary DECs, including ACKR3, CSF2RA, ACKR2, TNFSF10 and CCR7. Figure 3I showed the enriched KEGG pathways after GSEA. Proteoglycans in cancer, a pathway involved in extracellular matrix remodeling, and NOD-like receptor signaling pathway were activated, and cell adhesion molecules and endocytosis were suppressed in PGRMC2 KO compared to wild-type immortalized CTCs.
Lastly, 46 downregulated genes and 67 upregulated genes were seen in PGRMC2 KO CTCs compared to wild-type immortalized CTCs in response to mOS (Supplementary Fig. 4C). NOS2, NFATC1, BCL2, IL17RB and IL10RA were the top downregulated genes, while OAS2, IFI27, CD70, CXCL2 and MX1 were the top upregulated genes. There were 24 downregulated genes and 4 upregulated genes in primary DECs. Figure 3J shows the enriched KEGG pathways after GSEA. Of note was the suppression of pathways involving cell shape maintenance (cell adhesion molecules and regulation of actin cytoskeleton) and NK cell-mediated cytotoxicity.
Extracellular matrix remodeling and downregulation of cell adhesion molecules are seen in epithelial-mesenchymal transition, therefore we immunostained PGRMC2 KO CTCs with vimentin. As shown in Fig. 3K, there is increased vimentin expression by PGRMC2 KO CTCs, suggesting cellular transition to mesenchymal phenotype. Given that accumulation of amnion mesenchymal cells promotes localized inflammation at the fetal membranes12 and transcriptomic data signifies activation of inflammatory pathways in response to maternal insults, we determined if the increase in chorion mesenchymal phenotype is associated with elevated pro-inflammatory mediators (Fig. 3L–M, Supplementary Fig. 5). In control conditions or in response to either LPS, pIC or mOS, there was significant increase in IL-6 and TNF-ɑ (Fig. 3L) in PGRMC2 KO CTCs compared to wild-type immortalized CTCs. There was a further significant increase in IL-6 and TNF-ɑ levels in response to LPS compared to control. The absence of PGRMC2 interestingly also affected the primary DEC response (Fig. 3M). In response to pIC, there was an observed increase in decidual IL-6, IL-10 (Supplementary Fig. 5F), TNF-ɑ (Supplementary Fig. 5G) and GM-CSF compared to wild-type counterpart. This significant increase in decidual IL-6 was also seen in response to pIC compared to control. Elevated decidual GM-CSF was also seen in response to LPS compared to their wild-type counterparts (Fig. 3M). No significant changes in cytokine levels were seen in primary DECs in response to mOS.
Consistent with transcriptomic analysis, baseline secretion of sHLA-G decreased in PGRMC2 KO CTCs (Fig. 3N), implying a regulatory role for PGRMC2 in HLA-G production. Interestingly, HLA-G expression decreased only in the absence of PGRMC2 but not PGRMC1 (Fig. 3O, Supplementary Fig. 6), which signifies PGRMC2, not PGRMC1, as a specific upstream regulator of HLA-G expression. On the other hand, baseline progesterone production increased in PGRMC2 KO CTCs compared to wild-type immortalized CTCs (Fig. 3P) in control conditions or in response to pIC, likely a result of autocrine or paracrine feedback to compensate for decreased PGRMC2 expression. The overall inflammation and loss of immunotolerance in primary DEC resulted in significant immune cell migration into PGRMC2 KO CTCs in the presence of LPS (Fig. 3Q).
In summary, the absence of PGRMC2 resulted in the activation of pathways involved in extracellular matrix degradation and physical barrier disruption in CTCs in favor of an escape to a mesenchymal phenotype, thus compromising fetal membrane integrity. In response to maternal insults, activation of inflammatory pathways was observed in addition to continued fetal membrane compromise, leading to immune cell infiltration or chorionitis.
HLA-G regulates baseline refractoriness of CTCs to inflammation
HLA-G is a non-classical major histocompatibility complex-I strongly expressed in the cell membranes and cytoplasmic regions of CTCs (Fig. 4A). Given that HLA-G is an important and unique immunomodulator regulated by PGRMC2, we also successfully generated HLA-G-knockout immortalized CTCs without changes in overall cell morphology (Fig. 4B, C). This knockout resulted in 124 differentially expressed inflammation and immunity genes in CTCs (Fig. 4D): 41 downregulated and 83 upregulated. Along with HLA-G, the top downregulated genes were ACKR3, CCR7, NOS2, CSF3R and ID2, while the top upregulated genes were IFI44, IL24, CD70, CXCL10 and C3. In response to HLA-G knockout, gene expression also changed in primary DECs, with 35 downregulated and 124 upregulated genes. GSEA portrayed suppression of virion – human immunodeficiency virus and activation of neuroactive ligand-receptor interaction in HLA-G KO CTCs (Fig. 4E). Enrichment of only two KEGG pathways due to HLA-G knockout shows that (1) only a few downstream pathways were regulated by HLA-G, or (2) HLA-G is one of the end-effector molecules. The latter enriched pathway is of significance since it includes prostanoid and relaxin signaling, both of which are important processes in the initiation of labor2. Conversely, activation of bacterial invasion of epithelial cells and various cancer pathways and suppression of Th17 cell differentiation were observed in primary DECs (Fig. 4F), the latter of which is important in the maintenance of pregnancy40. Many cancer cells utilize the immunomodulatory properties of HLA-G41; the absence of HLA-G leads to immune recognition, hence the enrichment of cancer pathways.

A Immunohistochemistry of human fetal membranes, with HLA-G staining brown. B Phase-contrast images of WT and PGRMC2 KO CTC. Scale bar, 100 µm. C Western blot analysis of HLA-G expression in WT and HLA-G KO CTCs. Beta-actin serves as control. D Volcano plots of genes involved in inflammation and immunity in HLA-G KO CTC and DEC compared to WT in control conditions. Significantly downregulated genes (–log10 adj. p-value > 1, log2 fold change < –1) are shown in blue dots, while significantly upregulated genes (–log10 adj. p-value > 1, log2 fold change > 1) are shown in red dots. GSEA dot plots of top significantly-enriched KEGG pathways in (E) HLA-G KO CTC and (F) corresponding DEC. Circle size denotes number of enriched genes in a pathway. Gene ratio denotes the quotient between number of enriched genes and total number of genes in the said pathway. Color denotes p value. G Venn diagram of common differentially expressed genes in HLA-G KO CTC compared to wild-type CTC and their corresponding DEC in various conditions. GSEA ridge plots of top significantly-enriched KEGG pathways in HLA-G KO CTC and their enrichment scores (x-axis) in response to (H) LPS, (I) pIC and (J) mOS. Peak corresponds to enrichment score; enrichment score below 0 denotes suppressed pathways, while enrichment score above zero denotes activated pathways. Color denotes p-value. Measurement of (K) chorionic IL-6, IL-10, TNF-ɑ and GM-CSF, (L) decidual GM-CSF, (M) sHLA-G and (N) progesterone production in CDi-on-chip (at least n = 4 devices from one placenta per treatment group). Data are presented as mean ± SEM. Statistics: Multiple student’s t-tests (two-tailed). O Quantification of CD45+ cell migration from DEC into CTC (n = 5 devices from one placenta per treatment group). Data are presented as median (2nd quartile; solid line) and 1st and 3rd quartiles (dashed lines). Statistics: Multiple Mann Whitney U tests (two-tailed).
Figure 4G shows the differentially expressed genes in HLA-G KO CTCs and primary DECs across various treatments. In PGRMC2 KO CTCs, 28 downregulated genes were seen10 in treatments other than control, of which 5 were unique to LPS, 1 to pIC, and 12 to mOS. Similarly, 28 upregulated genes were observed in non-control groups, of which 6 were unique to LPS, 4 to pIC, and 12 to mOS. This implies that when HLA-G is absent in immortalized CTCs, the baseline response drastically changes even in control conditions. Of the downregulated genes in primary DECs, 29 were unique to the control group, 13 to the mOS group, and 6 were common to both the control and mOS groups. For upregulated genes, 117 were unique to the control group, 1 to the mOS group, and 6 were shared between the control and mOS groups. This signifies that the baseline primary DEC also substantially changed in the absence of HLA-G interacting with, and of sHLA-G reaching the primary DEC and cell line derived innate immune cells from immortalized CTCs. However, in the presence of infectious stimulants, the primary DEC response did not change any further.
Looking into stimulant-specific responses, 40 downregulated genes and 74 upregulated genes were seen in HLA-G KO CTCs compared to wild-type immortalized CTCs in response to LPS (Supplementary Fig. 7A). NOS2, CCR7, HLA-G, ID2 and CSF3R were the top downregulated genes, while TLR4, TGB2, CD70, IL24 and LTB were the top upregulated genes. No differentially expressed genes were seen in the primary DECs. Figure 4H shows the enriched KEGG pathways after GSEA. Common in all three treatments was the activation of the overall inflammatory process as indicated by TNF signaling, IL-17 signaling and NF-kappa B signaling, and other inflammatory conditions (Fig. 4H–J). This signifies a highly dysregulated inflammatory phenotype characterized by the production of pro-inflammatory cytokines and activation of innate immune signaling pathways exhibited by immortalized CTCs in the absence of HLA-G. Of note, apelin and insulin signaling pathways and ubiquitin-mediated proteolysis, important in homeostatic metabolic activities, were suppressed, suggesting a shift of physiologic processes to favor inflammatory activity.
