
Abstract
Rice (Oryza sativa L.) endosperm is essential to provide nutrients for seed germination and determine grain yield. RNA editing, a post-transcriptional modification essential for plant development, unfortunately, is not fully characterized during rice endosperm development. Here, we perform systematic analyses to characterize RNA editome during rice endosperm development. We find that most editing sites are C-to-U CDS-recoding in mitochondria, leading to increased hydrophobic amino acids and changed structures of mitochondrial proteins. Comparative analysis of RNA editome reveals that CDS-recoding sites present higher editing frequencies with lower variabilities and their resultant recoded amino acids tend to exhibit stronger evolutionary conservation across many land plants. Furthermore, we classify mitochondrial genes into three groups, presenting distinct patterns in terms of CDS-recoding events. Besides, we conduct genome-wide screening to detect pentatricopeptide repeat (PPR) proteins and construct PPR-RNA binding profiles, yielding candidate PPR editing factors related to rice endosperm development. Taken together, our findings provide valuable insights for deciphering fundamental mechanisms of rice endosperm development underlying RNA editing machinery.
Similar content being viewed by others


Introduction
Rice (Oryza sativa L. subsp. Geng (Japonica) cv. Nipponbare) is a staple crop feeding more than half the world’s population1 as well as a model plant for genetic studies and molecular breeding2,3. The development of rice endosperm, rich in starch granules and protein bodies4, determines grain production and quality5. Rice endosperm development is roughly divided into four stages: (i) coenocyte (1–2 days after flowering (DAF) or pollination; the pollination occurs at or slightly before flowering6,7), (ii) cellularization (3–5 DAF), (iii) storage product accumulation (6–20/21 DAF), and (iv) maturation (21/22–30 DAF)4,8. Among these stages, it is at stage III that accumulation of storage products and programmed cell death (PCD) simultaneously occur and play crucial roles in endosperm development8,9.
Plant mitochondria provide energy for endosperm development and are related to PCD10,11. For rice, its mitochondrial genome contains 53 protein-coding genes, falling into four categories: (i) complexes composing the mitochondrial electron transport chain (mETC), including complex I (NADH dehydrogenase), complex III (cytochrome c reductase), complex IV (cytochrome c oxidase) and complex V (adenosine triphosphate (ATP) synthase), (ii) cytochrome (Cyt) c biogenesis proteins, (iii) ribosomal proteins, and (iv) other proteins. Furthermore, mETC tightly couples with ATP synthesis; besides, it has been found that complexes I and III in the mETC produce reactive oxygen species (ROS) related to PCD12,13. In mitochondria, two c-type cytochromes, namely, Cyt c and c1, participate in multiple biological processes; Cyt c transfers electrons from complex III to IV14 and likely regulates PCD15,16, and Cyt c1 is integrated into complex III14. In addition, MatR, a maturase-related protein, is assigned to the “other proteins” group and is required for the splicing of Group II introns in mitochondria17,18. Ribosomal proteins are responsible for translating mRNAs encoded by mitochondrial genomes and regulating protein synthesis, such as complexes in the mETC19,20.
RNA editing, as one of the most important post-transcriptional modifications, creates RNA products that differ from their DNA templates. RNA editing presents unique patterns of editing types in land plants, namely, mainly cytidine-to-uridine (C-to-U) alterations in RNA transcribed from mitochondrial and plastid DNA, termed as mitochondrial RNA editing21,22,23,24 and plastid RNA editing25,26, respectively. Importantly, pentatricopeptide repeat (PPR) proteins, including P-type and PLS-type (that involves four classes, viz., PLS, E (E1 and E2), E+, and DYW), specifically bind RNA depending on PPR codes and mediate C-to-U RNA editing27, playing a key role as RNA editing factors28. Specifically, P-type PPR proteins can mediate RNA editing by involving in the formation of E/E+PPR editosome29,30,31,32,33,34, or directly binding RNA35,36,37,38. Among PLS-type PPR proteins, PLS-class (that does not contain E, E+ and DYW domains) mediates RNA editing39,40 yet with unclear regulatory mechanisms, E-class and E+-class (that do not contain DYW domain) mediate RNA editing by recruiting DYW domain-containing proteins41, and DYW-class mediates RNA editing by utilizing DYW domain to implement the deamination of cytidine to uridine42. Over the past decades, valuable efforts have been devoted to investigating RNA editing via PPR proteins during plant endosperm development. Evidence has been accumulated that RNA editing in mitochondrial transcripts is important for endosperm development43,44,45,46,47,48,49,50,51 by affecting functions of complexes composing the mETC, Cyt c biogenesis proteins, ribosomal proteins and MatR. Specifically, RNA editing in mitochondrial genes regulated by PPR proteins is related to rice endosperm development, such as editing in nad7 mediated by SMK152, atp6, cob, cox3, nad1, nad9, rpl16 and rps12 by EMP553, cox2, cox3, ccmC, nad2 and nad4 by OGR154, and multiple mitochondrial genes (such as nad9) by cooperation of P-type FLO22 and DYW-class DYW335. As a result, RNA editing in mitochondrial genes as well as PPR editing factors has been identified in wild-type and mutant rice endosperm at specific developmental stages (for details see the Plant Editosome Database55), yet lacking comprehensive analyses of RNA editome during rice endosperm development.
Toward this end, here we collect rice endosperms across five successive developmental stages, mainly including the process of grain filling (stage III of rice endosperm development), and perform whole-genome re-sequencing and strand-specific RNA sequencing (RNA-seq). Based on these sequencing data, we systematically profile RNA editome, analyze RNA editing features in terms of location, editing frequency & variability, evolutionary conservation and structure, identify PPR genes in rice genome and construct PPR-RNA binding profiles, which collectively provide insightful clues for better understanding rice endosperm development from RNA editing machinery.
Results
Profiling RNA editome during rice endosperm development
To systematically investigate RNA editing events during rice endosperm development, we obtain both genome re-sequencing and strand-specific RNA-seq data from rice endosperm derived from five successive developmental stages (3, 6, 9, 12, and 15 DAF). After strict quality control, sequence alignment, and filtration (for details see Methods), we identify a total of 369 RNA editing sites across the five developmental stages (Supplementary Data 1). Specifically, the primary RNA editing type in rice mitochondria is C-to-U (298) and the rest of types are G-to-A (65) and U-to-C (1), whereas in plastids the only type is C-to-U (5), revealing the dominance of C-to-U editing during rice endosperm development (Fig. 1A and Supplementary Data 2). Furthermore, we find that these C-to-U editing sites identified in endosperm are different from those in seedling, presumably suggesting the tissue-specificity of RNA editing (for details see Supplementary Fig. 1A, B). In addition, our procedure for identification of RNA editing sites, by comparison with a previous study56, is more effective to obtain high-confidence editing sites (for details see Supplementary Fig. 1C, D).

A RNA editing sites in rice mitochondria and plastids. 369 RNA editing sites in mitochondria (364) and plastids (5) are categorized by editing types, which are color-coded, with pink for C-to-U (mitochondria), blue for C-to-U (plastids), green for G-to-A (mitochondria) and purple for U-to-C (mitochondria), respectively. B Distribution of 369 RNA editing sites at 3, 6, 9, 12, and 15 DAF (n = 5). Bars with pink, green, blue, and purple denote C-to-U (mitochondria), G-to-A (mitochondria), C-to-U (plastids), and U-to-C (mitochondria) RNA editing types, respectively. C Distribution of RNA editing events and their corresponding RNA-seq coverage in rice mitochondria. The outer circle denotes rice mitochondrial genes; genes outside the circle are on the positive strand and oriented clockwise, while genes inside the circle are on the negative strand and oriented anticlockwise. The gray circles, from outer to inner denote RNA-seq coverage (Bar plot, C: Coverage) and frequency (Heatmap, E: Editing) of RNA editing sites at 3, 6, 9, 12, and 15 DAF. D Distribution of editing frequency of 298 C-to-U editing sites. Components of the box plot are: center line, median value; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; error bars, the highest and lowest values excluding outliers. E Distribution of 298 C-to-U editing sites in different sequence regions. Mit mitochondria, Plt plastids.
