
Abstract
Identifying anti-inflammatory peptides (AIPs) and antimicrobial peptides (AMPs) is crucial for the discovery of innovative and effective peptide-based therapies targeting inflammation and microbial infections. However, accurate identification of AIPs and AMPs remains a computational challenge mainly due to limited utilization of peptide sequence information. Here, we propose PepNet, an interpretable neural network for predicting both AIPs and AMPs by applying a pre-trained protein language model to fully utilize the peptide sequence information. It first captures the information of residue arrangements and physicochemical properties using a residual dilated convolution block, and then seizes the function-related diverse information by introducing a residual Transformer block to characterize the residue representations generated by a pre-trained protein language model. After training and testing, PepNet demonstrates great superiority over other leading AIP and AMP predictors and shows strong interpretability of its learned peptide representations. A user-friendly web server for PepNet is freely available at http://liulab.top/PepNet/server.
Similar content being viewed by others

Introduction
Bacterial infections and inflammation play a crucial role in human health and safety. Bacterial infections, including skin infections, urinary tract infections1, and gastrointestinal infections, can cause serious pathological consequences. Inflammation is a basic physiological body response to injury or infection, intended to protect tissues and promote repair. However, when inflammation persists for a long time without control, it may develop into chronic inflammation2,3, which is strongly associated with a variety of diseases, including neurodegenerative diseases, cardiovascular diseases, cancers, and autoimmune diseases. For bacterial infections, antibiotics are commonly used as a treatment, effectively killing or inhibiting the growth and reproduction of bacteria4. However, the overuse of antibiotics has led to the emergence of bacterial resistance, posing major global health problems due to the spread of infections caused by drug-resistant pathogens1. For chronic inflammation, nonsteroidal anti-inflammatory drugs (NSAIDs)5,6, corticosteroids, and immunosuppressants, which inhibit the immune system and reduce inflammation levels, are currently the main treatment methods7. However, similar to antibiotics, overuse of them often results in many adverse reactions and can induce drug resistance. Therefore, there is an urgent need to discover and rationally design effective antimicrobial and anti-inflammatory drugs.
Antimicrobial peptides (AMPs) and anti-inflammatory peptides (AIPs) have been demonstrated to be less drug-resistant and offer broader applications and advantages in the treatment of bacterial infections and inflammation8,9. AMPs can penetrate bacterial cell membranes, disrupt their membrane structures, or interact directly with biomolecules inside bacteria, leading to bacterial death10. On the other hand, AIPs can inhibit the production and release of inflammatory mediators and reduce tissue inflammatory response11. Therefore, it is crucial to effectively identify antibacterial and anti-inflammatory peptides with biological activity for the development of drug candidates. Traditional experimental methods for identifying antibacterial and anti-inflammatory peptides are time-consuming, expensive, and labor-intensive1,12,13. Hence, computational methods, especially traditional machine learning-based methods, and the currently popular deep learning-based methods have received widespread attention for their ability to identify antibacterial and anti-inflammatory peptides rapidly and efficiently with high throughput.
The machine learning methods, AMP Scanner Vr.114, AIPStack15, and PPTPP16 can efficiently identify the AMPs or AIPs via random forests17. However, the performance of traditional ML-based methods, such as SVM (supporting vector machine)18 and RF (random forests)17, is seriously influenced by the handcraft features. In recent years, deep learning-based methods have demonstrated superior performance in identifying AMPs and AIPs, owing to their unique network architectures and strong learning abilities14. Prominent deep learning models, including convolutional neural networks (CNN)19, recurrent neural networks (RNN)20, and transformer21 have shown success in this domain. CNNs are able to extract local features from peptide sequences, while RNNs are specialized in recurrently encoding sequential information in a peptide sequence. Long short-term memory (LSTM)22, bidirectional long short-term memory (Bi-LSTM)23, and gated recurrent unit (GRU)24, are three variants of RNN proposed to solve the problem of gradient disappearance and to capture the long-term dependence of the peptide sequences. Transformer enables capturing relationships between different residues in various high-dimensional feature sub-spaces. For instance, AMPlify25 employs Bi-LSTM26 and multi-head scale dot-product attention in transformer (MHSDPA)27, AMP Scanner Vr.214 leverages CNN and LSTM, TriNet28 employs CNN, Bi-LSTM, and the encoder in Transformer for AMP recognition, and AMP-BERT29 is a deep learning model that fine-tunes the bidirectional encoder representations from transformers (BERT) architecture. AIP_MDL30, another method, employs GRU, CNN, and attention mechanism for AIP recognition.
While existing deep learning methods have achieved much success in predicting AMPs and AIPs, they still exhibit many drawbacks, including feature extraction and network architecture. In terms of feature extraction, current deep learning-based methods typically employ hand-designed features, such as amino acid composition (AAC)31, adaptive skip-gram of dipeptide composition (ASDC)32, and physicochemical properties. However, these handcrafted features often fail to fully capture the intricate patterns and relationships hidden in peptide sequences, potentially overlooking crucial information pertinent to antimicrobial or anti-inflammatory activities. In terms of network architecture, CNNs fail to extract global or long-range dependencies in peptide sequences, and RNNs are prone to gradient vanishing or explosion33. LSTM and GRU models are designed to handle long-dependency problems and have many parameters, which may lead to overfitting and inefficient training for short peptide sequences33. By contrast, simpler architectures might be more effective and computationally efficient for short peptide sequences. Therefore, fully extracting function-related peptide features by combining a reasonable and interpretable deep learning model is crucial for predicting AMPs and AIPs.
In this study, we introduce PepNet, an interpretable deep learning framework for predicting peptides with antimicrobial or anti-inflammatory activities via a pretrained protein large language model to extract function-related high-dimensional peptide sequence features (see Fig. 1 for the workflow of PepNet). For a given peptide, PepNet first extracts original features, including amino acid types and physicochemical properties, as well as high-dimensional features that contain more informative and generalized sequence information from a pretrained protein large language model. The original features are encoded by a specially designed residual dilated convolution block to capture the spaced neighboring information, and the pre-trained features, along with the encoded original features, are fed into a specially developed residual Transformer block to capture the global sequence information by combining all the positional information from the peptide sequence. Finally, an average pool operation is applied to obtain the peptide representation for the classification of peptide activities.