In response to pIC, 30 downregulated genes and 72 upregulated genes were seen in HLA-G KO CTCs compared to wild-type immortalized CTCs (Supplementary Fig. 7B). CCR7, CSF3R, NOS2, ACKR3, and HLA-G were the top downregulated genes, while CSF2, MMP9, CSF3, CD70 and NOD2 were the top upregulated genes. No differentially expressed genes were seen in primary DECs. Figure 4I shows the enriched KEGG pathways after GSEA. Similar to that observed in LPS treatment, pathways involved in housekeeping (central carbon metabolism in cancer, ubiquitin-mediated proteolysis, and adherens junction) were suppressed in HLA-G KO compared to wild-type immortalized CTCs following pIC treatment.
Lastly, 46 downregulated genes and 72 upregulated genes were seen in HLA-G KO CTCs compared to wild-type immortalized CTCs in response to mOS (Supplementary Fig. 7C). CCR7, NOS2, ACKR3, CSF3R and ID2 were the top downregulated genes, while TNFRSF9, CD70, LTB, CXCL2 and CXCL8 were the top upregulated genes. Unlike in LPS and pIC treatments, there were 19 downregulated genes and 8 upregulated genes in primary DECs, which signifies a differential response to mOS in the decidua when HLA-G was knocked out. Figure 4J shows the enriched KEGG pathways after GSEA. Of note was the suppression of neurotrophin signaling in HLA-G KO compared to wild-type immortalized CTCs. Lower neurotrophin levels (in particular, nerve growth factor) in cord blood are associated with preterm delivery42, thus highlighting a major difference between infectious inflammation and sterile inflammation arising from oxidative stress.
We then determined if the cytokine response was consistent with transcriptomic findings (Fig. 4K–L, Supplementary Fig. 8). In control conditions, there was a significant increase in TNF-ɑ and GM-CSF (Fig. 4K) in HLA-G KO CTCs compared to wild-type immortalized CTCs. A 16-fold increase in IL-6 (Fig. 4K) and a 15-fold increase in IL-8 (Supplementary Fig. 8A) were also observed. In response to either LPS, pIC, or mOS, there was a significant elevation in IL-8, TNF-ɑ, and GM-CSF in HLA-G KO CTCs compared to wild-type immortalized CTCs. IL-6 and IL-10 in response to LPS, and IL-10 in response to pIC were also significantly higher in HLA-G KO CTCs compared to wild-type (Fig. 4K). In addition, 16-fold and 37-fold increases in IL-6 were also observed in pIC and mOS groups, but these were not statistically significant. In all three stimulants, except for the significant decrease in IL-10 in mOS compared to control in HLA-G KO CTCs, the observed baseline elevated levels of cytokines were not statistically significant when compared to their corresponding control groups in HLA-G KO CTCs. This was contrasted with PGRMC2 KO CTCs in which a significant increase in IL-6 and TNF-ɑ levels was seen in response to LPS compared to control. These data are consistent with gene expression data that demonstrate activation of various inflammatory pathways in HLA-G KO CTCs, thus supporting the notion of highly dysregulated inflammatory phenotype. However, the magnitude of inflammation seen was surprising, as many pro-inflammatory cytokines were elevated at least 15-fold. The observed increase in cytokine levels was likely a result of a lack of autocrine and paracrine regulation of cytokine levels via sHLA-G43. There was also an observed change in decidual response, with decidual GM-CSF increasing compared to wild-type counterpart upon treatment with LPS or pIC (Fig. 4L), consistent with its role in fetal membrane weakening44.
As expected, there was a significant decrease in sHLA-G secretion in HLA-G KO CTCs regardless of treatment groups (Fig. 4M). There was no significant change in progesterone production compared to their corresponding treatment groups in wild-type immortalized CTCs (Fig. 4N). Notably, HLA-G knockout resulted in statistically significant immune cell infiltration in response to LPS compared to control (Fig. 4O). Whether the significant immune cell infiltration observed in PGRMC2 KO in response to LPS was primarily due to the absence of HLA-G requires further investigation. In summary, the absence of HLA-G disturbed the observed refractoriness of CTCs to maternal insults by activating various inflammatory pathways.
Discussion
The CDi is established by the fusion of amniotic and chorion sacs forming the amniochorion or fetal membranes with the decidua parietalis, thus restructuring the uterine cavity and forming another maternal-fetal interface distinct from the placental disc-decidua basalis interface45. Towards the end of gestation, the CDi overlies 70% of the intrauterine cavity lining46. Given its proximity not only to the internal os of the cervix but also its direct apposition to maternal tissues, this interface experiences constant exposure from infectious agents, other exogenous exposures, and oxidative stress from maternal lifestyle factors. Yet infections or lifestyle behaviors do not always result in immune cell infiltration and collapse of fetal membrane integrity, specifically premature activation of labor, highlighting the barrier function of the CDi.
The decidua is composed of decidual stromal cells and resident immune cells. Decidual immune cells consist mostly of T cells, although substantial amounts of innate immune cells such as NK cells, macrophages, and neutrophils are also present47. Decidual stromal cells also have immunomodulatory properties: they express HLA-A and HLA-B which are important in NK cell education48, and produce prolactin which helps promote immune tolerance and regulate innate immune responses49. Opposite the decidua is the multi-layered chorion composed of CTCs. Unlike amnion epithelial cells which can easily transition into amnion mesenchymal cells in the presence of a stressor, CTCs are resistant to EMT50. The persistence of epithelioid phenotype by CTCs is important to maintain the intercellular connections between chorion trophoblast cells, thus contributing to structural integrity. A mesenchymal transition of CTC contributing to localized inflammation can be structurally and functionally detrimental to the barrier function of the chorion and prevent these stressors or stressor induced changes to reach the amnion which is highly vulnerable to transition under pressure. Unlike other barrier cell types, CTCs are unique as they can produce immunomodulatory molecules without stimulation51. Among the mediators produced by CTCs are progesterone18, non-classical major histocompatibility complexes class I antigens HLA-E, HLA-F and HLA-G52, soluble components of HLA to buffer immune response and immune cell infiltration, and a mix of pro- and anti-inflammatory cytokines including IL-1021. Since no extracellular matrix separates the chorion and the decidua, these CTC-derived mediators can modulate the decidual immune microenvironment. We theorize that these cellular and immunobiological factors provide the barrier functions that make the chorion a “great wall” to prevent excessive inflammation that may lead to premature activation of labor due to constant maternal exposure to various exogenous factors.
In the presence of a stimulant, whether infection or oxidative stress, resident immune cells in the decidua become activated, and more leukocytes are recruited via the production of chemokines. This shifts the local microenvironment into a pro-inflammatory phenotype1. During the acute phase, innate immune cells induce inflammation via Toll-like receptor53, TNF54, and NFκB55 signaling pathways. CTCs withstand these insults by acting as a structural and immunobiological barrier, containing inflammation to the decidual side. This is supported by the absence of epithelial-mesenchymal transition by CTCs to maintain epithelioid phenotype, baseline secretion of immunomodulatory molecules IL-10, sHLA-G, and progesterone, and lack of substantial immune cell infiltration into the chorion observed in our study. By precluding the propagation of inflammation to fetal tissues, the chorion layer prevents premature activation of fetal inflammatory response, especially from fetal membrane cells, which is vital in promoting parturition56. Failure to resolve inflammation, excessive inflammation, or immune dysregulation can lead to premature decidual activation, which is implicated in the pathophysiology of spontaneous preterm birth2. While this can be maternal exposure-dependent (due to superinfection, high microbial load, and conditions of extreme oxidative stress such as smoking or pollution), having a history of previous preterm birth as a strong risk factor for preterm birth57 suggests a possible collapse of barrier functions at the CDi as the culprit. To test this, we knocked out two barrier functions we identified in our previous studies: PGRMC225 and HLA-G20.