For each rice endosperm developmental stage, the number of C-to-U editing sites in mitochondria is consistently larger in contrast to other editing types (Fig. 1B). Furthermore, we capture a whole view of RNA editing events in rice mitochondrial genome across the five developmental stages (Fig. 1C). We find that the average editing frequency of all 298 C-to-U sites in mitochondria varies from 2% to 99.8% at the five developmental stages, with the median at 69.8% (Fig. 1D). Besides, the distribution of the distance between two neighboring editing sites shows that C-to-U editing sites tend to be located within 50 bp (Supplementary Fig. 2). Among these 298 C-to-U editing sites in rice mitochondria, we observe that there are 253 sites located in coding sequence (CDS) regions (214 for CDS-recoding and 39 for CDS-synonymous), which are more prevalent compared to those in intronic (73), intergenic (12) and pseudogenic (8) regions (Fig. 1E). Since one gene may be overlapped with other genes in the plant mitochondrial genome, notably, we find that among these 73 intronic editing sites, 43 are located in CDS (35 for CDS-recoding and 8 for CDS-synonymous) regions and 5 are in pseudogenic regions (Fig. 1E and Supplementary Data 1). Unlike the diversity of C-to-U editing distribution, the 65 G-to-A editing sites in mitochondria are located in intronic (23, 35.38%) and intergenic (42, 64.62%) regions and the 5 C-to-U editing sites in plastids are only in CDS regions (2 for CDS-recoding and 3 for CDS-synonymous) (Supplementary Data 1). Taken together, it is clearly shown that CDS-recoding sites are more ubiquitous in mitochondria during rice endosperm development.
Amino acid biochemical properties changed by RNA editing
Studies have shown that RNA editing is functionally important by affecting amino acid biochemical properties57,58. In our study, among these 214 CDS-recoding sites, we find that 88 (41.12%) and 126 (58.88%) are located at the first and second codon positions, respectively. Contrastingly, among these 39 CDS-synonymous sites, most (36, 92.31%) occur at the third codon position producing plenty of U-ending codons, except 3 sites (7.69%; Leu-to-Leu, 2 for CUG-to-UUG and 1 for CUA-to-UUA) at the first codon position (Supplementary Data 3 and 4). Accordingly, we identify 8 codon alteration types with more than 10 editing events: UCA-to-UUA (Ser-to-Leu), CGG-to-UGG (Arg-to-Trp), UCG-to-UUG (Ser-to-Leu), UCU-to-UUU (Ser-to-Phe), CCA-to-CUA (Pro-to-Leu), UCC-to-UUC (Ser-to-Phe), CGU-to-UGU (Arg-to-Cys), and UUC-to-UUU (Phe-to-Phe) (Fig. 2A and Supplementary Data 4). Considering the relative frequency of amino acids corresponding to these 253 sites in CDS regions (214 for CDS-recoding and 39 for CDS-synonymous) after RNA editing, Leu (94, 37.30%) and Phe (55, 21.83%) are relatively dominant (Fig. 2A and Supplementary Data 4). Regarding the amino acid biochemical properties, we find that nearly all amino acids after RNA editing on these 253 sites in mitochondria are hydrophobic except Ser and Thr (Fig. 2A); the percentage of hydrophobic amino acids is dramatically increased from 37.70% to 86.90% by most CDS-recoding editing sites (188/214) (Supplementary Data 4).

A Amino acid and codon alterations of 253 editing sites in CDS regions. Red and gray arrows denote codon alterations with more and less than 10 editing events, respectively. Edited RNA bases are underlined in codons. Amino acids in membrane structures and water drops denote hydrophobic and hydrophilic amino acids, respectively. Asterisks denote stop codons. Ala Alanine, Arg Arginine, Cys Cysteine, Gln Glutamine, His Histidine, Ile Isoleucine, Leu Leucine, Met Methionine, Phe Phenylalanine, Pro Proline, Ser Serine, Thr Threonine, Trp Tryptophan, Tyr Tyrosine, Val Valine. B Normalized CDS-recoding site counts in different grouped genes/proteins/complexes as well as their corresponding hydrophobic amino acid counts before and after editing. Purple bars denote normalized CDS-recoding site counts in protein-coding genes. Numbers in the left bars denote total normalized CDS-recoding site counts in different types of proteins or complexes (complex I, III, IV, V, Cyt c biogenesis proteins, ribosomal proteins and other proteins from up to down). Normalized counts of hydrophobic amino acids corresponding to editing sites before and after editing in protein-coding genes are shown as blue and red bars, respectively. Numbers in the right bars denote normalized hydrophobic amino acid counts after editing in different types of proteins or complexes. Normalized editing site counts (or normalized hydrophobic amino acid counts) in protein-coding genes (or proteins, or complexes) is denoted as “CDS-recoding site counts (or hydrophobic amino acid counts)/total length of CDS in the corresponding protein-coding gene (or protein, or complex) × 1000”. C Recoded amino acids caused by RNA editing with mapped positions in the 3D structure of apocytochrome b (COB). The abbreviation used is: IMM, inner mitochondrial membrane.
In terms of 53 rice mitochondrial protein-coding genes assigned to 7 groups encoding complex I, complex III, complex IV, complex V, Cyt c biogenesis proteins, ribosomal proteins, and other proteins, we investigate the distribution of these 214 CDS-recoding sites as well as the corresponding amino acid hydrophobicity (Fig. 2B). Notably, editing at 64 of these 214 CDS-recoding sites has been greatly affected in mutant endosperm of O. sativa, Zea mays and A. thaliana43,44,45,48,50,51,52,54,59 (for details see Supplementary Data 5), suggesting the critical significance of the rest CDS-recoding sites for endosperm development. Among these 53 genes, nearly half (25 genes) harbor at least one CDS-recoding site (Fig. 2B). Furthermore, we detect RNA editing hotspots that feature more CDS-recoding sites and more hydrophobic amino acids after editing. We find that three gene groups, encoding Cyt c biogenesis proteins (normalized CDS-recoding sites: 19.16, normalized hydrophobic amino acids caused by CDS-recoding editing: 14.87), complex III (normalized CDS-recoding sites: 12.56, normalized hydrophobic amino acids caused by CDS-recoding editing: 10.89) and IV (normalized CDS-recoding sites: 7.66, normalized hydrophobic amino acids caused by CDS-recoding editing: 7.66) (Fig. 2B), are RNA editing hotspots, including a bunch of previously reported editing sites that can be affected in endosperm mutants43,47,48,49,50,52,54,60,61 (for details see Supplementary Data 5). In plastids, atpA-1148 is the only CDS-recoding site that presents higher editing frequencies (0.838 on average) (Supplementary Data 1 and 6), which has been reported to be critical for ATP synthesis62.
