
Abstract
According to WHO, plague, caused by Yersinia pestis, has resurged since 2000. Inner Mongolia, harboring a quarter of China’s plague foci, has accounted for 80% of national plague cases in the past five years. Despite its pivotal role in Chinese plague epidemiology, the genetic diversity and transmission dynamics of Y. pestis in this region remain under-investigated. Our analysis of 585 Y. pestis strains from Inner Mongolia (1948–2021) revealed three primary lineages, with 2.MED3 being predominant. We further delineated seven sub-phylogroups in 2.MED3, with 2.MED3.1.2 and 2.MED3.1.4 showing recent dominance. These two subgroups reveal dual transmission patterns: localized short-distance spread and long-distance dispersals over 300 km. Xilingol League is highlighted as a key source and reservoir for Y. pestis, predominantly spreading from central-eastern to southwestern Inner Mongolia, including occasional reverse transmissions. These findings enhance understanding of Y. pestis diversity and transmission in Inner Mongolia, aiding in enhanced surveillance and control measures.
Similar content being viewed by others


Introduction
Plague, caused by the bacterium Yersinia pestis1, is a severe infectious disease, predominantly spread through flea bites and respiratory droplets. It manifests in several clinical forms1, notably bubonic, pneumonic, and septicemic plague. Historically responsible for three pandemics, plague has claimed over 100 million lives1,2,3. Recent decades have witnessed a significant resurgence in plague cases worldwide, raising substantial concern within the global health community4.
There are 12 identified natural plague foci in China, of which the Inner Mongolia Autonomous Region is particularly significant, containing four principal foci5,6: Marmota sibirica plague focus, Microtus brandti plague focus, Spermophilus dauricus plague focus, and Meriones unguiculatus plague focus. Since 2019, Inner Mongolia has become a hotspot for plague outbreaks. A pronounced incident in November 2019, where two individuals contracted pneumonic plague from the Meriones unguiculatus plague focus, led to the quarantine of 447 individuals in Beijing, triggering widespread concern7. Over the following years, the region and neighboring Mongolia have also reported multiple cases8,9,10,11,12, including fatalities and the spread to nearby provinces such as the Ningxia Hui Autonomous Region (NX). Surveillance data from 2018 to 2021 confirms ongoing plague transmission among wildlife13,14, highlighting the region’s pivotal role in public health monitoring and genomic epidemiological studies of plague.
Understanding the genetic diversity and transmission dynamics of Y. pestis is essential for the development of effective surveillance and control strategies. Previous research on Y. pestis in Inner Mongolia has primarily utilized molecular genetic techniques like differential fragment analysis (DFR)15, multiple loci variable-number tandem-repeat analysis (MLVA)16,17, and clustered regularly interspaced short palindromic repeats (CRISPR)18, targeting specific genomic regions. While these methods provide valuable insights, their resolution is limited by the absence of whole-genome variation analysis. Moreover, the pursuit of in-depth genomic studies on the population structure and spread patterns of Y. pestis in the region has been impeded by a scarcity of publicly accessible whole-genome data and insufficient sample representativeness7,8,9,19.
To address these gaps, we analyze 585 whole-genome sequences of Y. pestis collected from Inner Mongolia over more than seven decades of surveillance, from 1948 to 2021. Our study reveals the bacterium’s genetic diversity, spatiotemporal distribution, and transmission dynamics in the region, offering novel insights into its evolutionary trajectories.
Results
Genetic diversity and spatiotemporal distribution of Y. pestis in Inner Mongolia
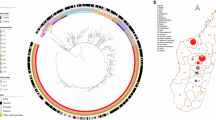
In our comprehensive analysis of 585 Y. pestis genomes from Inner Mongolia and 1436 from other regions or countries (See “Methods” and Supplementary Data 1, 2), we identified 8246 SNPs (See Supplementary Data 3) and employed maximum likelihood (ML) methods to reconstruct their phylogenetic relationships. According to the phylogeny, we observed a pronounced dominance of three primary phylogroups in Inner Mongolia: 2.MED3 (74.53%, 436/585), 0.PE4 (12.99%, 76/585), and 2.ANT3 (11.45%, 67/585), with occasional occurrences in the 1.IN2 (0.68%, 4/585), 0.ANT1 (0.17%, 1/585), and 1.ORI1 (0.17%, 1/585) phylogroups (Fig. 1).