As previously mentioned, fetal membranes do not have nuclear progesterone receptors, thus functional progesterone withdrawal in fetal membranes needs to be mediated by PGRMC18. In particular, PGRMC2 is downregulated during the onset of labor18. Unlike PGRMC1, less information is available regarding the function of PGRMC2 in gestational tissues58. In amnion mesenchymal cells, progesterone induces MET through PGRMC2 in a c-MYC-dependent manner and reduces accumulation of mesenchymal cells in the extracellular matrix region of the fetal membranes25. MET in chorion cells to maintain epithelioid phenotype is likely mediated through the same pathway, but its refractoriness to EMT unlike amnion epithelial cells is likely due to endogenous progesterone production by CTCs18, facilitating autocrine and paracrine regulation of cellular transition. In the absence of PGRMC2, the balance in cellular transition becomes dysregulated, favoring the persistence of mesenchymal phenotype, which was supported by an increase in vimentin expression by immortalized CTCs in our study. Whether the observed increase in the frequency of chorion mesenchymal cells was a result of EMT or the persistence of mesenchymal phenotype by spontaneously transitioned CTCs requires further scrutiny. Importantly, our transcriptomic analysis also showed suppression of adherens junction and cell adhesion molecules which are important in maintaining structural barrier integrity19. Taken together, both findings suggest disruption of the physical barrier function of the CTCs in the absence of PGRMC2.
In addition to barrier disruption, there was also dysregulation of gene expression of genes involved in inflammation and immunity, mostly affecting leukocyte migration and recruitment (Supplementary Fig. 9). Unlike in HLA-G KO immortalized CTCs where they were highly inflammatory even at control conditions based on pro-inflammatory cytokine levels, production of IL-6, IL-8, TNF-ɑ and GM-CSF in PGRMC2 KO immortalized CTCs still increased in response to LPS. Interestingly, we also observed decreased HLA-G expression and sHLA-G secretion with PGRMC2 knockout. Progesterone is known to enhance the expression of HLA-G via the binding of the progesterone receptor complex to the progesterone response element (PRE) sequence in HLA-G promoter59,60. However, CTCs do not express nuclear progesterone receptors, hence, the observed HLA-G downregulation is likely due to non-classical regulation of HLA-G expression involving PGRMC2, which is yet to be determined. Since PGRMC2 knockout does not eliminate the production of HLA-G and secretion of sHLA-G, this could explain the lower levels of pro-inflammatory cytokines observed compared to HLA-G KO immortalized CTCs. Therefore, PGRMC2 acts as a key regulator of inflammation directly by contributing to constitutive HLA-G expression, reducing pro-inflammatory cytokines, preventing immune cell infiltration, and indirectly by preventing the persistence of inflammatory chorion mesenchymal phenotype to maintain physical barrier function.
Most studies about HLA-G have focused on expression by extravillous trophoblasts in the placenta, which are of a different cell type than CTCs19. Because of this, most of what we know about HLA-G is about its role in immunomodulation61, spiral artery remodeling62, trophoblast invasion43 during implantation and placentation, and pregnancy complications linked to these processes such as recurrent pregnancy loss63 and preeclampsia64. To our knowledge, this is the first study that utilized a gene knockout method to confirm the functional role of HLA-G in fetal membrane cells rather than placental cells65. In the absence of HLA-G, maternal LPS treatment caused immune cell infiltration into the chorion. HLA-G acts by direct binding to one of its receptors (KIR2DL4, ILT-2, CD160)66 or secretion of soluble isoform (sHLA-G) to the decidual compartment where these mediators interact with resident immune cells20. This interaction results in NK cell inhibition and reprogramming, macrophage modulation, controlled release of IL-6 and IL-8, and direct T cell inhibition67. Without the immunomodulatory activity of HLA-G, decidual immune cells become activated. However, the decidual response is only one part: the absence of HLA-G also resulted in CTCs exhibiting a highly dysregulated inflammatory phenotype characterized by activation of inflammatory pathways and excessive production of pro-inflammatory cytokines. Given that soluble HLA-G regulates trophoblast function (in this case, extravillous trophoblast invasion) via autocrine signaling43, it is possible that sHLA-G also plays an autocrine role in modulating inflammation in CTCs. Currently, there is a lack of epidemiological or clinical studies providing a strong link between fetal membrane HLA-G and susceptibility to ascending intrauterine infection65. Since most of the studies look into HLA-G expression and sHLA-G concentration in placental villous tissues52,68, amniotic fluid69, and maternal blood70, investigating the relationship between decreased fetal membrane HLA-G levels and subsequent inflammation of the fetal membranes (chorioamnionitis) is worth pursuing further. Regardless, overwhelming immune cell infiltration similar to that observed in chorioamnionitis was not seen due to the absence of immune cell recruitment from the bloodstream and fetal immune response with our model71. Overall, these results suggest that HLA-G from CTCs, both in its membrane-bound or soluble form, acts as a frontline immunomodulator at the CDi, acting not only on immune cells to modulate inflammation and maintain tolerance, but also to guarantee autocrine and paracrine regulation of cytokine production by CTCs.
Recent advances in biomedical engineering have allowed us to recreate intercellular interactions in a microphysiological system via microfluidics technology. Various stages of pregnancy were designed and modeled using this technology from implantation72 to parturition37. Nevertheless, there is a major limitation intrinsic to the design of the microphysiological system. Ideally, to accurately reflect the fetal membrane architecture in vivo, the microphysiological system should follow a vertical design (that is, two vertically positioned culture chambers separated by a porous membrane32. This design would allow the reconstruction of a structural barrier phenotype. However, difficulty in imaging through the membrane and complicated microfabrication steps to ensure consistent sealing of the microfluidic device render the utilization of such a design challenging. While opting for a planar design loses the direct intercellular interactions provided for by a vertical design, intercellular communication via soluble mediators and extracellular vesicles between the two cell chambers is still possible with the current planar design32. As such, this design does not hinder the investigation of the immunobiological barrier functions of the chorion.
In our CDi-on-chip, we were able to put together in one system immortalized CTCs, primary DECs, and cell line-derived innate immune cells and view how interactions between cell types and compartments led to certain responses and phenotypes. In particular, the presence of immune cells, albeit derived from immune cell lines, allowed a better understanding of inflammatory and immune processes than immune cell-less models, which was a major limitation of our previous microphysiological systems. Nonetheless, the addition of immune cells also had its limitations. We only included innate immune cells, which comprise less than 20% of the immune cells found in decidua parietalis (i.e., decidual layer that lines the fetal membranes) at term prior to labor (Fig. 1C). This was primarily due to the kind of stimulants and the duration of the insult (48 h), in which inflammation and innate immunity both play major roles before adaptive immunity kicks in53. Our model does not consider the contribution and synergy the fetal and maternal adaptive immune systems provide, in particular T cells which are the most abundant immune cell subpopulation in the decidua parietalis. These T cells are particularly important in maintenance of tolerance during pregnancy73. While the collapse of the “Great Wall” of CTCs at the chorio-decidual interface may explain how these insults lead to premature labor activation in primigravid pregnancies, the relevance of the maternal adaptive system (in particular, T cells) becomes more apparent in subsequent pregnancies due its interaction with fetal components during and after pregnancy (i.e., fetal microchimerism)74,75. To get a more complete picture of decidual immune homeostasis as part of future direction of the study, other immune cell subpopulations can be added in the existing CDi model.
Overall, we conclude that PGRMC2 and HLA-G are key regulators of inflammation at the CDi, with differing roles as seen in Fig. 5. PGRMC2 serves as a critical upstream regulator of HLA-G expression and mesenchymal-epithelial transition, ensuring the chorion’s structural integrity. Both membrane and soluble HLA-G, on the other hand, serve as frontline immunomodulatory molecules, interacting directly with immune cells or indirectly via autocrine and paracrine regulation of cytokine production. We believe that our findings would contribute to the elucidation of the regulatory mechanisms of the CDi and improve our understanding of human pregnancy and parturition.

PGRMC2 acts as a critical molecule regulating chorion integrity by maintaining the epithelioid phenotype of CTCs. It also mediates immune homeostasis either via direct activation of inflammatory pathways or via acting as an upstream regulator of HLA-G expression. HLA-G, by itself or via the action of soluble HLA-G, also modulates inflammation possibly via autocrine or paracrine signaling, although the exact mechanism remains unknown. This image is created with BioRender.com.