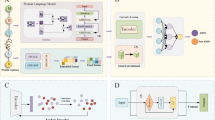
A The overall flowchart of the PepNet model. B The residual dilated convolution block. The residue type and physicochemical property features are passed through three residual dilated convolution layers, where the dilation rate of each layer increases sequentially (e.g., d = 1, 2, 4). Finally, the feature vectors, after the residual connections, are passed through a fully connected layer to derive the output of the residual dilated convolution block. C The residual transformer block. Firstly, the features extracted from the pre-trained protein language model are embedded via a fully connected layer and are then combined with the outputs of the residual dilated convolution block. Subsequently, the combined features are fed into the residual Transformer block to attend to all-positional information across the entire peptide sequence.
PepNet was trained and tested on anti-inflammatory and antimicrobial peptide datasets and demonstrated superior performance over other leading predictors in identifying both AMPs and AIPs. For instance, its F1-score and MCC values are 4.2% and 8.4% higher than those of the second-best model on AMP test set and 14.0% and 33.2% higher than those of the second-best model on AIP test set, respectively. Furthermore, by visualizing the learned representations of the residual dilated convolution block and the residual Transformer block, PepNet shows strong interpretability, particularly on the AMP test set. For the convenience of users, we have developed a user-friendly web server for PepNet to facilitate online predictions.
Results
Overview of the PepNet framework
PepNet predicts peptides with anti-inflammatory or antibacterial activity by taking the peptide sequence as input and calculating the probability of anti-inflammatory or antibacterial activity. Its main framework comprises the following four parts (see Fig. 1): (1) extracting diverse peptide features, (2) bi-channel feature encoding via the residual dilated convolution block, (3) residue representation learning by the residual Transformer block, and (4) peptide-wise binary prediction generation.
For a given peptide sequence, PepNet extracts the one-hot encoding of the amino acid types, physicochemical properties, and high-dimensional embedding features derived from the pre-trained protein language model34, resulting in a feature matrix X of shape L × D, where L represents the fixed length of the peptide sequence and D denotes the dimension of the extracted features. In cases where the length of the peptide sequence is less than L, zero-padding is employed; conversely, if the length exceeds L, truncation is applied. The physicochemical properties contain eight amino acid indices and six specific amino acid properties. The original features, including the one-hot encoding and the physicochemical properties, are encoded via the residual dilated convolution block to capture the information of multi-order neighbors for each amino acid in the sequence based on the residual dilated convolution block layers. Inspired by the TCN35 block, we construct three dilated convolution layers that progressively expand the receptive field and capture information from the increasingly spaced sequence neighbors. Subsequently, the encoded original features, along with the features derived from the pre-trained protein language models are fed into a residual transformer block for capturing the global sequence information. The transformer encoder and decoder modules can extract information from all positions in the peptide sequence and calculate the dependencies between different positions within the peptide sequence. Finally, the learned sequence features pass through an average pooling, and the representation of the peptide sequence is generated, which is then fed into a multilayer perceptron (MLP) for the classification of peptide activities.
Comparison with other leading predictors
In this section, we compare the performance of PepNet with other state-of-the-art AMP or AIP predictors. The AMP prediction models participating in the comparison include AMPlify25, AMP Scanner Vr.1 (RF Precision)14, AMP Scanner Vr.1 (Earth precision)14, AMP Scanner Vr.214, AMP Scanner Vr.2 (retrained with our data)14, TriNet (retrained with our data)28, and AMP-BERT29. The AIP prediction models compared are AIPStack15, PPTPP (class feature)16, PPTPP (probability feature)16, PPTPP (Fusion feature)16, AIP_MDL30, and TriNet (retrained with our data)28. Furthermore, the Fast version of PepNet without pre-trained features (detailed in the section “Utilization of PepNet via an online web server”) is also compared. Five commonly used evaluation metrics are applied in this study to evaluate the performance of the models, namely Matthews correlation coefficient (MCC), accuracy (ACC), precision, recall, and F1-score. A detailed description and formula for each metric can be found in Supplementary Note 1.
Performance comparison on AMP prediction
The results of the performance comparison on the AMP test set are presented in Table 1 and Fig. 2. Based on the comparison results, we find that PepNet exhibits the best performance on the AMP test set, outperforming all other compared methods. The values of accuracy, recall, precision, F1-score, and MCC for PepNet are 0.950, 0.954, 0.947, 0.951, and 0.901, respectively. Furthermore, the improvement rates achieved by PepNet relative to the other methods are: 3.3–22.3%, 4.4–21.2%, 1.6–29.7%, 3.5–21.3%, and 7.3–60.3% in terms of accuracy, recall, precision, F1-score, and MCC. Specifically, the recall, F1-score, MCC value of PepNet are largely improved and achieve 4.4%, 3.5%, and 7.3% higher than the second-best model. Moreover, PepNet is the only model with all five evaluation metrics above 0.9, indicating its strong ability to accurately identify AMPs.

This figure displays the performance of PepNet and other compared methods on the AMP test set, where the performance of PepNet is shown in red on the far right.
Performance comparison on AIP prediction
The performance of the compared methods on the AIP test set is presented in Table 2 and Fig. 3, which show that the performance of PepNet on the AIP test set is again the best among all the methods that are evaluated. In detail, the values of accuracy, recall, precision, F1-score, and MCC of PepNet are 0.819, 0.940, 0.705, 0.806, and 0.666, respectively. In addition, relative to other compared methods, the improvement rates of PepNet for accuracy, recall, F1-score and MCC are 8.2–30.6%, 28.6–889.5%, 14.0–374.1%, and 33.2–276.3%, respectively. The recall, F1-score, and MCC of PepNet are greatly improved and are respectively 28.6%, 14%, and 33.2% higher than the second-best model, respectively. Although the precision of PepNet is slightly lower than that of PPTPP (Fusion feature) and AIP_MDL, its other metrics are considerably higher, resulting in PepNet achieving the best overall performance. Furthermore, given that recall and precision are two trade-off metrics, PepNet achieves notably higher recall than other algorithms, even with a relatively small decrease in precision, indicating that PepNet is more sensitive in identifying true positives.

This figure displays the performance of PepNet and other compared methods on the AIP test set, where the performance of PepNet is shown in red on the far right.
In summary, PepNet demonstrates significantly better performance compared to state-of-the-art methods on both AMP and AIP identification. The high values observed in the F1-score and MCC indicate that PepNet achieves a high standard of accuracy and consistency in peptide function prediction.