A maximum likelihood tree was constructed based on 8246 SNP sites identified from the core genome (present in 95% of strains) of 2021 Y. pestis genomes, including 585 genomes from Inner Mongolia and 1436 from other regions or countries. The innermost ring denotes the phylogroup, followed by the isolation location (Geographic area), the isolation time (Year). The phylogroups associated with Inner Mongolia strains are highlighted in shaded areas, with colors and labels corresponding to each group. To expand the definition of the rest of the tree, the very long branch containing the Angola strain (0.PE3) was truncated.
Our investigation into the spatial and temporal distributions of these leading phylogroups revealed distinct clustering patterns (Supplementary Fig. 1). Specifically, the 0.PE4 group is primarily found in the Xilingol League (Xlglm), a region characterized by the Microtus brandti plague focus, which also encompasses zones overlapping with the Meriones unguiculatus plague focus, while 2.ANT3 is mainly localized to the Spermophilus dauricus plague focus. Importantly, the 2.MED3 phylogroup, currently the most widespread, spans from central to southwestern Inner Mongolia, particularly within the plague foci of Meriones unguiculatus and Microtus brandti (Fig. 2 and Supplementary Fig. 2). Moreover, while 2.ANT3 dominated the Inner Mongolia’s Y. pestis population until 1970, the 2.MED3 group has since taken precedence, with all strains isolated since 2018 exclusively belonging to 2.MED3, confirming its dominance in the region (Supplementary Fig. 3).

The map outlines the administrative subdivisions at the county level within the region (sourced from https://datav.aliyun.com/portal/school/atlas/area_selector), with varying fill colors indicating distinct natural plague foci. Circles on the map, differentiated by fill color, denote the three major phylogenetic groups, while their size reflects the number of strains. An inset provides a global context, highlighting the location of the Inner Mongolia Autonomous Region.
Genetic diversity in Inner Mongolia’s dominant 2.MED3 phylogroup
With the 2.MED3 lineage being the predominant Y. pestis population in Inner Mongolia, our study analyzed 775 sequences from this lineage, encompassing 436 strains from Inner Mongolia and others from adjacent regions. Utilizing CO92 as both outgroup and reference, we detected 536 SNPs, enabling us to delineate the phylogenetic structure of the 2.MED3 lineage into seven subgroups: from 2.MED3.1.1 to 2.MED3.1.6, plus 2.MED3.2 (Fig. 3a and Supplementary Fig. 4a). The intra-subgroup SNP genetic distance averaged below 11 SNPs (Fig. 3b). Notably, over half of the Inner Mongolian isolates fell within the 2.MED3.1.2 subgroup (55.73%), while the remaining isolates were distributed among the 2.MED3.1.4 (16.97%), 2.MED3.1.5 (13.53%), and 2.MED3.1.6 (11.24%) subgroups. In comparison, the 2.MED3.1.1 and 2.MED3.1.3 subgroups were scarcely represented, with merely 7 and 4 strains, respectively. Notably, 2.MED3.1.2 and 2.MED3.1.4 are the predominant circulating subgroups in Inner Mongolia from 2018 to 2021 (Supplementary Fig. 4b).

a A maximum likelihood phylogenetic tree, derived from 536 SNPs within the core genome of 775 Y. pestis strains (436 from Inner Mongolia and 339 from adjacent regions), uses the CO92 chromosome as a reference and outgroup. The tree, visualized through GrapeTree, features circles whose sizes correspond to strain counts and color intensities that reflect isolation date—darker shades denote more recent isolations. b A ridge plot illustrates the pairwise SNP genetic distances among strains within the 2.MED3 subgroups. Median distances are highlighted by white vertical lines.
