
Abstract
Fusarium graminearum (F. graminearum) and its derivative mycotoxin deoxynivalenol (DON) are highly concerned with food safety and sustainability worldwide. Although several transcription factors (TFs) had been elucidated, molecular mechanism participates in DON biosynthesis regulation remains largely unrevealed. Here, we first characterized a zinc finger-contained TF in F. graminearum, FgSfp1, which is indispensable for DON production since its depletion resulting in a 95.4% DON yielding reduction. Interestingly, contrast to previous knowledge, all TRI-cluster genes were abnormally upregulated in ΔFgSfp1 while Tri proteins abundance rationally decreased simultaneously. Further evidence show FgSfp1 could coordinate genetic translation pace by manipulating ribosomal biogenesis process. Specifically, FgSfp1-depletion leads to ribosome biogenesis assembly factor (RiBi) expression attenuation along with DON precursor acetyl-CoA synthase reduction since FgSfp1 actively interacts with RNA 2’-O-methylation enzyme FgNop1 revealed by Bi-FC. It subsequently influences mRNA translation pace. In conclusion, we elucidated that the FgSfp1 orchestrates DON biosynthesis via participating RNA posttranscriptional modification for ribosomal RNA maturation, offering insights into the DON biosynthesis regulation. Ultimately, this TF might be a key regulator for DON contamination control in the whole food chain.
Similar content being viewed by others

Introduction
A principal concern in the food industry worldwide is the presence of plant pathogens. A prominent example being F. graminearum, that frequently causes devastating Fusarium head blight (FHB) disease in cereal crops, thereby leading to a decrease in crop quality and contamination of food with a variety of mycotoxins1,2,3. Mycotoxins, as a group of fungal-derived secondary metabolites, pose significant challenges to food security and safety globally4. Deoxynivalenol (DON) is a type B trichothecene mycotoxin ubiquitously produced by F. graminearum and other Fusarium genera; it is the most highly abundant mycotoxin detected in the major crops throughout the world and hence causes serious economic problems5,6,7. The consumption of DON not only leads to devastating crop mycotoxin contamination, but also has short-term and long-term toxic effects on animals and humans8,9. Considered the negative impact DON imparts on higher organisms, it is imperative to decipher the enzymatic mechanisms and regulation of the DON biosynthesis pathway. Previous studies have defined a fundamental scheme of this intricate catalyzation process10,11. Genes that encode DON biosynthetic enzymes form three unique clusters that are distributed on different parts of the F. graminearum genome. The core TRI gene cluster is located at chromosome II, the TRI1 and TRI16 cluster is embedded in chromosome I and the single gene TRI101 locus exists on chromosome IV. The DON biosynthesis process begins with the precursor molecule farnesyl pyrophosphate (FPP), which is derived from acetyl-CoA via the mevalonate pathway12,13,14. Through the catalytic processes executed by enzymes encoded by TRI genes, DON is finally generated in the cytosol of F. graminearum1,10. The DON biosynthesis process is governed by various of environmental factors. Pathogens can adjust their gene expression program in response to adverse living conditions, like improper growth pH, nutrient deficiencies, or competition with other organisms within their ecological niche15.
Transcription factors (TFs) are a group of proteins that can recognize and bind specific DNA sequences to promote the downstream genetic transcription and elongation process and ultimately complete DNA sequence decoding. Previous work has shown that most TFs in F. graminearum contain a DNA binding domain (DBD). Examples include Zn (II)2Cys6, C2H2 zinc finger, GATA, bHLH, and B-ZIP16,17. These crucial TFs have been deeply investigated, especially for their role in regulating TRI genes. For instance, the C2H2 zinc finger TF Tri6, located inside the TRI-cluster gene region, has been identified as a crucial regulator of TRI-cluster gene expression, binding to their DNA promoters at the TNAGGCCT motif sequence18. Moreover, recent studies reported pathogens like F. graminearum require finely tuned systems that enable them to sense environmental conditions switch and identify potential threats to adjust their gene expression pattern. For instance, sterol biosynthesis controls TF FgSR19. The nitrogen resource-sensing TF FgAreA20 and FgAreB21, pH-dependent TF FgPacC3022. Considering the complex composition of TFs and the regulatory complexity of the gene expression pattern under different environmental conditions, it is crucial to understand the mechanisms underlying DON biosynthesis regulation mediated by various TFs. Current studies have successfully established several genetic strategies to confer wheat trichothecenes detoxifying traits by transferring glutathione S-transferase (GST) genes, such as Fhb7 and Fhb1, cloned from Triticeae E genome23,24. Another perspective in managing DON contamination is uncovering the internal DON biosynthesis regulatory mechanisms to identify potential TFs responsible for DON production. Hopefully, investigation of DON biosynthesis regulatory mechanisms could help in developing feasible solutions to eliminate food threats from mycotoxins produced by pathogens.
Although casual association between environmental factors affecting F. graminearum metabolism pattern switch by TFs and mycotoxin DON biosynthesis have been established over the past decade, mechanisms behind this complicated relationship mostly unrevealed. The target-of-rapamycin (TOR) protein is vastly existed and highly conserved in both eukaryotes and prokaryotes. TOR signaling is a nutrient-sensing pathway that regulates cell growth by orchestrating several anabolic processes, namely via ribosome biogenesis, translation initiation25,26,27, and nutrient import28,29, allowing cells adapting to environment alternation. It has been proposed that TOR inhibition in cells could be an effective strategy to extend lifespan when extracellular growth conditions are suboptimal. The regulation of TOR signaling pathway in relation to mycotoxin biosynthesis in F. graminearum is poorly poorly understood. A direct effector of FgTOR (locus tag FGSG_08133) is FgSch9 (locus tag FGSG_00472). Notably, deletion of FgTOR in F. graminearum leads to DON deficiency, however, the mechanism(s) underpinning this phenomenon is unknown30,31. Therefore, identifying the downstream effectors or transcription factors (TFs) that are governed by FgTOR could provide insight into how DON levels are regulated in F. graminearum. Previous studies have successfully constructed a TF mutant library encompassing 600 putative TFs in F. graminearum that are related to multiple morphological observations including hyphae growth, mycotoxin production, pathogenesis, and stress responses. Interestingly, among the reported TFs, we noticed that a gene (locus tag FGSG_11799) that is predicted to contain two C2H2 zinc finger domains, and its deletion leads to a reduction in DON production. BLASTp analysis revealed that this gene is the counterpart of Sfp1 (Split-finger protein 1) in Saccharomyces cerevisiae, which is considered as a downstream TF of the TOR signaling pathway in response to extracellular stresses32,33,34. Previous studies depicted that Sfp1 forms a complex with other TFs, binding to promoter regions of ribosomal protein genes to control ribosome biosynthesis35,36. However, it is unknown whether there is a link between the regulatory activity of FGSG_11799 and the TRI-cluster genes involved in DON biosynthesis in F. graminearum. Furthermore, the mechanism underpinning the decision-making process fungus undergoes when confronted with adverse living environment, as well as its unique pattern of balancing growth rate and controlling secondary metabolism also remains undefined.
Hence in the current study, we investigated the role of the identified FGSG_11799 gene, i) to examine the DON production capability and morphological characteristics of this mutant strain, the phenotypes with the significantly dramatic DON reduction well confirmed. ii) to perform a series of combinatorial approaches, such as complement strain construction, omics-analysis, and interaction network screening. The aim being to identify to uncover the key downstream effectors responsible for DON biosynthesis and elucidate the regulatory mechanisms of upstream signaling pathways related to TRI-cluster genes at both transcriptional and translational levels. iii) Based on the experimental data obtained, establish a hypothetical mechanism illustrating how the FGSG_11799 protein functions as a downstream effector orchestrating DON biosynthesis and anabolic activities in F. graminearum. In total, obtaining a transcription factor would be well helpful for profound understanding of DON biosynthesis in F. graminearum, supporting with the valuable references for DON control strategies in potentially appliable practice of food production.
Overall, understanding the role of this TF would be useful in understanding the underlying mechanisms of DON biosynthesis in F. graminearum, providing valuable insights for developing DON control strategies for the ultimate benefit of maximizing food production.
Results
A two-C2H2 zinc finger domain-comprised protein, FgSfp1, is indispensable for mycotoxin production, virulence, and hyphae growth in F. graminearum
To characterize the protein encoded by locus tag FGSG_11799 in F. graminearum, we initially predicted the structural characteristics of the FGSG_11799 amino acid sequence by using Pfam37 and SMART38 platforms. The results revealed that the gene locus tag FGSG_11799 was located on chromosome I, encoding a 287 amino acid protein that contains two typical TF structure motifs: C2H2-zinc finger (ZF) domains (Fig. 1a). Notably, this protein shares 41.8% identity with its ortholog protein Sfp1 in Saccharomyces cerevisiae, and accordingly, we titled this protein FgSfp1. To explore the functionality of FgSfp1 in F. graminearum, we primarily constructed locus FGSG_11799 deletion mutant (ΔFgSfp1, Table 1). The ΔFgSfp1 mutant was successfully obtained by synthesizing its flanking DNA fragments fused with selective genes and corresponding complementary strain ΔFgSfp1-C strain was constructed by using plasmid containing FgSfp1-mCherry cassette under the control of its native promoter. Accordingly, the genotype of ΔFgSfp1 was confirmed by PCR, transcriptomic results and quantitative PCR assays (Supplementary Fig. 1a), indicating that there is no detectable expression of FgSFP1 in constructed mutants. These two C2H2 zinc finger domains are highly conserved and exhibit remarkable similarity across different fungal organisms, as displayed by amino acid sequence alignment and phylogenetic analysis (Supplementary Fig. 1b; Supplementary Fig. 2). Colony morphology of the ΔFgSfp1 mutant displayed growth defects and decreased pigmentation compared with wild-type PH-1 strains (WT) incubated with potato dextrose agar (PDA) medium for 24 h, 48 h and 72 h (Fig. 1b). We also observed that FgSFP1 deletion led to a reduction in conidia production (Supplementary Fig. 1d). Collectively, these data indicate that FgSfp1 plays an important role in mycelia growth and conidiation processes, impaired pigmentation also indicates FgSfp1 participates in secondary metabolism regulation in physiological environment.

