
Abstract
The crosstalk between adipose tissue and the liver is finely controlled to maintain metabolic health. Yet, how adipose tissue controls toxic free fatty acid overflow into the liver remains incompletely understood. Here, we show that adipocyte activating transcription factor 3 (ATF3) was induced in human or mouse obesity. Adipocyte Atf3−/− (Atf3Adi−/−) mice developed obesity, glucose intolerance, and metabolic dysfunction-associated steatohepatitis (MASH) in chow diet, high-fat diet, or Western diet-fed mice. Blocking fatty acid flux by inhibiting hepatocyte CD36, but not the restoration of hepatic AMPK signaling, prevented the aggravation of MASH in Atf3Adi−/− mice. Further studies show that the loss of adipocyte ATF3 increased lipolysis via inducing adipose triglyceride lipase, which in turn induced lipogenesis and inflammation in hepatocytes. Moreover, Atf3Adi−/− mice had reduced energy expenditure and increased adipose lipogenesis and inflammation. Our data demonstrate that adipocyte ATF3 is a gatekeeper in counteracting MASH development under physiological and pathological conditions.
Similar content being viewed by others


Introduction
Obesity-related metabolic disorders, such as metabolic dysfunction-associated steatotic liver disease (MASLD) (formerly named nonalcoholic fatty liver disease (NAFLD)), diabetes, and hyperlipidemia, may affect about 300 million people worldwide1. As a widely recognized metabolic and endocrine organ in the body, adipose tissue plays a crucial role in maintaining glucose and lipid homeostasis through its subtle functions at both organ and systemic levels. Adipose tissue can be functionally classified as energy-storing white adipose tissue (WAT) and thermogenic brown adipose tissue (BAT). WAT browning, as well as BAT activation, has arisen as a potentially valuable strategy to treat obesity and its related metabolic disorders.
Activating transcription factor 3 (ATF3) is a member of the ATF/cAMP response element-binding protein family of transcription factors, which binds to the consensus sequence TGACGTCA in gene promoters to regulate gene expression2,3. ATF3 is induced by lipopolysaccharide (LPS) and suppresses inflammatory genes by inhibiting toll-like receptor 4 (TLR4) signaling in macrophages4,5. Macrophage ATF3 is also shown to mediate the anti-inflammatory effects of HDL6. Recent studies show that hepatocyte ATF3 protects against atherosclerosis by regulating lipoprotein and bile acid metabolism7 and prevents diet-induced metabolic dysfunction-associated steatohepatitis (MASH) via interaction with hepatocyte nuclear factor 4α8. Nonetheless, the role of adipocyte ATF3 in MASH development has not been investigated.
Adipose tissue is mainly composed of adipocytes which can be classified into white, beige, and brown adipocytes. WAT stores energy and is also an endocrine organ, while BAT generates heat to meet our physiological needs. Adipose tissue dysfunction may cause obesity, diabetes, dyslipidemia, and MASLD9,10. MASLD is a spectrum of liver diseases, including metabolic dysfunction-associated steatotic liver (MASL) and MASH11,12, which may further progress to liver cirrhosis and liver cancer. The global prevalence of MASLD is estimated at 24%, with the highest rates reported in South America and the Middle East, followed by Asia, the USA, and Europe13. MASH is projected to increase by up to 56% in the next 10 years due to the growing obesity epidemic14. So far, how adipocytes prevent the development of MASLD remains to be fully understood.
In this report, we show that ATF3 is highly induced in adipocytes of obese individuals and obese mice. ATF3 ablation in adipocytes caused obesity and MASLD in chow diet, high-fat diet (HFD) or high fat/high cholesterol diet (Western diet)-fed mice. Blocking fatty acid flux by inhibition of hepatocyte CD36, but not restoration of hepatic AMPK signaling, prevented the aggravation of MASLD in Atf3Adi−/− mice. Further studies show that loss of adipocyte ATF3 increased lipolysis via inducing adipose triglyceride lipase, which in turn induced lipogenesis and inflammation in hepatocytes. In addition, loss of adipocyte ATF3 reduced energy expenditure and induced lipogenesis and inflammation in adipose tissue. Our data indicate that adipocyte ATF3 is a key gatekeeper in preventing MASH under physiological and pathophysiological conditions.
Results
Adipocyte ATF3 is induced in obese humans and mice with genetic or diet-induced obesity
We have previously shown that hepatocyte ATF3 plays a key role in regulating lipid and lipoprotein metabolism and liver function7,8. To investigate whether adipocyte ATF3 played a role in metabolic homeostasis, we investigated Atf3 expression in WAT and BAT of mice with genetic or diet-induced obesity (DIO). Atf3 mRNA levels were markedly increased in WAT of db/db mice, ob/ob mice, high-fat diet (HFD)-fed mice and Western diet (WD)-fed C57BL/6 mice (Fig. 1a). Consistent with the changes in mRNA levels, ATF3 protein levels were induced by ≥3-fold in WAT (Fig. 1b, c) and in BAT (Supplementary Fig. S1a, b) of these mice. In adipose tissue isolated from lean (BMI < 25 kg/m2) versus obese (BMI > 35 kg/m2) individuals, ATF3 mRNA levels were increased by 2.2-fold (Fig. 1d). To investigate whether ATF3 expression was elevated in adipocytes, we isolated adipocytes and stromal vascular fractions (SVF) from lean and obese mice; the data showed that Atf3 mRNA and protein levels were induced in adipocytes and SVF of obese mice compared to lean mice (Fig. 1e–j and Supplementary Fig. S1c). Fasting/refeeding did not affect Atf3 expression in adipose tissue (Supplementary Fig. S1d). Taken together, these data demonstrate that adipocyte ATF3 is induced in obese humans and genetically obese or DIO mice.

a Atf3 mRNA levels in white adipose tissue (WAT) of db/db mice, ob/ob mice, high-fat diet (HFD)-fed mice, or Western diet (WD)-fed mice and their control mice (n = 7–8 per group). b, c Western blot assays were performed (b), and protein levels were quantified (c). d ATF3 mRNA levels in adipose tissue of lean versus obese individuals (n = 6–8). e–j Atf3 mRNA and protein levels in white adipocytes (e–g) and stromal vascular fraction (SVF) (h–j) of male C57BL/6 mice that had been fed a chow (lean) or Western diet (obese) for 4 months (n = 4). k–m 3T3-L1 cells were treated with DMEM containing IBMX/Dex/Insulin for 2 days, and the media were then changed to DMEM containing Dex/Insulin every 2 days until fully differentiated. After culture in DMEM without FBS for 3 h, the cells were treated with palmitate at indicated concentrations for 24 h. mRNA (k) (n = 6) and protein (l, m) levels (n = 3) were determined. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (a, c–e, g, h, j) or one-way ANOVA (k, m). *P < 0.05, **P < 0.01 versus controls. Additional supporting data can be found in Supplementary Fig. S1.