Transmission dynamics in Inner Mongolia’s dominant 2.MED3 phylogroup
Given the significant presence and recent dominance of the 2.MED3.1.2 and 2.MED3.1.4 subgroups within Inner Mongolia’s Y. pestis strains, our study delved into their transmission dynamics across Inner Mongolia and its bordering provinces, including Ningxia (NX), Shaanxi (SX), and Hebei (HB), as well as Zamyn-Uud (ZMWD), Mongolia, from 1970 to 2021. Analyzing 389 strains of the 2.MED3.1.2 subgroup, we identified a strong temporal signal (correlation coefficient = 0.8691, R squared = 0.7554) (Supplementary Fig. 5a), indicating a well-defined lineage evolution over time. Our Bayesian phylogeographic analysis estimated time to most recent common ancestor (tMRCA) of this subgroup around 1966 (95% HPD, 1962–1969), with Xlglm in central-eastern Inner Mongolia identified as the probable origin (location probability = 76.2%) (Fig. 4a). Additionally, our analysis also identifies Xlglm as a pivotal hub within the 2.MED3.1.2 subgroup’s transmission network, marked by the highest migration activity, with seven instances of inward migration and eight of outward migration (Fig. 4b, c). Focusing on the eight outward spread events, we found that it initiated with its southward movement from Xlglm to Kangbao County, HB, through three individual transmissions between 1969 and 1989 (Fig. 4a, b). The tMRCA of Y. pestis strains from Kangbao County dates back to 1969 (95% HPD, 1969–1970), aligning with its recognition as a plague focus in 197120. Our result implies that the emergence of the plague focus in Kangbao County likely resulted from strains introduced from Xlglm. In parallel, Y. pestis spread eastward from Xlglm to Chifeng City (Cfs) in 1968 (95% HPD, 1966–1969), and southwestward to Ulanqab City (Wlcbs) in 1976 (95% HPD, 1973–1979). Remarkably, a similar dispersal from Xlglm to Wlcbs was observed again 43 years later, in 2019 (95% HPD, 2018–2019). Similar to Wlcbs, Bayannur City (Bynes) also experienced two introductions from Xlglm, occurring respectively in 1983 (95% HPD, 1980–1986) and 2019 (95% HPD, 2018–2019), with the first instance further spreading to NX.

a The maximum clade credibility tree of the Y. pestis 2.MED3.1.2 subgroup, based on 143 SNPs, with nodes color-coded by sampling locations. The inset in the top-left corner shows the posterior probabilities for the inferred source locations. Geographic abbreviations for strain isolation locations are detailed as follows: Uppercase abbreviations denote provinces, including the Inner Mongolia Autonomous Region (NMG) and neighboring provinces of Ningxia Hui Autonomous Region (NX), Shaanxi Province (SX), and Hebei Province (HB) in China, along with Zamyn-Uud (ZMWD) in Mongolia’s Dornogovi Province. Lowercase abbreviations denote cities within Inner Mongolia, including Xilingol League (Xlglm), Ulanqab City (Wlcbs), Chifeng City (Cfs), Baotou City (Bts), Hohhot City (Hhhts), BayanNur City (Bynes), Erdos City (Eedss). b Transmission dynamics of the 2.MED3.1.2 subgroup, with the direction of spread indicated by arrows. Dashed and solid lines differentiate between long-distance and short-distance transmissions, respectively. Region labels are shaded with colors corresponding to those in panel (a). Color-coded lines represent the timeline of spread, with darker colors denoting more recent events. An additional inset in the bottom-right provides a full map of Inner Mongolia and adjacent provinces. c Migration histogram for the 2.MED3.1.2 subgroup, showing both inward and outward migrations for highlighted regions.
Further analysis of the transmission dynamics revealed two distinct patterns for Y. pestis strains in the 2.MED3.1.2 subgroup: localized short-distance spread through adjacent territories (i.e., spillover of Y. pestis between neighboring areas) and long-distance leapfrog dispersal, characterized by the appearance of genetically identical strains in non-adjacent cities separated by distances greater than several hundred kilometers (>300 km in this study). We identified 23 instances of localized spread and seven significant long-distance dispersals within the 2.MED3.1.2 strains, showing the overall direction of transmission was from the central-eastern to the southwestern parts of Inner Mongolia, alongside several reverse spread events. Notably, among the seven long-distance dispersal events, four prominently drove the spread from the central-eastern regions to the southwest. The remaining instances included one dispersal from NX to Wlcbs and two from Bynes to Xlglm and Wlcbs, showing leapfrog dispersals from the southwest back to the central-eastern parts (Fig. 4b).
For the sub-dominant group 2.MED3.1.4, we examined 73 strains from it and constructed a ML tree based on 104 core genome SNPs. Notably, similar to the 2.MED3.1.2 subgroup, the 2.MED3.1.4 subgroup also shows a temporal signal (Supplementary Fig. 5b) and exhibits similar dissemination patterns, predominantly spreading from central-eastern to southwestern Inner Mongolia, accompanied by a few instances of reverse transmission. Phylogeographic analysis traced the subgroup’s tMRCA to around 1943 (95% HPD, 1916–1965), with the geographically adjacent Wlcbs and Xlglm identified as potential origins (location probabilities of 42% and 38%, respectively) (Supplementary Fig. 6). One of the two parallel sub-branches, 2.MED3.1.4.1, spread from Xlglm through Wlcbs, Bts, and Bynes to Eedss, indicating a spread from central-eastern Inner Mongolia toward the southwest. Meanwhile, another parallel sub-branch 2.MED3.1.4.2, originating in Wlcbs in 1975 (95% HPD, 1966–1983), spread southwest to NX, and later a reverse migration occurred in 1991 (95% HPD, 1986–1995), extending toward Eedss and finally reaching Wuhai (Whs).