a Amino acid structure illustration encoded by the locus tag FGSG_11799, displaying positions of its two C2H2 zinc finger domains using SMART protein database and Pfam protein database. Predicted amino acid sequence structure visualization model of FgSfp1 were built with swiss-model (https://swissmodel.expasy.org), two zinc finger domains were zoomed in. b Colony morphology differences among wild-type PH-1, FgSFP1 deletion mutant strain (∆FgSfp1) and its complemented transformant (∆FgSfp1-C) on PDA incubated at 28 °C for 24 h, 48 h and 72 h. The results are from three independent experiments. c DON production of wild-type PH-1, ∆FgSfp1, and ∆FgSfp1-C was determined by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis. Deletion of FgSFP1 leads to dramatic DON production decrease. The cultures on PDA were harvested and extracted after incubation at 28 °C for 7 days. Data presented are mean ± standard errors from three biological replicates (n = 3, p = 0.0001). d Wild-type PH-1, ∆FgSfp1, and ∆FgSfp1-C strains exhibit different virulence on wheat coleoptiles. Wheat coleoptiles were infected with 2 μL conidia suspension. Representative images were captured at 7 days after conidia inoculation. Arrows indicate lesion areas on wheat coleoptiles. Three independent experiments were performed. e Virulence test of PH-1, ∆FgSfp1 and ∆FgSfp1-C strains on wheat head. The morphology of infected wheat head was recorded at 14 day-post-inoculation (dpi). Sterile water inoculated as negative control (Mock). f First line, MALDI-TOF imaging of DON (m/z 295.153) in negative ionization mode on wheat kernel infected by wild-type PH-1, ∆FgSfp1, and ∆FgSfp1-C and harvested at 7 dpi, respectively. Scale bar, 1 mm. The Jet color scale indicates the range of total ion current-normalized intensity. Second line, MALDI-TOF imaging of DON (m/z 295.153) on spike sections showed in (e), the wheat seed samples with palea were embedded with 15% gelatin. Scale bar, 1 mm.
Next, we compared mycotoxin producing abilities of F. graminearum in the presence and absence of FgSfp1. We capitalized on liquid chromatography with tandem mass spectrometry (LC-MS/MS) to determine whether the ΔFgSfp1 mutant exhibits DON production deficiency. After incubating the strains in PDA medium for 7 days, we harvested the cultures and performed DON extraction using established methods39. The LC-MS/MS data revealed that DON production dropped by 95.465% in the ΔFgSfp1 mutant, decreasing from 351.794 mg/kg to 15.954 mg/kg on average (Fig. 1c). We also detected the yield of another mycotoxin produced by F. graminearum PH-1, zearalenone (ZEN). LC-MS/MS result shows no significant change between wild type strain and ΔFgSfp1 strain (Supplement Fig. 1h). This result indicates that deletion of the FgSFP1 has relatively exclusive impacts on DON production process. Notably, it has been reported that DON production is an indispensable prerequisite for F. graminearum infection40. Therefore, we detected the virulence alternation followed by DON reduction in ΔFgSfp1 mutant. Wheat coleoptiles inoculation assay showed that infection lesions caused by ΔFgSfp1 conidia were significant allayed compared with the wild-type strain PH-1 (Fig. 1d). Meanwhile, point inoculation of conidial suspension on flowering wheat head also showed same outcome of mild lesions infecting with ΔFgSfp1 strain (Fig. 1e; Supplementary Fig. 1c). These observations suggest that virulence of PH-1 is impaired after conducting deletion of FgSfp1. Combined with the observed morphology changes, these results highlight an indispensable role for FgSfp1 in mycelia growth, virulence, and mycotoxin DON biosynthesis in F. graminearum. During the storage stage of harvested cereal grains, two critical conditions needs to be carefully regulated to prevent spoilage is humidity and temperature. We stimulated proper circumstances of pathogen-contaminated wheat kernels by inoculating conidia of WT and ΔFgSfp1 on irradiating-sterilization grains of wheat cultivar Y158. The auxiliary of High mass resolution matrix-assisted laser desorption/ionization MS-time of flight (MALDI-TOF) imaging allows us presenting DON accumulation and distribution differences in wheat kernel sections at high resolution caused by WT and mutant strains (Fig. 1f; Supplementary Fig. 1f). As shown in Fig. 1, we observed high level of DON signals (m/z 295.153) in WT strain infected group which mainly appears at kernel periphery region and accumulates in the central area of wheat grains. However, ΔFgSfp1 infection exhibits significant DON signal attenuation compared to WT (Supplementary Fig. 1e). Collectively, the observation of above experiments indicates that two-C2H2 zinc finger domain-contained protein FgSfp1 is highly related to the hyphae growth, microbial pathogenesis and DON biosynthesis in F. graminearum.
Moreover, a few putative paralogs of FgSfp1 were predicted using Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Basic Local Alignment Search Tool (BLASTp) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). We analyzed the amino acid sequence of these candidates that showed only one of the paralog genes, locus tag FGSG_09410 on chromosome IV, its protein has similar C2H2 domains and shares 35.6% identity with FgSfp1, accordingly, we constructed genetic deletion mutants and quantified DON production. The LC-MS/MS data indicates FGSG_09410 deletion exhibits 42.89% of DON yield reduction (Supplementary Fig. 1g), which is less significant than ΔFgSfp1. Therefore, we suggest that FGSG_09410 protein has relatively lower impact on mycotoxin biosynthesis than FgSfp1 in F. graminearum, despite sharing the similar zinc finger domains.
Subcellular localization assay shows that FgSfp1 is localized in both the nucleus and cytoplasm in F. graminearum
To determine the subcellular localization of FgSfp1 in vivo, we constructed a FgSfp1-enhanced green fluorescent protein (eGFP) fusion protein and successfully expressed it in the wild-type F. graminearum PH-1 strain. Positive colonies were identified by PCR. The mycelia of positive colonies were incubated in yeast extract peptone dextrose medium (YEPD) at 28 °C and 180 rpm for 48 h. After co-staining with the nuclear-exclusive binding dye 4’,6-diamidino-2-phenylindole (DAPI), the mycelia were then imaged using a confocal laser scanning microscope. It was revealed that FgSfp1 co-localized with DAPI, being mainly distributed throughout the nucleus (Fig. 2a; Supplementary Fig. 3a). Noteworthy, the GFP signal was observed in the cytoplasm simultaneously, co-localization analysis confirmed GFP signals are not totally overlapped with DAPI. These data indicate that a portion of the FgSfp1 protein was localized in the cytoplasm. These data indicate that FgSfp1 might translocate from the cytoplasm into the nucleus and serve as a TF responsible for DON production in Fusarium graminearum.

a Wild-type PH-1 strain bearing FgSfp1-GFP constructs were incubated in yeast extract peptone dextrose medium (YEPD) at 28°C, 180 rpm for 24 h. The GFP fluorescent signal was detected by confocal laser scanning microscope (both). The nucleus of F. graminearum was co-stained with nuclear staining 4′-6-diamidino-2-phenylinodle (DAPI) and the images were captured at the same field with GFP signal channel (middle lanes). Scale bar = 40 μm. Fluorescence intensity and colocalization analysis by using imageJ software (line scan plots) represent fluorescent signals distributed in both nucleus and cytoplasm area. Horizontal axis indicates the distance. Results are from three independent experiments. b, c RNA-Seq data shows that the individual transcript levels of TRI genes are differentially expressed in wild-type PH-1 and ∆FgSfp1 strains. Depicted by bar plots (FPKM: Fragments Per Kilobase of exon model per Million mapped fragments). The numeric values are log2(FPKM + 1). d Relative mRNA expression levels of TRI5, TRI1, TRI101 in the ∆FgSfp1 mutant compared with wild-type PH-1, detected by qRT-PCR assay. Line bars in each column represent mean ± s.d. (n = 3). FgTUBULIN (locus tag FGSG_09530) was used as an internal loading control. e Relative Tri protein expression levels quantified by proteomics analysis. The results represented by bar plot. Tri protein relative abundance decreased in the ∆FgSfp1 mutant (red) compared with the wild-type PH-1 strain (blue). The data was analyzed with MaxQuant. Line bars in each column represent mean ± s.d. (n = 9). Relative Tri protein expression levels quantified by proteomics analysis. The results represented by bar plot. Tri protein relative abundance decreased in the ∆FgSfp1 mutant (red) compared with the wild-type PH-1 strain (blue). The data was analyzed with MaxQuant. Line bars in each column represent mean ± s.d. (n = 9).