Methods
Immunohistochemistry of the fetal membranes
Immunohistochemistry (IHC) was performed to stain for the presence of vimentin (rabbit 1:300; Cat# ab92547; Abcam, Cambridge, UK), cytokeratin-18 (mouse 1:300; Cat# ab668; Abcam, Cambridge, UK), PGRMC2 (rabbit 1:800; Cat# PA559465; ThermoFisher Scientific, Waltham, MA, USA), and HLA-G (mouse 1:50; Cat# sc-21799; Santa Cruz Biotechnology, Dallas, TX, USA) within fetal membranes. Formalin-fixed, paraffin-embedded sections of human fetal membranes delivered at term via cesarean section at John Sealy Hospital, Galveston, TX, USA were deparaffinized with xylene and ethanol in various concentrations. Antigen retrieval was performed by placing slides in a citrated buffer (2.95 g tri-sodium citrate in 1000 mL deionized water adjusted to a pH of 6.0 and 500 µL of Tween 20) and using the 2100 Antigen Retriever system (Aptum Biologics, Southampton, UK). Slides were placed into 3% hydrogen peroxide blocking reagent (Abcam, Cambridge, UK) followed by washing with Tris-buffered saline (TBS) (Corning, Corning, NY, USA) 3 times for 5 min. Universal blocking buffer (50 mL phosphate buffered saline [PBS], 500 mg bovine serum albumin [BSA], 50 mg cold fish gelatin, and 250 µL Triton X-100) was added to the slides for 1 h in a humidified chamber followed by washing thrice with TBS for 5 min. Tissue sections were treated with primary antibodies overnight at 4 °C then washed 4 times with TBS for 5 min. Secondary antibody (Biotinylated goat anti-rabbit IgG, ab64256; Abcam, Cambridge, UK) was added for 30 min at 22 °C, then washed thrice with TBS for 5 min. Slides were treated with streptavidin peroxidase (Vector Laboratories, Newark, CA, USA) for 30 min at 22 °C, then washed thrice with TBS for 5 min. Color development with 3, 3-diaminobenzidine (DAB) substrate (Cat# 550880; BD, Franklin Lakes, NJ, USA) or 3-amino-9-ethylcarbazole (AEC) substrate (Cat# 551015; BD, Franklin Lakes, NJ, USA) was optimized for each antibody and used for the same time for each replicate. Slides were washed twice with deionized water 2 times for 5 min, then counterstained with hematoxylin (Volu-sol, Salt Lake City, UT, USA) for 30 s followed by washing with deionized water for 5 min. Slides were placed in Bluing Reagent (ScyTek, Logan, UT, USA) for 20 s followed by washing with deionized water for 5 min. The slides were mounted with Acrymount (Statlab, McKinney, TX, USA), and microscopy was performed with a Keyence BZ-X810 microscope (Keyence, Itasca, IL, USA). Sections with 1× PBS in place of primary antibody served as negative control.
Primary decidual cell culture
The primary decidual cells used in the study was isolated from the deidentified placenta from a non-laboring woman delivered at term by cesarean section at John Sealy Hospital and processed within 30 min at the University of Texas Medical Branch at Galveston, TX, USA. Primary human decidual cells were isolated from decidua parietalis attached to the fetal membranes according to protocol by Radnaa et al.34 (Supplementary Fig. 10). A 5 × 5 cm piece of the fetal membranes was gently rinsed in pre-warmed saline solution. The amnion was separated from the chorion, and the decidua parietalis layer was dissected from the chorion using a sterile scalpel. Decidual tissue was minced and digested in digest buffer I [0.125% trypsin and 0.02% DNase-I in Hanks’ Balanced Salt Solution (HBSS)] for 30 min at 37 °C. The digested tissue was centrifuged at 350 g, 10 min at room temperature, and the resulting pellet was resuspended in digest buffer II (0.125% trypsin, 0.02% DNase-I and 0.2% collagenase type IA in HBSS). After incubation for 1 h at 37 °C, the digestion process was neutralized with equal volume of complete Dulbecco’s Modified Eagle Media [DMEM/F-12 supplemented with 10% heat-inactivated fetal bovine serum (hi-FBS) and 1% penicillin-streptomycin (P/S)]. The mixture was filtered through four layers of sterile gauze, and the cells were centrifuged at 300 g, 10 min at room temperature. The cell pellet was resuspended in 4 mL serum-free DMEM/F-12, and decidua cells were added on top of 4–40% OptiPrepTM gradient in 50 mL conical tube prepared according to manufacturer’s instructions. The gradient mixture was centrifuged at 350 g for 35 min at room temperature, and the cells were collected and washed with 4 volumes of serum-free DMEM/F-12. The cell mixture was centrifuged at 300 g for 10 min at room temperature, and the decidua cells were resuspended and maintained in complete DMEM/F-12 media at 37 °C, 5% CO2 in the Menon Laboratory, The University of Texas Medical Branch at Galveston, TX, USA.
Cell lines and culture conditions
Immortalized chorion trophoblast cells from term cesarean fetal membranes generated by Radnaa et al. were used in the study34. Promyelocytic cell line HL-60 (CCL-240), monocytic cell line THP-1 (TIB-202), natural killer cell line NK-92 (CRL-2407) and choriocarcinoma cell line BeWo (CCL-98) were obtained from ATCC (Manassas, VA, USA). No misidentified cell lines as identified by International Cell Line Authentication Committee were used. CTCs were maintained in DMEM/F-12 supplemented with 1% P/S, 1% amphotericin B, 0.2% hi-FBS, 0.3% bovine serum albumin (BSA), 1X insulin-transferrin-selenium-ethanolamine, 0.8 mM valproic acid, 0.01 mM β-mercaptoethanol, 5 µM Y27632, 2 µM CHIR99021, 1 µM SB431542, 0.5 µM A83-01, 1.5 µg/mL L-ascorbic acid and 10 µg/mL epithelial growth factor; primary DECs in DMEM/F-12 supplemented with 10% hi-FBS and 1% P/S; HL-60 cells in Iscove’s Modified Dulbecco Medium (IMDM) supplemented with 20% hi-FBS and 1% P/S; THP-1 cells in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% hi-FBS, 1% P/S and 50 µM β-mercaptoethanol; NK-92 cells in Minimum Essential Medium (MEM) α with nucleosides supplemented with 12.5% heat-inactivated horse serum (hi-HS), 12.5% hi-FBS, 1% P/S and 20 ng/mL recombinant improved sequence human interleukin-2 (IL-2; equivalent to at least 100 units/mL); and BeWo cells in DMEM/F-12 supplemented with 15% hi-FBS and 1% P/S. The cell lines from ATCC were authenticated by the manufacturer. The cell cultures were frequently checked in the lab for their morphological features. Cell cultures were routinely tested for Mycoplasma contamination via commercial nucleic acid amplification test.
Differentiation of HL-60 into neutrophil-like cells
Around 5 days before seeding of immune cells into microfluidic devices, HL-60 cells were collected via centrifugation at 300 g for 5 min at 20 °C, and 100,000–200,000 cells were seeded into IMDM supplemented with 20% hi-FBS, 1% P/S, and 1.3% dimethylsulfoxide (DMSO) in 25 cm2 cell culture flask. The final DMSO concentration used was based on ref. 76. The cells were incubated at 37 °C, 5% CO2 for 120 h, with media change every 2 days. Differentiated HL-60 cells exhibited a loss of uniformity in cell shape and the presence of membrane projections under a phase-contrast microscope (Supplementary Fig. 1D).
Differentiation of THP-1 into macrophage-like cells
Around 3 days before seeding of immune cells into microfluidic devices, THP-1 cells were collected via centrifugation at 300 g for 5 min at 20 °C, and 1–2 million cells were seeded into RPMI 1640 supplemented with 10% hi-FBS, 1% P/S, 50 µM β-mercaptoethanol, and 20 ng/mL phorbol 12-myristate 13-acetate (PMA) in 25 cm2 cell culture flask. The final PMA concentration used was based on ref. 77. The cells were incubated at 37 °C, 5% CO2 for 72 h, with media change every 2 days. Differentiated THP-1 cells became adherent and more elongated under a phase-contrast microscope (Supplementary Fig. 1E).
Phenotypic conversion of NK-92 into decidual NK-like cells
Co-culture of NK-92 and BeWo cells to obtain decidual NK-like cells was based on ref. 78. Around 4 days before seeding of immune cells into microfluidic devices, BeWo cells were trypsinized for 4 min with 0.25% trypsin-EDTA. Detached BeWo cells were collected via centrifugation at 300 g for 5 min at 20 °C, and 500,000 cells were seeded into MEM α with nucleosides supplemented with 12.5% hi-HS, 12.5% hi-FBS and 1% P/S in 25 cm2 cell culture flask. To allow cell attachment, the culture flask was incubated overnight at 37 °C, 5% CO2. The following day, media in the flask containing BeWo cells were replaced with MEM α with nucleosides supplemented with 12.5% hi-HS, 12.5% hi-FBS, and 1% P/S, and 500,000 NK-92 cells were seeded in the same flask (Supplementary Fig. 1F). The cells were incubated at 37 °C, 5% CO2 for 96 h, with media change every 2 days. An increase in CD16179 and KIR2DL480 expression and a decrease in NKG2D81 expression via Western blot analysis confirmed phenotypic conversion (Supplementary Fig. 1G).
Preparation of HLA-G and PGRMC2 knockout CTCs
HLA-G and PGRMC2 genes were knocked out in immortalized CTCs via CRISPR/Cas9 gene editing technology (Synthego, Redwood City, CA, USA). In summary, 1.3:1 sgRNA: Cas9 ribonucleoprotein complex solution (7.8 pmol sgRNA, 6 pmol Cas9, LipofectamineTM Cas9 Plus Reagent in Opti-MEMTM I reduced serum medium) and transfection solution (LipofectamineTM CRISPRMAXTM reagent in Opti-MEMTM I reduced serum medium) were combined with 80,000 CTCs in 500 µL CTC medium. The resulting cell mixture was split into two wells of a 24-well plate and incubated at 37 °C, 5% CO2 for 24 h. The medium was then replaced with regular CTC medium, and the cells were maintained under same conditions until confluency. One well was used for propagation and succeeding experiments, while the other well was used to confirm knockout via Western blot analysis.