Robustness and generalization ability of PepNet
In addition to the two AMP and AIP datasets, we added five AMP datasets with different activities (antibacterial, antifungal, antiviral, anticancer, and anti-mammalian cells) collected from iAMPCN36 and three AIP datasets (aip_data1, aip_data2, and aip_data3) collected from AIPStack15, BertAIP37, and IF-AIP38 to compare the performance of PepNet with other predictors (see Supplementary Tables 1–8) and demonstrate its robustness and generalization ability. As most of these datasets are unbalanced, the ACC metric is unable to measure the performance of a method and therefore, we removed it in performance comparison. As shown in Supplementary Tables 1–8, PepNet consistently demonstrates the best overall performance on all the added datasets compared to other methods. Additionally, as the proportion of positive and negative samples varies across different datasets, we found that PepNet exhibits better performance in datasets with more balanced positive and negative samples. In particular, for the unbalanced antifungal and anti-mammalian-cells datasets, AMP-BERT29 is unable to learn the attributes of positive samples and all the peptides are predicted as non-AMPs.
Considering that many antimicrobial or anti-inflammatory peptides can be toxic, we added a function of predicting toxicity by training the model on a toxic peptide dataset collected from ATSE39 and compared it with ClanTox40, ToxinPred-RF41, ToxinPred-SVM41, ATSE39, and ATSE’s variants (Only-GNN and Only-CNN_BiLSTM). As shown in Supplementary Table 9 and Supplementary Fig. 1, we find that PepNet shows the best overall performance.
Ablation studies on PepNet
In this study, PepNet employs a sequence-based feature extraction method and incorporates advanced deep learning techniques to enhance the accuracy and robustness for the predictions for both AMPs and AIPs. In this section, we delve into a detailed analysis of each constituent of the model to elucidate its role and validate its contribution, particularly focusing on components responsible for feature extraction and sequence information processing. Through a series of ablation experiments, we systematically investigate the impact of altering individual model components or hyperparameters, adhering to a methodology where only one component or hyperparameter is modified at a time. According to the architecture of the PepNet framework, the ablation experiments examine the impact of feature selection, the contribution of the residual dilated convolution block, the contribution of the residual Transformer block, and the approach of pooling amino acid features into a peptide feature.
Impact of feature selection on PepNet
To investigate the contribution of features to PepNet, we trained and tested PepNet by removing one-hot features, physicochemical property features, and pre-trained features, respectively. As illustrated in Fig. 4 (see detailed results in Supplementary Tables 10, 11), the exclusion of each of the three distinct features clearly influences the performance of PepNet, leading to a reduction of 1.9–4.5% and 3.7–8.7% in F1-score and MCC on the AMP test set, and 7.7–10.2% and 17.7–21.5% in F1-score and MCC on the AIP test set. Specifically, the pre-trained features, which are typically obtained by a protein language model trained on large-scale protein datasets, show the highest contribution to PepNet on both the AMP and AIP test sets and the removal of this feature results in large decreases of 4.5% and 10.2% in F1-score, and 8.7% and 21.5% in MCC, respectively on AMP and AIP test sets, compared to using all features. Notably, the physicochemical properties also contribute significantly to PepNet on the AIP test set, resulting in a decrease of 9.9% in F1-score and 21.2% in MCC after removing it.

This figure displays the performance of PepNet under different ablation experiments in terms of accuracy (A), recall (B), precision (C), F1-score (D), and MCC (E) on the AMP and AIP test sets. In each figure, the letters A1–A4 represent feature ablation experiments of PepNet by using the amino acid type one-hot encoding, amino acid physicochemical properties, pre-trained features derived from the large protein language model, and the combination of them; B1–B5 represent the residual dilated convolution block ablation experiments of PepNet by removing the residual dilated convolution block, removing the residual connection operation within the block, and substituting the dilated convolution layer with Bi-LSTM, LSTM, and GRU layers, respectively, and B6 represents the residual dilated convolution block applied by PepNet; C1–C6 represent the residual Transformer block ablation experiments of PepNet by removing the residual dilated convolution block, removing the residual connection operation within the block, and with 1–4 Transformer layers in the block; D1 and D2 represent the maximum and average pooling strategies on PepNet.
Contribution of the residual dilated convolution block to PepNet
The residual dilated convolution block is responsible for capturing spaced neighbors information in peptide sequences and is a key component for dynamically understanding the distribution of amino acids in a sequence. To explore the impact of the residual dilated convolution block on PepNet, we conducted experiments by altering its architecture as follows: (1) removing the residual dilated convolution block entirely, (2) removing the residual connection operation within the block, and (3) substituting the dilated convolution layer with Bi-LSTM, LSTM, or GRU, respectively. As illustrated in Fig. 4 (see detailed results in Supplementary Tables 10-11), the exclusion of the residual dilated convolution block has a great impact on PepNet, leading to a performance reduction of 4.9% and 9.2% in F1-score and MCC on the AMP test set, and 10.5% and 24.2% on the AIP test set. However, the removal of residual connection leads to a decrease of 1.7% and 3.0% in F1-score and MCC on the AMP test set, and 6.3% and 14.4% on the AIP test set. Specifically, when replacing the dilated convolution with Bi-LSTM, LSTM, or GRU, the performance decreases by 3.9–4.0% and 7.1–8.1% in F1-score and MCC on the AMP test set, and 7.0–8.8% and 16.4–19.8% on the AIP test set, indicating that the dilated convolution layers effectively capture global spaced-neighbor information, which is important for identifying AMPs and AIPs.
Contribution of the residual Transformer block to PepNet
The residual Transformer block is tasked with attending to key positional amino acid information while also capturing comprehensive positional details throughout the whole peptide sequence. In order to investigate the influence of the residual Transformer block on PepNet, we also conducted experiments by excluding the residual Transformer block, removing the residual connection operation within the block, and changing the hyperparameter for the number of Transformer layers in the block. As shown in Fig. 4 (see detailed results in Supplementary Tables 10, 11), removing the residual Transformer block results in a notable degradation of PepNet’s performance, particularly observed in the AIP test set, where both F1-score and MCC demonstrate substantial declines of up to 21.7% and 54.8%, respectively. It is noteworthy that during the experiments involving variations in the hyperparameters of Transformer layers, we found that the performance of PepNet declines as the number of layers increases. This phenomenon may be attributed to the increased model complexity caused by the increased number of layers, potentially leading to overfitting or inadequate training to effectively support deeper network architectures. Although this effect is less pronounced on the larger AMP training set, in contrast to that of AIP, it remains present. These findings indicate that when designing Transformer-based models, the choice of the number of layers must be carefully calibrated to the specific task and data characteristics to avoid unnecessary complexity and performance loss.