Transcriptomic data combined with proteomics data analysis shows that loss of FgSfp1 leads to transcriptional-translational conflict of TRI-cluster genes
Previous studies have demonstrated that the DON biosynthetic pathway in F. graminearum begins with the conversion of precursor farnesyl pyrophosphate (FPP) into trichodiene (TDN) by a trichodiene synthase encoded from the TRI5 gene10. This substrate then enters through a series of catalytic steps involving enzymes encoded by TRI-cluster genes to finally produce the mycotoxin DON11. To further elucidate whether FgSfp1 controls the expression of DON biosynthetic genes, we sought to profile the RNA-sequencing (RNA-Seq) approach and transcriptomic analysis to evaluate TRI genes transcript changes in wild-type PH-1 and ΔFgSfp1 strains. Surprisingly, we observed an upregulation for all TRI genes, contrasting the decreased DON production phenotype observed in the ΔFgSfp1 mutant compared to wild-type PH-1 (Fig. 2b, c). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) further validated that relative mRNA expression levels of TRI5, TRI1, and TRI101 increased in the absence of FgSfp1 (Fig. 2d). These data indicate that mRNA level of DON biosynthesis enzymes encoded by TRI-cluster genes is upregulated in the absence of FgSfp1. These results highlight a contradictory between DON yield and the transcriptional pattern of the TRI-cluster genes in F. graminearum. Previous work reported that a reduction in DON production was always accompanied with TRI-cluster gene downregulation. This phenotypic and transcriptional inconsistency prompted us to further investigate the explanation for this abnormal phenomenon.
Given that the process of gene decoding can be divided into transcription and translation processes. According to central dogma, these two independent and interlaced activities ensure the genetic information decodes into functional proteins with high precision. Both biological processes are catalyzed by specific enzymes and related proteins that are involved in complicated regulatory networks. However, protein abundance does not always the reflect abundance of the corresponding mRNA41,42. This prompted us to determine protein abundance differences in the ΔFgSfp1 mutant versus PH-1 strain. We performed label-free proteomics experiments using LC-LTQ Orbitrap Velos Pro (Thermo Fisher Inc.) to determine relative protein expression levels in ΔFgSfp1 mutant and wild-type PH-1 strains. Using the Swiss-Prot database, we aligned more than 1300 proteins (Supplementary Fig. 4a; Supplementary Data 4–5). Enrichment analysis of Gene Ontology (GO) for downregulated proteins indicated differentially expressed genes are mainly enriched in small molecule metabolic process, carboxylic acid metabolic process, ribose phosphate biosynthetic process (Supplementary Fig. 4b–e). Upregulated proteins were predominantly enriched in steroid metabolic process and steroid biosynthetic process (Supplementary Fig. 4d). Notably, we determined protein abundance of enzymes involved in the DON biosynthesis pathway and identified the aligned TRI-cluster gene-encoded proteins from the database. The results revealed that FGSG_00071 (Tri1), FGSG_03534 (Tri3), FGSG_03535 (Tri4), FGSG_03537 (Tri5), FGSG_03540 (Tri11), FGSG_07896 (Tri101) protein levels were relatively decreased in the ΔFgSfp1 mutant compared to WT (Fig. 2e). Among them, the coverage of Tri5, Tri3, Tri4 and Tri11 protein in ΔFgSfp1 is absent, indicating their expression abundance is too low to be detected by LC-MS (Supplementary Data 1). These data suggest that the DON production deficiency observed in the ΔFgSfp1 mutant strain is primarily attributed to the decreased abundance of Tri proteins involved in the DON biosynthesis pathway. Although when we examined the protein expression levels of enzymes involved in the mevalonate pathway, which is responsible for producing the DON biosynthetic precursor FPP, we found that most of the enzyme expression levels remained relatively unchanged compared to the wild-type PH-1 strain (Supplementary Fig. 4b). However, mRNA levels displayed a tendency to be expressed at higher quantities in the ΔFgSfp1 mutant compared to wild-type PH-1 strain (Supplementary Fig. 4a).
Partial restoration of DON levels in the ΔFgSfp1 mutant after the addition of exogenous acetyl-CoA indicates that FgSfp1 participates in acetyl-CoA anabolism pathway regulation in F. graminearum
The transcriptome analysis also shows there were 2588 differentially expressed genes (DEGs) comparing WT to FgSfp1 deletion strains, of which 1098 DEGs were upregulated whereas 1490 DEGs were downregulated. The RNA-Seq data indicates that FgSfp1-deprivation impacts on various catalytic processes in F. graminearum PH-1. The deficiency of FgSfp1 leads to alterations in synthetic and metabolic pathways, which could have implications for mycotoxin biosynthesis and other cellular processes in the fungus. The KEGG pathway enrichment analysis of the RNA-Seq data showed that a subset of downregulated DEGs were enriched in starch and sucrose metabolism and the glycolysis/ gluconeogenesis pathway in the ΔFgSfp1 mutant. Glycolysis is a highly efficient metabolic process in eukaryotic organisms, sustaining essential physiological activities and providing energy for various cellular metabolic pathways14. It consists of a series of metabolic components that not only contribute to metabolism but also participate in the biosynthesis of secondary metabolites that enable fungi to thrive in challenging environments. Acetyl-coenzyme A (acetyl-CoA) is an essential metabolic intermediate produced from the catabolism of various fuels; it serves multiple purposes in eukaryotic organisms, such as entering the tricarboxylic acid (TCA) cycle to sustain its activity and acting as precursor for the de novo synthesis of amino acids, fatty acids, and sterols13. In F. graminearum, acetyl-CoA plays a role in the mevalonate pathway, working as a precursor for intermediates like farnesyl pyrophosphate (FPP) that enter the DON biosynthesis pathway11,12. Hence, we hypothesized that the reduction in DON production in the absence of FgSfp1 may be due to a lack of original reaction substrates, such as acetyl-CoA. Therefore, we propose a hypothesis that the reduction of acetyl-CoA synthetases leads to a reduction in the DON biosynthetic precursor acetyl-CoA, ultimately resulting in a reduction in DON biosynthesis activity. To test this, we conducted a feeding assay by exogenously adding the biosynthetic precursor of FPP, acetyl-CoA, to the PDA medium at concentrations of 0 mg L−1, 5 mg L−1, and 50 mg L−1. The results showed that the supplementation of 5 mg L−1 and 50 mg L−1 acetyl-CoA could increase DON accumulation in the ΔFgSfp1 mutant (Fig. 3a), indicating that exogenously feeding the DON biosynthetic precursor can partially rescue deficiencies in DON production. These results highlight a role for acetyl-CoA as a key building block for manufacturing DON and that the reduced abundance of this molecule under FgSfp1 depletion is an important factor contributing to a decrease in DON levels.

a Examination of DON yield in the ΔFgSfp1 mutant strain in PDA medium supplemented with 0 mg/L, 5 mg/L, 50 mg/L acetyl-CoA, respectively. Line bars in each column represent standard errors of three independent experiments. b Colony morphology of FGSG_00330 knockout mutant (ΔFGSG_00330) and FGSG_01743 knockout mutant (ΔFGSG_01743) strains in PDA medium incubated at 28 °C for 3 days. No significant differences in growth rate and colonial diameter in the two mutants compared with wild-type PH-1. c DON production in ΔFGSG_00330 and ΔFGSG_01743 detected by LC-MS/MS. Cultures were harvested after being inoculated on PDA medium and incubated at 28 °C for 7 days. Line bars in each column represent three biological replicates, mean ± s.d. (n = 3). The P values were calculated using a one-way ANOVA test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. d FPKM of FGSG_00330 and FGSG_01743 in wild-type PH-1 and ΔFgSfp1 mutant strains attained by RNA-Seq data. The numeric values are log2(FPKM + 1). Line bars in each column represent three biological replicates, mean ± s.d. (n = 3). e Relative expression of acetyl-CoA synthase genes, FGSG_00330 and FGSG_01743, mRNA levels in ΔFgSfp1 mutant compared with wild-type PH-1 examined by qRT-PCR assay. The mycelia were incubated in TBI medium for 3 days and collected for total RNA extraction. Line bars in each column represent mean ± s.d. (n = 3), FgTUBULIN (locus tag FGSG_09530) was used as an internal loading control. f Proteomics data show relative expression levels of FGSG_00330 and FGSG_01743 are different in wild-type PH-1 and ΔFgSfp1 mutant strains. Line bars in each column represent three biological replicates, mean ± s.d. (n = 3).
Acetyl-CoA pool accumulation in cytosol mainly relies on the transportation process from mitochondrial acetyl-CoA and other two ubiquitous metabolic pathways: glutamine reductive carboxylation and acetyl-CoA synthetase short-chain family 2 (ACSS2) to convert acetate into acetyl-CoA. ACSS2 becomes the main source of acetyl-CoA when the glycolysis pathway is impaired13,43. Interestingly, we discovered two acetyl-CoA synthetases, namely locus tag FGSG_00330 and FGSG_01743, in F. graminearum showed different expression patterns in ΔFgSfp1 by analyzing transcriptomic data. We next performed gene knock-out assays to acquire two genetic deletion strains of FGSG_00330 and FGSG_01743. There are no significant changes in colonial diameter in the FGSG_00330 and FGSG_01743 knock-out mutants after 3 days of incubation on PDA plates, suggesting that these mutations do not impact on colony growth (Fig. 3b). However, when we utilize LC-MS/MS to quantify DON accumulation in the two acetyl-CoA synthetase deletion strains, result shows that the FGSG_00330 deletion induced an 87.85% reduction in DON production, while the FGSG_01743 knock-out mutant had a 65.82% reduction in DON yield (Fig. 3c). This observation implies that these acetyl-CoA synthetases both have regulatory effects on the DON biosynthetic process, but FGSG_00330 shows more significant correlation with DON production. Next, we looked at the expression level changes of acetyl-CoA synthetase FGSG_00330 and FGSG_01743 from the RNA-Seq data in the ΔFgSfp1 strain. Expression levels of FGSG_00330 were elevated whereas FGSG_01743 expression levels were downregulated (Fig. 3d). This finding was further validated by qRT-PCR assay, which showed consistent results with the RNA-Seq data analysis (Fig. 3e). However, the differences in mRNA expression levels did not correspond to protein abundance in the proteomics data. Protein levels of FGSG_00330 in the ΔFgSfp1 mutant were reduced compared to wild-type PH-1, while there were no changes in FGSG_01743 protein levels (Fig. 3f).