Discussion
Through extensive whole-genome sequencing and genomic epidemiological analysis of Y. pestis strains from long-term surveillance in Inner Mongolia, we have elucidated the genomic diversity of Y. pestis in the region. Our research uncovers significant temporal and geographic clustering among the dominant clades in Inner Mongolia, namely the 2.MED3, 2.ANT3, and 0.PE4 phylogroups. The 2.ANT3 and 0.PE4 clades, in particular, were mainly found in the natural plague foci of Spermophilus dauricus and Microtus brandti, with a marked prevalence from 1949 to 1970. Post-1970, the 2.MED3 lineage emerged as the leading phylogroup in Inner Mongolia, predominantly occurring within the Meriones unguiculatus natural plague focus (Supplementary Fig. 2). Based on previous studies19, the divergence between the 2.MED and 2.ANT phylogroups occurred approximately 340–675 years ago, with the current-dominant 2.MED3 lineage in Inner Mongolia dating back to a common ancestor only 150–385 years ago, indicating ongoing diversification and turnover in the region’s Y. pestis populations. The common ancestor of the three main lineages dates back to 1216–2954 years ago, indicating a long history of Y. pestis activity in Inner Mongolia, likely due to the local natural environment conducive to its survival. The replacement of 2.ANT3 and 0.PE4 phylogroups by 2.MED3 strains suggests a shift in the activity of natural plague foci, as well as a population shift in the primary reservoir toward Meriones unguiculatus. This phenomenon is likely attributable to a combination of genomic mutations, ecological changes, host and vector dynamics, and human interventions. Further investigations into local natural plague foci are necessary to elucidate these factors.
The 2.MED3 lineage, dominant in Inner Mongolia, comprises seven sub-phylogroups, with six identified in the region. Before 2010, the dominant subgroups varied during different time periods (see Supplementary Fig. 4b), but notably, 2.MED3.1.2 repeatedly emerged as the dominant subgroup. After 2010, especially from 2018 to 2021, four sub-phylogroups faded, leaving 2.MED3.1.2 and 2.MED3.1.4 widely distributed in the natural focus of Meriones unguiculatus and sparsely present in the natural plague focus of Microtus brandti. The reasons for this population shift in the natural plague foci of Inner Mongolia remain unclear. Our analysis of subgroup-specific variations for 2.MED3.1.2 and 2.MED3.1.4 identified four SNPs with no clear impact on Y. pestis survival or virulence and no signs of natural selection, suggesting genetic drift as the main driver of population shifts (see “Methods” section and Supplementary Table 1).
Although the variable clock rate and slow substitution rate likely contribute to the generally weak temporal signals observed across the majority of contemporary phylogroups of Y. pestis19,21, high sampling density and extended temporal coverage may facilitate the detection of temporal signals in some Y. pestis populations, such as the 2.MED3.1.2 and 2.MED3.1.4 sub-phylogroups in our study. This enables us to perform BEAST phylogeographic analysis. The results suggest that the Xlglm is the likely source of the 2.MED3 lineage in Inner Mongolia and the origin of its two prominent circulating sublineages, 2.MED3.1.2 and 2.MED3.1.4. Moreover, this region has been the starting point for three significant long-distance transmissions. For instance, historical records identify Damao Banner in Bts as a plague focus since 197020, with our analysis tracing its earliest strains back to Xlglm from 1964 (95% HPD, 1955–1970), suggesting Xlglm’s strains initiated the plague, thereby establishing a stable focus. The southwestern part of the Xlglm, which borders the natural plague foci of Mongolia, encompasses extensive desert grasslands that provide a suitable habitat for Meriones unguiculatus22, facilitating the persistent prevalence of animal plague. The wide range of plague foci permits broad spatial transmission and continuous circulation among local hosts, which likely contributes to the region’s role as the origin of plague in Inner Mongolia. Our findings highlight the importance of focusing on surveillance and management of zoonotic plague outbreaks within the natural focus of the Xlglm as a priority for future plague prevention and control efforts.