Pooling operations of amino acid features
The pooling operation is employed to downscale and extract crucial features, which represents a pivotal step in the generation of the final sequence representation. Maximum pooling tends to concentrate more on capturing the most salient signals, while average pooling offers a more comprehensive vector of sequence features. To evaluate the impact of different pooling strategies on PepNet, we replaced average pooling with maximum pooling. As shown in Fig. 4 (see detailed results in Supplementary Tables 10, 11), maximum pooling results in a 2.6% decrease in F1-score and a 5.5% decrease in MCC on the AMP test set, along with an 11.2% decrease in F1-score and a 24.5% decrease in MCC on the AIP test set for PepNet. This indicates that solely focusing on the maximum features of all amino acids fails to adequately characterize the entire peptide sequence, leading to large information loss.
Interpretability of the PepNet model
To deeply understand the learning mechanism of PepNet in the detection of antibacterial and anti-inflammatory activities, we explore what it learns in different ways. For instance, what are the contributions of the pre-trained features to the classification? What does the residual dilated convolution block learn? What does the residual Transformer block learn? In this study, we apply the t-SNE, a machine learning algorithm commonly used for dimensionality reduction and visualization of high-dimensional data in a lower-dimensional space, to perform a detailed visualization of the high-dimensional feature representations learned by PepNet. By projecting the learned peptide representation learned by PepNet into a reduced-dimensional space, it is easy to find the similarity between positive or negative samples and the degree of their distinguishability. Moreover, we illustrate the interpretability of the PepNet model by exploring whether it is perceiving cationic and amphiphilic properties in AMPs. The visualization results are presented in Fig. 5 and Supplementary Fig. 2.

This figure displays the visualization of the 2D t-SNE projections of the original features (A), pre-trained features (B), processed features by the residual dilated convolution block (C), the input (D) and the output (E) features of the residual Transformer block.
The original and the pre-trained features influence the learning process in different manners
The original features, containing the one-hot encoding of the amino acid type and the physicochemical properties, are strongly related to the activities of a peptide, while the pre-trained features derived from a large protein language model are much richer, more informative, and more generalized. According to the 2D t-SNE projections of the original features and pre-trained features on the AMP test set (Fig. 5A, B), the separation of AMPs (red) and non-AMPs (blue) is clearer under the pre-trained features compared to the original features, indicating the important role of the pre-trained features in identifying AMPs.
The residual dilated convolution block is learning the spaced neighboring information
The residual dilated convolution block starts learning with the original features as input and outputs the spaced neighboring information in peptide sequences. By comparing the t-SNE scatter plots of the input and output features of the residual dilated convolution block (Fig. 5A, C), it is clearly observed that the boundaries between positive and negative samples are blurred in the unprocessed original features, whereas the clustering of the samples improves after the residual dilated convolution block. However, the boundary between the two categories is still not very clear. Visualization of the data before and after the residual dilated convolution block demonstrates a reduction in category overlap, indicating that the residual dilated convolution block is able to capture the key characteristics from the original features by aggregating the spaced-neighboring information, which is effective for AMP detection.
The residual Transformer block is learning the global information in the peptide
The residual Transformer block takes the output of the residual convolution block and the embedding of the pre-trained features as input, and produces global information in the peptide sequence as output. By comparing the t-SNE scatter plots of the input and output features of the residual Transformer block (Fig. 5D, E), a clear cluster segmentation of positive and negative samples within the AMP test set is evident. The clear distinction between the two categories of samples suggests that the representations learned by the residual Transformer block exhibit different characteristics among positive and negative samples and that the residual Transformer block significantly improves the recognition of AMPs by capturing comprehensive positional information across the peptide sequence.
The feature visualization analysis of each state of PepNet on the AIP test set also reveals a similar phenomenon (Supplementary Fig. 2). The t-SNE visualization results on the two test sets collectively substantiate the significance and influence of the various components in PepNet. This multi-stage feature visualization analysis not only deepens our understanding of the working mechanism of the PepNet model but also points the way to further model optimization and application practice.
PepNet perceives cationic and amphiphilic properties in AMPs
Cationic amphiphilic sequences often adopt an α-helical structure in hydrophobic environments such as cell membranes. These sequences are crucial components in many biologically active peptides due to their ability to interact with and disrupt biological membranes, making them valuable in antimicrobial therapies. In this study, we use the charge at pH 7.0 and the grand average of hydropathy (GRAVY) to measure the cationic and amphiphilic properties of peptides. The charge and GRAVY distributions in the AMP and AIP training sets are shown in Fig. 6A. Since PepNet is a data-driven deep learning model, it learns the distributions of training sets to predict the distributions of test sets. Therefore, whether these characteristics are perceived by the model depends on the distributions of the two characteristics on the training sets. Based on the AMP and AIP datasets used in this study, we illustrate the true distributions of cationic and amphiphilic properties in the AMP and AIP test sets (see Fig. 6B) and their distributions as predicted by PepNet (see Fig. 6C). It can be observed that antimicrobial peptides contain more cationic amphiphilic sequences than non-antimicrobial peptides, especially the cationic sequences, which is not applicable to the AIP dataset. Additionally, we find that PepNet accurately perceives the distributions of positive and negative samples for both the charge and GRAVY. Moreover, we visualize the final representation learned by PepNet using the t-SNE tool, colored with charge and GRAVY scores (see Fig. 6D), which clearly shows that PepNet perceives the cationic properties of peptides in the AMP dataset. Due to the slight difference in GRAVY distributions between AMPs and non-AMPs, it is hard to directly discriminate whether PepNet perceives the amphiphilic properties from the t-SNE visualization, which again coincides with the visualization result. Moreover, we can observe from Fig. 6D that the differences in both the charge and GRAVY distributions between AMPs and non-AMPs are much larger than those between AIPs and non-AIPs, which indicates that both the cationic and amphiphilic properties contribute more to antimicrobial than to anti-inflammatory properties.

A The true distributions of positive and negative samples in AMP and AIP training sets. B The true distribution of positive and negative samples in AMP and AIP test sets. C The predicted distributions of positive and negative samples by PepNet in AMP and AIP test sets. D The illustration of the final representation learned by PepNet, with points colored by positive and negative scores of charge and GRAVY.