FgSfp1 controls the ribosome biogenesis pace in F. graminearum
Given the lack of correlation between mRNA and protein expression levels of TRI genes and ACSS FGSG_00330, we considered the possibility that FgSfp1 might function as a TF for regulating ribosomal protein genes in F. graminearum. Ribosome biosynthesis as the most energy-consuming process in eukaryotes requires prodigious dedication of all three types of RNA polymerase for transcription events of ribosomal constitution and fine-tuning regulators, including ribosomal protein genes (RPGs), ribosome biogenesis (RiBi) genes35,44,45. In Saccharomyces cerevisiae, approximately 200 different assembly factors (encoded by RiBi genes) are involved in the maturation of pre-rRNA assembled with ~80 ribosomal proteins (RPs), playing crucial roles in pre-rRNA modifications and ribosome maturation46,47. Previous studies have shown that Sfp1, the homolog of FgSfp1 in Saccharomyces cerevisiae, binds to the promoter regions and forms a complex with other TFs, thereby controls the transcription of RPGs and RiBi48. To determine the consequences of FgSFP1 deletion on ribosome biosynthesis process, we aimed to monitor de novo protein synthesis in ΔFgSfp1 and WT. Puromycin incorporation were decreased in FgSfp1-deficient strain compared with WT (Fig. 4a). We also performed a click chemistry approach to testify nascent peptides synthesis decrease in ΔFgSfp1 by incorporating azide-labeled Met mimetic azidohomoalanine (AHA). After visualization AHA-containing nascent protein with alkyne tetramethylrhodamine (TAMRA), fluorescent signal exhibits its lower newly protein synthesis rate in ΔFgSfp1 (Fig. 4b). These results indicate FgSfp1-deficiency leads to nascent protein synthesis inhibition in F. graminearum. It prompts us to determine the reason behind impaired ribosomal efficiency caused by FgSfp1-deficiency. Primarily, we hypothesized the ribosomal proteins might decreased under FgSFP1 knockout given that Sfp1 related with RPGs transcription initiation. Surprisingly, there was no significant RPGs expression difference, neither transcriptional nor translational levels, in the ΔFgSfp1 mutant and wild-type PH-1 (Supplementary Data 2). However, in our transcriptomics data, we identified downregulated DEGs enriched in the ribosomal biogenesis pathway in eukaryotes (Fig. 4c, Table 2). The mRNA expression levels of majority assembly factors were downregulated in ΔFgSfp1 mutant compared with wild-type PH-1 (Fig. 4d, e), and this was supported by results attained from qRT-PCR analysis (Fig. 4f). These results suggest that FgSfp1 regulates the transcription of ribosome assembly factors to dictate the pace of ribosome biogenesis in F. graminearum. This regulatory mechanism differs from that of Sfp1 in Saccharomyces cerevisiae, underscoring the divergent roles of Sfp1 homologs in different organisms.

a Puromycin immunoblot of PH-1 and ∆FgSfp1. GAPDH was used as internal control. b Click-iT assay was performed to testify the nascent protein synthesis efficiency in WT and ∆FgSfp1. c Bubble plot showed KEGG pathways significantly enriched in metabolic pathways from down-regulated DEGs in ∆FgSfp1 compared with wild-type PH-1. d, e RNA-Seq data shows that the individual transcript levels of DEGs rich in ribosome biosynthesis wild-type PH-1 and ∆FgSfp1 strains. Depicted by bar plots (FPKM: Fragments Per Kilobase of exon model per Million mapped fragments). The numeric values are log2(FPKM + 1). f Relative mRNA expression levels of DEGs rich in ribosome biosynthesis listed in (c), detected by qRT-PCR assay. Line bars in each column represent mean ± s.d. (n = 3). FgTUBULIN (locus tag FGSG_09530) was used as an internal loading control. g Wild-type PH-1 strain bearing FgNop1-GFP constructs were incubated in yeast extract peptone dextrose medium (YEPD) at 28 °C, 180 rpm for 48 h. The GFP fluorescent signal was detected by confocal laser scanning microscope (both). The nucleus of F. graminearum was co-stained with DAPI and the images were captured at the same field with GFP signal channel (middle lanes). Scale bar = 10 μm. Fluorescence intensity and colocalization analysis by using imageJ software (line scan plots) represent fluorescent signals distributed in both nucleus and cytoplasm area. Horizontal axis indicates the distance. Results are from three independent experiments. h Y2H assay determined that FgNop1 interacts with and FgSfp1. Yeast cells co-expressing FgNop1-pGADT7 and FgSfp1-pGBKT7 plasmids were plated on selective plates. Results are from three independent experiments. i Bi-FC assay confirmed that FgSfp1 interacted with FgNop1 in F. graminearum by YFP fluorescence detection with a confocal laser scanning microscope. Wild-type PH-1 co-expressing FgSfp1-CYFP and FgNop1-NYFP fusion proteins were incubated in YEPD medium for 48 h and observed using a confocal laser scanning microscope. The fungal nucleus was simultaneously stained by DAPI. DIC, differential interference contrast. Scale bar = 10 μm. FgSfp1-YFPC construct and YFPN empty vector co-transformation strain were used as negative control.
Previous studies have outlined that the transcription of ribosomal protein genes (RPGs) is controlled by a core set of transcription factors (TFs) that form a regulatory complex. These TFs include High Mobility Group B (HMGB) protein Hmo149, Ifh1, Fhl1, and Rap135. To determine the correlation between DON biosynthesis and these components of regulatory complex in F. graminearum, we successfully generated knockout mutants of their orthologs in the PH-1 strain. Subsequently, we appraised the impact of these mutations on DON biosynthesis by measuring DON levels using LC-MS/MS. There was a 38.55% reduction in DON levels in the FgFhl1 mutant, 72% in FgIfh1 mutant, 65.36% in Rap1 mutant, and 45.84% in Hmo1 mutant (Supplementary Fig. 7). These results indicate that these subunits affect DON biosynthesis, all at differing levels of magnitude. It is noteworthy to mention that none of these individual knockout mutants resulted in a DON reduction as substantial as that observed in the ΔFgSfp1 mutant, which indicates existence of distinct mechanisms behind FgSfp1 controlling the ribosomal biogenesis.
FgSfp1 can directly interact with subunits of RNA posttranscriptional modification factor FgNop1 involving in RNA 2′-O-methylation
To further determine the additional regulatory mechanism of FgSfp1 orchestrating TRI-cluster gene expression, we intended to investigate interacting proteins with FgSfp1. Therefore, we applied a yeast two hybrid (Y2H) library system to screen the potential candidates, using FgSfp1as a bait protein. We constructed a F. graminearum cDNA library wherein the activation domain in the pGADT7 plasmid was used as prey while the PCR-synthesized FgSfp1 DNA sequence fused with the binding domain of GAL4 promoter was used as bait. After screening over 400 library prey colonies, positive yeast colonies were generated and sequenced. A total of 160 positive interactors were discovered in the screening result, which is listed in the Supplementary Data 3. Among those positive hits, we observed that FgSfp1 could directly interact with a high conserved protein, fibrillarin (FBL or FgNOP1, locus tag FGSG_01870), which is an only known methyltransferase responsible for rRNA 2′-O-methylation essential in the ribosome biogenesis as a catalytic constituent of the box C/D small nucleolar ribonucleoprotein (snoRNP) complex. Site-specific 2′-O-methylation of target RNAs is a widespread post-transcriptional RNA modification process found in various eukaryotic organisms50,51. Interestingly, as shown in Fig. 4e, the relative mRNA level of FBL (locus tag FGSG_01870) was decreased in the ΔFgSfp1 mutant. One-to-one Y2H and Bi-FC assays provide further validation that FgSfp1 interact with FgNop1 and co-localized in nucleus of F. graminearum (Fig. 4g-i; Supplementary Fig. 6a). It has been reported that FBL interacts with NOP56 before they assembled for pre-mature snoRNP biogenesis, which is indispensable for box C/D snoRNP to complete rRNA 2′-O-methylation52. Collectively, we raise a notion that FgSfp1 contributes to the ribosome biogenesis process by intervening RiBi gene transcription. FgSfp1 controls the ribosome translation pace not only by regulating rRNA 2′-O-methyltransferase fibrillarin protein expression levels but also directly binding with FgNop1 enhancing assembly of snoRNP to promote rRNA maturation. This would ultimately facilitate ribosome biogenesis process and ensure an efficient translation rate.