To address why adipose ATF3 was induced in obesity, we treated 3T3-L1 cells with 100 μM palmitic acid (PA), oleic acid (OA) or linoleic acid (LA). Interestingly, only PA highly induced Atf3, Il1b, and Timp1 expression (Supplementary Fig. S1e). Moreover, PA dose-dependently induced Atf3 mRNA and protein levels in 3T3-L1 cells (Fig. 1k–m). Excessive PA is known to induce endoplasmic reticulum (ER) stress in adipocytes15. ER stress is a known inducer of ATF3 expression3. Thus, palmitate may stimulate ATF3 expression by inducing ER stress. Given that palmitate is readily available in adipocytes, the induction of ATF3 may have a physiological or pathological consequence.
The loss of adipose ATF3 causes steatohepatitis
To explore the role of adipocyte ATF3 in metabolic homeostasis, floxed Atf3 (Atf3fl/fl) mice were cross-bred with adipoq-Cre mice (JAX; stock # 028020) to generate control (Atf3fl/fl) mice and adipocyte-specific Atf3−/− (Atf3Adi−/−) mice. When these mice were fed a Western diet (WD) for 20 weeks, Atf3Adi−/− mice had increased body and liver weight (Supplementary Fig. S2a) while food intake, plasma leptin or insulin levels were unchanged (Supplementary Fig. S2b–d). Atf3Adi−/− mice had elevated plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) (Fig. 2a), and free fatty acids (FFA) (Fig. 2b), and a reduction in plasma adiponectin levels (Fig. 2c). In the liver, Atf3Adi−/− mice had elevated levels of triglycerides (TG), FFAs, hydroxyproline (Fig. 2d–f), reactive oxygen species (ROS) (Supplementary Fig. S2e), and increased NAFLD activity score (NAS) (Fig. 2g). Histological staining showed that Atf3Adi−/− mice had increased lipid accumulation and fibrosis (Fig. 2h). Gene expression studies showed that Atf3Adi−/− mice had elevated hepatic mRNA levels of sterol regulatory element-binding protein 1c (Srebp1c), fatty acid synthase (Fasn), acetyl-coA carboxylase (Acc), cluster of differentiation 36 (Cd36), tumor necrosis factor alpha (Tnfa), alpha smooth muscle actin (a-Sma/Acta2), collagen type I alpha 1 chain (Col1a1), Col1a2, and interleukin 1 beta (Il1b) (Fig. 2i). Consistent with the changes in mRNA levels and plasma adiponectin levels, hepatic SREBP1 and ACC protein levels were significantly induced whereas hepatic phosphorylated AMP-activate protein kinase (P-AMPK) levels were reduced by ~50% (Fig. 2j, k). Thus, the data in Fig. 2 suggest that adipocyte ATF3 is important for preventing WD-induced steatohepatitis, likely by regulating hepatic fatty acid influx and lipogenesis.

Aft3fl/fl mice and Atf3Adi−/− mice were fed a western diet (WD) for 20 weeks (n = 7 per group). a Plasma transaminase levels. b Plasma free fatty acid (FFA) levels. c Plasma adiponectin levels. d Hepatic triglyceride (TG) levels. e Hepatic FFA levels. f Hepatic hydroxyproline levels. g NAFLD activity score (NAS). h Hematoxylin (H&E) staining (top panel), oil red O (ORO) staining (middle panel), and picrosirius red staining (bottom panel). i Hepatic mRNA levels. j Western blot assays of hepatic proteins. k Relative protein levels. Scale bars in (h): 50 μm. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (a–g, i, k). *P < 0.05, **P < 0.01 versus controls. Additional supporting data can be found in Supplementary Figs. S2–4.
The loss of adipose ATF3 causes obesity
In addition to inducing steatohepatitis, loss of adipocyte Atf3 also increased body fat content (Fig. 3a) and adipocyte size (Fig. 3b, c). Consistent with these phenotypic changes, Atf3Adi−/− mice had reduced energy expenditure during the day or night time (Fig. 3d, e). In addition, Atf3Adi−/− mice displayed glucose intolerance (Fig. 3f, g). There was no change in insulin insensitivity (Fig. 3h). At protein expression levels, loss of adipocyte Atf3 reduced phosphorylated AKT protein kinase (P-AKT) levels in the liver, ATF3 and uncoupled protein 1 (UCP-1) in BAT (Fig. 3i, j). The reduction in UCP1 expression in BAT may account for the reduced energy expenditure in Atf3Adi−/− mice. In addition, Atf3Adi−/− mice had elevated levels of TNFα, IL1β and MCP1 proteins in the plasma (Fig. 3k) and mRNA in adipose tissue (Fig. 3l). Loss of adipocyte ATF3 also induced some other inflammatory, fibrogenic or lipogenic genes (e.g., Ccl5, Mmp2, Mmp9, Col3a1, Srebp1c) in WAT or BAT (Supplementary Fig. S2g–j). Transient transfection studies showed that ATF3 highly induced Ucp1 promoter activity (Supplementary Fig. S2k), suggesting that ATF3 directly regulates Ucp1 gene transcription.

Aft3fl/fl mice and Atf3Adi−/− mice were fed a western diet (WD) for 20 weeks (n = 7 per group). a Body fat content. b H&E staining of epididymal white adipose tissue (eWAT) and brown adipose tissue (BAT). c eWAT adipocyte size. d Energy expenditure over a 48-h period. e Energy expenditure during the full day, day, or night time. f Glucose tolerance test (GTT). g The area under the curve (AUC) of GTT. h Insulin tolerance test (ITT). i Western blot assays of hepatic (top panel) or BAT (bottom panel) proteins. j Quantification of protein levels. k Plasma cytokine levels. l Gene expression in adipose tissues. Scale bar in (b): 50 mm. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (a, c, g, j–l) or two-way ANOVA (d–f, h). *P < 0.05, **P < 0.01 versus controls. Additional supporting data can be found in Supplementary Figs. S2–4.
To further test the role of loss of adipocyte ATF3 in insulin resistance, we also injected insulin into WD-fed Atf3fl/fl mice and Atf3Adi−/− mice. The data of Supplementary Fig. S3a–f showed that Atf3Adi−/− mice had reduced P-AKT levels in the liver, WAT, and skeletal muscle, indicating that loss of adipocyte ATF3 causes insulin resistance in these tissues.
The data presented in Figs. 2 and 3 were collected from mice fed a Western diet for 20 weeks. When mice were fed a WD for only 10 weeks, similar results were collected (Supplementary Fig. S4a–j). Taken together, the data of Fig. 3 and Supplementary Fig. S2–4 demonstrate that the loss of adipocyte ATF3 induces WD-induced obesity likely via reducing energy expenditure.