Structural effects provoked by RNA editing
Considering the possible effect of RNA editing in protein structure58,63, we map these 214 CDS-recoding sites to three-dimensional (3D) structures of edited gene products. Notably, we observe that within mitochondria, most CDS-recoding sites (167/188) featuring hydrophobic amino acids are situated in/ near the inner membrane and/or the interior of proteins, characterized by a hydrophobic lipid environment (Supplementary Fig. 3 and Supplementary Data 7), suggesting that editing at these sites plays a crucial role in preserving the insertion of membrane-proteins, ensuring protein stability, and subsequently influencing assemblies and activities of mitochondrial complexes/proteins, as previously elucidated58. Especially, cytochrome b (cob), encoding apocytochrome b (COB), a subunit of complex III in mitochondria that is one of RNA editing hotspots, possesses 15 CDS-recoding sites, among which 13 produce hydrophobic amino acids, conforming well with its hydrophobic environment (Fig. 2C and Supplementary Data 7). Likewise, in plastids, atpA-1148 and ndhG-347 causing hydrophobic amino acids are in/near the inner mitochondrial membrane (IMM) and/or buried in the interior of proteins (Supplementary Fig. 3 and Supplementary Data 7).
To further investigate the effect of these CDS-recoding sites on protein 3D structure, we align 3D structures of mitochondrial proteins before and after editing (for details see Methods and Data availability). Obviously, we detect a significant difference (Root Mean Square Deviation, RMSD > 2.5Å64) in 3D structures of proteins encoded by atp6, ccmB, ccmC, ccmFc, ccmFn, cob, cox2, mat-r, nad5, nad6, orf183, orf288, orfX, rpl2, rps1, rps4 and rps19 (Supplementary Fig. 4), indicating that CDS-recoding editing in these genes greatly affects protein 3D structures. Notably, the RNA editing hotspots identified above, all present significant changes in 3D structures, especially ccmB (RMSD = 30.90 Å) and ccmFc (RMSD = 25.06 Å) (Supplementary Fig. 4). On the contrary, the rest mitochondrial protein-coding genes (atp9, nad1, nad2, nad4, nad4L, nad7, rps7, and rps13) delicately change their 3D structures with RMSD < 2.5 Å (Supplementary Fig. 4), implying that CDS-recoding editing probably fine-tunes these proteins’ structures.
We also find that two intronic editing sites at nad7-i3-D5-22 (position 22 at domain 5 (D5) in the 3rd intron of nad7, genomic position at 87,186) and ccmFc-i-D5-26 (position 26 at D5 in the only intron of ccmFc, genomic position at 332,291) affect D5’s structure (Supplementary Fig. 5), which is essential for intron splicing65. Specifically, the two RNA editing sites are likely to produce GAAA sequence, which is located in the apical loops of RNA secondary structure models of edited nad7 intron 3 D5 and ccmFc intron D5 (Supplementary Fig. 5) and important for interaction activities of D566,67.
RNA editing frequency, variability and evolutionary conservation
We next investigate the editing frequency and variability of these 298 C-to-U sites in mitochondria across the five developmental stages. For each stage, it is consistently observed that CDS-recoding sites present significantly higher editing frequencies than CDS-synonymous, intronic and intergenic ones (Fig. 3A). Strikingly, all CDS-recoding and CDS-synonymous sites in cob, encoding a subunit of complex III and belonging to the editing hotspot, feature higher editing frequencies at each developmental stage (Supplementary Fig. 6). As one site may present variable editing frequencies at different developmental stages, we further examine the editing variability of these sites. We find that CDS-recoding sites also possess lower editing variability than other sites (Fig. 3B), indicating the significant role of their relative constant editing in rice endosperm development. Specifically, we notice that most CDS-recoding sites in atp6, ccmC, cox2, nad4L, nad6, orf183 as well as ribosomal protein genes, present not only higher editing frequencies but also lower editing variabilities across the five developmental stages (Supplementary Fig. 6), clearly showing the critical significance of RNA editing in altering amino acids to yield functional proteins for rice endosperm development.

A Editing frequencies of 247 C-to-U sites (outside of overlapped genes) in CDS (179 for CDS-recoding and 31 for CDS-synonymous), intronic (25) and intergenic (12) regions during five rice endosperm development stages. In addition, editing frequency of three sites in pseudogenic regions are not compared with that of CDS-recoding sites because of few sites. The Wilcoxon test was used to calculate the statistical significance (p-value, with ***<0.001, and ****<0.0001). Components of the box plot are: center line, median value; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; error bars: the highest and lowest values excluding outliers. B Editing variabilities of these 247 C-to-U sites across five stages during rice endosperm development. The Wilcoxon test was used to calculate the statistical significance (p-value, with **<0.01, and ***<0.001). Components of the box plot are: center line, median value; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; error bars: the highest and lowest values excluding outliers. C Correlation between editing frequency of CDS-recoding sites and evolutionary conservation of amino acids corresponding to these CDS-recoding sites. Spearman’s correlation coefficient is calculated for the correlation. The fitted curve is implemented by the generalized linear model. Points in different colors denote CDS-recoding sites in mitochondrial genes encoding different proteins or complexes. Based on the density distribution of conservation of amino acids (Supplementary Fig. 7), thresholds of high and low conservation were set at 0.50 and 0.15, respectively. Similarly, thresholds of high and low editing frequencies were set at 0.89 and 0.10, respectively, according to the density distribution of editing frequencies of these CDS-recoding sites (Supplementary Fig. 8).
RNA editing is believed to restore evolutionarily conserved amino acids68. In other words, for one CDS-recoding site, its recoded amino acid via RNA editing may be restored to the amino acid that is relatively conserved in multiple species. To explore the conservation of amino acids corresponding to these 214 CDS-recoding sites, we retrieve a large collection of protein sequences in accordance with these 214 sites from 122 land plants (for details see Methods), falling into 6 different clades, namely, liverworts (12 species; 189 CDS-recoding sites), mosses (25 species; 192 CDS-recoding sites), hornworts (5 species; 82 CDS-recoding sites), lycophytes (3 species; 109 CDS-recoding sites), ferns (2 species; 107 CDS-recoding sites), and seed plants (75 species; 198 CDS-recoding sites) (Supplementary Data 8 and 9). Accordingly, we find that these CDS-recoding sites present a positive correlation between amino acid conservation and editing frequency (R = 0.31, p = 9.4e−06) (Fig. 3C; for details see Supplementary Fig. 9), which, albeit the correlation coefficient is not so strong, is observed in nearly all clades except hornworts and ferns (Supplementary Fig. 10).
We further classify these editing sites into 7 groups in terms of gene products as mentioned above. We find that editing sites in genes encoding Cyt c biogenesis proteins exhibit the positive correlation between amino acid conservation and editing frequency (Supplementary Fig. 11). Particularly, cob (encoding a subunit of complex III), cox2 (encoding a subunit of complex IV) and nad4L (encoding a subunit of complex I) tend to possess more conserved amino acids and higher editing frequencies (Fig. 3C). On the contrary, it is also believed that RNA editing enriches proteomic diversity69. Notably, ccmC, ccmFn, cox2, rps1, and rps7 harbor a few CDS-recoding sites with higher editing frequencies but less conserved amino acids, indicating the enriched proteomic diversity in these genes (Fig. 3C).
It is worth noting that RNA editing can provoke initiation and stop codons. Here we find that these CDS-recoding editing events lead to one initiation codon (nad4L-2, ACG to AUG) and three stop codons (atp9-223, CGA to UGA; ccmFc-1306, CGA to UGA; and rps1-505, CAA to UAA) (Supplementary Data 6). Specifically, nad4L-2, converting ACG into the canonical initiation codon AUG, has lower editing frequency during rice endosperm development and higher conservation in its encoded amino acid across 117 land plants (Fig. 3C and Supplementary Data 10). In addition, atp9-223 and ccmFc-1306 present higher conservation across land plants, leading to stop codons at the end of corresponding gene products (Fig. 3C; Supplementary Data 6 and 10). Contrastingly, RNA editing at rps1-505 resulting in a stop codon is observed only in rice and absent in other land plants, accordingly reducing the length of rps1 products by 4 amino acids.