The nutrient-sensing transcription factor FgSfp1 interacts with FgSch9 as a downstream effector of FgTOR signaling pathway
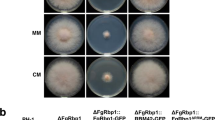
Using Y2H screening assay, we also detected FgSch9 (locus tag FGSG_00472) as a putative interactor for FgSfp1 (Fig. 5a). FgSch9 is the ortholog of Sch9 in Saccharomyces cerevisiae and serves as a downstream kinase in the TOR signaling pathway30, which controls various aspects of cellular metabolism, including amino acid biosynthesis and glucose homeostasis25. Bi-FC experiment determines the interaction between FgSch9 and FgSfp1 in F. graminearum. The Y2H assay also confirmed that FgSfp1 can interact with FgSch9 kinase and with FgMaf1— an additional protein known to interact with FgSch9 and related to DON biosynthesis (Fig. 5b, c). To determine whether a relationship exists between FgSfp1 and FgTOR, we used a specific TOR signaling pathway inhibitor to block this signal transduction process, observing the alternation of FgSfp1. The result shows a FgSfp1 localization shift after the 12 h-treatment of rapamycin (Fig. 5d). These data indicate FgSfp1 function as a downstream transcription factor of FgTOR signaling pathway in mycelia growth-suitable condition, activating by the kinase FgSch9 through phosphorylation then translocating into nucleus. This observation is consistent with Sfp1 in Saccharomyces cerevisiae32. Furthermore, we inoculated ΔFgSfp1 and wild-type PH-1 mycelia in complete medium (CM) supplemented with a gradient concentration of rapamycin to test their sensitivity to the FgTOR inhibitor. The data showed that the ΔFgSfp1 mutant exhibited an increased sensitivity to rapamycin compared to wild-type PH-1, even at low concentrations (Fig. 5e, f; Supplementary Fig. 8), indicating that FgSfp1 acts as a downstream TF of the FgTOR signaling pathway. However, the precise mechanism by which FgTOR regulates mycotoxin biosynthesis in complex living environments remains unclear. A hypothesis we postulated was that FgSfp1 acts as a central mediator, receiving signals from FgTOR and regulating the mycotoxin biosynthesis process in F. graminearum. Y2H screening assay shows there are two downstream proteins FgSch9 and FgMaf1 could also interact with FgSfp1. Taken together, these findings support the notion that FgSfp1 functions as a downstream transcription factor of the FgTOR signaling pathway. FgSfp1 would bind to the promoter sequence of downstream target genes after translocating into the nucleus when it is activated by FgTOR signaling pathway, finally orchestrating the cellular process of mycotoxin DON biosynthesis in F. graminearum.

a Y2H assay determined that FgSfp1 interacts with and FgSch9. Yeast cells co-expressing FgSch9-pGADT7 and FgSfp1-pGBKT7 plasmids were plated on selective plates. Experiments were conducted successfully three times independently. b Bi-FC assay confirms that FgSfp1 interacted with FgSch9 in F. graminearum by YFP fluorescence detection with a confocal laser scanning microscope. Wild-type PH-1 co-expressing FgSfp1-NYFP and FgSch9-CYFP fusion proteins were incubated in YEPD medium for 48 h and observed using a confocal laser scanning microscope. The fungal nucleus was simultaneously stained by DAPI. DIC, differential interference contrast. Scale bar = 10 μm. c Y2H assay determined that FgSfp1 interacts with and FgMaf1. Yeast cells co-expressing FgMaf1-pGADT7 and FgSfp1-pGBKT7 plasmids were plated on selective plates. Experiments were repeated three times independently. d Rapamycin-treatment assay demonstrates FgTOR signaling pathway inhibition leads to FgSfp1 retaining at cytoplasm observed with fluorescent microscope. Conidia of FgSfp1-GFP overexpression strain were used for 12 h rapamycin treatment. Final concentration at 120 µg/mL, same volume of solvent was added as mock group. Experiments were repeated for three times independently. e Different colony morphology of wild-type PH-1 and ∆FgSfp1 mutant strains on complete medium (CM) supplemented with a gradient concentration of rapamycin incubated at 28 °C for 4 days. f Inhibition rate of rapamycin on PH-1 and ∆FgSfp1 calculated from colonies diameter measurements from (b). The P-values were evaluated using t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Discussion
F. graminearum is a principal phytopathogen that secretes the contaminating mycotoxin DON, resulting in huge yield losses in cereal grains worldwide. Thus, efficient control strategies are urgently needed to be implemented to mitigate the economic losses caused by this epidemic. Uncovering the effectors involved in DON biosynthesis in F. graminearum is a promising means for developing such strategies. Despite previous studies having identified key TFs such as TRI6, TRI10, FgAREA, FgCreA, PacC, etc.1,18,22 that play crucial roles in DON biosynthesis, the generic pathway and overall control mechanisms for DON biosynthesis are still enigmatic. In this study, we focused our attention on TF FgSfp1, that contains two highly conserved C2H2 zinc finger domains and is implicated in hyphal growth, virulence, conidia development, and mycotoxin biosynthesis in F. graminearum. Strikingly, our data revealed that mRNA levels of TRI-cluster genes, that are key participants in DON biosynthesis, were abnormally up-regulated in the ΔFgSfp1 mutant, as confirmed by qRT-PCR and RNA-Seq analysis, yet this upregulation did not correspond to a significant increase in DON production. This discrepancy contrasts with previous reports where DON yield reduction was consistently associated with a decrease in TRI-cluster gene expression. Despite this, the current consensus does point out that mRNA and protein abundance are not strongly correlated, particularly during dynamic transitions. This inspired us to quantify Tri protein expression levels between ΔFgSfp1 mutant and wild-type PH-1 strains. LFQ proteomics data shows Tri proteins were decreased in the ΔFgSfp1 mutant consistent with DON reduction phenomenon. These data suggest that in the absence of FgSfp1, the mRNAs of the TRI-cluster genes are unable to be efficiently manufactured into the corresponding proteins. We speculated that this is due to impairments in ribosome biogenesis and/or function.
In eukaryotes, 2′-O-methylation of rRNA in ribosome biogenesis processing is a crucial step to determine the function of the RNA molecule. The exact function of this RNAs posttranscriptional modification remains cryptical, although it has been elucidated that box C/D snoRNP abundance alternation would impact on rRNA-methylation level and, consequently, switch cellular mRNA initiation modes. Specifically, the posttranscriptional modifications of rRNAs increase influence translational fidelity whereas promote the mode of translation initiation (cap-dependent or internal ribosome entry site, IRES). High levels of NOP1 are followed with rRNA methylation pattern change, translation fidelity impairments, and increased IRES-dependent (cap-independent) translation initiation of key cancer genes53. Considering the impact of Nop1 abundance on genes expression pattern, we raised one possible theory that FgSfp1 deficiency influences TRI-cluster mRNA translation in Fusarium graminearum is due to IRES-dependent translation decrease by FgNop1-downregulation. Emerging evidence has shown that methylation on mRNA by NOP1 increases their stability protecting from degradation. Recent study has also reported that NOP1 is also capable of methylating histone H2A at glutamine 104 (H2AQ104me), this specific modification changes 35 rDNA transcription process, finally regulating ribosome biogenesis54. Given that FgNop1 participates in several different target methylation resulting in various consequences, whether the translation process regulation carried out from FgSfp1-FgNop1 interaction is a result of histone modification alternation needs to be further elucidated.
The Y2H library screening system allowed us to identify putative proteins that interact with FgSfp1 in F. graminearum. Among the positive candidates (Supplementary Data 3), large subunit ribosomal proteins L35Ae (locus tag FGSG_01278), L15e (locus tag FGSG_00845), L18e (locus tag FGSG_09866) and small subunit ribosomal proteins, locus tags FGSG_00634 and FGSG_10755. These findings indicate that FgSfp1 plays a direct role in the regulatory process of ribosome biosynthesis. We identified the interaction between FgSfp1 and DNA-directed RNA polymerase II subunit RPB4, implying that FgSfp1 may be involved in recruiting RNA polymerase II and forming the transcription complex that initiates gene transcription processes. Another group of putative interactors with FgSfp1 is ribosomal subunits. The exact purpose of FgSfp1 binding to these subunits and the consequences of this interaction are not clear, although it provides preliminary evidence that FgSfp1 may directly participate in ribosome biogenesis. Collectively, these data suggest that FgSfp1 regulates the transcription process of ribosome assembly factors rather than RPGs. It also directly interacts with NOP1 involved in pre-mRNA maturation processing. Consequently, FgSfp1 plays a crucial role in ensuring the functionality of cellular ribosomes and maintaining normal translation processes. Finally, last intriguing putative candidate is transcription initiation factor TFIIE subunit beta (FGSG_08619) (Table 3). We also have predicted this interaction by utilizing STRING database (functional protein association networks) (https://cn.string-db.org). This implies that FgSfp1 associates with RNA polymerase II and functions as a constitutive member of a transcription complex, offering us another distinct insight into the mechanism of FgSfp1 controlling the transcription process in F. graminearum.
Notably, FgSfp1 interacts with downstream effectors of FgTOR, FgSch9 and FgMaf1. In Saccharomyces cerevisiae, Ser/Thr kinase Sch9 of the AGC family member is a direct substrate of TORC1. Their interaction activates downstream effectors by phosphorylation events28. Based on this, we postulated that FgSfp1 could also be activated by FgSch9, leading to its translocation from the cytoplasm into the nucleus, where it forms a transcription complex that binds to the promoter regions of downstream target genes involved in ribosome biogenesis regulation. Inhibition of the FgTOR signaling pathway by rapamycin treatment, or environmental stress, in F. graminearum triggers a nutrient stress response in the pathogen. Under such conditions, the function of FgSfp1 may be hindered, causing a slowdown in ribosome assembly and the subsequent biosynthesis of secondary metabolites, such as DON mycotoxin. This regulatory mechanism executed by FgSfp1 could be a strategy employed by F. graminearum to adapt its internal metabolic patterns and ensure the sustainability of its basic biological activities.