The loss of adipose ATF3 causes metabolic dysfunctions independent of dietary conditions
To investigate whether adipocyte ATF3-regulated metabolic homeostasis was dependent on a specific diet, we also fed Atf3Adi−/− mice and their control mice with a chow diet or HFD. Loss of adipocyte Atf3 did not affect food intake or energy expenditure on a chow diet (Fig. S5a–c). On either a chow diet or an HFD, Atf3Adi−/− mice displayed elevated plasma ALT, AST and FFA levels and reduced plasma adiponectin levels (Fig. 4a–d). Atf3Adi−/− mice also had increased hepatic TG, FFA and hydroxyproline levels (Fig. 4e–g), and increased glucose intolerance (Fig. 4h). Histological staining showed that Atf3Adi−/− mice had aggravated liver steatosis and fibrosis (Fig. 4i, j), and increased fat content and adipocyte size (Fig. 4k–m). We also collected other biochemical, histological or functional parameters in chow- or HFD-fed mice as we did for the Western diet-fed mice. The data of Figs. S5–8 show that the results from chow- or HFD-fed mice were similar to those observed in Western diet-fed mice. The unchanged Ucp1 expression in chow-fed mice (Supplementary Fig. S6g) was consistent with unchanged energy expenditure in these mice (Supplementary Fig. S5b). These data indicate that adipocyte ATF3 regulates metabolic functions independently of dietary conditions.

Aft3fl/fl mice and Atf3Adi−/− mice were fed a chow diet or a high-fat diet (HFD) for 16 weeks (n = 7–8 per group). a Plasma ALT levels. b Plasma AST levels. c Plasma adiponectin levels. d Plasma free fatty acid (FFA) levels. e Hepatic triglyceride (TG) levels. f Hepatic FFA levels. g Hepatic hydroxyproline levels. h The area under the curve (AUC) of GTT. i Oil red O staining. j Picrosirius red staining. k Fat content. l H&E staining of eWAT. m eWAT adipocyte size. Scale bars in (i, j, l): 50 mm. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (a–h, k, m). *P < 0.05, **P < 0.01. Additional supporting data can be found in Supplementary Figs. S5–8.
The loss of adipocyte ATF3 aggravates MASH and obesity independently of the AMPK pathway
The data of Figs. 2 and 4 show that Atf3Adi−/− mice have reduced plasma adiponectin levels and reduced hepatic AMPK activity. Since the adiponectin-AMPK pathway plays an important role in promoting glucose utilization and fatty acid oxidation (FAO)16,17 and may prevent obesity-related metabolic dysfunction18, we explored whether restoration of the AMPK pathway would fully or partially normalize obesity and MASH in Atf3Adi−/− mice. Atf3Adi−/− mice and their control mice were fed an HFD for 12 weeks, followed by i.p. injection of AICAR, an AMPK activator. Activation of AMPK (Fig. 5a) reduced plasma ALT and AST levels (Fig. 5b, c), hepatic TG, FFA, and hydroxyproline levels (Fig. 5d–f), and steatosis or fibrosis (Fig. 5g, h) to similar extents in both Atf3Adi−/− mice and their control mice.

a–k Aft3fl/fl mice and Atf3Adi−/− mice were fed a high-fat diet (HFD) for 12 weeks and then injected i.p. with either saline (vehicle) or 5-Aminoimidazole-4-carboxamide-1-b-4-ribofuranoside (AICAR, 500 mg/kg/day) for 4 weeks (n = 7–8 per group). a Western blot assays of WAT proteins (top panel) or hepatic proteins (bottom panel). b Plasma ALT levels. c Plasma AST levels. d Hepatic triglyceride (TG) levels. e Hepatic free fatty acid (FFA) levels. f Hepatic hydroxyproline levels. g H&E staining of liver sections. h Picrosirius red staining of liver sections. i Body fat content (%). j eWAT weight. k H&E staining of eWAT. Statistical analysis was performed using two-way ANOVA (b–f, i, j). *P < 0.05, **P < 0.01. l–u Aft3fl/fl mice and Atf3Adi−/− mice were i.v. injected with AAV8-TBG-Null or AAV8-TBG-AMPKa, then fed a western diet (WD) for 16 weeks (n = 7 per group). l–n Hepatic levels of rAmpka mRNA (l) and proteins (m, n). o Plasma ALT levels. p Plasma AST levels. q Plasma FFA levels. r Hepatic TG levels. s Hepatic FFA levels. t Hepatic hydroxyproline levels. u H&E staining (top panel) and Picrosirius red staining (bottom panel) of liver sections. v Body fat content (%). w liver-to-body weight ratio (%). Scale bars in (g, h, k, u): 50 mm. Statistical analysis was performed using one-way ANOVA (l, m–t, v, w). *P < 0.05, **P < 0.01. Additional supporting data can be found in Supplementary Figs. S5–8.
To complement the study involving AICAR, we overexpressed constitutively AMPKα in hepatocytes of Atf3Adi−/− mice by i.v. injection of AAV8-TBG-AMPKα. These mice and their control mice were then fed a Western diet for 16 weeks. As shown in Fig. 5l–w, overexpression of hepatocyte AMPKα (Fig. 5l–n) failed to correct the MASLD (Fig. 5o–u) or obesity (Fig. 5v, w) phenotypes in Atf3Adi−/− mice. Thus, our pharmacological or genetic approaches indicate that the loss of adipocyte ATF3 aggravates obesity and MASH development independently of the AMPK pathway.
The loss of adipocyte ATF3 induces steatohepatitis by promoting hepatocyte fatty acid uptake
In addition to secreting adipokines, adipose tissue is the major source of plasma non-esterified fatty acids (NEFA/FFA), which play an important role in the pathogenesis of MASLD9,19. CD36 plays a key role in fatty acid uptake by hepatocytes. Loss of hepatocyte CD36 prevents the development of MASLD20,21. Thus, we generated an AAV expressing Cd36 shRNA specifically in hepatocytes (AAV8-TBG-shCd36). AAV8-TBG-shCd36 and its control AAV (AAV8-TBG-shScr) were i.v. injected into Atf3Adi−/− mice or Atf3fl/fl mice. Inhibition of hepatocyte Cd36 in Atf3Adi−/− mice did not affect body weight or plasma FFA levels (Fig. 6a, b), but normalized liver weight (Fig. 6c), plasma ALT and AST levels (Fig. 6d, e), and hepatic TG, FFA and hydroxyproline levels (Fig. 6f–h). At gene expression levels, inhibition of hepatocyte Cd36 in Atf3Adi−/− mice normalized hepatic expression of lipogenic genes (Srebp1c, Fasn, Acc), pro-inflammatory genes (Tnfa, Il6, Il1b), and fibrogenic genes (Col1a2, a-Sma/Acat2) (Fig. 6i–l). These biochemical changes were further confirmed by H&E staining and picrosirius red staining of liver sections (Fig. 6m). Thus, the data of Fig. 6 demonstrate that loss of adipocyte ATF3 induces the development of steatohepatitis via promoting fatty acid flux into hepatocytes.