RNA editing pattern and classification of mitochondrial genes
Since CDS-recoding editing sites predominate and present important effects on biochemical properties and protein structures during rice endosperm development, we further investigate their editing patterns during the five developmental stages. Based on unsupervised clustering, we group the 214 CDS-recoding sites into five clusters, namely, C1~C5, with each cluster presenting a distinctive RNA editing pattern, containing 26, 17, 25, 43, and 103 sites, respectively (Fig. 4A). We find that the average editing frequency is on the increase from C1 to C5 (with 0.08 in C1, 0.28 in C2, 0.50 in C3, 0.74 in C4, and 0.92 in C5, respectively), and their corresponding editing variability is gradually on the decrease (with τ value at 0.85, 0.44, 0.25, 0.15 and 0.03, respectively; lower τ value indicates more relatively constant editing frequencies). Clearly, C5 features higher editing frequency and lower editing variability, whereas C1 features lower editing frequency and higher editing variability.

A Clustering of 214 CDS-recoding sites in five developmental stages of rice endosperm, leading to five clusters (C1~C5) that show distinct patterns by factoring both editing frequency (“f”) and variability (“τ”). In each cluster, the red triangle denotes the average editing frequency in each development stage. Components of the box plot are: center line, median value; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; error bars: the highest and lowest values excluding outliers; points, outliers. B Classification of edited mitochondrial protein-coding genes based on the clustering patterns of these 214 CDS-recoding sites.
To further investigate 25 mitochondrial genes containing CDS-recoding sites, we classify these genes, by factoring C1~C5, into Groups I, II, and III which contain 3, 9, and 13 genes, respectively. Specifically, Group I harbors editing events present in C1~C4 yet absent in C5, Group II features C5-specific editing events, and Group III contains editing events spanning broadly from C1~C5 (Fig. 4B). In particular, in Group I, nad4 encoding a subunit of complex I contains CDS-recoding events only from C4. Of note, cob encoding a component of complex III and cox2 encoding a component of complex IV, belonging to Group II, are RNA editing hotspots. Similarly, four genes (ccmB, ccmC, ccmFc and ccmFn) encoding Cyt c biogenesis proteins in Group III are also RNA editing hotspots, with diverse CDS-recoding events covering all five clusters (C1~C5) except ccmC. Furthermore, since editing frequencies are increased or decreased in mutant endosperms (Supplementary Data 5), regulating RNA editing in genes from Groups I and III is potential to affect functions of mitochondrial complexes/proteins and further improve rice endosperm development.
Construction of PPR-RNA binding profiles
To explore which PLS-type PPR proteins regulate C-to-U RNA editing during rice endosperm development according to the distinctive mechanism of PPR-RNA binding, we identify 480 PPR genes in the rice genome (Fig. 5A and Supplementary Data 11). To further identify potential RNA editing factors, we predict the subcellular localization of PPR proteins and PPR-RNA binding (for details see Methods) and obtain a total of 156 PLS-type PPR proteins as potential editing factors, namely, 149 in mitochondria and 7 in plastids (Supplementary Data 12 and 13). To further verify the confidence of these 156 predicted PPR proteins, we perform protein sequence alignments and find that 49 present higher similarity with extant PPR editing factors experimentally discovered in A. thaliana, Glycine max, Gossypium hirsutum, Hordeum vulgare and Z. mays (for details see Supplementary Data 14), indicating that the predicted PPR proteins are valuable candidates for experimental validation.

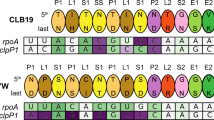
A A total of 480 PPR genes were identified in the rice genome, containing P-type PPR and PLS-type PPR (including PLS, E (E1 and E2), E+, and DYW class PPR) genes. B, C Motif arrangement of LOC_Os12g17080 and LOC_Os11g10740 and their binding to the upstream sequence of cox2-167 and nad7-836, respectively. The combination of the fifth and last amino acids in each motif (P1, L1, S1, P2, L2 and S2) denotes a PPR code, specifically binding a RNA base. The cytidine (C) in red is the edited RNA base.
Meanwhile, we construct PLS-type PPR-RNA binding profiles for these 156 predicted PPR proteins (Supplementary Data 13 and 15). Based on binding affinities of PPR codes for RNA bases, we find that LOC_Os12g17080 and LOC_Os11g10740 tend to bind RNA sequences upstream of the top 10 editing sites (Supplementary Data 13). Among these top 10 sites, notably, cox2-167 (ranking 3rd) (Fig. 5B) and nad7-836 (ranking 6th) (Fig. 5C) have been experimentally validated to affect endosperm development in rice52,54, implying the great potential of our predicted PPR-RNA binding profiles in studying RNA editing machinery during rice endosperm development. Furthermore, we investigate PPR gene expression during rice endosperm development and find that PPR (especially PLS-type PPR) genes extensively exhibit low expression (Supplementary Data 16) in conformity with previous findings70,71,72, indicating that expression is not a proper indicator for screening PPR RNA editing factors.
Discussion
Here, we profiled RNA editome in mitochondria during rice endosperm development and performed systematic analyses to characterize RNA editing sites, demonstrating that CDS-recoding editing is conserved in evolution and development, increases the proportion of hydrophobic amino acids located in/near the mitochondrial inner membrane and/or interior of proteins, and changes 3D structures of mitochondrial proteins. Moreover, we classified mitochondrial protein-coding genes containing CDS-recoding editing events into three groups based on editing frequency and variability, and constructed PPR-RNA binding profiles to yield a high-confidence collection of candidate PPR editing factors. Based on these results, we present a model of RNA editing in mitochondria for rice endosperm development (Fig. 6): RNA editing mediated by one PPR protein changes RNA and amino acid sequences and affects complex/protein formation to maintain ATP synthesis, protein synthesis, and PCD involving genes that encode complexes (complex I, III, IV and V) in the mETC, Cyt c biogenesis proteins and ribosomal proteins, which together sustain rice endosperm development.

Editing regulated by one PPR protein changes RNA and amino acid sequences, regulates formations and functions of complexes (complex I, III, IV and V), Cyt c biogenesis proteins and ribosomal proteins to ensure ATP synthesis, PCD and protein synthesis, which together maintain rice endosperm development. Different gene categories containing CDS-recoding editing events are color-coded, with complexes (complex I, III, IV and V) for brown, Cyt c biogenesis proteins for pink, ribosomal proteins for gray, and other proteins for green. The yellow dashed line denotes CcmB, CcmFc and CcmFn are related to Cyt c1 biogenesis, while the question mark means that the role of CcmC in Cyt c1 biogenesis is uncertain. Complex II is encoded by two nuclear genes and is not found to contain RNA editing events in rice. AA amino acid, PCD programmed cell death, IMS intermembrane space, IMM inner mitochondrial membrane, CCBPs cytochrome c biogenesis proteins, ROS reactive oxygen species, Cyt cytochrome.