Utilization of PepNet via an online web server
For the convenience of users in using our PepNet tool, we develop a user-friendly web server for online prediction of peptide sequences as antimicrobial peptides (AMPs) or anti-inflammatory peptides (AIPs). Based on different user requirements, we introduce two running modes: a Fast and a Standard version of the interface (Fig. 7A). The performance of these modes can be seen in Tables 1 and 2. The Fast version utilizes a trained model that does not rely on pre-trained features, facilitating quick predictions. Users can simply upload a FASTA file containing multiple peptide sequences, select the desired model type (AMP or AIP), and obtain predictions promptly. Conversely, due to our limited equipment, the Standard version requires users to submit a FASTA file alongside the corresponding pre-trained feature storage file, generated in H5PY format by ProtT5-XL-U50. Upon submission, the application generates a prediction result page (Fig. 7B) where users can view the prediction outcome for each submitted peptide sequence, including the peptide sequence, the predicted score, and the classification result. Additionally, users have the option to download the result file for further analysis. This web server provides a convenient and efficient platform for researchers to predict the antimicrobial or anti-inflammatory activity of peptide sequences. In addition, we provide an online web server for toxicity prediction.

A The interface of the online web server of PepNet. B The result interface of the application displays a result table containing the peptide sequence, the predicted probability, and the binary classification result. The rows highlighted in red indicate peptides predicted as positive. In addition, users can download the result file from the top of the table.
Discussion
Existing methods for the identification of AMPs and AIPs are often limited by a strong reliance on their handcraft features and their model architectures27,42. With the advent of deep learning, some studies have begun to employ convolutional neural networks (CNNs), recurrent neural networks (RNNs), and other sequence-based learning models, which are capable of capturing local patterns and long-distance dependencies of sequences14,43. However, they still face the challenge of insufficient understanding of complex sequence relationships and fail to capture the function-related sequence patterns.
PepNet is a sequence-based deep learning model for predicting AIPs and AMPs. It generates representative sequence representations to classify peptide sequences by combining the type and physicochemical properties of amino acids, and pre-trained features from a large protein language model. The residual dilated convolution block employs the variant dilated coefficients in the convolution to capture the spaced-neighboring information in peptide sequences. The residual Transformer block employs an all-positional attention mechanism to capture global information of all positions in a peptide sequence. These two modules empower PepNet to capture the intrinsic sub-sequences and complex biological relationships among amino acids, thereby facilitating a deeper understanding of the biological functions inherent in peptides.
After an assessment of PepNet’s performance and a comparative analysis with other prominent prediction methods on several challenging datasets, PepNet demonstrated superior accuracy in predicting AIPs and AMPs, outperforming all comparative methods based on widely accepted criteria. Furthermore, PepNet has not only been demonstrated to improve the accuracy in predicting AMPs and AIPs but also exhibits strong model interpretability, explaining the biological significance of its black-boxes. The superiority of PepNet may stem from the following several key factors: (1) Utilization of high-dimensional pre-trained features derived from extensive protein data training, facilitating the capture of essential and diverse biological information, thereby enhancing prediction accuracy. (2) Application of the dilation convolution layers, specifically three layers with distinct dilated coefficients in the residual dilated convolution block, enabling the progressive expansion of the receptive field of amino acids in peptide sequences at intervals without increasing parameters, thereby enriching multi-order neighboring information and improving model performance. (3) Incorporation of an all-positional attention mechanism from the Transformer model, facilitating comprehensive analysis of peptide sequences and enhancing feature capture comprehensiveness. (4) Implementation of residual connections in both the residual dilated convolution block and the residual Transformer block, preserving the original feature memory after three-layer dilated convolution and maintaining dual-channel output feature memory after Transformer processing. PepNet introduces an innovative deep learning strategy in sequence feature extraction and global information integration, which greatly improves the accuracy of the identification of AMPs and AIPs.
Despite the large improvement achieved by PepNet in identifying AIPs and AMPs, further optimization is necessary to address the remaining challenges in AIP or AMP prediction studies. These challenges include: (1) Limited data availability. With the limited peptide sequence data, especially the AIP data, PepNet may fail to capture the entire underlying patterns and variations in the data, limiting its generalization abilities on unseen examples. Therefore, the production of more data for training is eager for this research area. (2) Loss of information due to fixed sequence lengths. In PepNet, to facilitate batch learning, we pad sequences with zeros if they are shorter than the predefined length and truncate sequences that exceed the predefined length. However, this process may result in the loss of peptide sequence information. (3) Resource consumption of the attention mechanism. The attention mechanism typically costs additional computational resources, particularly when handling long sequences or large-scale datasets. Moreover, the attention mechanism may lead to the model over-relying on a few salient features, thereby increasing the risk of overfitting. Based on the above issues, future work will be devoted to the implementation of these improvement options in order to build more robust and reliable models that can better serve the task of AMP and AIP prediction.
Methods
Data preparation
In this study, both the AIP and AMP datasets are collected from previous studies25,30. For the AIP data, the researchers30 collected the peptides from the Immune Epitope Database44 (IEDB) as the positive samples (AIPs) that can induce the anti-inflammatory cytokines in human and mouse T-cell assays, and those that fail to induce the negative samples. Anti-inflammatory cytokines include IL-1045 (suppresses pro-inflammatory cytokine production, protects against autoimmune and allergic diseases), IL-446 (central to the TH2 immune shift, promotes M2 macrophage differentiation), IL-1347,48 (similar to IL-4, alleviates autoimmune inflammation such as inflammatory bowel disease), IL-2249,50 (from TH17 cells, essential for protection and regeneration of intestinal tissue), TGFβ47 (maintains immune balance, suppresses T and B cell activity) and IFN-α/β51,52 (antiviral with anti-inflammatory properties via the JAK-STAT pathway)47,53, etc. For the AMP data, the researchers25 collected the peptides from the Anti-microbial Peptide Database54 (APD3) and Database of Anuran Defense Peptides55 (DADP) as positive samples (AMPs), and those known not to have any antimicrobial activities (antimicrobial, antibiotic, antibacterial, antiviral, antifungal, antimalarial, antiparasitic, anti-protist, anticancer, defense, defensin, cathelicidin, histatin, bacteriocin, microbicide, fungicide) as the negative samples. There are 4194 and 8346 non-redundant samples in the AIP and AMP datasets for training and testing. As with the two corresponding studies25,30, both the AIP and AMP datasets are split into training and test sets with a radio of 3:1 and 4:1, respectively. The training set is further split into training and valid sets with a radio of 4:1. Finally, the AIP training, valid, and testing sets contain 2516, 629, and 1049 samples, while the AMP training, valid, and testing sets consist of 5340, 1336, and 1670 samples, respectively. Furthermore, we collect five additional AMP datasets with different activities (antibacterial, antifungal, antiviral, anticancer, and anti-mammalian cells) from iAMPCN36 and three additional AIP datasets (aip_data1, aip_data2, and aip_data3) from AIPStack15, BertAIP37, and IF-AIP38. These datasets include both balanced and unbalanced sets, which helps demonstrate the generalization ability of the model.