Aft3fl/fl mice and Atf3Adi−/− mice were i.v. injected with AAV8-TBG-shScr or AAV8-TBG-shCd36 and then fed a western diet (WD) for 16 weeks (n = 7 per group). a Body weight. b Plasma FFA levels. c Liver weight. d Plasma ALT levels. e Plasma AST levels. f Hepatic triglyceride (TG) levels. g Hepatic FFA levels. h Hepatic hydroxyproline levels. i Western blot assays. j Quantification of protein levels. k, l Hepatic mRNA levels. m Hematoxylin-eosin (H&E) staining (top panel) and Picrosirius red staining (bottom panel) of liver sections. Scale bars in (m): 50 μm. shScr scramble shRNA. Data are expressed as mean ± SEM. Statistical analysis was performed using one-way ANOVA (b–h, j–l) or two-way ANOVA (a). *P < 0.05, **P < 0.01 versus controls.
Adipocyte ATF3 inhibits lipolysis via ATGL in vivo
The finding that Atf3Adi−/− mice have elevated plasma FFA levels (Figs. 2b and 4d) suggests that ATF3 may inhibit lipolysis. The data of Fig. 7a show that Atf3Adi−/− mice had elevated mRNA levels of genes involved in adipogenesis [CCAAT/enhancer-binding protein alpha (Cebpa), peroxisome proliferator-activated receptor gamma1 (Pparg1), Pparg2], lipogenesis [fatty acid synthase (Fasn), acetyl-CoA carboxylase (Acc), diacylglycerol transferase 2 (Dgat2), stearoyl-CoA desaturase 1 (Scd1)], and lipolysis [adipose triglyceride lipase (Atgl)]. Western blot assays showed that ATF3 protein levels were reduced by 78% while ATGL and phosphorylated hormone-sensitive lipase (HSL) levels were increased by 2.5-fold and 1.5-fold, respectively (Fig. 7b, c). ATGL and HSL catalyze the first and second steps of lipolysis in adipocytes, respectively, by hydrolyzing triglycerides into FFAs and glycerol. Consistent with the changes in lipolytic genes, isoproterenol, a β1/β2 adrenergic receptor agonist that activates protein kinase A (PKA) to induce lipolysis, significantly stimulated the release of both FFA and glycerol from the control adipocytes (P < 0.001), which was further enhanced when adipocyte Atf3 was deficient (P < 0.05) (Fig. 7d, e). Atglistatin, an ATGL inhibitor, significantly inhibited FFA and glycerol release from both the control and Atf3-deficient adipocytes in the presence or absence of isoproterenol (P < 0.001) (Fig. 7d, e). There was no difference in the FFA or glycerol release between the two genotypes under the basal, unstimulated conditions (Fig. S9a, b). Transient transfection studies using wild-type (WT) and mutant Atgl promoters showed that ATF3 inhibited the Atgl promoter activity by binding to an ATF3 binding site located at −272 bp (Fig. 7f, g). Chromatin immunoprecipitation (ChIP) assays further confirmed that ATF3 bound to the Atgl promoter in vivo (Fig. 7h). Thus, the data of Fig. 7f–h suggest that Atgl is a direct downstream target of ATF3. Taken together, the data of Fig. 7 indicate that adipocyte ATF3 inhibits lipolysis via ATGL in vivo.

a–c Aft3fl/fl mice and Atf3Adi−/− mice were fed a Western diet (WD) for 20 weeks (n = 7 per group). a mRNA levels in WAT. b, c Protein levels in WAT. d, e Lipolysis was performed in the presence of isoproterenol (10 μM) and/or Atglistatin (20 μM) for 2 h (n = 6 per group). d Released free fatty acid (FFA) levels. e Released glycerol levels. f, g Transient transfections were performed by co-transfecting pGL3 (vector) or pGL3-Atgl with CMV-Null or CMV-ATF3 into 3T3-L1 cells (n = 10). After 36 h, relative luciferase activities (RLU) were determined and normalized to beta-galactosidase activity. WT or Mut refers to wild-type or mutant Atgl promoter. h ChIP assays were performed using 3T3-L1 cell lysates overexpressing ATF3-Flag (n = 8). After pulldown by a Flag or IgG (control) antibody, the DNA enrichment in the Atgl promoter was determined by qRT-PCR. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (a, c, h) or two-way ANOVA (d–g). *P < 0.05, **P < 0.01. Additional supporting data can be found in Supplementary Fig. S9.
ATF3 inhibits adipogenesis and lipolysis in 3T3-L1 cells
To better understand the role of ATF3 in adipose biology, we overexpressed human ATF3 or knocked down Atf3 expression in 3T3-L1 preadipocytes, which were then differentiated into mature adipocytes in the presence of a hormonal cocktail containing insulin and dexamethasone (DEX). Overexpression of human ATF3 significantly inhibited genes involved in adipogenesis [Cebpa, Pparg1, Pparg2, perilipin 1 (Plin1)], lipogenesis (Acc, Fasn, Dgat1, Dgat2, Scd1) and lipolysis (Atgl) (Fig. S10a–c). Consistent with the changes in gene expression, overexpression of human ATF3 significantly inhibited lipid droplet formation (Fig. 8a), de novo lipogenesis (Fig. 8b), and the release of FFA (Fig. 8c) and glycerol (Fig. 8d) stimulated by isoproterenol, indicating that overexpression of human ATF3 inhibits adipogenesis and lipolysis. There was no change in FFA or glycerol release under the basal, unstimulated conditions (Fig. S10d, e).

a–d 3T3-L1 cells were infected with Ad-Empty or Ad-ATF3 and then differentiated to adipocytes in the presence of the hormonal cocktail (n = 6). Oil red O staining (a) and de novo lipogenesis in the presence of 14C-acetic acid (b) were performed. The 2-h release of free fatty acid (FFA) (c) or glycerol (d) in the presence of isoproterenol (10 μM) and/or atglistatin (20 μM) was analyzed. e–h 3T3-L1 cells were infected with Ad-shLacZ or Ad-shAtf3 and then differentiated to adipocytes in the presence of the hormonal cocktail (n = 6). Oil red O staining (e) and de novo lipogenesis in the presence of 14C-acetic acid (f) were performed. The 2-h release of FFA (g) or glycerol (h) in the presence of isoproterenol (10 μM) and/or atglistatin (20 μM) was analyzed. Scale bars in (a, e): 50 mm. Data are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed, unpaired t-test (b, f) or two-way ANOVA (c, d, g, h). *P < 0.05, **P < 0.01. Additional supporting data can be found in Supplementary Figs. S10, 11.