CDS-recoding editing in genes encoding complexes (complex I, III, IV, and V) in the mETC and encoding Cyt c biogenesis proteins, is important for ATP synthesis during rice endosperm development. For instance, cox2-167, an editing site identified in the gene encoding complex IV that is the last electron acceptor in the mETC and crucial for ATP formation, has been documented to present aberrant RNA editing leading to abnormal rice endosperm54. Strikingly, there is a body of related evidence supported by several studies on maize endosperm development: (i) absence of editing at cob-908 (also identified in this study) results in deficiency of complex III, decrease of ATP production and reduction of maize endosperm growth43, (ii) loss of editing at ccmB-43 (also identified in this study) affects the maturation of Cyt c and c1, and biogenesis of complex III48, and (iii) abolishment of editing at ccmFn-1553 in maize (corresponding to ccmFn-1514 identified in this study) results in the loss of CcmFN protein (a subunit of heme lyase complex) and affects cytochrome c maturation, mitochondrial oxidative phosphorylation47, which, as a result, affects maize endosperm development.
In addition, attention should be paid to CDS-recoding editing in genes that encode other mETC complexes, such as complex I and V, because, besides ATP synthesis, complex I is related to formation of ROS that is the crucial signaling molecule regulating PCD12,13. Consistently, abolishment of editing at nad2-26 and nad5-1916 (also identified in this study) affects PCD and maize endosperm development50. Moreover, CDS-recoding editing events in nad4L (nad4L-110 and nad4L-179) encoding a subunit of complex I and atp9 (atp9-191 and atp9-212) encoding a subunit of complex V (also identified in this study), are closely associated with mutant maize endosperm31,49. In addition, abolishment of editing at nad7-836 affects the structural stability and activity of NAD7 (a subunit of complex I) and further influences respiration rate, starch biosynthesis, and maize endosperm development52.
CDS-recoding editing in ribosomal proteins is important for protein synthesis during rice endosperm development. Particularly, it has been reported that editing at rps4-335 (in maize, A. thaliana, and rice in this study), converting Pro into Leu, is related to the maturation of Cyt c, the assembly of complex I, III, and V, and affects protein translation and endosperm development48. In addition, ribosomal protein S1, S7 and S13 are essential for protein synthesis73,74,75 and CDS-recoding editing events in rps1 (rps1-107 and rps1-377), rps7 (rps7-277 and rps7-332) and rps13 (rps13-56, rps13-100, rps13-256 and rps13-287) (also identified in this study) are affected in mutant maize endosperm31,47,51,59,76, implying that these editing events play an important role in protein synthesis during rice endosperm development.
CDS-recoding editing in mitochondrial genes encoding other proteins may also be critical for rice endosperm development. Notably, 3D structures of genes mat-r, orf183, orf288 and orfX are significantly changed after RNA editing, especially for orf288 (RMSD = 24.57 Å) and mat-r (RMSD = 16.77 Å) (Supplementary Fig. 4), implying the importance of editing in these genes for rice endosperm development. Specifically, it has been reported that abnormal editing at mat-r-86, mat-r-92, mat-r-295, and mat-r-296 is potentially associated with mutant endosperm of maize (also identified in this study)31,44. In addition, it has been found that abolishment of editing at one site in mat-r can reduce the splicing efficiency of nad4 Introns 1 and 3 and further affect maize endosperm development46. However, orfX, orf183, and orf288, similar to a chimeric cytoplasmic male sterility-associated gene orf31277, have not been well studied, requiring further investigations on uncovering molecular mechanisms and functions of RNA editing.
Methods
Whole–genome re-sequencing and RNA-seq
DNA was extracted from leaves and endosperm (at 3 DAF) of the wild-type Nipponbare (Japonica) by the DNA extraction kit (DP305) from HUAYUEYANG Biotechnology, BEIJING CO.,LTD. RNA was extracted from endosperms at 3, 6, 9, 12, and 15 DAF by the RNA extraction kit (GK0416) produced by HUAYUEYANG Biotechnology, BEIJING CO.,LTD. Sequencing libraries were prepared parallelly by the KAPA Stranded mRNA-seq Library Preparation kit (KK8421) and the Epicentre Ribo-Zero Magnetic Kit (Plant Seed/Root, MRZSR116), which were sequenced on the Illumina HiSeq 2500.
Read mapping and gene expression quantification
Trimmomatic78 was used to eliminate low-quality reads and remove adapters. Cleaned reads were mapped to the mitochondria genome (NC_011033.1)56, the chloroplast genome (NC_001320.1)79 and the nuclear genome (Os-Nipponbare-Reference-IRGSP-1.0)80 by HISAT281 with the parameter “--rna-strandness FR” to infer strand-specific information. The mapped reads were used to quantify expression with FPKM (Fragments Per Kilobase of exon per Million fragments mapped) for genes and transcripts by stringTie82.
Identification of RNA editing sites and estimation of editing frequency
To detect editing sites, matched RNA and genomic DNA were analyzed by the REDItoolDnaRna.py script in REDItools83. Single nucleotide polymorphisms (SNPs) were identified by comparing the re-sequenced DNA sequence to the reference genome and were excluded. The parameters “-e -E -d -D” were used to remove multiple mapped reads caused by gene transfers as well as duplicated reads. Low-quality bases (quality score <25) and low-mapping sites (quality score <25) were removed. DNA-RNA mismatches featuring multiple types were excluded, and only those supported by both RNA and DNA sequencing with base coverages greater than 30 and variations supported by at least 3 reads were kept. RNA editing sites identified at five developmental stages were mutually confirmed. RNA editing sites in cox1, nad2, nad7 and rpl2 were corrected by annotations from NCBI RefSeq under accession number DQ167400.1.
Detecting the variability of editing frequency during rice endosperm development
The τ value84 was adopted to estimate the variability of editing frequency across different developmental stages. For an editing site, smaller τ value means relative constant editing frequencies across different conditions and larger τ value signifies variable editing frequencies.
Correlation between editing frequency and evolutionary conservation
To investigate the correlation between the editing efficiency of CDS-recoding sites and the evolutionary conservation of corresponding recoded amino acids, alterations of amino acids caused by editing were obtained from mitochondrial genes among land plants in RefSeq85 and GenBank86. The correlation in orf183 and orf288 was filtered due to few plants containing RNA editing sites in these two genes. Amino acids recoded by ccmFn-37, ccmFn-38, mat-r-295, mat-r-296, rps19-163, and rps19-164 were filtered because of more than one editing site in the corresponding codons. Multiple sequence alignment conducted by ClustalW87 in MEGA1188 was used to calculate the evolutionary conservation of amino acids corresponding to RNA editing sites. These amino acids existing in more than 80% species were kept because of gaps in the multiple sequence alignment. The correlation between the editing frequency and the conservation of amino acids was analyzed by Spearman’s correlation and fitted by the generalized linear model (GLM).
RNA secondary structure prediction
To functionally investigate editing sites in intronic regions of mitochondrial genes, editing sites in sequence-conserved domain 5 (D5) in introns were selected. D5 in rice mitochondrial genes was searched by aligning with D5 of Pylaiella littoralis89, and D5 containing editing sites was kept. The secondary structure of D5 before and after RNA editing was predicted by MFold90 (http://www.unafold.org/Dinamelt/applications/quickfold.php).
Mapping CDS-recoding sites to protein 3D structures
To investigate locations of CDS-recoding sites on 3D structures of proteins, experimentally verified protein 3D structures in PDB91 (https://www.rcsb.org) were used with priority. Protein sequences were aligned with those in other plants with 3D annotations in PDB, and CDS-recoding sites were mapped to the 3D structures. Given that some annotations of protein 3D structures are not available in PDB, mapping of CDS-recoding sites to protein 3D structures was conducted based on annotations in Uniprot92 (https://www.uniprot.org).