Western blot analysis
Cells were lysed with 1× radioimmunoprecipitation assay buffer (RIPA; 50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, and 1.0 mM EDTA pH 8.0, 0.1% SDS) supplemented with 1× HaltTM protease and phosphatase inhibitor cocktail (ThermoScientific, Rockford, IL, USA) and 1 mM phenylmethylsulfonyl fluoride. The cell lysates were vortexed for 10 s, sonicated for 30 s, and centrifuged at 12,000 g for 20 min at 4 °C. The resulting supernatant was collected, and protein concentrations were determined using PierceTM BCA Protein Assay Kit (ThermoScientific, Rockford, IL, USA). About 20 µg total protein was loaded per well into 10% SDS-polyacrylamide gel, and the gel was subjected to electrophoresis. Proteins in the gel were transferred to methanol-activated polyvinylidene fluoride membranes using Trans-Blot® Turbo™ Transfer System (Bio-Rad Laboratories, Hercules, CA, USA), and the membranes were blocked in 5% non-fat dry milk in 1× Tris-buffered saline with 0.1% Tween 20 (TBST) buffer for 1 h. The primary antibodies diluted in 5% non-fat dry milk in 1× TBST were added, and the membranes were incubated overnight with rocking at 4 °C except for β-actin (which was incubated for 1 h with rocking at room temperature, then directly imaged). The following primary antibodies were used: β-actin (mouse 1:20,000; Cat# ab49900; Abcam, Cambridge, UK), HLA-G (rabbit 1:1000; Cat# ab283260; Abcam, Cambridge, UK), PGRMC2 (rabbit 1:500; Cat# PA559465; ThermoFisher Scientific, Waltham, MA, USA), CD161 (mouse 1:1000; Cat# ab233785; Abcam, Cambridge, UK), G9P/KIR2DL4 (rabbit 1:500; Cat# ab154386; Abcam, Cambridge, UK), NKG2D (rabbit, 1:500; Cat# ab96606; Abcam, Cambridge, UK). The membranes were washed thrice with 1× TBST, and incubated with appropriate secondary antibodies for 2 h (either HRP-linked donkey anti-rabbit [Cat# NA934] or HRP-linked sheep anti-mouse [Cat #NA931]; both 1:10,000; Cytiva, Marlborough, MA, USA). Proteins were visualized using Clarity™ Western ECL Substrate (Bio-Rad Laboratories, Hercules, CA, USA) with ChemiDoc™ Imaging System (Bio-Rad Laboratories, Hercules, CA, USA).
Preparation of treatments
To prepare cigarette smoke extract (CSE) concentrate, smoke from a single lit commercial cigarette (unfiltered CamelTM, R.J. Reynolds Tobacco, Winston-Salem, NC, USA) was infused into 25 mL serum-free DMEM/F-12 media, and passed through a 0.22 μm Steriflip® filter unit (MilliporeSigma, Burlington, MA, USA). CSE concentrate was stored at 4 °C for up to 48 h only. Stock lipopolysaccharide (LPS) solution was prepared by dissolving 1 mg lipopolysaccharides from Escherichia coli O55:B5 (Sigma Aldrich, Burlington, MA, USA) in 1 mL ultrapure water, and stored at –20 °C. On the other hand, stock poly(I:C) (pIC) solution was prepared by dissolving 10 mg polyinosinic–polycytidylic acid potassium salt (Sigma Aldrich, Burlington, MA, USA) in 1 mL ultrapure water, and stored at –80 °C.
Chorio-decidual interface (CDi)-on-chip design and device preparation
A two-chamber microfluidic device previously described by Richardson et al.33, and validated for modeling chorio-decidual immune interface by Richardson et al.20 was used in this study. In summary, the master mold was fabricated using a two-step photolithography process involving photosensitive epoxy SU-8 (MicroChem, Westborough, MA, USA) and was coated with (tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane (United Chemical Technologies, Bristol, PA, USA) to form the 24-array microchannel layer (height: 5 µm, length: 600 µm, width: 30 µm) and the cell culture chamber layer (height: 500 µm). Microfluidic devices were fabricated using soft lithography technique wherein polydimethylsiloxane (PDMS) polymer (10:1 mixture, Sylgard 184; DowDuPont, Midland, MI, USA) was poured onto the mold and cured at 85 °C for 60 min. One inner chamber reservoir and two outer chamber reservoirs were punched out from the device using a 4-mm biopsy punch. The devices were washed with 70% isopropyl alcohol and air-dried, followed by removal of dust and other particles using adhesive tape. The cleaned devices were bonded to MAS Adhesive Microscope Slides (Cat# 5077W90; Matsunami Glass, Bellingham, WA, USA) via oxygen plasma treatment (Harrick Plasma, Ithaca, NY, USA) for 90 s, and the patency of the microchannels were inspected under a light microscope. Before cell seeding, the devices were sterilized with 70% ethanol for 15 min under a biosafety cabinet, then washed with pre-warmed culture media (CTC media for inner chamber, DEC media for outer chamber).
Cell seeding and treatments in CDi-on-chip
The primary decidua parietalis cells used were isolated from one placenta. The wild-type and knockout CTCs used were from the same immortalized CTCs generated by Radnaa et al. (2022). The same cell passage number was used in all experiments. Immortalized CTCs and primary DECs were detached from their respective cell culture flasks via trypsinization at 37 °C for 8 min and 3 min, respectively. The cells were further manually dissociated via tapping the flasks vigorously, and the cell mixtures were collected via centrifugation at 350 g for 5 min. Immortalized CTCs (30,000 cells in 80 µL media per device) and primary DECs (65,000 cells in 160 µL media per device) were prepared and loaded into inner and outer chambers, respectively. To allow cell attachment, the devices were placed in sterile Nunc™ OmniTray™ single-well plates (ThermoFisher Scientific, Waltham, MA, USA), and the plates were incubated at 37 °C, 5% CO2 overnight. The following day, differentiated THP-1 cells were detached via trypsinization at 37 °C for 5 min, while suspensions of differentiated HL-60 and decidualized NK-92 cells were directly transferred to 15 mL conical tubes. Cells were collected via centrifugation at 350 g for 5 min, and a mixture of immune cells (20,600 NK-92 cells, 4,700 THP-1 cells, 1,700 HL-60 cells per device) was prepared. Experimental treatment (1:50 dilution CSE to mimic maternal oxidative stress (mOS), 100 ng/mL LPS, or 5 µg/mL pIC) in IL-2-deficient NK-92 media) was combined with immune cell mixture totaling 160 µL per device, and the resulting mixture were seeded into DEC chamber. The medium in CTC chamber was also replaced with fresh CTC medium. The devices were placed back in sterile single-well plates, and phase-contrast images were captured using a Nikon Eclipse TS100 microscope (Nikon, Tokyo, Japan) at 40×, 100× and 200× magnification to determine overall cell morphology. The plates were incubated at 37 °C, 5% CO2 for 48 h.
Timelapse imaging of immune cell trafficking in CDi-on-chip
To monitor immune cell trafficking from primary DECs into immortalized CTCs, timelapse imaging of the microchannels was performed under controlled conditions (37 °C, 5% CO2) provided by Stage Top Incubation System STXF (Tokai Hit, Bala Cynwyd, PA, USA) using timelapse module in Keyence BZ-X810 microscope (Keyence, Itasca, IL, USA) at a rate of 1 frame every 30 min for 24 h.
Immunofluorescence staining of cells in CDi-on-chip
After 48 h of treatment, cells in both inner and outer chambers of the devices were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Triton-X in 1× PBS for 10 min, and blocked with 3% BSA in 1× PBS for 30 min. The cells were incubated overnight with the following primary antibodies: CD45 (mouse 1:200; Cat# ab30470; Abcam, Cambridge, UK), vimentin (rabbit, 1:300; Cat# ab92547; Abcam, Cambridge, UK). The following day, the cells were washed with 1× PBS and incubated with appropriate secondary antibodies diluted in 1× PBS for 1 h (either AlexaFluor™ 488-conjugated donkey anti-rabbit; 1:1000; Abcam, Cambridge, UK or AlexaFluor™ 594-conjugated goat-anti mouse; 1:1000; ThermoFisher Scientific, Waltham, MA, USA). The devices were stained with NucBlue Fixed ReadyProbes reagent (ThermoFisher Scientific, Waltham, MA, USA), and washed with 1× PBS before viewing under Keyence BZ-X810 microscope (Keyence, Itasca, IL, USA). Image stitching module was utilized to capture fluorescent images of the entirety of the inner chamber as well as the microchannels. Decidual cells in the outer chamber and transitioned chorion mesenchymal cells in the inner chamber appeared as green, elongated cells, chorion trophoblast cells in the inner chamber appeared as cluster of nuclei without cytoplasmic staining, while immune cells appeared as round magenta or white cells. Immune cell migration from DEC into CTC was quantified manually by an independent assessor blinded to the experimental groups.