While there are instances of transmission from the southwest to the middle east, the predominant spread of the plague in Inner Mongolia moves from the middle east to the southwest. This trend has generated several transmission waves, extending the plague from grasslands to more populated southern areas, posing a public health risk. Such pattern underscores the necessity of strengthening plague surveillance across Inner Mongolia.
In Inner Mongolia, plague dissemination exhibits a coexistence of localized short-distance and long-distance leapfrog transmission modes. Existing reports indicate that Y. pestis primarily spreads via vectors like fleas to nearby susceptible animals within natural foci, with transmission distance constrained by the animals’ mobility, typifying short-distance transmission4,17. For instance, originating from Xlglm, the plague expands into neighboring regions such as Wlcbs and HB and subsequently extends to Bts, Hhhts, and Bynes, illustrating a progressive spread to the surrounding area. Interestingly, this study revealed several instances of long-distance leapfrogging transmission in Inner Mongolia and its surrounding regions. Previous research by Girard et al. defined long-distance transmission as the spread of Y. pestis over significant distances (>10 km) and across habitats unsuitable for host animals23. In our study, we observed instances of strain diversity consistency or ancestral-descendant relationships over distances exceeding 300 km between non-adjacent regions within a short period, indicating that Y. pestis can achieve far-reaching cross-regional spread. Contrary to the “source-sink” theory, long-distance plague transmission in Inner Mongolia demonstrates that strains reaching new areas can proliferate and disperse extensively. For example, in events where strains transmitted from Xlglm to Bts and from Bynes to NX, they established, spread, and created local breeding populations.
Some scholars suggest that long-distance transmission of Y. pestis may be associated with human activities and migratory birds24,25,26,27. Our study identified thirteen long-distance transmission events from 1943 to 2021, with eight of these occurring within a time span of less than five years. We hypothesize that advancements in modern transportation have potentially facilitated the expansion of this ancient pathogen. For instance, railway and highway construction, along with afforestation for sand control and roadside protection, may alter the landscape, creating optimal habitats for rodents and thus increasing the risk of plague spread along transportation routes28,29. Additionally, the travel of infected patients can rapidly disseminate plague to remote locations. Notably, in 2019 and 2022, instances in Inner Mongolia involved individuals infected with the plague traveling across provincial borders for treatment, introducing the bacterium into new regions7. Despite the lack of subsequent secondary cases or wildlife reintroduction due to effective isolation and control measures, these incidents emphasize the notable public health implications of long-distance plague spread from Inner Mongolia. The repeated occurrence of such events challenges the effectiveness of traditional localized “rodent control and source elimination” strategies in blocking plague transmission. This situation necessitates further investigation into the routes and mechanisms of Y. pestis’s long-distance dispersal from Inner Mongolia and surrounding areas, aiming to provide scientific support for rigorous containment efforts.
In addition, early efforts at plague surveillance in China during the 1950s and 1960s were hindered by limited resources. Additionally, the unavailability of some strains due to unsuccessful revival or other reasons likely resulted in biased sampling in Inner Mongolia and adjacent provinces, potentially influencing the understanding of Y. pestis transmission dynamics. For example, this sampling bias might affect the estimated number of inward and outward migrations based on BEAST phylogeographic analysis30. It may also overlook populations that rapidly emerged and then went extinct, as well as misestimate the tMRCA of populations21. In future studies, a more comprehensive sampling set would help to further investigate the diversity and transmission patterns of Y. pestis populations with higher resolution.
Observations from this study, highlighting population shifts, a trend of spread from the central-eastern to the southwestern regions, and instances of long-distance leapfrog dispersals within a limited time span, suggest complex interactions between the plague pathogen and its natural environment. Advancing our understanding of Y. pestis’s adaptive evolution and the triggers for its outbreaks requires future research focused on collecting ecological data and conducting integrated ecological-evolutionary investigations. Such an informed approach is crucial for developing and refining effective plague management strategies.
Methods
Strain collection
Our dataset integrates 2021 Y. pestis genomes (Supplementary Data 1, 2), comprising 213 newly sequenced genomes from the Inner Mongolia Autonomous Region, 1030 globally diverse strains from the National Center for Biotechnology Information (NCBI) GenBank and SRA databases, and 778 strains from Inner Mongolia and seven adjacent provinces (Ningxia, Gansu, Shaanxi, Hebei, Jilin, Liaoning, Heilongjiang), publicly accessible through the China National Microbiology Data Center (NMDC, https://nmdc.cn/en). Specifically, 585 genomes were collected in Inner Mongolia between 1948 and 2021, spanning 10 cities and 31 counties.