In contrast, knockdown of Atf3 in 3T3-L1 cells induced genes involved in adipogenesis (Cebpa, Pparg1, Pparg2, Plin1, Plin2), lipogenesis (Fasn, Dgat1, Dgat2, Scd1) and lipolysis (Atgl) (Fig. S11a–c). Atf3 knockdown also induced lipid droplet formation (Fig. 8e), de novo lipogenesis (Fig. 8f), and lipolysis in the presence of isoproterenol (Fig. 8g, h), but did not affect lipolysis under the basal conditions (Fig. S11d, e). Given that adipocyte ATF3 inhibits lipolysis (Fig. 7), the inhibition of adipogenic and lipogenic programs may help to prevent fat accumulation in adipose tissue.
Adipocyte ATF3 inhibits lipogenesis and inflammation in hepatocytes
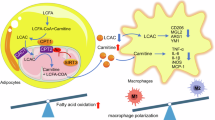
Our data have indicated that FFAs, but not adiponectin, released from adipose tissue of Atf3Adi−/− mice promote the development of MASLD. To confirm the in vivo findings, we treated primary hepatocytes with conditioned media of fat pads isolated from Atf3Adi−/− versus Atf3fl/fl mice (Fig. 9a). Stimulation of lipolysis in Atf3−/− adipocytes significantly induced lipogenic genes (Srebp1c, Acc, Fasn) and inflammatory genes (Mcp1, Tnfa) in hepatocytes (Fig. 9b). Furthermore, palmitic acid, oleic acid, or linoleic acid was sufficient to induce the lipogenic genes (Srebp1c, Fasn, Acc) and inflammatory genes (Tnfa, Il1b, Mcp1) in primary hepatocytes (Fig. 9c). Thus, the data of Fig. 9, together with those in Figs. 6–8, demonstrate that loss of adipocyte ATF3 induces the development of MASLD via inducing ATGL-mediated lipolysis (Fig. 9d).

a, b Fat pads from Aft3fl/fl mice or Atf3Adi−/− mice were treated with vehicle or isoproterenol (10 μM) for 2 h. The conditioned media were then used to treat primary hepatocytes for 24 h (a). b mRNA levels of Srebp1c, Acc, Fasn, Mcp1, and Tnfa were determined (n = 6 per group). c Primary hepatocytes were treated with bovine serum albumin (BSA; control) or 100 mM palmitic acid, oleic acid, or linoleic acid for 24 h, and mRNA levels were quantified (n = 6). d A model for ATF3 in white adipose tissue (WAT) or brown adipose tissue (BAT) to inhibit the development of MASLD. Adipocyte ATF3 is induced in obesity, which in turn prevents MASLD via blocking FFA flux to hepatocytes by inhibiting ATGL-mediated lipolysis. ATF3 also inhibits lipogenesis and inflammation in WAT and induces thermogenesis in BAT, thus preventing obesity and insulin resistance (IR). Data are expressed as mean ± SEM. Statistical analysis was performed using two-way ANOVA (b) or one-way ANOVA (c). *P < 0.05, **P < 0.01.
Discussion
Obesity is a risk factor for many metabolic disorders, including MASLD. Interestingly, not all obese individuals develop MASLD or insulin resistance. Thus, it is intriguing to investigate how adipocytes prevent obesity-related metabolic disorders. In this report, we show that ATF3 is induced in adipose tissue of obese mice and in subcutaneous adipocytes of humans with severe obesity. Ablation of ATF3 in adipocytes induces obesity, and MASLD in chow, HFD, or Western diet-fed mice, suggesting that adipocyte ATF3 is a gatekeeper in preventing MASH development under both physiological and pathological conditions (Fig. 9d).
Adipocytes are known to secrete numerous hormones, such as leptin, adiponectin and resistin. Adiponectin plays an important role in regulating lipid and glucose metabolism and inflammation via binding to adiponectin receptors22. AMPK is required for many adiponectin functions in many tissues, including stimulation of glucose utilization and FAO and preventing obesity-related metabolic disorders16,17,18,22. Atf3Adi−/− mice have reduced plasma adiponectin levels and tissue AMPK activation. However, restoration of AMPK activation in these mice by AICAR fails to attenuate metabolic disorders. Moreover, hepatocyte-specific expression of constitutively active AMPK fails to prevent the development of MASLD. These data indicate that loss of adipocyte ATF3 induces obesity and MASH independent of the AMPK pathway.
Adipose tissue-derived FFAs can promote lipogenesis, immune cell accumulation, inflammation, and fibrogenesis in the liver9,23, and induce insulin resistance24. When fatty acid flux into hepatocytes is blocked by AAV8-mediated inhibition of CD36, loss of adipocyte ATF3 fails to promote the development of MASLD, suggesting that Atf3Adi−/− mice develop MASLD due to increased fatty acid flux from adipose tissue to the liver. Adipose tissue releases FFAs into circulation via lipolysis, a highly regulated biochemical process that mobilizes TG stores. ATGL catalyzes the first and rate-limiting step of lipolysis. Loss of adipocyte ATGL results in reduced adipocyte lipolysis and serum lipids, increased systemic glucose tolerance, insulin tolerance, and hepatic insulin signaling, a marked reduction in hepatic steatosis and immune cell infiltration, and unchanged body weight or fat mass by 24 weeks of age25. Specific inhibition of ATGL by Atglistatin reduces adipose lipolysis, weight gain, insulin resistance, and MASLD in HFD-fed mice26. In contrast, global inactivation of ATGL increases adipose mass and TG accumulation in multiple tissues (e.g., heart) in mice27 and humans28. These data suggest that adipocyte ATGL may harbor a therapeutic potential for treating obesity-associated metabolic disorders26,28. Our data show that ATF3 inhibits ATGL expression via direct binding to its promoter and that loss of adipocyte ATF3 induces adipose ATGL expression and lipolysis, elevated plasma FFA levels, and MASLD. Thus, our data are consistent with those observed in adipocyte-specific Atgl−/− (AtglAdi−/−) mice. Moreover, we show that Atglistatin completely prevents FFA release from adipose tissue of Atf3Adi−/− mice, suggesting that the loss of adipocyte ATF3 promotes adipose lipolysis. Interestingly, ATF3 also mildly induces the phosphorylation of HSL, and the underlying mechanisms remain to be explored.