In conclusion, we identified a TF, FgSfp1, which acts as a downstream effector of the FgTOR signaling pathway and plays a key role in controlling mycotoxin DON biosynthesis process in F. graminearum. We have demonstrated that FgSfp1 controls the translation process of TRI-cluster genes by regulating the pace of ribosome assembly, thereby influencing the abundance of TRI-cluster proteins involved in DON production. Moreover, our experiments identified the interaction between FgSfp1 and the only known methyltransferase for RNA, FgNop1. As a crucial constituent of box C/D snoRNP complex responsible for rRNA posttranscription 2′-O-methylation modification, FgSfp1 could intervene the rRNA maturation process in charge of cellular translational pace, finally controls gene expression pattern. On the contrary, blocking the FgTOR signaling pathway with rapamycin treatment leads to FgSfp1 inactivation. Consequently, mRNA of TRI-cluster genes accumulates owing to the impediment in ribosome assembly. Interestingly, we observed an inconsistent relationship between the transcriptional and translational levels of the TRI-cluster genes in F. graminearum. Our findings suggest that the production of DON is tightly linked to extracellular nutrient conditions, which are evaluated and relayed through the signaling pathway governed by FgSfp1. This precise regulatory behavior holds significant importance for pathogens in their ability to combat challenging living conditions and adapt to adversity (See Fig. 6). Overall, the data obtained on FgSfp1 as a regulatory TF provides valuable information for achieving the ultimate objective of controlling DON levels through the regulation of DON biosynthesis and in understanding the pathogenesis of F. graminearum.

The grey circle represents FgSfp1-deficiency condition. At nutrient-sufficient conditions suitable for mycelia growth, FgSfp1 can be activated by FgTOR pathway, binding to the promoter region provoke downstream RiBi gene transcription. Meantime, FgSfp1 controls the pre-rRNA maturation rhythm interacting with a main component for rRNA 2′-O-methylation facilitating ribosomal biogenesis, FgNop1. On the contrary, FgSfp1 deprivation leads to ribosome biogenesis pace slowdown, transcripts of TRI-cluster genes translation process will be impeded. FgSfp1 orchestrates DON biosynthesis activities by coordinating the expression of ribosome assembly factor genes and directly interacting with RNA posttranscriptional 2′-O-methylation modification factor FgNop1, functions as a pivotal executor in Fusarium graminearum.
Methods
Strains, culturing conditions, and virulence determination
Fusarium graminearum PH-1 wild-type strain (NRRL 31084) was used as a parental strain for transformation experiments. Wild-type strain and transformants were inoculated on Potato dextrose agar (PDA) medium for hyphal growth and morphological examination. For RNA and protein extraction, each strain was grown in liquid trichothecene biosynthesis-inducing (TBI) medium. Conidia production was completed in the carboxymethyl cellulose (CMC) liquid medium (15 g carboxymethyl cellulose, 1 g yeast extract, 0.5 g MgSO4, 1 g NH4NO3, 1 g KH2PO4 and 1 L water) shaken at 180 rpm, 25 °C for 5 days. For virulence tests, seeds of wheat cultivar Sumai3 were sown in sterile water for 3 days. The upper part (2–3 mm) of the wheat coleoptiles was removed, and a 5 μL aliquot of a conidial suspension (1 × 105) was added. The samples were then incubated for an additional 3 days.
Strain construction
The deletion mutant strains ΔFgSfp1, ΔFGSG_09410, ΔFGSG_01743, ΔFGSG_00330, ΔFgHmo1, ΔFgIfh1, ΔFgFhl1, ΔFgRap1 were generated using the homologous recombination strategy. Double-joint PCR of the 5’ and 3’ flanking regions, approximately 1 kb in length, were amplified using primers listed in Table S1. Next, these flanking sequences were fused with selective gene hygromycin resistance cassette (HPH) by second-round PCR. Subsequently, the obtained constructs were transformed into the protoplasts of the wild-type strain PH-1 using a polyethylene glycol (PEG)-mediated protoplast transformation method. To obtain protoplasts, the fresh mycelia were harvested from YEPD (1% yeast extract, 2% peptone, 2% dextrose in 1 L water) cultures and digested with 0.02 g driselase (D9515, Sigma, MO, USA), 0.2 g lysozyme (A002880, Sangon Biotech, Shanghai, China) and 0.2 g snailase (A600870, Sangon Biotech, Shanghai, China). The transformed strains were screened on PDA media containing 100 μg mL−1 hygromycin B and potential mutants were identified by PCR assays using relevant primers in Table S1. The genotype of the FgSfp1 deletion mutant was further confirmed by quantitative PCR assays and RNA-Seq data (Supplementary Fig. 2a, b). All primers are listed in Supplementary Table 1.
To construct FgSfp1-GFP fusion cassettes, the FgSfp1 open reading frame (ORF) without the stop codon, along with its native promoter, was amplified from PH-1 genomic DNA. The resulting amplicons were co-transformed with XhoI-digested pFL2 into the yeast strain XK1-25. The FgSfp1-GFP fusion vector was recovered from the yeast transformant using a yeast plasmid extraction commercial kit (DP112, TianGen, Beijing, China) and then transferred into DH5α strain for amplification. Using the same strategy, FgSfp1-mCherry fusion cassettes were also constructed for obtaining complemented strain (ΔFgSfp1-C). Each recombination plasmid was transformed into wild-type PH-1 strain or the corresponding mutant strain. All primers are listed in Supplementary Table 2.
Deoxynivalenol extraction and DON production analysis
For the quantification of DON production, each strain was cultured in PDA medium at 28 °C for 7 days until no moisture remained. Mycelium sample was crushed and placed into a 50 mL centrifuge tube, adding 10 ml of acetonitrile: H2O (80/20, v/v) reagent. The mixture was shaken for 10 mins and then underwent ultrasonication for 1 h. Next, adjust the solvent composition until acetonitrile (ACN): H2O (v/v) reaches 20:80. The samples were then centrifuged at 13200 rpm for 30 min and the supernatant was filtered through 0.22 μm filters. DON production in each sample was determined using liquid chromatography with tandem mass spectrometry (LC-MS/MS). An Accela 1250 UPLC system (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a TSQ Vantage TM (Thermo Fisher Scientific, San Jose, CA, USA) triple-stage quadrupole mass spectrometer in positive electrospray ionization (ESI+) mode was used for DON detection. Chromatographic separation process was performed on an Agilent Extend C18 column (150 mm × 4.6 mm, 3.5 μm). The parameters used were as follows: mobile phase A (water containing 5 mM ammonium acetate) and mobile phase B (methanol) gradient elution with a flow rate of 0.35 mL/min at 30 °C. The injection volume was 5 μL. Each experiment was repeated three times independently.
Virulence determination on wheat coleoptile and spikelet
Fresh conidia of each strain were harvested from CMC medium and adjusted the concentration of conidia suspension to 105 conidia mL−1. Five microliters aliquot of each strain conidia suspension were inoculated onto coleoptile sections after cutting off 2 mm of the tip region. Images of brown lesions on each group were captured at seven days post-inoculation (dpi). For Ten wheat coleoptile replicates were used for each strain, experiment was repeated for four times. Ten microliters aliquot of each suspension was used for single-floret injection into a in the middle spikelet of a wheat head of wheat cultivar Y158 at anthesis. Visible infections are recorded at fourteen dpi. Seven spikelet replicates were used for each strain.
MALDI-TOF imaging analysis
Ten grams irradiating-sterilization grains of wheat cultivar Y158 mixing with 10 mL sterile water were infected by conidia of PH-1 and mutant strains. After incubating at 28 °C for 7 days, contaminated kernels were used for MALDI-TOF imaging. For damaged wheat spikelet samples, wheat kernels covered with its palea were selected for embedding and section to maintain integrity. Samples were flash frozen by liquid nitrogen after embedded with 10% Gelatin (Sangon, China) solution. Cryosection of embedded wheat seeds was undertaken with cryostat (CM1950 Leica, Germany) setting at 10 µm thickness and thaw-mounted on dedicated glass slides (8237001, Bruker, Germany). The slides then were fixed in the instrument metal holder and scanned with a flat-bed scanner (EPSON V850 Pro, EPSON, USA), dehydrated for 30 min, and then coated with 1,6-diphenyl-1,3,5-hexatriene (D208000, Sigma) supersaturated solution in 90% (v/v) acetonitrile (ACN) and 0.1% (w/v) trifluoroacetic acid (TFA) matrix to a density of approximately 3 µg mm−2 using a sublimation apparatus (TM-Sprayer, HTX technology). Matrix solution was prepared by dissolving saturated 1,6-diphenyl-1,3,5-hexatriene in 90% acetonitrile and 0.1% trifluoroacetic. Mass spectrometry data were obtained using a rapifleX MALDI Tissuetyper by Bruker Daltonics equipped with a smart-beam Nd:YAG 355-nm laser. The laser spot size was 5 μm and the spatial resolution was set at 50 μm. The number of laser shots was 200 per pixel, scanning from left to right. Raw Data processing was acquired by Flexcontrol (version 4.2) and Fleximaging (version 6) over a mass range of m/z 100–1000. The spectrometer Laser energy was set at 50%. SCILS Lab (version 20221 Premium 3D) was used to process and visualize the mass spectrometry data. MALDI mass spectra were normalized with the total ion current. The signal intensity of deoxynivalenol was displayed.