Protein 3D structure prediction and alignment
To investigate effects of CDS-recoding editing on protein 3D structures, ColabFold93(https://colab.research.google.com/github/sokrypton/ColabFold) was used to predict 3D structures of each protein before and after RNA editing, and hypothetical 3D structures were aligned and visualized by SuperPose94 (http://superpose.wishartlab.com). Root Mean Square Deviation (RMSD) greater than 2.5 Å was considered a significant difference of protein 3D structures64.
Prediction of candidate PLS-type PPR editing factors and PPR-RNA binding
PPRFinder95,96 (https://ppr.plantenergy.uwa.edu.au) was used to search PLS-type PPR genes and motifs. PPR codes were predicted by PPRCODE97 (https://colab.research.google.com/github/YaoYinYing/PPRCODE_Guideline/blob/master/PPRCODE.ipynb). Motif gaps in PLS-type PPR genes were filled with “inferred motifs”95, and “##” is taken as pseudo-PPR codes of inferred motifs. PPR proteins with fewer than six motifs are filtered. Furthermore, subcellular localization of PPR proteins was predicted by TargetP 2.098 (https://services.healthtech.dtu.dk/services/TargetP-2.0), WoLF PSORT99 (https://wolfpsort.hgc.jp), iPSORT100 (https://ipsort.hgc.jp), Plant-mPLoc101 (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi), Predotar102 (https://urgi.versailles.inra.fr/predotar) and the reported subcellular localization predicted by TargetP 1.1103,104. For each PPR protein, the top 10% predictions generated by TargetP 2.0 and WoLF PSORT, and the top 20% predictions generated by Predotar were retained, respectively. Only those predictions with the same subcellular localizations implemented by at least two aforementioned tools were kept for subsequent analysis. The binding of PPR-RNA was predicted and scored based on PPRmatcher105. The binding score greater than or equal to 1 in mitochondria and 2 in plastids is retained. BLASTP (specific parameters: -evalue 1e-5 -max_hsps 1) was used to detect the similarity of PPR genes and RNA editing factors106.
Statistics and reproducibility
The statistical significance of differences between RNA editing frequencies/variabilities of sites in different genomic regions was evaluated by the two-sided Wilcoxon’s rank-sum test. The p value was adjusted by the Bonferroni correction. A p value less than 0.05 was considered statistically significant. Correlation between editing frequency of CDS-recoding sites and evolutionary conservation of amino acids was calculated by the Spearman’s correlation coefficient and the fitted curve was implemented by Generalized Linear Models. 214 CDS-recoding sites in five developmental stages of rice endosperm were clustered by the K-means clustering algorithm. Five rice endosperm samples were collected across five successive developmental stages (3, 6, 9, 12, and 15 DAF). The sample at each stage was used to conduct RNA-seq (polyA and Ribo-Zero). In addition, RNA editing sites identified at five developmental stages were mutually confirmed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the source data can be found in the “Supplementary Information” file and the “Supplementary Data 1-16” file associated with the manuscript. All other data in this manuscript are available from the corresponding author (or other sources, as applicable) on reasonable request. The raw re-sequencing genome of rice and RNA-seq data of rice endosperm at 3, 6, 9, 12, and 15 DAF were deposited in the Genome Sequence Archive (GSA: CRA008750)107 in the National Genomics Data Center (NGDC), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences108. The RNA-seq data of rice seedling was obtained from GSA (CRA011887)109. Data of 3D structures of proteins encoded by mitochondrial genes before and after RNA editing were deposited in the Open Archive for Miscellaneous Data (OMIX: OMIX006521)107.
Code availability
Computational scripts for RNA editing analysis are available at GitHub (https://github.com/Bioinfo-Ming/RiceEndospermRNAEditing) and Zenodo110. Trimmomatic (version 0.35), HISAT2 (version 2.2.1), stringTie, REDItools (version 1.0.3) (specific parameters: -c 30,30 -e -E -d -D -u -U -l -L -V), ClustalW, MEGA11, MFold (version 2.3), ColabFold, SuperPose, PPRFinder, PPRCODE, TargetP (version 2.0), WoLF PSORT, iPSORT, Plant-mPLoc, Predotar (version 1.04), PPRmatcher (https://github.com/ian-small/PPRmatcher), and BLASTP (BLAST 2.12.0+) (specific parameters: -evalue 1e-5 -max_hsps 1) are used for data analysis in our study.
References
Zhao, M., Lin, Y. & Chen, H. Improving nutritional quality of rice for human health. Theor. Appl. Genet. 133, 1397–1413 (2020).
Zhou, S. et al. Transcriptional and post‐transcriptional regulation of heading date in rice. N. Phytologist 230, 943–956 (2021).
Chen, H., He, H., Zhou, F., Yu, H. & Deng, X. W. Development of genomics-based genotyping platforms and their applications in rice breeding. Curr. Opin. Plant Biol. 16, 247–254 (2013).
An, L. et al. Embryo-endosperm interaction and its agronomic relevance to rice quality. Front. Plant Sci. 11, 587641 (2020).
Zhou, S.-R., Yin, L.-L. & Xue, H.-W. Functional genomics based understanding of rice endosperm development. Curr. Opin. Plant Biol. 16, 236–246 (2013).
Hoshikawa, K. Anthesis, fertilization and development of caryopsis. In Science of the Rice Plant. Vol. I. Morphology, T. Matsuo and K. Hoshikawa, eds (Tokyo: Nobunkyo), pp. 339–376. (1993).
Matsui, T., Kobayasi, K., Yoshimoto, M., Hasegawa, T. & Tian, X. Dependence of pollination and fertilization in rice (Oryza sativa L.) on floret height within the canopy. Field Crops Res. 249, 107741 (2020).
Wu, X., Liu, J., Li, D. & Liu, C. M. Rice caryopsis development II: Dynamic changes in the endosperm. J. Integr. Plant Biol. 58, 786–798 (2016).
Domínguez, F. & Cejudo, F. J. Programmed cell death (PCD): an essential process of cereal seed development and germination. Front. Plant Sci. 5, 366 (2014).
Møller, I. M., Rasmusson, A. G. & Van Aken, O. Plant mitochondria–past, present and future. Plant J. 108, 912–959 (2021).
Farooq, M. A., Zhang, X., Zafar, M. M., Ma, W. & Zhao, J. Roles of reactive oxygen species and mitochondria in seed germination. Front. Plant Sci. 12, 781734 (2021).
Blokhina, O. & Fagerstedt, K. V. Reactive oxygen species and nitric oxide in plant mitochondria: origin and redundant regulatory systems. Physiologia Plant. 138, 447–462 (2010).
Wu, J. et al. Deficient plastidic fatty acid synthesis triggers cell death by modulating mitochondrial reactive oxygen species. Cell Res. 25, 621–633 (2015).
Giegé, P., Grienenberger, J. & Bonnard, G. Cytochrome c biogenesis in mitochondria. Mitochondrion 8, 61–73 (2008).
Kim, M. et al. Mitochondria-associated hexokinases play a role in the control of programmed cell death in Nicotiana benthamiana. Plant Cell 18, 2341–2355 (2006).
Elena‐Real, C. A. et al. Proposed mechanism for regulation of H2O2‐induced programmed cell death in plants by binding of cytochrome c to 14‐3‐3 proteins. Plant J. 106, 74–85 (2021).
Sultan, L. D. et al. The reverse transcriptase/RNA maturase protein MatR is required for the splicing of various group II introns in Brassicaceae mitochondria. Plant Cell 28, 2805–2829 (2016).
Brown, G. G., Colas des Francs-Small, C. & Ostersetzer-Biran, O. Group II intron splicing factors in plant mitochondria. Front. Plant Sci. 5, 35 (2014).