For the sequence length distribution, we presented the length distribution of all the datasets used in this study in Supplementary Fig. 3. From the results, we found that the lengths of AMPs and AIPs mainly distribute within 50AA. For the percent identity shared amongst and between classes, we used the Clustal Omega56 (clustalo) tool for global sequence alignment to calculate the percent identity of pairwise sequences within positive samples, within negative samples, and between positive and negative samples for each dataset. We showed the percent identity of all the datasets used in this study in Supplementary Figs. 4 and 5, which demonstrates that the percent identity of AMP and AIP datasets distributes differently, but primarily distributes in the intervals [0, 40%] and [0, 30%], respectively.
The PepNet architecture
In this section, we provide a detailed description of the PepNet model framework, which comprises five main components: extraction of sequence features, encoding of the original features via the residual dilated convolution block, transfer learning of the pre-trained features via the embedding layer, learning the residue representations by the residual Transformer block, and generation of peptide-wise binary prediction.
Extraction of sequence features
For a given peptide sequence, we extract two kinds of features pertaining to the amino acids: the original features, including the amino acid types and physicochemical properties, and the pre-trained features derived from a large protein language model34. The first features consist of a one-hot encoding of amino acid types (20 standard amino acids) along with 14 physicochemical properties of amino acids. These properties encompass eight amino acid indices (namely BLAM930101, BIOV880101, MAXF760101, TSAJ990101, NAKH920108, CEDJ970104, LIFS790101, and MIYS990104, selected as representatives from over 500 amino acid indices available in the AAindex database57 using a consensus fuzzy clustering method58), as well as six specific physicochemical attributes (atomicity, polarity, polarizability, net charge, hydrophobicity, propensity for β-sheet conformations). The second features comprise 1024-dimensional sequence embeddings for each amino acid generated by ProtT5-XL-U5034, a widely utilized pre-trained protein language model based on the transformer architecture. In order to encode peptide sequences in the same neural network, PepNet constructs a feature matrix X of shape L×(34 + 1024) for each peptide, where L is a free hyperparameter representing a fixed peptide sequence length (in this study, L = 40). In cases where the length of the peptide sequence is less than L, zero-padding is employed; conversely, if the length exceeds L, truncation is applied. Zero-padding introduces noises, and truncation leads to information loss. Therefore, selecting the appropriate sequence length, L is crucial. As the lengths of AMPs and AIPs are primarily within 50 AA, we selected the optimal L value by conducting experiments with L values of 30, 40, and 50 (see Supplementary Table 12). Based on the results in the table, we selected L = 40.
Encoding of the original features via the residual dilated convolution block
The original features are encoded by the residual dilated convolution block, which contains three dilated convolution layers of hidden size d/2, progressively expanding the receptive field and capturing information from the 2m + 1 spaced sequence neighbors. Suppose the input feature matrix is Xin, the dilated convolution operation F on the element Xiin of the sequence features is defined as
where 2m + 1 is the kernel size (i.e. the number of neighbors), k is the dilation factor (k = 1,2,4 in the three dilated convolution layers, respectively), \(w\in {R}^{(2m+1)\times {D}^{{{ {in}}}}\times {D}^{{{ {out}}}}}\) is the learnable parameters, \(b\in {R}^{{D}^{{{ {out}}}}}\), i = 1,2,…,L. Thus, the residual dilated convolution block can be summarized as follows:
where Xoriginal is the original feature matrix. Then, the outputs Xe1 from three convolution layers are then residual connected with the input original features, followed by a multilayer perceptron (MLP) as follows:
where cat represents the concatenation operation, d is the hidden size.
Transfer learning of the pre-trained features via the embedding layer
For the pre-trained features generated by ProtT5-XL-U5034, we reuse the embedding features through an MLP for the specific AIP or AMP classification tasks. Suppose the pre-trained feature matrix is Xe0 of shape L × 1024, then
where d is the hidden size.
Learning the residue representations by the residual Transformer block
For the encoded sequence features Xe2 and the reused embedding features Xe3, PepNet first incorporates them through a concatenate and MLP operations as follows:
where the cat represents the concatenate operation, d is the hidden size.
After that, the combined feature matrix Xe4 is fed into the residual Transformer block for capturing the global sequence information. The residual Transformer block comprises a residual Transformer containing an encoder–decoder structure, where the encoder maps the input sequence representations Xe4 to Z, and the decoder integrates the Xe4 and Z to produce output sequence representations Xe5. The encoder is structured with N identical layers, each containing two sub-layers: a multi-head self-attention layer, and a position-wise fully connected feed-forward network. N is the free hyperparameter which is set to 1 in this study. A residual connection59 around both sub-layers is employed, followed by layer normalization60. Similar to the encoder, the decoder is structured with N identical layers, each containing three sub-layers: two multi-head self-attention layers, and a position-wise fully connected feed-forward network. N is the free hyperparameter which is set to 1 in this study. The additional multi-head attention layer is implemented at the output layer of the encoder and the first multi-head layer. The core component of the Transformer is the multi-head self-attention, which can effectively capture the relationship between amino acid residues over long distances, facilitating the extraction of representation information from specific peptide. The process of the multi-head self-attention can be summarized as follows.
where Q, K, and V are the queries, keys, and values in the multi-head self-attention layer, cat is the concatenate operation, h is the number of parallel attention heads (in this study, h = 4), WQ, WK, WV, Wo, are the learnable weights applied to Q, K, V, and the multi-head outputs, Attention(q, k, v) is the attention mechanism which is performed as follows:
where q, k, v are the queries, keys, and values in the self-attention mechanism. The process of the position-wise fully connected feed-forward network is conducted as follows.
where X is the input feature matrix. Thus,
where Tencoder and Tdecoder are the encoder and decoder modules in Transformer.
Furthermore, a residual connection is implemented between the incorporated features Xe4 and the output presentations of the Transformer block Xe5.
where cat represents the concatenate operation.
In addition, the Transformer model, different from recurrent neural networks20 (RNN) which inherently possess sequential order, simultaneously processes all residue information within a peptide sequence. Therefore, it is imperative to incorporate positional encodings for delineating sequence features. In PepNet, we use sine and cosine functions on different feature dimensions as follows.
where pos is the position of the amino acid in the peptide sequence, i is the dimension, and d is the hidden size.