Microscopic examinations
To observe the localization of FgSfp1, the FgSfp1-GFP fusion cassettes were transformed into the wild-type F. graminearum PH-1 strain. The fluorescent signal was observed using an Olympus FV1200 confocal laser scanning microscope (Olympus, Japan). For sample preparation, conidia of FgSfp1-eGFP mutant were inoculated in TBI liquid medium at 25 °C on a shaker (180 rpm) for 72 h. The mycelia were collected, mounted on glass slides, stained with DAPI (nuclear specific dye 4′-6-diamidino-2-phenylinodle) (P0131, Beyotime, Shanghai, China), and sealed with cover glasses. The microscopy parameters were adjusted as follows: the laser excitation wavelength was set at 405 nm for DAPI (blue pseudo-color), and at 488 nm for GFP signal (green pseudocolor).
RNA-Seq and data processing
The wild-type strain PH-1 and corresponding conidia were inoculated in TBI liquid medium and incubated for 3 days at 25°C with shaking at 180 rpm. The RNA extraction procedure described above in the qRT-PCR assay was followed. The purity and integrity of the extracted RNA were assessed by agarose gel electrophoresis and 2100 Bioanalyser (Agilent). The concentration of total RNA was determined using a Nanodrop spectrophotometer. For transcriptome analysis, a library was constructed using 1 μg of total RNA with the TruSeqTM RNA sample preparation Kit from Illumina (San Diego, CA). RNA samples were sequenced by Illumina NovaSeq 6000 Sequencer (2 × 150 bp read length) using the standard procedure of Majorbio Bio-Pharm Technology Co. Ltd. For RNA-Seq data analysis. Reads were mapped to F. graminearum PH-1 (NRRL 31084) reference genome information and downloaded on NCBI using HISAT255. FPKM values were calculated using StringTie56 with Refgene annotation from the UCSC genome browser. Genes with FPKM values ≥1 were considered expressed. Read counts were generated using HTSeq-count57. Differentially expressed genes (DEGs) were calculated using the R package DESeq258. Genes were considered differentially expressed if the DESeq2 p-value was < 0.1 and log2 fold change was ≥1. The clusterProfiler was used to analyze GO terms and KEGG pathways.
qRT-PCR assay
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assay was performed to evaluate mRNA levels of TRI genes. Corresponding conidia of each strain were inoculated in the TBI liquid medium for 3 days at 25 °C with shaking at 180 rpm. The mycelium samples were collected three-layer gauzes then rapidly frozen and pulverized to a fine powder with liquid nitrogen. Total RNA extraction and reverse transcription experiments were performed using RNAiso Plus reagent (9109, Takara, Beijing, China) and PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (RR047A, Takara, Beijing, China) following the manufacturer’s instructions. All qRT-PCR primers are listed in Supplementary Table 3. The 2−ΔΔCt method was utilized to analyze the relative expression levels of each target gene. The experiment was independently repeated three times.
Intercellular protein extraction and proteomics analysis
To prepare protein samples, mycelia of each strain were harvested and ground in liquid nitrogen after being cultured in TBI liquid medium for 3 days at 25 °C. Subsequently, 0.1 g of biomass powder was transferred to a 2 mL microcentrifuge tube containing 1 ml of ice-cold lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) Triton X-100) and 10 μL of 100X protease inhibitor cocktail (K1007, APE × BIO, USA) was added. The samples were placed on ice for 10 mins after three times vortex. Then, the tubes were centrifuged at 4 °C, 13,200 rpm for 30 mins. The supernatant was transferred into a new tube and protein concentration was quantified using a BCA kit (20201ES76, Yeasen, China). Sample preparation for label-free proteomics was conducted according to the standard procedure published by Cold Spring Harbor59. In brief, we performed filter-aided sample preparation (FASP) for protein sample purification. Protein digestion of the samples was completed by using PierceTM Trypsin Protease, MS-Grade (90057, Thermo, USA) at 37 °C overnight. The digested samples were loaded onto an LC-LTQ Orbitrap Velos Pro (Thermo Fisher Inc.). Raw data was collected and analyzed with MaxQuant software.
Bimolecular fluorescence complementation (Bi-FC) assay
For the Bi-FC assay, fusion constructs of FgCreA-YFPN (yellow fluorescent protein C-terminal), FgNop1-YFPN, and FgSch9-YFPN cassettes were generated by cloning the full-length of the corresponding genes into the pHZ65 vector that carries NYFP and hygromycin-resistance marker. The FgSfp1-CYFP DNA fragment was cloned into the pHZ68 plasmid using the same strategy. Once the constructs were successfully generated, FgCreA-NYFP (pHZ65) or other gene-YFPN (pHZ65) plasmids and FgSfp1-CYFP (pHZ68) were co-transformed into the wild-type PH-1 strain using the polyethylene glycol (PEG) mediated protoplast transformation method as described earlier. Proper amounts of transformant mycelia were placed onto glass slides, stained with DAPI (nuclear specific dye 4′-6-diamidino-2-phenylinodle) (P0131, Beyotime, Shanghai, China) and sealed with a cover glass. The fluorescent signals were observed using an Olympus FV1200 confocal microscope (Olympus, Japan).
Yeast two hybrid (Y2H) assay and library screening
For the Y2H assay, the coding sequence of each target gene was amplified from cDNA of the PH-1 strain. The FgSfp1 cDNA was constructed into the pGBKT7 Gal4-baiting plasmid (630442, Clontech, Mountain View, CA, USA), while target genes were fused with the pGADT7 Gal4-pray plasmid (630442, Clontech, Mountain View, CA, USA). The two successfully constructed plasmids were co-transformed into the Y2HGold yeast strain (630498, Clontech, Mountain View, CA, USA) following the yeast protocols handbook (Clontech, Mountain View, CA, USA). Transformants were plated onto synthetic defined (SD) medium lacking leucine (L), tryptophan (T) (DDO) and incubated at 30°C for 2-3 days until yeast colonies appeared. Co-transformation of pGADT7-T (containing SV40 large T-antigen) and pGBKT7-53 (containing murine p53) were used as the positive control and plasmid pairs pGADT7-T and pGBKT7-Lam (lamin) served as the negative control. All primers are listed in Supplementary Table 4.
Puromycin incorporation assay
Conidia of PH-1 strain and ΔFgSfp1 mutant were inoculated into YEPD medium for 47.5 h at 180 rpm, 25 °C. Cultures were treated with 20 µM puromycin (ST551, Beyotime, China) and incubated for another 30 min. Mycelia was harvested by three-layer gauze then rapidly frozen and pulverized to a fine powder with liquid nitrogen. Total proteins were extracted with lysis buffer and 10 μL of protease inhibitor cocktail (K1007, APE × BIO, USA). The lysates were centrifuged at 12,000 × g same with protein extraction protocol. To check de novo protein synthesis, western immunoblotting of puromycin was performed with puromycin monoclonal antibody (MABE343, Merck, USA, 1:1000 dilution) and mouse monoclonal anti-GAPDH antibody (EM1101, HuaBio, China, 1:5000 dilution) as a reference. Secondary antibody Goat polyclonal anti-rabbit IgG-HRP (#LF102, EpiZyme, 1:1000 dilution) or Goat polyclonal anti-mouse IgG-HRP (#LF101, EpiZyme, 1:1000 dilution), respectively, chemiluminescence was exposed and imaged by Tanon 4600 (Tanon, China).
Nascent synthesized protein analysis
To assess the ability of nascent protein synthesis in PH-1 and its mutant, a copper-catalyzed azide–alkyne cycloaddition reaction was performed for newly synthetic protein labeling and visualization. De novo synthesis of proteins in PH-1 and ΔFgSfp1 was monitored by Click-iT® protein reaction system (C10276, Thermo Fisher, USA) according to manufacturer’s protocol. Final concentration to 1 × 104 conidia/mL of each strain was inoculated to YEPD medium for 48 h with 180 rpm at 26 °C, the hyphae were collected and reserved into methionine-free MM adding azide-containing Met mimetic azidohomoalanine (AHA) (C10102, Thermo Fisher, USA) to a final concentration of 50 µM. After incubating for another 6 h, total protein extraction of samples was performed using protocols described above. The incorporation of AHA enabled fluorescence visualization of AHA-containing proteins upon coupling to the alkyne-containing fluorophore (alkyne tetramethylrhodamine, TAMRA) (T10182, Thermo Fisher, USA). A total 20 µg of protein samples were used for SDS-PAGE gel analysis. Images were captured with automatic chemiluminescence image analysis system (5200, Tanon, China). The experiments were repeated for 3 times independently.
Acetyl-CoA feeding assay
Conidia of ΔFgSfp1 mutant strain were inoculated into PDA medium embedded with acetyl coenzyme A sodium of 0 mg L−1, 5 mg L−1, 50 mg L−1. For acetyl-CoA addition, concentration of 50 mg mL−1 stock solution (A27773, ABCone, China) was prepared with deionized water. Mycelia samples were harvested and processed after incubating at 28 °C for 7 days with deoxynivalenol extraction method described above. DON detection was also performed with LC-MS/MS. Data analyzed with Prism 8 software. The experiments were repeated for 3 times independently.
Rapamycin-treatment assay
Conidia of F. graminearum PH-1 FgSfp1-GFP overexpression strain was inoculated into YEPD medium incubating at 26 °C, 180 rpm. Final concentration at 120 µg mL−1 of rapamycin (V900930, Sigma, Germany) dissolved with absolute ethanol was added into cultures after incubating for 24 h. Same volume of solvent was added into control group. Within or without 12 h rapamycin treatment, the mycelia were collected and observed with fluorescent microscopy (RVL-100-G, Echo Revolve, U.S.A).