Although the loss of adipocyte ATF3 induces adipose lipolysis, AtglAdi−/− mice are mildly obese. Mild obesity may result from increased lipogenesis and reduced energy expenditure that overcome the fat loss from increased lipolysis in AtglAdi−/− mice. ATF3 induces UCP1 and inhibits SREBP1C by directly regulating their promoter activities. In agreement with our observations, ST32db, a synthetic compound that can induce ATF3, inhibits diet-induced obesity 29,30. It will be interesting to investigate whether ST32db exerts its anti-obesity effects via adipocyte ATF3 and whether It also inhibits MASH development.
ATF3 has been well known for its anti-inflammatory property by directly inhibiting NF-κB activity31 and TLR4 signaling4. Thus, ATF3 in macrophages or hepatocytes is shown to inhibit the inflammatory response8,32. Consistent with these findings, the loss of ATF3 in adipocytes induces inflammatory genes. As a result, plasma inflammatory cytokines are increased in Atf3Adi−/− mice.
Adipose ATF3 is induced in obesity, which is consistent with a previous report33. Interestingly, hepatic ATF3 is reduced in MAFLD and obesity, likely resulting from increased hepatic miR-149 expression8. Different metabolic cues or regulators may be responsible for the differential expression of ATF3 in adipose tissue versus the liver. Interestingly, Kim et al. showed that hepatic ATF3 mRNA level was mildly elevated in induced in MASH patients34 but ATF3 protein levels were not checked. They found ATF3 protein levels were elevated in ZDF obese rats34. However, no positive controls were used to validate the ATF3 antibody. Further, they showed that i.v. injection of an ATF3 siRNA caused improved glucose tolerance, hepatic steatosis, and inflammation34. These results are not supported by other studies that utilized genetic approaches. Zhu et al. show that global Atf3−/− mice had increased inflammation and liver damage35. Liu et al. showed that global Atf3−/− mice developed liver steatosis36. We showed that hepatocyte-specific loss of ATF3 aggravated MASLD, whereas adeno-associated virus-mediated overexpression of ATF3 ameliorated MASLD8. Thus, the data published by Kim et al. are not supported by several independent groups. ATF3 siRNAs used by Kim et al. might have off-target effects, leading to opposite conclusions.
Both hepatic and adipose ATF3 protect against MASLD/MASH, but the mechanisms of action are different. In the liver, ATF3 protects against MASLD/MASH via hepatocyte nuclear factor 4α (HNF4α)8. HNF4α plays an important role in protection against MASLD/MASH via reducing lipotoxicity by inducing hepatic lipolysis, fatty acid oxidation, and bile acid synthesis from cholesterol37. In contrast, adipose ATF3 prevents MASLD/MASH by preventing FFA influx into hepatocytes by inhibiting adipose ATGL.
In summary, we have demonstrated that adipocyte ATF3 plays a key role in preventing MASH development by inhibiting adipose lipolysis. Given that adipocyte ATF3 also inhibits adipogenesis and inflammation and induces thermogenesis, targeting adipocyte ATF3 may represent an approach to treat MASH and other obesity-associated metabolic disorders.
Methods
Mice, diets and human adipose tissue
C57BL/6J mice (stock # 000664), ob/ob mice (stock # 000632) and db/db mice (stock # 000697) were purchased from the Jackson Laboratories (JAX; Bar Harbor, ME). The Western diet (WD) containing 42% kcal from fat/0.2% cholesterol was purchased from Envigo (stock # TD.88137). The high-fat diet (HFD) containing 60% kcal from fat was purchased from Research Diets (stock # D12492). Some C57BL/6J mice were fed an HFD or Western diet for 20 weeks. Floxed Atf3 (Atf3fl/fl) mice, which were described previously7, were cross-bred with adipoq-Cre (JAX; stock # 028020) to generate control (Atf3fl/fl) mice and adipocyte-specific Atf3−/− (Atf3Adi−/−) mice. All mice received human care and were housed at 20–22 °C in a temperature-controlled room with a 12-h light/dark cycle and free access to water and food. Unless otherwise stated, 8-week-old male mice were used and were fasted for 5–6 h before euthanasia. All the animal experiments were approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University (NEOMED). We have complied with all relevant ethical regulations for animal use. Deidentified human adipose tissue was provided by the Cooperative Human Tissue Network (CHTN). The use of human tissues was approved by the Institutional Review Board at NEOMED.
Chemicals
Dexamethasone (DEX, D4902), bovine serum albumin (BSA, cat # A3803), dimethyl sulfoxide (DMSO, cat # D2438), 3-Isobutyl-1-methylxanthine (IBMX, cat # I5879), insulin (cat # I9278), isoproterenol (cat # I5627) and atglistatin (cat # sml1075) were purchased from Sigma. 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR, A611700) was purchased from Toronto Research Chemicals Ins.
In vivo AICAR treatment
Atf3fl/fl and Atf3Adi−/− mice were fed an HFD for 12 weeks. The mice were then randomized into four groups (n = 7–8 mice per group), and received intraperitoneal (i.p.) injection of 500 mg/kg AICAR or saline once a day for 4 weeks.
Body composition analysis and energy expenditure
Body fat content was measured by EchoMRI-700 (EchoMRI, LLC, Houston, TX). Oxygen expenditure and physical activity were determined in a Comprehensive Lab Animal Monitoring System (CLAMS) using Columbus Instruments hardware and Oxymax software (Columbus Instruments, Columbus, OH), as detailed previously 38. The data were analyzed using the web-based CalR program39.
Adenoviruses and adeno-associated viruses
Adenoviruses expressing human ATF3 with C-terminal Flag and His tags (Ad-ATF3) were purchased from Vigene (VH804537). Adenoviruses expressing a shRNA against mouse or human ATF3 (Ad-shATF3) were generated using the BLOCK-iT Adenoviral RNAi Expression System following the manufacturer’s instructions (Invitrogen). Adenoviruses were purified using cesium chloride gradient centrifugation. AAV-TBG-shCd36 was constructed by cloning a Cd36 shRNA sequence (AAGCTATTGCGACATGATTAA) on a miR-155 scaffold40 into an AAV-TBG empty vector (Penn Vector Lab at University of Pennsylvania, Pennsylvania, PA, USA). AAV-TBG-rAMPKα was constructed by cloning constitutively active AMPKα141 into an AAV-TBG empty vector (Penn Vector Core). All AAVs were packaged and produced in Vector Biolabs (Malvern, PA, USA).
Differentiation of 3T3-L1 preadipocytes
3T3-L1 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. On day 1, differentiation was induced in DMEM containing 10% FBS, 0.5 mM IBMX, 1 μM DEX, and 1.7 μM insulin. Adenoviruses were also added to over-express or knock down Atf3 expression. On day 3, the differentiation medium was replaced with a maintenance medium containing 10% FBS, 1 μM DEX, and 1.7 μM insulin. The maintenance medium was changed every 2 days until the cells were utilized for experimentation.