Generation of peptide-wise binary prediction
For the learned residue presentation matrix Xe6, we first average-pool them into a sequence representation, which can then be classified by using an MLP.
where score is the probability ranging from 0 to 1, where a score closer to 1 indicates a higher likelihood that the input peptide belongs to the positive class, and a score closer to 0 indicates a higher likelihood that the input belongs to the negative class.
Statistics and reproducibility
In this study, all the experiments, including validation experiments, ablation experiments, and interpretation experiments, were conducted based on the AMP and AIP datasets61. The AMP dataset was collected from AIP_MDL30, comprising 2516, 629, and 1049 samples in the training, validation, and test sets, respectively. The AIP dataset was collected from AMPlify25, comprising 5340, 1336, and 1670 samples in the training, validation, and test sets, respectively. The datasets used for reproducing all the presented results can be accessed at https://zenodo.org/records/1322351661, and the source code files for reproducing and evaluating PepNet are available at https://zenodo.org/records/1373425862. The numerical data used to generate the main figures can be found in the Supplementary Data 1 file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data used for training and testing the models and for reproducing all the presented results in this study can be available at https://zenodo.org/records/1322351661. The numerical data used to generate the main figures can be found in the Supplementary Data 1 file.
Code availability
PepNet is implemented by Python using the PyTorch framework. All supporting source codes can be downloaded from https://zenodo.org/records/1373425862.
References
Fjell, C. D., Hiss, J. A., Hancock, R. E. & Schneider, G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11, 37–51 (2012).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Serhan, C. N. & Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 (2005).
Alanis, A. J. Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res. 36, 697–705 (2005).
Day, R. O. & Graham, G. G. Non-steroidal anti-inflammatory drugs (NSAIDs). BMJ 346, f3195 (2013).
Bindu, S., Mazumder, S. & Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem. Pharmacol. 180, 114147 (2020).
Klaassen, C. D. et al. Principles of toxicology and treatment of poisoning. In The Pharmacological Basis of Therapeutics 11th edn (Goodman & GilmanÕs), 1739–1752 (McGraw Hill, Columbus, OH, USA, 2006).
Gupta, S., Sharma, A. K., Shastri, V., Madhu, M. K. & Sharma, V. K. Prediction of anti-inflammatory proteins/peptides: an insilico approach. J. Transl. Med. 15, 1–11 (2017).
Hof, W. V. T., Veerman, E. C., Helmerhorst, E. J. & Amerongen, A. V. N. Antimicrobial peptides: properties and applicability. Biol. Chem. 382, 597–619 (2001).
Andreu, D. & Rivas, L. Animal antimicrobial peptides: an overview. Pept. Sci. 47, 415–433 (1998).
Yuan, L., Zhang, F., Shen, M., Jia, S. & Xie, J. Phytosterols suppress phagocytosis and inhibit inflammatory mediators via ERK pathway on LPS-triggered inflammatory responses in RAW264. 7 macrophages and the correlation with their structure. Foods 8, 582 (2019).
Zhang, L. & Falla, T. J. Antimicrobial peptides: therapeutic potential. Expert Opin. Pharmacother. 7, 653–663 (2006).
Mahlapuu, M., Håkansson, J., Ringstad, L. & Björn, C. Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 6, 235805 (2016).
Veltri, D., Kamath, U. & Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018).
Deng, H. et al. Prediction of anti-inflammatory peptides by a sequence-based stacking ensemble model named AIPStack. Iscience 25, (2022).
Zhang, Y. P. & Zou, Q. PPTPP: a novel therapeutic peptide prediction method using physicochemical property encoding and adaptive feature representation learning. Bioinformatics 36, 3982–3987 (2020).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Hearst, M. A., Dumais, S. T., Osuna, E., Platt, J. & Scholkopf, B. Support vector machines. IEEE Intell. Syst. Appl. 13, 18–28 (1998).
Xie, L. & Yuille, A. Genetic CNN. in 2017 IEEE International Conference on Computer Vision (ICCV) 1388–1397 (IEEE, Venice, 2017). https://doi.org/10.1109/ICCV.2017.154.
Rumelhart, D. E., Hinton, G. E. & Williams, R. J. Learning representations by back-propagating errors. Nature 323, 533–536 (1986).
Vaswani, A. et al. Attention is all you need. Advances in neural information processing systems. 30, 5998–6008 (2017).
Yu, Y., Si, X., Hu, C. & Zhang, J. A review of recurrent neural networks: LSTM cells and network architectures. Neural Comput. 31, 1235–1270 (2019).
Zhou, P. et al. Attention-Based Bidirectional Long Short-Term Memory Networks for Relation Classification. in Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers) 207–212 (Association for Computational Linguistics, Berlin, Germany, 2016). https://doi.org/10.18653/v1/P16-2034.
Dey, R. & Salem, F. M. Gate-variants of Gated Recurrent Unit (GRU) neural networks. in 2017 IEEE 60th International Midwest Symposium on Circuits and Systems (MWSCAS) 1597–1600 (IEEE, Boston, MA, 2017). https://doi.org/10.1109/MWSCAS.2017.8053243.
Li, C. et al. AMPlify: attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genom. 23, 77 (2022).
Shahid, F., Zameer, A. & Muneeb, M. Predictions for COVID-19 with deep learning models of LSTM, GRU and Bi-LSTM. Chaos Solitons Fractals 140, 110212 (2020).
Yan, J. et al. Recent progress in the discovery and design of antimicrobial peptides using traditional machine learning and deep learning. Antibiotics 11, 1451 (2022).
Zhou, W. et al. TriNet: a tri-fusion neural network for the prediction of anticancer and antimicrobial peptides. Patterns 4, 100702 (2023).
Lee, H., Lee, S., Lee, I. & Nam, H. AMP‐BERT: prediction of antimicrobial peptide function based on a BERT model. Protein Sci. 32, e4529 (2023).
Guan, J. et al. Predicting anti-inflammatory peptides by ensemble machine learning and deep learning. J. Chem. Inf. Model. 63, 7886–7898 (2023).
Roy, S., Martinez, D., Platero, H., Lane, T. & Werner-Washburne, M. Exploiting amino acid composition for predicting protein–protein interactions. PloS one 4, e7813 (2009).