Statistics and reproducibility
Statistics and graphs were prepared using Prism v8 (GraphPad), standard statistical analyses were also performed using R. The type of what statistical tests to use and test results were described in the figure legends, with p-values of <0.05 considered statistically significant. No experimental samples were excluded from statistical analysis. Data represented the results of three independent replicate experiments unless mentioned specifically, and sample sizes were indicated in figure legends. RNA-Seq data and proteomics data are for principal component analysis and calculating Pearson correlation coefficient.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-Seq data have been deposited to Gene Expression Omnibus (GEO) with access number GSE262607. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD051048. All data that support the findings of this study are available in the article and Supplementary Data files. Source data for Figs. 2–4 can be found in Supplementary Data 1 and 5.
References
Chen, Y., Kistler, H. C. & Ma, Z. Fusarium graminearum trichothecene mycotoxins: biosynthesis, regulation, and management. Annu Rev. Phytopathol. 57, 15–39 (2019).
Xu, X. & Nicholson, P. Community ecology of fungal pathogens causing wheat head blight. Annu Rev. Phytopathol. 47, 83–103 (2009).
Alkadri, D. et al. Natural co-occurrence of mycotoxins in wheat grains from Italy and Syria. Food Chem. 157, 111–118 (2014).
Keller, N. P. Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180 (2019).
Merhej, J., Richard-Forget, F. & Barreau, C. Regulation of trichothecene biosynthesis in Fusarium: recent advances and new insights. Appl. Microbiol. Biotechnol. 91, 519–528 (2011).
Lee, H. J. & Ryu, D. Worldwide occurrence of mycotoxins in cereals and cereal-derived food products: public health perspectives of their co-occurrence. J. Agric Food Chem. 65, 7034–7051 (2017).
Mishra, S., Srivastava, S., Dewangan, J., Divakar, A. & Kumar Rath, S. Global occurrence of deoxynivalenol in food commodities and exposure risk assessment in humans in the last decade: a survey. Crit. Rev. Food Sci. Nutr. 60, 1346–1374 (2020).
Schaarschmidt, S. & Fauhl-Hassek, C. The fate of mycotoxins during secondary food processing of maize for human consumption. Compr. Rev. Food Sci. Food Saf. 20, 91–148 (2021).
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439 (2019).
Alexander, N. J., Proctor, R. H. & McCormick, S. P. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev. 28, 198–215 (2009).
Seong, K. Y. et al. Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 72, 354–367 (2009).
Mullen, P. J., Yu, R., Longo, J., Archer, M. C. & Penn, L. Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731 (2016).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Bar-Even, A., Flamholz, A., Noor, E. & Milo, R. Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat. Chem. Biol. 8, 509–517 (2012).
Gardiner, D. M., Kazan, K. & Manners, J. M. Nutrient profiling reveals potent inducers of trichothecene biosynthesis in Fusarium graminearum. Fungal Genet. Biol. 46, 604–613 (2009).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Lee, T. I. & Young, R. A. Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013).
Nasmith, C. G. et al. Tri6 is a global transcription regulator in the phytopathogen Fusarium graminearum. PLoS Pathog. 7, e1002266 (2011).
Liu, Z. et al. A phosphorylated transcription factor regulates sterol biosynthesis in Fusarium graminearum. Nat. Commun. 10, 1228 (2019).
Ma, T. et al. Plant defense compound triggers mycotoxin synthesis by regulating H2B ub1 and H3K4 me2/3 deposition. N. Phytol. 232, 2106–2123 (2021).
Jian, Y. et al. Interplay of two transcription factors for recruitment of the chromatin remodeling complex modulates fungal nitrosative stress response. Nat. Commun. 12, 2576 (2021).
Gu, Q. et al. Inhibition of histone acetyltransferase GCN5 by a transcription factor FgPacC controls fungal adaption to host-derived iron stress. Nucleic Acids Res. 50, 6190–6210 (2022).
Wang, H. et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368, eaba5435 (2020).
Li, G. et al. Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat. Genet. 51, 1106–1112 (2019).
Wullschleger, S., Loewith, R. & Hall, M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006).
Gonzalez, A., Hall, M. N., Lin, S. C. & Hardie, D. G. AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 31, 472–492 (2020).
Blenis, J. TOR, the gateway to cellular metabolism, cell growth, and disease. Cell 171, 10–13 (2017).
Urban, J. et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674 (2007).
Powers, T. TOR signaling and S6 kinase 1: yeast catches up. Cell Metab. 6, 1–2 (2007).
Gu, Q. et al. Protein kinase FgSch9 serves as a mediator of the target of rapamycin and high osmolarity glycerol pathways and regulates multiple stress responses and secondary metabolism in Fusarium graminearum. Environ. Microbiol. 17, 2661–2676 (2015).
Yu, F. et al. The TOR signaling pathway regulates vegetative development and virulence in Fusarium graminearum. N. Phytol. 203, 219–232 (2014).
Marion, R. M. et al. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl Acad. Sci. USA 101, 14315–14322 (2004).
Lempiainen, H. et al. Sfp1 interaction with TORC1 and Mrs6 reveals feedback regulation on TOR signaling. Mol. Cell 33, 704–716 (2009).
Albert, B. et al. A molecular titration system coordinates ribosomal protein gene transcription with ribosomal RNA synthesis. Mol. Cell 64, 720–733 (2016).
Zencir, S., Dilg, D., Rueda, M. P., Shore, D. & Albert, B. Mechanisms coordinating ribosomal protein gene transcription in response to stress. Nucleic Acids Res. 48, 11408–11420 (2020).
Lee, T. I. R. N. et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804 (2002).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015).
Sun, W. et al. A reliable liquid chromatography-tandem mass spectrometry method for simultaneous determination of multiple mycotoxins in fresh fish and dried seafoods. J. Chromatogr. A 1387, 42–48 (2015).
Jansen, C. et al. Infection patterns in barley and wheat spikes inoculated with wild-type and trichodiene synthase gene disrupted Fusarium graminearum. Proc. Natl Acad. Sci. USA 102, 16892–16897 (2005).
Liu, Y., Beyer, A. & Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 (2016).
Buccitelli, C. & Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644 (2020).
Li, Z. et al. Acetyl-CoA synthetase 2: a critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab. 33, 78–93.e77 (2021).
Abraham, K. J. et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 585, 298–302 (2020).
Shore, D., Zencir, S. & Albert, B. Transcriptional control of ribosome biogenesis in yeast: links to growth and stress signals. Biochem. Soc. Trans. 49, 1589–1599 (2021).
Bassler, J. & Hurt, E. Eukaryotic ribosome assembly. Annu Rev. Biochem 88, 281–306 (2019).
de la Cruz, J., Karbstein, K. & Woolford, J. L. Jr. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu Rev. Biochem. 84, 93–129 (2015).
Albert, B. et al. A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. Elife 8, e45002 (2019).
Kasahara, K., Ki, S., Aoyama, K., Takahashi, H. & Kokubo, T. Saccharomyces cerevisiae HMO1 interacts with TFIID and participates in start site selection by RNA polymerase II. Nucleic Acids Res. 36, 1343–1357 (2008).
Abou Assi, H. et al. 2’-O-Methylation can increase the abundance and lifetime of alternative RNA conformational states. Nucleic Acids Res. 48, 12365–12379 (2020).
Falaleeva M., Welden J. R., Duncan M. J., Stamm S. C/D-box snoRNAs form methylating and non-methylating ribonucleoprotein complexes: Old dogs show new tricks. Bioessays 39, 10.1002 (2017).
Yi, Y. et al. A PRC2-independent function for EZH2 in regulating rRNA 2’-O methylation and IRES-dependent translation. Nat. Cell Biol. 23, 341–354 (2021).
Marcel, V. et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330 (2013).
Tessarz, P. et al. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505, 564–568 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Christopher, J. A. et al. Subcellular proteomics. Nat. Rev. Methods Prim. 1, 1–7 (2021).
Acknowledgements
We would like to thank Wei-Jie He at College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, for conidia infection assay conduction. We also thank Zheng Yan, Qianqian Chu, and Zi Li from Institutional Center for Shared Technologies and Facilities of SINH, CAS for technical assistance. The work was financially supported by the National Science Fund for Distinguished Young Scholars of China (grant No. 32025030) to A.W.; Young Scientists Fund of the National Natural Science Foundation of China (grant No. 32001809) to D.Y.
Author information
Authors and Affiliations
Contributions
A.W. and D.Y. conceived coordinated and supervised the project. S.S. performed most experiments, collected, and analyzed data, prepared figures. M.G. performed nascent peptides detection assay and M.T. conducted MALDI-TOF samples preparation and data collection. Z.Y., S.S. and W.S. performed proteome samples preparation, Z.Y. operated the Orbitrap Velos mass spectrometer and collected proteomic data. S.S. prepared the first draft of the manuscript. A.W. and D.Y. provided insights during the studies, A.W. helped in manuscript preparation, revision and edition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, S., Yu, D., Guo, M. et al. The transcription factor FgSfp1 orchestrates mycotoxin deoxynivalenol biosynthesis in Fusarium graminearum. Commun Biol 7, 1584 (2024). https://doi.org/10.1038/s42003-024-07265-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07265-4
This article is cited by
-
Mycotoxin-driven proteome remodeling reveals limited activation of Triticum aestivum responses to emerging chemotypes integrated with fungal modulation of ergosterols
npj Science of Plants (2026)
-
A conserved Lsm8–exosome module maintains RNA splicing fidelity to control fungal stress adaptation and virulence
Stress Biology (2026)