The 213 newly sequenced strains were collected from 2018 to 2021 during routine monitoring in Inner Mongolia, following the National Scheme of Plague Surveillance of China (http://www.gov.cn/yjgl/2005-08/30/content_28245.htm) and the Inner Mongolia Scheme of Plague Surveillance (https://www.nmg.gov.cn/zwgk/zfgb/2020n/202016/202009/t20200903_307068.html). Positive samples were identified using indirect hemagglutination assays from dead rodents, biological specimens from individuals with clinically suspected plague, and routinely captured rodents along with their associated fleas. These positive samples were then processed to isolate Y. pestis strains.
Whole-genome sequencing and assembly
Stored Y. pestis strains were cultured on blood (rabbit) LB medium at 28 °C for 24 h. After three passages, their DNA was extracted using the Qiagen DNeasy Blood & Tissue Kit (No. 69506). Whole-genome sequencing was performed on the Illumina Novaseq 6000 and Ion GeneStudio S5 Plus platforms, yielding whole-genome data for 213 Y. pestis strains, with 193 having paired-end sequencing data (average depth of 222X) and 20 having single-end data (average depth of 62X). Raw sequencing data underwent quality control and trimming using Trimmomatic (v0.38) software to remove adapters and low-quality reads (average quality score <Q20)31. Paired-end data assembly was performed using shovill software (v1.0.1, https://github.com/tseemann/shovill), while single-end data assembly was conducted using SPAdes software (v3.13.0)32.
Variant calling and annotation
Whole-genome sequences of each Y. pestis strain were aligned to the reference genome utilizing the MUMmer software package (v3.23)33. For most analyses, the Y. pestis CO92 genome served as the reference, with the exception of in-depth phylogenetic analyses of subgroups 2.MED3.1.2 and 2.MED3.1.4, where strain 74002 from subgroup 2.MED3.2 was employed. We utilized the “core_ope” pipeline (available at https://sourceforge.net/projects/coreope/) to build a relaxed core genome (present in 95% of the strains in this study) from the MUMmer alignments. Repetitive regions identified using Tandem Repeats Finder (v4.07b)34 and BLAST (v2.12.0+, https://blast.ncbi.nlm.nih.gov/Blast.cgi) on the reference genome were removed from the core genome. The resulting non-repetitive core genome was used for SNP calling. The identified SNPs were further validated by aligning trimmed reads to reference genomes using BWA (v0.7.12)35 and calling variants with GATK’s HaplotypeCaller module (v4.2.4)36,37,38. This process yielded a high-quality SNP matrix, confirmed by both the assembly sequence and trimmed reads, requiring a sequencing depth greater than 10X and an allele frequency above 90% for inclusion in subsequent analyses.
Additionally, small insertions and deletions (Indels, shorter than 50 bp), were identified by aligning trimmed reads to the CO92 reference genome via BWA, followed by calling with GATK’s HaplotypeCaller. Different samples were aggregated using GATK’s CombineGVCFs and GenotypeGVCFs modules. Only Indels located in the core genome (excluding repetitive regions) were considered, while those supported by sequencing depth <10X and allele frequency <80% were excluded.
The SNP and Indel datasets were integrated and annotated using SnpEff software (v5.0e) against the CO92 reference to identify mutation types, gene names, and functions39. Variants unique to dominant subgroups (2.MED3.1.2 and 2.MED3.1.4), present in all strains of a specific subgroup but absent in others, were extracted. No indels unique to either subgroup 2.MED3.1.2 or 2.MED3.1.4 were identified.
Phylogenetic analysis and genetic distance calculation
Concatenated SNPs were used to create fasta sequences for analysis. Phylogenetic trees were then constructed with IQ-Tree software (v1.6.5)40, employing the GTR+G model with fast bootstrap set to 1000, and visualized using iTOL (https://itol.embl.de/) and Grapetree (v1.5.0)41. Pairwise SNP distances among subgroups were calculated using snp-dists (v0.8.2, https://github.com/tseemann/snp-dists) and visualized using ggplot2 in R (v4.1.3) to generate ridge plots42.