Multiplex cytokine detection via bead-based immunoassay
Custom MILLIPLEX® Human Cytokine/Chemokine/Growth Factor Panel A kit (Cat# HCYTA-60K-06; EMD Millipore, Burlington, MA, USA) detecting GM-CSF, IL-1β, IL-6, IL-8, IL-10, TNF-α was performed according to the manufacturer’s instructions. Briefly, media from the CTC or DEC chamber were combined with assay buffer and fluorescent-coded magnetic beads in each well. After overnight incubation and washing, detection antibodies and subsequently streptavidin-phycoerythrin were added to each well. Following rounds of incubation and washing, the beads were resuspended in xMAP® Sheath Fluid PLUS (MilliporeSigma, Burlington, MA, USA), and the plate was run on Luminex® 200TM instrument (Austin, TX, USA). To calculate analyte concentrations, the median fluorescent intensity data were analyzed using a 5-parameter logistic curve-fitting method.
Determination of soluble HLA-G levels via sandwich enzyme-linked immunosorbent assay (ELISA)
Soluble HLA-G (sHLA-G) ELISA kit (Cat# RD194070100R; BioVendor, Asheville, NC, USA) was performed according to the manufacturer’s instructions. 1:20 dilution of media from the CTC chamber was added to each well. After overnight incubation and washing, conjugate solution and subsequently substrate solution were added to each well. Absorbance was measured at 450 nm using a microplate reader, and sample concentrations were determined by correlating the sample absorbance to the standard curve by linear regression analysis.
Determination of progesterone levels via competitive ELISA
Progesterone competitive ELISA kit (Cat# EIAP4C21; ThermoFisher Scientific, Waltham, MA, USA) was performed according to the manufacturer’s instructions. Briefly, 1:5 dilution of media from the CTC chamber was combined with horseradish peroxidase (HRP)-conjugated progesterone and mouse anti-progesterone antibody in each goat anti-mouse IgG-coated well. After incubation and washing, 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate was added to each well. Absorbance was measured at 450 nm using a microplate reader, and sample concentrations were determined by correlating the sample absorbance to the standard curve by linear regression analysis.
Targeted RNA sequencing and analysis
RNA extraction from cells in CDi-on-chip
To isolate and purify total RNA from cells from each chamber of the device, RNeasy® Plus Mini Kit (QIAGEN, Hilden, Germany) was performed according to the manufacturer’s instructions. Cells were trypsinized from each chamber, and the cells were disrupted and homogenized. The homogenized lysate was transferred to a genomic DNA Elimination spin column, and the sample was centrifuged at 8000 g for 30 s. Equal volume of 70% ethanol was added to the flow-through, and the sample was transferred to an RNeasy spin column. The spin column was washed successively with 700 µL Buffer RW1 and two rounds of Buffer RPE, with centrifugation at 8000 g for 15 s in between every round. After the last round of Buffer RPE, the spin column was dried by centrifugation at 8000 g for 2 min, and RNA was eluted from the column by centrifugation at 8000 g for 1 min with 30 µL RNase-free water. RNA yield was measured using NanoDrop™ Eight Spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA), while RNA quality was assessed using Agilent RNA 6000 Pico Kit ran on Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Samples with RNA integrity number (RIN) > 7 were used for cDNA library preparation.
Next-generation targeted RNA-sequencing of inflammation and immunity genes
To create cDNA libraries, QIAseq Targeted RNA Human Inflammation & Immunity Transcriptome Panel (Cat# 333005 RHS-005Z-96, QIAGEN, Hilden, Germany) was performed according to manufacturer’s instructions. Using 30 ng RNA as input, first-strand cDNA was synthesized, which was then tagged with unique molecular identifiers. After molecular barcoding, the reaction was cleaned up with magnetic bead-based purification method provided, and the eluted amplicons underwent two-stage polymerase chain reaction enrichment for library construction and indexing. The reaction was cleaned up again, and the quality of cDNA libraries were assessed using Agilent High Sensitivity DNA Kit ran on Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). After library quantification using Invitrogen™ Qubit™ 1X dsDNA High Sensitivity (HS) Assay kit (ThermoFisher Scientific, Waltham, MA, USA), the purified libraries were normalized and pooled into a single library for sequencing. Single-end 150 bp sequencing was done on an Illumina MiSeq sequencing platform (Illumina, San Diego, CA, USA) using QIAseq Read 1 Primer 1 custom primer. The reads that passed Illumina quality control filtering were used as raw data for further bioinformatics analysis.
RNA-seq reference genome and transcriptome preparation
The QIASeq RNA Panels hg38 was utilized as the reference dataset: this included the reference sequence data based on RefSeq Genome Reference Consortium Human Build 38 patch release 9 (GRCh38.p9), target sequence regions as defined by the RHS-005Z, and a list of adapters to be trimmed. The target sequence regions were created by extracting the panel targets from the reference sequence, concatenating exons of multi-exon targets into single target sequences and prepending 12 ambiguous “N” nucleotides to the 5’ region to account for unique molecular identifiers (UMIs).
RNA-seq data import and quality control
Raw sequencing data in FASTQ format were imported into the CLC Genomics Workbench (version 23.0.3, QIAGEN Bioinformatics, Hilden, Germany), a comprehensive bioinformatics platform that provides an integrated suite of tools for the analysis of next-generation sequencing data. The quality of the sequencing reads was assessed using the built-in quality control tools, including the calculation of Q-scores, GC content, and sequence length distribution. Sequencing data were initially trimmed using the “Trim Reads v2.9” module. Reads containing nucleotides below the quality threshold of 0.05 (using the modified Richard Mott algorithm) and those with two or more unknown nucleotides or sequencing adapters were trimmed out to ensure the accuracy of downstream analyses. Reads shorter than 60 bases or longer than 150 bases, as well as broken read pairs were excluded from the analysis.
RNA-seq expression quantification
After the reads were mapped, RNA expression was quantified using the “Quantify QIAseq RNA v1.3” module. Reads that map to the target regions were removed if the UMI does have the expected length of 12 bases. Additionally, UMI merging to account for polymerase chain reaction (PCR) and sequencing errors was performed82. UMIs that have a count of ≤2 or lower than 5% of the maximum observed UMI count and with up to 1 mismatch and a count that is 6-fold smaller to another UMI were merged. Abundances were quantified as transcripts per kilobase million (TPM), reads per kilobase million (RPKM), relative RPKM, and UMI counts.
RNA-seq differential expression analysis
Differential gene expression analysis was performed as part of the “RNA-Seq Analysis” module. The gene expression count was modeled by a separate Generalized Linear Model (GLM), and statistical significance was evaluated using the Wald Test. This test is based on the negative binomial distribution and considers the biological variability between samples. P-values were adjusted for multiple hypothesis testing using False Discovery Rate. Differentially expressed genes (DEGs) were defined as those with an adjusted p value ≤ 0.1 and an absolute fold change value ≥ 2. Principal component analysis was performed using the function prcomp(), and heatmaps of differentially expressed genes were generated using the function pheatmap(). Overrepresentation analysis was done separately for downregulated and upregulated genes using the function enrichKEGG() and visualized using the function cnetplot() in clusterProfiler 4.083. Gene set enrichment analysis was done using the function gseKEGG() and visualized using the functions ridgeplot() and dotplot() in clusterProfiler 4.083. Data were analyzed and visualized using RStudio version 2023.06.1 + 524 (Posit, Boston, MA, USA).
Ingenuity pathway analysis
Following the identification of DEGs, the Ingenuity Pathway Analysis (version 24.0, Bioinformatics, Hilden, Germany) software was employed to investigate the biological functions and pathways associated with these genes. A core expression analysis was performed on the imported DEGs. The expression log ratio was used to calculate the directionality (z-scores) of identified features. The core analysis provides diseases and biological functions associated with the DEGs based on the Ingenuity Knowledge Base (direct and indirect relationships), a comprehensive repository of biological interactions and functional annotations. Interaction and causal networks were generated.
Statistics and reproducibility
The n numbers are indicated in figure legends, where appropriate. All experiments were performed at least two times. Values identified as outliers by Dixon’s Q test were excluded from statistical analysis. Multiple unpaired two-tailed t-tests were employed to determine if the differences in cytokine and other soluble mediators between control and experimental treatment conditions were statistically significant. Multiple two-tailed Mann-Whitney U test was done to determine whether the number of migrating CD45+ cells differed in various experimental treatment conditions. P-values of 0.05 or less were considered statistically significant. Data were analyzed and visualized using GraphPad Prism 9.5.0 (GraphPad Software, Boston, MA, USA).
