
Abstract
The ancestry composition and Sinicisation process of the descendants of the immigrants of Hu people living in ancient China are largely unknown due to the lack of genetic evidence. Tang Dynasty people in Fudamen cemetery (傅大门) excavated from Shandong province in eastern China are believed to be related to the descendants of Hu people, as some of the individuals with the surnames An (安). The genetic origin of the Fudamen population requires genetic clarification using ancient DNA data. Here we successfully obtain genome-wide SNP data for 17 Tang dynasty individuals from Fudamen cemetery. Based on autosomal data, although all Fudamen individuals show high levels of middle reaches of Yellow River-related ancestry as previously published historical period Shandong populations, 2 Fudamen individuals require ~5% Western Eurasian/Central Asian-related ancestry to describe their ancestry composition best. To the best of our knowledge, it is the first evidence of such ancestry in Shandong, the eastern part of today’s China. Moreover, the admixture pattern is also reflected by the presence of both west and east Eurasian-specific mtDNA and Y chromosomal haplogroups in Fudamen people. The estimated admixture time is also consistent with periods when Sogdians and other non-Han populations were active in ancient China. These genomic findings suggest that intermarriage with Han Chinese involved the Sinicization process of the Hu people.
Similar content being viewed by others

Introduction
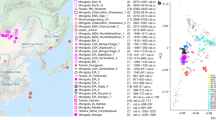
The Tang dynasty (唐代, 618–907 AD) stood out as one of the most prosperous dynasties in imperial Chinese history. With the rapid development of the Tang Dynasty’s economy and its open attitude towards foreign cultures, the famous Silk Road, which linked ancient China with the Western regions, experienced its most flourishing age1. Hu people (胡人) (a term used in the Tang Dynasty to refer to various ethnic minority tribes in Northwest China, Central Asia, and Western Eurasia), such as Sogdians (粟特人) from Central Asia, migrated along the Silk Road and entered ancient China for trade. Recent ancient DNA investigations updated the complex demographic history of Central Asians, who experienced dynamic human migrations and admixture since the Bronze Age2,3. For example, Bronze Age people from Xinjiang had already displayed the Eastern and Western admixture patterns. In southern Xinjiang, the Bronze Age Tarim Basin people from Xiaohe sites, harboured high proportions of Ancient Northern Eurasian-related ancestry (represented by Russia_AfontovaGora3), and the remaining ancestry was represented by Ancient Northeast Asian (ANA)-related ancestry3. The northern Xinjiang people were highly structured. There were four main ancestries in northern Xinjiang: ANA-related ancestry, Bactria-Margiana Archaeology Complex (BMAC)-related ancestry, Western Steppe herder-related ancestry, and local Tarim Basin-related ancestry2,3. Compared with the Bronze Age Xinjiang people, the Iron Age and historical Xinjiang people received additional gene flows related to central Asians and East Asians2. Historical records also documented the marriage between Han and non-Han people4,5,6. A recent ancient DNA study observed that all historical era individuals but two genetic outliers in Hexi Corridor were the direct descendants of Central Plain-related ancestry (represented by Late Neolithic middle Yellow River millet farmer-related ancestry (i.e., YR_LN published in ref. 7)). These two genetic outliers sampled from the western part of Hexi Corridor carried ~30–50% Western Eurasian/Central Asian-related ancestry and ~50–70% Central Plain-related ancestry, providing direct evidence of the eastward migration of people with Western Eurasian/Central Asian-related ancestry to the western part of Hexi Corridor and admixed with Hexi Corridor people8. There are detectable genetic influences from Western Eurasian/Central Asian-related people in the gene pool of Tang Dynasty people from the Central Plain (~1–2%) and Guanzhong Plain (~3–15%), the heart place of ancient China9,10. However, whether Western Eurasian/Central Asian-related ancestry had intruded in other areas of ancient China remains poorly understood as few ancient genomes have been studied.