Isolation of adipocytes and stromal vascular fraction
Mouse primary preadipocytes, stromal vascular fraction (SVF) and macrophage were isolated from white adipose tissue (WAT) as previously described42.
De novo lipogenesis
3T3-L1 cells were grown in serum-free DMEM in a 6-well plate for 2 h and then incubated for 2 h in the presence of 0.5 μCi 14C-acetic acid (PerkinElmer)/10 μM cold acetic acid. After washing twice with PBS, cells were collected in 0.1 N HCl. Lipids were extracted in 500 μl of 2:1 chloroform/methanol (v/v). The radioactivity in the lipid layer was determined by scintillation counting and normalized to protein levels.
Lipolysis assay
Lipolysis in WAT was measured using the method described previously43. About 50–60 mg epididymal fat pads or 3T3-L1 cells were incubated for 2 h at 37 °C in 2% fatty acid-free BSA with or without 10 μm isoproterenol and/or 20 μM atglistatin. After inactivation at 65 °C for 10 min, glycerol release was determined using a kit from Sigma (cat # MAK117-1KT), and free fatty acids (FFAs) were determined using reagents from Fujifilm Wako Chemicals (cat # 995-34791/991-34891). The basal lipolysis (0 h) and indued lipolysis rates (at 2 h) were normalized to proteins.
Transient transfection and luciferase assays
pGL3-Atgl (−975 to 71 bp), a general gift of Dr. Liangyou Rui44, or pGL3-Ucp1 (−2.3 kb to +94 bp), a general gift of Dr. Laurent Vergnes45, was co-transfected with CMV-beta-galactosidase (CMV-β-gal), CMV-Null or CMV-ATF3 into 3T3-L1 cells using Lipofectamine 3000 (ThermoFisher, cat # L3000015). After 36 h, luciferase activity was measured using GloMax Navigator with Dual Injectors (Promega, cat # GM2010), and normalized to β-gal activity.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was carried out using a ChIP assay kit (cat # 17-295, Millipore) according to the manufacturer’s protocol with minor modifications46. Briefly, 3T3-L1 cells were infected with Ad-Empty and Ad-ATF3-Flag. After 24 h, 3T3-L1 cells were washed in cold 1x PBS and then cross-linked with 1% formaldehyde. After sonication and preclearance with Protein A beads, sheared chromatin was then immunoprecipitated using IgG or an antibody against Flag (cat # F7425, Sigma). After elution, the precipitated DNA–antibody complex was digested with proteinase K. DNA was then extracted and used for qPCR analysis with primers flanking the ATF3-binding site in the Atgl promoter. The supernatant before immunoprecipitation of each sample was used as its own input control.
RNA extraction and mRNA levels
RNA was extracted using Trizol Reagent (ThermoFisher, cat # A33248), and mRNA levels were quantified by RT-qPCR using PowerUP SYBR Green Master Mix (ThermoFisher, cat # A25778) on a 7500 Real-Time PCR machine (Applied Biosystems). mRNA levels were normalized to Rplp0 (36b4).
Western blot assays
Western blot assays were performed using total tissue or cell lysates. Antibodies against acetyl-CoA carboxylase (ACC) (cat # 3662), phospho-AMP-activated protein kinase (P-AMPK) (cat # 2535), AMP-activated protein kinase (AMPK) (cat # 2603), phospho-hormone-sensitive lipase (P-HSL) (cat # 45804), hormone-sensitive lipase (HSL) (cat # 4107), adipose tissue triglyceride lipase (ATGL) (cat # 2439), uncoupling protein 1 (UCP1) (cat # 14670), phospho serine/threonine kinase 1 (P-AKT) (cat # 4051), or total AKT (cat # 9272) were purchased from Cell Signaling Technology (Boston, MA). Antibodies against activating transcription factor 3 (ATF3) (cat # NBP1-85816) or sterol regulatory element-binding protein 1 (SREBP-1) (cat # NB600-582) were purchased from Novus. Antibody against tubulin (cat # ab4074) and F4/80 (cat # ab6640) were purchased from Abcam. The CD36 antibody was purchased from Invitrogen (PA1-16813). The anti-tubulin antibody was used at a 1:5000 dilution, and other primary antibodies were used at a 1:1000 dilution. Western blot images were collected by the Amersham Imager 680. Quantification of immunoblots was performed using ImageJ.
Analysis of lipids, hydroxyproline, ROS, adiponectin, leptin, insulin, AST, and ALT
Liver tissue (100 mg) was homogenized in methanol, and lipids were extracted in chloroform/methanol (2:1 v/v) as described47. Hepatic triglyceride (TG) and cholesterol (chol) levels, as well as plasma ALT or AST levels, were measured using Infinity reagents from ThermoFisher Scientific. Plasma and hepatic total free fatty acids or plasma free cholesterol were determined using kits from Fujifilm Wako Pure Chemicals. Hepatic hydroxyproline (cat # STA675) and reactive oxygen species (ROS) (cat # STA-347) levels were measured using kits from Cell BioLabs (San Diego, CA, USA). Plasma adiponectin (cat # MRP300) and leptin (cat # MOB00B) levels were measured using kits from R&D Biosystems. Plasma insulin levels were measured using an ultra-sensitive mouse insulin ELISA kit (Crystal Chem, Inc., cat # 90080).
Oil Red O, hematoxylin and eosin, or picrosirius red staining, and NAFLD activity score (NAS)
Liver, WAT or BAT were fixed in 10% formalin and then embedded in optimum cutting temperature compound or paraffin. Oil Red O (ORO), hematoxylin and eosin (H&E), or picrosirius red staining was performed using the standard techniques. NAFLD activity score (NAS) was determined in a blinded manner according to the published criteria48.
Glucose or insulin tolerance tests
Glucose tolerance test (GTT) was performed after mice were fasted overnight, followed by i.p. injection of 2 g/kg glucose. Insulin tolerance test (ITT) was performed after mice were fasted for 6 h followed by i.p. injection of 1 U/kg insulin. Blood glucose levels were measured using a glucometer at the indicated time points.
Statistics and reproducibility
All data were expressed as mean ± SEM. Statistical significance was analyzed using an unpaired Student t-test, or one-way or two-way ANOVA with a post hoc Bonferroni test for multiple comparisons by Prism (GraphPad, CA). Differences were considered statistically significant at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated or analyzed during this study are included in this manuscript and its Supplementary information files. All other data are available from the corresponding author upon reasonable request.
References
Unamuno, X. et al. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Invest. 48, e12997 (2018).
Thompson, M. R., Xu, D. & Williams, B. R. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 87, 1053–1060 (2009).
Hai, T., Wolford, C. C. & Chang, Y. S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 15, 1–11 (2010).