Study approval
Anonymous discarded placental specimens used in the study did not meet definition of a human subject under Health and Human Services regulations, 45 CFR part 46.102, thus this study did not require approval or oversight from Institutional Review Board of The University of Texas Medical Branch at Galveston, TX, USA (UTMB 11-251). This study was also exempted for review by the University of the Philippines Manila Research Ethics Board (UPMREB 2022-0050-EX).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Targeted RNA-seq data have been deposited at Gene Expression Omnibus under accession number GSE251809, and are publicly available as of the time of publication. Source data, including uncropped Western blot images, are provided with this paper as Supplementary Data 1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
Gomez-Lopez, N., StLouis, D., Lehr, M. A., Sanchez-Rodriguez, E. N. & Arenas-Hernandez, M. Immune cells in term and preterm labor. Cell Mol. Immunol. 11, 571–581 (2014).
Vidal, M. S., Lintao, R. C. V., Severino, M. E. L., Tantengco, O. A. G. & Menon, R. Spontaneous preterm birth: involvement of multiple feto-maternal tissues and organ systems, differing mechanisms, and pathways. Front. Endocrinol. 13, 1015622 (2022).
Menon, R., Richardson, L. S. & Lappas, M. Fetal membrane architecture, aging and inflammation in pregnancy and parturition. Placenta 79, 40–45 (2019).
Menon, R. & Moore, J. J. Fetal membranes, not a mere appendage of the placenta, but a critical part of the fetal-maternal interface controlling parturition. Obstet. Gynecol. Clin. North Am. 47, 147–162 (2020).
Kammala, A. K. et al. Expression of CYP450 enzymes in human fetal membranes and its implications in xenobiotic metabolism during pregnancy. Life Sci. 307, 120867 (2022).
Aye, I. L., Paxton, J. W., Evseenko, D. A. & Keelan, J. A. Expression, localisation and activity of ATP binding cassette (ABC) family of drug transporters in human amnion membranes. Placenta 28, 868–877 (2007).
Ganguly, E. et al. Organic anion transporting polypeptide 2B1 in human fetal membranes: a novel gatekeeper for drug transport during pregnancy? Front. Pharm. 12, 771818 (2021).
Kammala, A., Benson, M., Ganguly, E., Richardson, L. & Menon, R. Functional role and regulation of permeability-glycoprotein (P-gp) in the fetal membrane during drug transportation. Am. J. Reprod. Immunol. 87, e13515 (2022).
Kammala, A. et al. Fetal membranes contribute to drug transport across the feto-maternal interface utilizing the breast cancer resistance protein (BCRP). Life 12, https://doi.org/10.3390/life12020166 (2022).
Lintao, R. C. V., Kammala, A. K., Vora, N., Yaklic, J. L. & Menon, R. Fetal membranes exhibit similar nutrient transporter expression profiles to the placenta. Placenta 135, 33–42 (2023).
Ramuta, T., Šket, T., Starčič Erjavec, M. & Kreft, M. E. Antimicrobial activity of human fetal membranes: from biological function to clinical use. Front. Bioeng. Biotechnol. 9, 691522 (2021).
Menon, R., Behnia, F., Polettini, J. & Richardson, L. S. Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes. Semin. Immunopathol. 42, 431–450 (2020).
Jacobs, S. O. et al. Characterizing the immune cell population in the human fetal membrane. Am. J. Reprod. Immunol. 85, e13368 (2021).
Poisner, A. M., Wood, G. W., Poisner, R. & Inagami, T. Localization of renin in trophoblasts in human chorion laeve at term pregnancy. Endocrinology 109, 1150–1155 (1981).
Van Meir, C. A. et al. Immunoreactive 15-hydroxyprostaglandin dehydrogenase (PGDH) is reduced in fetal membranes from patients at preterm delivery in the presence of infection. Placenta 17, 291–297 (1996).
King, A. E., Kelly, R. W., Sallenave, J. M., Bocking, A. D. & Challis, J. R. Innate immune defences in the human uterus during pregnancy. Placenta 28, 1099–1106 (2007).
Bukowski, R. et al. Onset of human preterm and term birth is related to unique inflammatory transcriptome profiles at the maternal fetal interface. PeerJ 5, e3685 (2017).
Lozovyy, V., Richardson, L., Saade, G. & Menon, R. Progesterone receptor membrane components: key regulators of fetal membrane integrity. Biol. Reprod. 104, 445–456 (2021).
Marsh B., Zhou Y., Kapidzic M., Fisher S., Blelloch R. Regionally distinct trophoblast regulate barrier function and invasion in the human placenta. Elife 11, https://doi.org/10.7554/eLife.78829 (2022).
Richardson, L. et al. A microphysiological device to model the choriodecidual interface immune status during pregnancy. J. Immunol. 210, 1437–1446 (2023).
Zaga-Clavellina, V. et al. In vitro secretion profiles of interleukin (IL)-1beta, IL-6, IL-8, IL-10, and TNF alpha after selective infection with Escherichia coli in human fetal membranes. Reprod. Biol. Endocrinol. 5, 46 (2007).
Merlino, A. et al. Nuclear progesterone receptor expression in the human fetal membranes and decidua at term before and after labor. Reprod. Sci. 16, 357–363 (2009).
Kumar, D. et al. Progesterone inhibits in vitro fetal membrane weakening. Am. J. Obstet. Gynecol. 213, 520.e1–9 (2015).
Feng, L. et al. Progesterone receptor membrane component 1 (PGRMC1) expression in fetal membranes among women with preterm premature rupture of the membranes (PPROM). Placenta 35, 331–333 (2014).
Richardson L. S., Taylor R. N., Menon R. Reversible EMT and MET mediate amnion remodeling during pregnancy and labor. Sci. Signal. 13, https://doi.org/10.1126/scisignal.aay1486 (2020).
Kapałczyńska, M. et al. 2D and 3D cell cultures - a comparison of different types of cancer cell cultures. Arch. Med. Sci. 14, 910–919 (2018).
Fortunato, S. J., Menon, R., Swan, K. F. & Lyden, T. W. Organ culture of amniochorionic membrane in vitro. Am. J. Reprod. Immunol. 32, 184–187 (1994).
Pereira, P. N. et al. Amnion formation in the mouse embryo: the single amniochorionic fold model. BMC Dev. Biol. 11, 48 (2011).
Carter, A. M. IFPA Senior Award Lecture: mammalian fetal membranes. Placenta 48, S21–S30 (2016).
Downs K. M., Rodriguez A. M. The mouse fetal-placental arterial connection: a paradigm involving the primitive streak and visceral endoderm with implications for human development. Wiley Interdiscip. Rev. Dev. Biol. 9, e362. https://doi.org/10.1002/wdev.362 (2020).
Chuva de Sousa Lopes, S. M., Roelen, B. A. J., Lawson, K. A. & Zwijsen, A. The development of the amnion in mice and other amniotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 377, 20210258 (2022).
Richardson, L., Kim, S., Menon, R. & Han, A. Organ-on-chip technology: the future of feto-maternal interface research? Front. Physiol. 11, 715 (2020).
Richardson, L., Jeong, S., Kim, S., Han, A. & Menon, R. Amnion membrane organ-on-chip: an innovative approach to study cellular interactions. FASEB J. 33, 8945–8960 (2019).
Radnaa, E. et al. Generation and characterization of human fetal membrane and decidual cell lines for reproductive biology experiments†. Biol. Reprod. 106, 568–582 (2022).
Kaur, G. & Dufour, J. M. Cell lines: valuable tools or useless artifacts. Spermatogenesis 2, 1–5 (2012).
Mosebarger, A. et al. Immune cells at the feto-maternal interface: comprehensive characterization and insights into term labor. J. Reprod. Immunol. 163, 104239 (2024).
Radnaa, E. et al. Extracellular vesicle mediated feto-maternal HMGB1 signaling induces preterm birth. Lab. Chip 21, 1956–1973 (2021).
Richardson, L. S., Kim, S., Han, A. & Menon, R. Modeling ascending infection with a feto-maternal interface organ-on-chip. Lab. Chip 20, 4486–4501 (2020).
Richardson, L. S. et al. Development of oxidative stress-associated disease models using feto-maternal interface organ-on-a-chip. FASEB J. 37, e23000 (2023).
Wang, W., Sung, N., Gilman-Sachs, A. & Kwak-Kim, J. T Helper (Th) cell profiles in pregnancy and recurrent pregnancy losses: Th1/Th2/Th9/Th17/Th22/Tfh cells. Front. Immunol. 11, 2025 (2020).
Martín-Villa, J. M. et al. HLA-G: too much or too little? Role in cancer and autoimmune disease. Front. Immunol. 13, 796054 (2022).
Sahay, A., Kale, A. & Joshi, S. Role of neurotrophins in pregnancy and offspring brain development. Neuropeptides 83, 102075 (2020).
McCormick, J., Whitley, G. S., Le Bouteiller, P. & Cartwright, J. E. Soluble HLA-G regulates motility and invasion of the trophoblast-derived cell line SGHPL-4. Hum. Reprod. 24, 1339–1345 (2009).