Ghifari, A. S., Saha, S. & Murcha, M. W. The biogenesis and regulation of the plant oxidative phosphorylation system. Plant Physiol. 192, 728 (2023).
Janska, H. & Kwasniak, M. Mitoribosomal regulation of OXPHOS biogenesis in plants. Front. Plant Sci. 5, 79 (2014).
Covello, P. S. & Gray, M. W. RNA editing in plant mitochondria. Nature 341, 662–666 (1989).
Hiesel, R., Wissinger, B., Schuster, W. & Brennicke, A. RNA editing in plant mitochondria. Science 246, 1632–1634 (1989).
Bonnard, G., Gualberto, J. M., Lamattina, L., Grienenberger, J. M. & Brennlcke, A. RNA editing in plant mitochondria. Crit. Rev. Plant Sci. 10, 503–524 (1992).
Takenaka, M., Jörg, A., Burger, M. & Haag, S. RNA editing mutants as surrogates for mitochondrial SNP mutants. Plant Physiol. Biochem. 135, 310–321 (2019).
Lu, Y. RNA editing of plastid-encoded genes. Photosynthetica 56, 48–61 (2018).
Chateigner-Boutin, A.-L. & Small, I. Plant RNA editing. RNA Biol. 7, 213–219 (2010).
Okuda, K. et al. Quantitative analysis of motifs contributing to the interaction between PLS‐subfamily members and their target RNA sequences in plastid RNA editing. Plant J. 80, 870–882 (2014).
Small, I. D., Schallenberg‐Rüdinger, M., Takenaka, M., Mireau, H. & Ostersetzer‐Biran, O. Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056 (2020).
Andrés-Colás, N. et al. Multiple PPR protein interactions are involved in the RNA editing system in Arabidopsis mitochondria and plastids. Proc. Natl Acad. Sci. 114, 8883–8888 (2017).
Guillaumot, D. et al. Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl Acad. Sci. 114, 8877–8882 (2017).
Wang, Y. et al. Maize PPR-E proteins mediate RNA C-to-U editing in mitochondria by recruiting the trans deaminase PCW1. Plant Cell 35, 529–551 (2023).
Wang, Y. et al. Multiple factors interact in editing of PPR-E+-targeted sites in maize mitochondria and plastids. Plant Commun. 5(5), 100836 (2024).
Yang, Y.-Z. et al. GRP23 plays a core role in E-type editosomes via interacting with MORFs and atypical PPR-DYWs in Arabidopsis mitochondria. Proc. Natl Acad. Sci. 119, e2210978119 (2022).
Yang, J. et al. Maize PPR278 functions in mitochondrial RNA splicing and editing. Int. J. Mol. Sci. 23, 3035 (2022).
Yang, H. et al. Rice FLOURY ENDOSPERM22, encoding a pentatricopeptide repeat protein, is involved in both mitochondrial RNA splicing and editing and is crucial for endosperm development. J. Integr. Plant Biol. 65, 755–771 (2023).
Lan, J. et al. Young Leaf White Stripe encodes a P‐type PPR protein required for chloroplast development. J. Integr. Plant Biol. 65, 1687–1702 (2023).
Leu, K.-C., Hsieh, M.-H., Wang, H.-J., Hsieh, H.-L. & Jauh, G.-Y. Distinct role of Arabidopsis mitochondrial P-type pentatricopeptide repeat protein-modulating editing protein, PPME, in nad1 RNA editing. RNA Biol. 13, 593–604 (2016).
Sun, J., Tian, Y., Lian, Q. & Liu, J.-X. Mutation of DELAYED GREENING1 impairs chloroplast RNA editing at elevated ambient temperature in Arabidopsis. J. Genet. Genomics 47, 201–212 (2020).
Zhang, H.-D. et al. PPR protein PDM1/SEL1 is involved in RNA editing and splicing of plastid genes in Arabidopsis thaliana. Photosynthesis Res. 126, 311–321 (2015).
Pyo, Y. J., Kwon, K.-C., Kim, A. & Cho, M. H. Seedling Lethal1, a pentatricopeptide repeat protein lacking an E/E+ or DYW domain in Arabidopsis, is involved in plastid gene expression and early chloroplast development. Plant Physiol. 163, 1844–1858 (2013).
Toma-Fukai, S. et al. Structural insight into the activation of an Arabidopsis organellar C-to-U RNA editing enzyme by active site complementation. Plant Cell 35, 1888–1900 (2023).
Yan, J., Zhang, Q. & Yin, P. RNA editing machinery in plant organelles. Sci. China Life Sci. 61, 162–169 (2018).
Sosso, D. et al. PPR2263, a DYW-subgroup pentatricopeptide repeat protein, is required for mitochondrial nad5 and cob transcript editing, mitochondrion biogenesis, and maize growth. Plant Cell 24, 676–691 (2012).
Dai, D. et al. Maize pentatricopeptide repeat protein DEK53 is required for mitochondrial RNA editing at multiple sites and seed development. J. Exp. Bot. 71, 6246–6261 (2020).
Wang, G. et al. E+ subgroup PPR protein defective kernel 36 is required for multiple mitochondrial transcripts editing and seed development in maize and Arabidopsis. N. Phytologist 214, 1563–1578 (2017).
Zang, J., Zhang, T., Zhang, Z., Liu, J. & Chen, H. DEFECTIVE KERNEL 56 functions in mitochondrial RNA editing and maize seed development. Plant Physiol. 194,1593–1610 (2024).
Sun, F. et al. Empty pericarp7 encodes a mitochondrial E-subgroup pentatricopeptide repeat protein that is required for ccmFN editing, mitochondrial function and seed development in maize. Plant J. 84, 283–295 (2015).
Yang, Y. Z. et al. The pentatricopeptide repeat protein EMP9 is required for mitochondrial ccmB and rps4 transcript editing, mitochondrial complex biogenesis and seed development in maize. N. Phytologist 214, 782–795 (2017).
Ding, S. et al. SMK6 mediates the C-to-U editing at multiple sites in maize mitochondria. J. Plant Physiol. 240, 152992 (2019).
Ren, R. C. et al. Pentatricopeptide repeat protein DEK40 is required for mitochondrial function and kernel development in maize. J. Exp. Bot. 70, 6163–6179 (2019).
Wang, Y. et al. Empty Pericarp21 encodes a novel PPR-DYW protein that is required for mitochondrial RNA editing at multiple sites, complexes I and V biogenesis, and seed development in maize. PLoS Genet. 15, e1008305 (2019).
Li, X. J. et al. Small kernel 1 encodes a pentatricopeptide repeat protein required for mitochondrial nad7 transcript editing and seed development in maize (Zea mays) and rice (Oryza sativa). Plant J. 79, 797–809 (2014).
Liu, Y.-J., Xiu, Z.-H., Meeley, R. & Tan, B.-C. Empty pericarp5 encodes a pentatricopeptide repeat protein that is required for mitochondrial RNA editing and seed development in maize. Plant Cell 25, 868–883 (2013).
Kim, S. R. et al. Rice OGR1 encodes a pentatricopeptide repeat–DYW protein and is essential for RNA editing in mitochondria. Plant J. 59, 738–749 (2009).
Li, M. et al. Plant editosome database: a curated database of RNA editosome in plants. Nucleic Acids Res. 47, D170–D174 (2019).
Notsu, Y. et al. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445 (2002).
Wu, C. S. & Chaw, S. M. Evolution of mitochondrial RNA editing in extant gymnosperms. Plant J. 111, 1676–1687 (2022).
Maldonado, M., Abe, K. M. & Letts, J. A. A structural perspective on the RNA editing of plant respiratory complexes. Int. J. Mol. Sci. 23, 684 (2022).