Temporal signal detection and phylogeographic analysis
We applied TempEst (v1.5.3) to detect temporal signals in strains from the 2.MED3.1.2 and 2.MED3.1.4 sub-phylogroups43, as these were the predominant subgroups in Inner Mongolia in recent years. Six strains from the 2.MED3.1.4 subgroup, identified as outliers through the Residuals module of TempEst software, were subsequently excluded from further analysis. Upon detecting temporal signals, BEAST software (v1.10.4) was utilized to analyze the spread dynamics and estimate the dates for the most recent common ancestors, based on concatenated SNP sequences alongside sampling dates and locations44. We used the GTR+Gamma model for nucleotide substitution, an uncorrelated relaxed clock for substitution rates45, a constant population size for tree priors, and Bayesian Stochastic Search Variable Selection (BSSVS) for inferring social networks. Independent Markov Chain Monte Carlo (MCMC) chains were executed for sub-phylogroup 2.MED3.1.2 over 600 million generations and for 2.MED3.1.4 over 300 million generations, with respective sampling frequencies of every 60,000 and 30,000 to guarantee chain convergence. Convergence was verified via Tracer (v1.7.2)46, with all key parameters achieving an effective sample size of over 200. TreeAnnotator (v1.10.4) was employed to generate maximum clade credibility (MCC) trees, excluding the initial 20% of states as burn-in for 2.MED3.1.2 and the initial 10% for 2.MED3.1.4. The resulting MCC trees were then visualized with FigTree (v1.4.4, http://tree.bio.ed.ac.uk/software/figtree/).
Statistics and reproducibility
We collected 2021 Y. pestis genomes, with the collection methods detailed in the “Strain Collection” section. Additionally, the software and models used for statistical analysis of these data are comprehensively explained in the “Methods” section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The whole-genome sequencing data from this study have been submitted to the China National Microbiology Data Center (NMDC, https://nmdc.cn/en) under the BioProject accession number NMDC10018743, with the strain background information available in Supplementary Data 1. The publicly available genomes were downloaded from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) and the NMDC under the BioProject accession number NMDC10018536, as detailed in Supplementary Data 2.
Code availability
No custom code was used in this study, and all software and their versions have been provided in “Methods”.
References
Perry, R. D. & Fetherston, J. D. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10, 35–66 (1997).
Achtman, M. et al. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl Acad. Sci. USA 96, 14043–14048 (1999).
Stenseth, N. C. et al. Plague: past, present, and future. PLoS Med. 5, e3 (2008).
Barbieri, R. et al. Yersinia pestis: the natural history of plague. Clin. Microbiol. Rev. 34, e00044–19 (2020).
Fang, X.-y et al. Ecological-geographic landscapes of natural plague foci in China VII. Typing of natural plague foci. Chin. J. Epidemiol. 33, 1144–1150 (2012).
Ben-Ari, T. et al. Identification of Chinese plague foci from long-term epidemiological data. Proc. Natl Acad. Sci. USA 109, 8196–8201 (2012).
Wang, Y. et al. Isolated cases of plague—Inner Mongolia-Beijing, 2019. China CDC Wkly. 1, 13–16 (2019).
Shen, X., Li, J., Fan, M., Xia, L. & Li, W. A reemergent case of bubonic plague—Inner Mongolia Autonomous Region, China, July, 2020. China CDC Wkly. 2, 549–550 (2020).
Gao, J. et al. Human plague case diagnosed in Ningxia tracked to animal reservoirs—Inner Mongolia Autonomous Region, China, 2021. China CDC Wkly. 3, 1109–1112 (2021).
National Disease Control and Prevention Administration. The Overview of Notifiable Infectious Diseases Nationwide in July 2022. http://www.nhc.gov.cn/jkj/s3578/202208/f470f0abf544436d901c2660b06f3911.shtml (2022).
National Disease Control and Prevention Administration. The Overview of Notifiable Infectious Diseases Nationwide in August 2023. https://www.ndcpa.gov.cn/jbkzzx/c100016/common/content/content_1706568814483075072.html (2023).
National Disease Control and Prevention Administration. The Overview of Notifiable Infectious Diseases Nationwide in 2020. http://www.nhc.gov.cn/jkj/s3578/202103/f1a448b7df7d4760976fea6d55834966.shtml (2020).
Han, B. et al. An epidemiological survey of animal plague in Meriones unguiculatus plague foci of Inner Mongolia Autonomous Region, China, 2018–2022. Chin. J. Vector Biol. Control 34, 697–702 (2023).