Kumar, D. et al. Decidual GM-CSF is a critical common intermediate necessary for thrombin and TNF induced in-vitro fetal membrane weakening. Placenta 35, 1049–1056 (2014).
Favaron, P. O., Carvalho, R. C., Borghesi, J., Anunciação, A. R. & Miglino, M. A. The amniotic membrane: development and potential applications—a review. Reprod. Domest. Anim. 50, 881–892 (2015).
Joyce, E. M. et al. In-vivo stretch of term human fetal membranes. Placenta 38, 57–66 (2016).
Sindram-Trujillo, A. P. et al. Comparison of decidual leukocytes following spontaneous vaginal delivery and elective cesarean section in uncomplicated human term pregnancy. J. Reprod. Immunol. 62, 125–137 (2004).
Sharkey, A. M. et al. Tissue-specific education of decidual NK cells. J. Immunol. 195, 3026–3032 (2015).
Flores-Espinosa, P. et al. Immunomodulatory role of decidual prolactin on the human fetal membranes and placenta. Front. Immunol. 14, 1212736 (2023).
Goldman, B., Radnaa, E., Kechichian, T. & Menon, R. Silencing P38 MAPK reduces cellular senescence in human fetal chorion trophoblast cells. Am. J. Reprod. Immunol. 89, e13648 (2023). Jan.
Yang L., et al. Innate immune signaling in trophoblast and decidua organoids defines differential antiviral defenses at the maternal-fetal interface. Elife 11, https://doi.org/10.7554/eLife.79794 (2022).
Hackmon R., et al. Definitive class I human leukocyte antigen expression in gestational placentation: HLA-F, HLA-E, HLA-C, and HLA-G in extravillous trophoblast invasion on placentation, pregnancy, and parturition. Am. J. Reprod. Immunol. 77, https://doi.org/10.1111/aji.12643 (2017).
Koga, K. & Mor, G. Toll-like receptors at the maternal-fetal interface in normal pregnancy and pregnancy disorders. Am. J. Reprod. Immunol. 63, 587–600 (2010).
Romero, R., Mazor, M., Manogue, K., Oyarzun, E. & Cerami, A. Human decidua: a source of cachectin-tumor necrosis factor. Eur. J. Obstet. Gynecol. Reprod. Biol. 41, 123–127 (1991).
Gómez-Chávez, F. et al. NF-κB and its regulators during pregnancy. Front. Immunol. 12, 679106 (2021).
Menon, R. Fetal inflammatory response at the fetomaternal interface: a requirement for labor at term and preterm. Immunol. Rev. 308, 149–167 (2022).
Glover, A. V. & Manuck, T. A. Screening for spontaneous preterm birth and resultant therapies to reduce neonatal morbidity and mortality: a review. Semin. Fetal Neonatal Med. 23, 126–132 (2018).
Pru, J. K. & Clark, N. C. PGRMC1 and PGRMC2 in uterine physiology and disease. Front. Neurosci. 7, 168 (2013).
Castelli, E. C. et al. Insights into HLA-G genetics provided by worldwide haplotype diversity. Front. Immunol. 5, 476 (2014).
Amodio, G. & Gregori, S. HLA-G genotype/expression/disease association studies: success, hurdles, and perspectives. Front. Immunol. 11, 1178 (2020).
Xu, X., Zhou, Y. & Wei, H. Roles of HLA-G in the maternal-fetal immune microenvironment. Front. Immunol. 11, 592010 (2020).
Rouas-Freiss, N. et al. Role of the HLA-G immune checkpoint molecule in pregnancy. Hum. Immunol. 82, 353–361 (2021).
Barbaro G., et al. HLA-G and recurrent pregnancy loss. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24032557 (2023).
Aisagbonhi, O. & Morris, G. P. Human leukocyte antigens in pregnancy and preeclampsia. Front Genet 13, 884275 (2022).
Tantengco, O. A. G. et al. Histocompatibility antigen, class I, G (HLA-G)‘s role during pregnancy and parturition: a systematic review of the literature. Life 11, https://doi.org/10.3390/life11101061 (2021).
Lin, A. & Yan, W. H. Perspective of HLA-G induced immunosuppression in SARS-CoV-2 infection. Front. Immunol. 12, 788769 (2021).
Ferreira, L. M. R., Meissner, T. B., Tilburgs, T. & Strominger, J. L. HLA-G: at the interface of maternal-fetal tolerance. Trends Immunol. 38, 272–286 (2017).
Dunk, C. E. et al. Human leukocyte antigen HLA-C, HLA-G, HLA-F, and HLA-E placental profiles are altered in early severe preeclampsia and preterm birth with chorioamnionitis. Am. J. Obstet. Gynecol. 227, 641.e1–641.e13 (2022).
Kusanovic, J. P. et al. Amniotic fluid soluble human leukocyte antigen-G in term and preterm parturition, and intra-amniotic infection/inflammation. J. Matern. Fetal Neonatal Med. 22, 1151–1166 (2009).
Krop, J. et al. Soluble HLA-G blood levels are not increased during ongoing pregnancy in women with a history of recurrent pregnancy loss. J. Reprod. Immunol. 153, 103665 (2022).
Cappelletti, M., Presicce, P. & Kallapur, S. G. Immunobiology of acute chorioamnionitis. Front. Immunol. 11, 649 (2020).
Park, J. Y. et al. A microphysiological model of human trophoblast invasion during implantation. Nat. Commun. 13, 1252 (2022).
Bonney, E. A. & Johnson, M. R. The role of maternal T cell and macrophage activation in preterm birth: cause or consequence? Placenta 79, 53–61 (2019).
Boddy, A. M., Fortunato, A., Wilson Sayres, M. & Aktipis, A. Fetal microchimerism and maternal health: a review and evolutionary analysis of cooperation and conflict beyond the womb. Bioessays 37, 1106–1118 (2015).
Green, E. S. & Arck, P. C. Pathogenesis of preterm birth: bidirectional inflammation in mother and fetus. Semin. Immunopathol. 42, 413–429 (2020).
Millius, A. & Weiner, O. D. Manipulation of neutrophil-like HL-60 cells for the study of directed cell migration. Methods Mol. Biol. 591, 147–158 (2010).
Starr, T., Bauler, T. J., Malik-Kale, P. & Steele-Mortimer, O. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS One 13, e0193601 (2018).
Liu, Y. et al. Changes of inhibitory receptors on NK-92 cells and HLA-G on BeWo cells with Toxoplasma gondii infection. Inflammation 36, 1440–1447 (2013).
Montaldo, E. et al. Human NK cells at early stages of differentiation produce CXCL8 and express CD161 molecule that functions as an activating receptor. Blood 119, 3987–3996 (2012).
Montaldo, E. et al. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A 83, 702–713 (2013).
Zhang, C., Zhang, J., Niu, J., Zhou, Z. & Tian, Z. Interleukin-12 improves cytotoxicity of natural killer cells via upregulated expression of NKG2D. Hum. Immunol. 69, 490–500 (2008).
Peng, Q., Vijaya Satya, R., Lewis, M., Randad, P. & Wang, Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genom. 16, 589 (2015).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Acknowledgements
This study was supported by UH3TR003283-03 (NIH/NICHD and NIH/NCATS) and UH2TR004117-01 (NIH/NCATS) to R.M. The funders had no role in the conceptualization, design, and conduct of the study, and data analysis and interpretation. R.C.V.L. was an MD-PhD in Molecular Medicine Program trainee supported by Department of Science and Technology—Philippine Council for Health Research and Development (DOST-PCHRD). We thank Dr. Enkhtuya Radnaa, Dr. Benjamin Bush and Mohamed Bettayeb for their assistance in this project.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.C.V.L., L.S.R., L.M.M.D., and R.M.; methodology: R.C.V.L., L.S.R., A.K.K., L.M.M.D., and R.M.; investigation: R.C.V.L., A.K.K., J.C., D.A.Y.Y., K.K., and G.G.; formal analysis: R.C.V.L., J.C., and D.A.Y.Y.; validation: L.S.R., J.C., D.A.Y.Y., K.K., and G.G.; resources: L.S.R., D.A.Y.Y., K.K., G.G., and R.M.; writing: R.C.V.L., L.S.R., L.M.M.D., and R.M.; supervision: L.S.R., A.K.K., L.M.M.D., and R.M.; funding acquisition: R.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Justin Padron and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lintao, R.C.V., Richardson, L.S., Kammala, A.K. et al. PGRMC2 and HLA-G regulate immune homeostasis in a microphysiological model of human maternal-fetal membrane interface. Commun Biol 7, 1041 (2024). https://doi.org/10.1038/s42003-024-06740-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06740-2
This article is cited by
-
Immunomodulatory effect of mesenchymal stromal cell overexpressing HLA-G1 in cell-based therapy for myocardial infarction
Stem Cell Research & Therapy (2026)