Ren, R. C. et al. The novel E-subgroup pentatricopeptide repeat protein DEK55 is responsible for RNA editing at multiple sites and for the splicing of nad1 and nad4 in maize. BMC Plant Biol. 20, 1–15 (2020).
Fan, K. et al. Maize defective kernel605 encodes a canonical DYW-Type PPR protein that edits a conserved site of nad1 and is essential for seed nutritional quality. Plant Cell Physiol. 61, 1954–1966 (2020).
Zhao, J. et al. EMP80 mediates the C‐to‐U editing of nad7 and atp4 and interacts with ZmDYW2 in maize mitochondria. N. Phytologist 234, 1237–1248 (2022).
Liu, X.-Y. et al. ZmPPR26, a DYW-type pentatricopeptide repeat protein, is required for C-to-U RNA editing at atpA-1148 in maize chloroplasts. J. Exp. Bot. 72, 4809–4821 (2021).
Yura, K. & Go, M. Correlation between amino acid residues converted by RNA editing and functional residues in protein three-dimensional structures in plant organelles. BMC Plant Biol. 8, 1–11 (2008).
Bolhuis, P. Sampling kinetic protein folding pathways using all-atom models. Lect. Notes Phys. 703, 393–433 (2006).
Xu, C. et al. DEK46 performs C‐to‐U editing of a specific site in mitochondrial nad7 introns that is critical for intron splicing and seed development in maize. Plant J. 103, 1767–1782 (2020).
Jestin, J. L., Dème, E. & Jacquier, A. Identification of structural elements critical for inter–domain interactions in a group II self‐splicing intron. EMBO J. 16(10), 2945–2954 (1997).
Costa, M. & Michel, F. O. Frequent use of the same tertiary motif by self‐folding RNAs. EMBO J. 14, 1276–1285 (1995).
Ichinose, M. & Sugita, M. RNA editing and its molecular mechanism in plant organelles. Genes 8, 5 (2016).
Duan, Y., Tang, X. & Lu, J. Evolutionary driving forces of A‐to‐I editing in metazoans. Wiley Interdiscip. Rev. RNA 13, e1666 (2022).
Wang, W., Wu, Y. & Messing, J. Genome-wide analysis of pentatricopeptide-repeat proteins of an aquatic plant. Planta 244, 893–899 (2016).
Lurin, C. et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103 (2004).
Loiacono, F. V. et al. Emergence of novel RNA-editing sites by changes in the binding affinity of a conserved PPR protein. Mol. Biol. Evol. 39, msac222 (2022).
Sheahan, T. & Wieden, H.-J. Ribosomal protein S1 improves the protein yield of an in vitro reconstituted cell-free translation system. ACS Synth. Biol. 11, 1004–1008 (2022).
Fargo, D. C., Boynton, J. E. & Gillham, N. W. Chloroplast ribosomal protein S7 of Chlamydomonas binds to chloroplast mRNA leader sequences and may be involved in translation initiation. Plant Cell 13, 207–218 (2001).
Cukras, A. R., Southworth, D. R., Brunelle, J. L., Culver, G. M. & Green, R. Ribosomal proteins S12 and S13 function as control elements for translocation of the mRNA: tRNA complex. Mol. Cell 12, 321–328 (2003).
Liu, R. et al. The DYW-subgroup pentatricopeptide repeat protein PPR27 interacts with ZmMORF1 to facilitate mitochondrial RNA editing and seed development in maize. J. Exp. Bot. 71, 5495–5505 (2020).
Takatsuka, A., Kazama, T. & Toriyama, K. Cytoplasmic male sterility-associated mitochondrial gene orf312 derived from rice (Oryza sativa L.) cultivar Tadukan. Rice 14, 1–11 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Hiratsuka, J. et al. The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. MGG 217, 185–194 (1989).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1–10 (2013).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Picardi, E. & Pesole, G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814 (2013).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2005).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–D42 (2012).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Robart, A. R., Chan, R. T., Peters, J. K., Rajashankar, K. R. & Toor, N. Crystal structure of a eukaryotic group II intron lariat. Nature 514, 193–197 (2014).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
UniProt: the Universal Protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Maiti, R., Van Domselaar, G. H., Zhang, H. & Wishart, D. S. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594 (2004).
Cheng, S. et al. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 85, 532–547 (2016).
Gutmann, B. et al. The expansion and diversification of pentatricopeptide repeat RNA-editing factors in plants. Mol. Plant 13, 215–230 (2020).
Yan, J. et al. Delineation of pentatricopeptide repeat codes for target RNA prediction. Nucleic Acids Res. 47, 3728–3738 (2019).
Armenteros, J. J. A. et al. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2, e201900429 (2019).
Horton, P. et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 (2007).
Bannai, H., Tamada, Y., Maruyama, O., Nakai, K. & Miyano, S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 18, 298–305 (2002).
Chou, K.-C. & Shen, H.-B. Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PloS One 5, e11335 (2010).
Small, I., Peeters, N., Legeai, F. & Lurin, C. Predotar: a tool for rapidly screening proteomes for N‐terminal targeting sequences. Proteomics 4, 1581–1590 (2004).
Chen, G., Zou, Y., Hu, J. & Ding, Y. Genome-wide analysis of the rice PPR gene family and their expression profiles under different stress treatments. BMC Genomics 19, 1–14 (2018).
Emanuelsson, O., Nielsen, H., Brunak, S. & Von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 (2000).
Gutmann, B., Millman, M., Vincis Pereira Sanglard, L., Small, I. & Colas des Francs-Small, C. The pentatricopeptide repeat protein MEF100 is required for the editing of four mitochondrial editing sites in Arabidopsis. Cells 10, 468 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 1–9 (2009).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteom. Bioinforma. 19, 578–583 (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res. 52, D18–D32 (2024).
Liu, K., Xie, B., Peng, L., Wu, Q. & Hu, J. Profiling of RNA editing events in plant organellar transcriptomes with high‐throughput sequencing. Plant J. 118, 345–357 (2024).
Ming. Bioinfo-Ming/RiceEndospermRNAEditing: First release of codes in rice endosperm RNA editing analyses (v1.0.0). Zenodo https://doi.org/10.5281/zenodo.13767801 (2024).
Acknowledgements
These authors would like to thank Lili Hao, Shuhui Song, Lina Ma, Shuai Jiang, Dong Zou, Lin Liu, Qianpeng Li, Guangyi Niu, Zhao Li, Xiaonan Liu, Yang Zhang, Tongtong Zhu, Xing Zheng, Rong Pan, Sicheng Luo, Wei Jing, Yue Qi, Xinyu Zhou, Yuxin Qin, Wenzhuo Cheng, Zhixiang Yuan and Zhenxian Han for discussions and advice. This work was supported by grants from National Natural Science Foundation of China [32030021] and International Partnership Program of the Chinese Academy of Sciences [153F11KYSB20160008].
Author information
Authors and Affiliations
Contributions
Z.Z., S.Y.W., and S.N.H. conceived, initiated and supervised this study. M.C. and L.X. wrote the manuscript. M.C., L.X., S.Y.W., X.Y.T., S.H.G., S.W., M.L., Y.S.Z., T.Y.X. and Y.C.C. analyzed the data. M.C., Y.Y.C, and Y.C. implemented the data curation. S.Y.W. provided the rice endosperm samples.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Toshifumi Tsukahara and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Hanyang Cai and David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, M., Xia, L., Tan, X. et al. Seeing the unseen in characterizing RNA editome during rice endosperm development. Commun Biol 7, 1314 (2024). https://doi.org/10.1038/s42003-024-07032-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07032-5