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 (2006).
Whitmore, M. M. et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J. Immunol. 179, 3622–3630 (2007).
De Nardo, D. et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 15, 152–160 (2014).
Xu, Y. et al. Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism. Nat. Metab. 3, 59–74 (2021).
Xu, Y. et al. Hepatocytic activating transcription factor 3 protects against steatohepatitis via hepatocyte nuclear factor 4α. Diabetes 70, 2506–2517 (2021).
Wang, X. et al. Recent advances in adipose tissue dysfunction and its role in the pathogenesis of non-alcoholic fatty liver disease. Cells 10, 3300 (2021).
Kahn, C. R., Wang, G. & Lee, K. Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Invest. 129, 3990–4000 (2019).
Rinella, M. E. et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 78, 1966–1986 (2023).
Rinella, M. E. et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 79, 1542–1556 (2023).
Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20 (2018).
Huang, D. Q., El-Serag, H. B. & Loomba, R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18, 223–238 (2021).
Yin, J. et al. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: implications for apoptosis and inflammation. Int. J. Mol. Med. 35, 932–940 (2015).
Yamauchi, T. et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 (2002).
Nawrocki, A. R. et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J. Biol. Chem. 281, 2654–2660 (2006).
Achari, A. E. & Jain, S. K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 18, 1321 (2017).
Delarue, J. & Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 10, 142–148 (2007).
Wilson, C. G. et al. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 157, 570–585 (2016).
Rada, P., Gonzalez-Rodriguez, A., Garcia-Monzon, C. & Valverde, A. M. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. 11, 802 (2020).
Wang, Z. V. & Scherer, P. E. Adiponectin, the past two decades. J. Mol. Cell Biol. 8, 93–100 (2016).
Harmon, D. B. et al. Adipose tissue-derived free fatty acids initiate myeloid cell accumulation in mouse liver in states of lipid oversupply. Am. J. Physiol. Endocrinol. Metab. 315, E758–E770 (2018).
Morigny, P., Houssier, M., Mouisel, E. & Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 125, 259–266 (2016).
Schoiswohl, G. et al. Impact of reduced ATGL-mediated adipocyte lipolysis on obesity-associated insulin resistance and inflammation in male mice. Endocrinology 156, 3610–3624 (2015).
Schweiger, M. et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 8, 14859 (2017).
Haemmerle, G. et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737 (2006).
Schreiber, R., Xie, H. & Schweiger, M. Of mice and men: the physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 880–899 (2019).
Ku, H. C., Chan, T. Y., Chung, J. F., Kao, Y. H. & Cheng, C. F. The ATF3 inducer protects against diet-induced obesity via suppressing adipocyte adipogenesis and promoting lipolysis and browning. Biomed. Pharmacother. 145, 112440 (2022).
Cheng, C. F. et al. Adipocyte browning and resistance to obesity in mice is induced by expression of ATF3. Commun. Biol. 2, 389 (2019).
Kwon, J. W. et al. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-κB. Sci. Rep. 5, 14470 (2015).
Jadhav, K. & Zhang, Y. Activating transcription factor 3 in immune response and metabolic regulation. Liver Res. 1, 96–102 (2017).
Qi, L. et al. Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 9, 277–286 (2009).
Kim, J. Y. et al. Activating transcription factor 3 is a target molecule linking hepatic steatosis to impaired glucose homeostasis. J. Hepatol. 67, 349–359 (2017).
Zhu, Q. et al. Loss of ATF3 exacerbates liver damage through the activation of mTOR/p70S6K/ HIF-1α signaling pathway in liver inflammatory injury. Cell Death Dis. 9, 910 (2018).
Liu, Y. F., Wei, J. Y., Shi, M. H., Jiang, H. & Zhou, J. Glucocorticoid induces hepatic steatosis by inhibiting activating transcription factor 3 (ATF3)/S100A9 protein signaling in granulocytic myeloid-derived suppressor cells. J. Biol. Chem. 291, 21771–21785 (2016).
Xu, Y. et al. Hepatocyte nuclear factor 4α prevents the steatosis-to-NASH progression by regulating p53 and bile acid signaling (in mice). Hepatology 73, 2251–2265 (2021).
Zhang, Y. et al. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol. Endocrinol. 26, 272–280 (2012).
Mina, A. I. et al. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666.e651 (2018).
Shan, Z. X. et al. A quick and efficient approach for gene silencing by using triple putative microRNA-based short hairpin RNAs. Mol. Cell Biochem. 323, 81–89 (2009).
Liu, X. et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 18, 431–442 (2016).
Cassim Bawa, F. N. et al. Adipocyte retinoic acid receptor α prevents obesity and steatohepatitis by regulating energy expenditure and lipogenesis. Obesity 32, 120–130 (2024).
Roy, D., Myers, J. M. & Tedeschi, A. Protocol for assessing ex vivo lipolysis of murine adipose tissue. STAR Protoc. 3, 101518 (2022).
Sun, C. et al. Adipose Snail1 regulates lipolysis and lipid partitioning by suppressing adipose triacylglycerol lipase expression. Cell Rep. 17, 2015–2027 (2016).
Vergnes, L., Lin, J. Y., Davies, G. R., Church, C. D. & Reue, K. Induction of UCP1 and thermogenesis by a small molecule via AKAP1/PKA modulation. J. Biol. Chem. 295, 15054–15069 (2020).
Li, Y. et al. Hepatic forkhead box protein A3 regulates ApoA-I (apolipoprotein A-I) expression, cholesterol efflux, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 39, 1574–1587 (2019).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005).
Acknowledgements
The work was supported by grants from the National Institutes of Health R01HL142086, R01DK118941, R01DK118805, and R01DK121548 to Y.Zhang.
Author information
Authors and Affiliations
Contributions
S.H. and Y.Zhang conceived and designed the studies and guided the interpretation of the results. Y.Zhang supervised the project. Y.Zhang and S.H. prepared the manuscript. F.N.C.B. and Y.Zhu performed a number of in vivo studies. X.P., H.W., and R.G. performed various studies. Y.X. generated some adenoviruses and adeno-associated viruses. All authors discussed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Takayoshi Suganami and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Soona Shin and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, S., Cassim Bawa, F.N., Zhu, Y. et al. Loss of adipose ATF3 promotes adipose tissue lipolysis and the development of MASH. Commun Biol 7, 1300 (2024). https://doi.org/10.1038/s42003-024-06915-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06915-x
This article is cited by
-
Cardiometabolic Index as a predictor of mortality in metabolic Dysfunction-Associated Steatotic Liver Disease
BMC Gastroenterology (2025)
-
Separable cryo-microneedle patches delivery with capsaicin integrated mesoporous dopamine for obesity treatment
Journal of Nanobiotechnology (2025)