Zhang, Z., Li, J., Liu, F., Liu, Y. & Zhang, J. An analysis of the epidemiological characteristics of human plague in Inner Mongolia Autonomous Region, China, 1950–2021. Chin. J. Vector Biol. Control 35, 69–73 (2024).
Li, Y. et al. Different region analysis for genotyping Yersinia pestis isolates from China. PLoS ONE 3, e2166 (2008).
Li, J. et al. Genetic source tracking of human plague cases in Inner Mongolia-Beijing, 2019. PLoS Negl. Trop. Dis. 15, e0009558 (2021).
Li, Y. et al. Genotyping and phylogenetic analysis of Yersinia pestis by MLVA: insights into the worldwide expansion of Central Asia plague foci. PLoS ONE 4, e6000 (2009).
Cui, Y. et al. Insight into microevolution of Yersinia pestis by clustered regularly interspaced short palindromic repeats. PLoS ONE 3, e2652 (2008).
Cui, Y. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl Acad. Sci. USA 110, 577–582 (2013).
Cong, X., Liu, Z. & Li, Q. Natural Plague Foci in China 1950–2014 (People’s Medical Publishing House, 2019).
Eaton, K. et al. Plagued by a cryptic clock: insight and issues from the global phylogeny of Yersinia pestis. Commun. Biol. 6, 23 (2023).
Feng, Y. et al. Epidemiological features of four human plague cases in the Inner Mongolia Autonomous Region, China in 2019. Biosaf. Health 2, 44–48 (2020).
Girard, J. M. et al. Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc. Natl Acad. Sci. USA 101, 8408–8413 (2004).
Vogler, A. J. et al. A decade of plague in Mahajanga, Madagascar: insights into the global maritime spread of pandemic plague. mBio 4, e00623–00612 (2013).
Esquivel Gomez, L. R. et al. Phylogenetic analysis of the origin and spread of plague in Madagascar. PLoS Negl. Trop. Dis. 17, e0010362 (2023).
Demidova, E. About the role of terrestrial and avian predators in the spread of plague. Proc. Rep. Conf. Irkutsk Anti-Plague Inst. Ulan-Ude 3, 41–42 (1958).
Dubyanskiy, V. M. & Yeszhanov, A. B. Ecology of Yersinia pestis and the epidemiology of plague. Adv. Exp. Med. Biol. 918, 101–170 (2016).
Wang, S., Jiang, X., Tan, A. & Fan, M. Analysis of the epidemic situation of plague among rodents along the Jining-Erlianhot Railway Line from 2010 to 2011. Dis. Surveill. Control 6, 295–293 (2012).
Wang, S., Chen, X., Li, X., Mi, Q. & Wu, L. Current status and prevention of animal plague on the Jining-Erlianhot Railway Line. Chin. J. Endemiol. 20, 176–177 (2005).
Liu, P., Song, Y., Colijn, C. & MacPherson, A. The impact of sampling bias on viral phylogeographic reconstruction. PLoS Glob. Public Health 2, e0000577 (2022).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 70, e102 (2020).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv https://doi.org/10.1101/201178 (2017).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Zhou, Z. et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 28, 1395–1404 (2018).
Wickham, H. (ed.) ggplot2: Elegant Graphics for Data Analysis, 157–175 (Springer, 2009).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst. Virus Evol. 2, vew007 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Acknowledgements
This research was funded by the Technology Project of Inner Mongolia Autonomous Region (2021ZD0006), the National Key Research and Development Plan (No. 2021YFC1200205) and the State Key Laboratory of Pathogen and Biosecurity (No. SKLPBS2215).
Author information
Authors and Affiliations
Contributions
Y.W., J.L. and Y.C. designed this study. F.L., Y.H., Z.W., and W.W. collected the data on Y. pestis. Y.G. and M.C. performed the DNA extraction and sequencing. X.Zuo, Y.W. and H.X. performed this analysis. Y.C., X.Zuo and Y.W. wrote this paper. Y.C., J.L., R.Y., H.F. and X.Zhang contributed valuable technical expertise.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Luke Grinham and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zuo, X., Liu, F., Hu, Y. et al. Genomic diversity and transmission patterns of Yersinia pestis in Inner Mongolia Autonomous Region, China. Commun Biol 7, 1480 (2024). https://doi.org/10.1038/s42003-024-07190-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07190-6