Jiang, M. et al. NeuroPpred-Fuse: an interpretable stacking model for prediction of neuropeptides by fusing sequence information and feature selection methods. Brief. Bioinform. 22, bbab310 (2021).
Shiri, F. M., Perumal, T., Mustapha, N. & Mohamed, R. A comprehensive overview and comparative analysis on deep learning models: CNN, RNN, LSTM, GRU. arXiv preprint arXiv:2305.17473 (2023).
Elnaggar, A. et al. Prottrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2021).
Bai, S., Kolter, J. Z. & Koltun, V. An empirical evaluation of generic convolutional and recurrent networks for sequence modeling. arXiv preprint arXiv:1803.01271 (2018).
Xu, J. et al. iAMPCN: a deep-learning approach for identifying antimicrobial peptides and their functional activities. Brief. Bioinform. 24, bbad240 (2023).
Xu, T., Wang, Q., Yang, Z. & Ying, J. A BERT-based approach for identifying anti-inflammatory peptides using sequence information. Heliyon 10, e32951 (2024).
Gaffar, S., Hassan, M. T., Tayara, H. & Chong, K. T. IF-AIP: a machine learning method for the identification of anti-inflammatory peptides using multi-feature fusion strategy. Comput. Biol. Med. 168, 107724 (2024).
Wei, L., Ye, X., Xue, Y., Sakurai, T. & Wei, L. ATSE: a peptide toxicity predictor by exploiting structural and evolutionary information based on graph neural network and attention mechanism. Brief. Bioinform. 22, bbab041 (2021).
Naamati, G., Askenazi, M. & Linial, M. ClanTox: a classifier of short animal toxins. Nucleic Acids Res. 37, W363–W368 (2009).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8, e73957 (2013).
Huan, Y., Kong, Q., Mou, H. & Yi, H. Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front. Microbiol. 11, 582779 (2020).
Almagro Armenteros, J. J., Sønderby, C. K., Sønderby, S. K., Nielsen, H. & Winther, O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33, 3387–3395 (2017).
Vita, R. et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Wolf, A. M., Wolf, D., Rumpold, H., Enrich, B. & Tilg, H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem. Biophys. Res. Commun. 323, 630–635 (2004).
Huang, X.-L. et al. Role of anti-inflammatory cytokines IL-4 and IL-13 in systemic sclerosis. Inflamm. Res. 64, 151–159 (2015).
Marie, C., Pitton, C., Fitting, C. & Cavaillon, J. Regulation by anti-inflammatory cytokines (IL-4, IL-10, IL-13, TGFβ) of interleukin-8 production by LPS-and/or TNFα-activated human polymorphonuclear cells. Mediat. Inflamm. 5, 334–340 (1996).
Opal, S. M. & DePalo, V. A. Anti-inflammatory cytokines. Chest 117, 1162–1172 (2000).
Sanjabi, S., Zenewicz, L. A., Kamanaka, M. & Flavell, R. A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 9, 447–453 (2009).
Mühl, H. Pro-inflammatory signaling by IL-10 and IL-22: bad habit stirred up by interferons? Front. Immunol. 4, 18 (2013).
Benveniste, E. N. & Qin, H. Type I interferons as anti-inflammatory mediators. Science’s STKE 2007, pe70–pe70 (2007).
Billiau, A. Anti-inflammatory properties of Type I interferons. Antivir. Res. 71, 108–116 (2006).
Commins, S. P., Borish, L. & Steinke, J. W. Immunologic messenger molecules: cytokines, interferons, and chemokines. J. Allergy Clin. Immunol. 125, S53–S72 (2010).
Wang, G., Li, X. & Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093 (2016).
Novković, M., Simunić, J., Bojović, V., Tossi, A. & Juretić, D. DADP: the database of anuran defense peptides. Bioinformatics 28, 1406–1407 (2012).
Sievers, F. et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Kawashima, S. et al. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202–D205 (2007).
Saha, I., Maulik, U., Bandyopadhyay, S. & Plewczynski, D. Fuzzy clustering of physicochemical and biochemical properties of amino acids. Amino Acids 43, 583–594 (2012).
He, K., Zhang, X., Ren, S. & Sun, J. Deep Residual Learning for Image Recognition. in 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) 770–778 (IEEE, Las Vegas, NV, USA, 2016). https://doi.org/10.1109/CVPR.2016.90.
Ba, J. L., Kiros, J. R. & Hinton, G. E. Layer normalization. arXiv preprint arXiv:1607.06450 (2016).
Jiyun Han, T. K. & Liu, J. PepNet: an interpretable neural network for anti-inflammatory and antimicrobial peptides prediction using a pre-trained protein language model [Data set]. Zenodo https://zenodo.org/records/13223516 (2024).
Jiyun Han, T. K. & Liu, J. PepNet: an interpretable neural network for anti-inflammatory and antimicrobial peptides prediction using a pre-trained protein language model [Code]. Zenodo https://zenodo.org/records/13734258 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China with code 2020YFA0712400, and the National Natural Science Foundation of China with code 62272268. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would also like to thank Qixuan Chen for his contribution to the AIPStack and PPTPP algorithm operation for this work.
Author information
Authors and Affiliations
Contributions
J.L. conceived and designed the experiments. J.H. and T.K. performed the experiments. J.H., T.K., and J.L. analyzed the data. J.H. and T.K. contributed reagents/materials/analysis tools. J.H., T.K., and J.L. wrote the paper. J.H. designed the software used in analysis. J.L. oversaw the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Mehmet Özbil and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors:Aylin Bircan and Laura Rodríguez Pérez. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, J., Kong, T. & Liu, J. PepNet: an interpretable neural network for anti-inflammatory and antimicrobial peptides prediction using a pre-trained protein language model. Commun Biol 7, 1198 (2024). https://doi.org/10.1038/s42003-024-06911-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06911-1
This article is cited by
-
eCBT-I dialogue system: a comparative evaluation of large language models and adaptation strategies for insomnia treatment
Journal of Translational Medicine (2025)
-
NeXtMD: a new generation of machine learning and deep learning stacked hybrid framework for accurate identification of anti-inflammatory peptides
BMC Biology (2025)
-
Antimicrobial peptide prediction based on contrastive learning and gated convolutional neural network
Scientific Reports (2025)
-
How Machine Learning Helps in Combating Antimicrobial Resistance: A Review of AMP Analysis and Generation Methods
International Journal of Peptide Research and Therapeutics (2025)
-
Omics-based profiling and therapeutic potential of natural components in pan-Shennongjia medicinal herbs
Chinese Medicine (2025)