In 2021, an archaeological cemetery of the Tang Dynasty, named Fudamen (傅大门), was discovered in the western region of today’s Shandong (Fig. 1A). One Fudamen individual 34 N was directly dated to 1282–1130 cal BP, corresponding to the Tang dynasty (i.e., radiocarbon dating). The Shandong Provincial Institute of Cultural Relics and Archaeology conducted a rescue excavation on the cemetery, where 35 tombs were uncovered. We display the photo of one tombstone unearthed in Fudamen cemetery in Fig. 1B, which belonged to the individual second_8S whose genome was studied in this study. The epitaph suggested that individual second_8S had the surname An (安). There were three origins for the Tang Dynasty people with the surname An11: (1) the descendants of Bukhara (安国) Sogdian people from today’s Uzbekistan who moved to ancient China since the Northern Wei Dynasty (386–534 AD). The Sogdians in China were known as the “Nine Surnames of Zhaowu” (昭武九姓), and An is one of them; (2) the descendants of Prince Anqing (安清) of the Parthian(安息) Empire from today’s northeast Iran. The Prince Anqing moved to ancient China in the Eastern Han Dynasty (25–220 AD); (3) the descendants of Xianbei (鲜卑) Anchi tribe (安迟部) whose surname was changed to An by Emperor Xiaowen (孝文帝) of Northern Wei (386–534 AD). Craniometric studies also showed that some individuals from Fudamen cemetery had some non-Han-related facial features, such as deep eyes and a tall nose bridge. These archaeological findings have raised curiosity about the population history of Fudamen cemetery. The populations associated with the ancestor of the surname An mentioned above were genetically distinguishable from the previously published historical Shandong people (represented by Shandong_HE published in ref. 12, who were genetically homogenous with Central Plain-related ancestry). Although no ancient genomes of Sogdian from central Asia or Parthians from Iran, the published genomes related to ancient Iranians and Central Asians belonged to the Western Eurasian-related lineages2,13,14. One present-day Central Asian population, Yaghnobis, settle along the Yaghnob River in northwest Tajikistan and are believed to be the only descendants of Sogdians as they speak a language of Old Sogdian origin. A genetic study based on genome-wide SNP data suggested that Yaghnobis people showed a closer genetic relationship to Tajik people from Tajikistan than to modern-day Iranians while harboured higher proportions of Neolithic Iranian-related ancestry and lower South Asian-related ancestry than Tajik people15. Xianbei people from northeast China (represented by AR_Xianbei_IA) was a mixture of ~85% ANA-related ancestry, ~13% Central Plain-related ancestry, and ~2% Western Eurasian-related ancestry7,16. Xianbei people from Mongolia (represented by Mongolia_IA_Xianbei) and Chinese Emperor Wu of Northern Zhou (represented by Xianbei_Wu_emperor) received more genetic influence from Central Plain (~14–32%) and Western Eurasian (~7–9%)-related lineages and carried less ANA-related ancestry (~61–77%)16,17 than AR_Xianbei_IA. Sinicization (from the prefix sino-, Chinese, relating to China) is the process by which non-Chinese societies or groups are acculturated or assimilated into Chinese culture, particularly the language, societal norms, culture, and ethnic identity of the Han Chinese—the largest ethnic group of China. The Sinicization process has been a core aim of archaeological and history studies for decades18. However, the Sinicization process of the Hu people was largely unknown from the genetic perspective due to the lack of ancient DNA data. Obtaining genetic data from Fudamen cemetery would aid in examining which non-Han people and to what extent they were involved in the genetic formation of Tang Dynasty Fudanmen people.

A The geographic location of newly sampled Shandong_H_Fudamen individuals and published representative populations in East Asia. Symbols as in (C). B The tombstone of Fudamen individual second_8S whose genome was studied in our study. The epitaph suggested that individual second_8S had the surname An. C Principal components analysis (PCA) under the genetic background of eastern Eurasian. The ancient individuals are projected on the PCs constructed by modern East Asian individuals using the “lsqproject: YES” option. Our newly generated Fudamen individuals were marked by black-filled shapes with red boundaries. See also Supplementary Data 5. D The ancestral composition of Eurasian individuals inferred by unsupervised ADMIXTURE for K = 3 (the lowest cross-validation error). See also Supplementary Data 7.
Here, we generated genome-level data from 17 individuals from Fudamen cemetery of the Tang Dynasty. 13 of 17 individuals were unrelated with each other. All but two individuals from the Fudamen cemetery of the Tany Dynasty were genetically homogenous with previously published historical Shandong. The other two individuals harboured ~95% historical Shandong-related ancestry and ~5% Western Eurasian/Central Asian-related ancestry. The Western Eurasian/Central Asian-related ancestry observed in two Fudamen individuals was consistent with the fact that one of the individuals had surname An whose ancestor could be linked to the Hu people. It suggested that the intermarriage with Han Chinese involved the Sinicization process of the Hu people.
Results
Ancient genomes from Fudamen cemetery
We collected archaeological samples from Fudamen and generated genome-wide SNP data from 17 ancient individuals by applying the shotgun sequencing strategy for fragments constructed by double-end strand libraries without uracil-specific treatment. All individuals displayed short sequence fragments (average length between 69.6 and 88.7 bases across genomes, after adaptor removal and collapsing) (Supplementary Data 1) with typical ancient DNA damage patterns at the 5’ and 3’ termini of the DNA fragments (Supplementary fig. 1). After removing samples with high levels of contamination (>5%, estimated using the mtDNA data for all individuals and X chromosomal data for biological males only) and a low number of SNPs on the 1240k panel (<20,000), 17 individuals were retained (Supplementary Data 1). We next conducted a kinship analysis to determine whether any Fudamen individuals were related to each other. Of the 17 individuals, 6 familial groups were identified (Supplementary Data 2 and Supplementary Note 1). We removed the individual with fewer SNP counts on the 1240k panel for each pair of related individuals, resulting in 13 unrelated individuals for further population genetics analysis (Supplementary Data 1). Across these 13 individuals, the human endogenous DNA rates ranged from 1.71% to 92.51% (Supplementary Data 1). The resulting depth of coverage of the autosomal SNPs on the 1240k panel ranged from 0.02 to 1.95× (average: 0.47×) (Supplementary Data 1). The number of SNPs covered at least once on the 1240k panel ranged from 24,017 to 1,024,585 (Supplementary Data 1). We further merged our newly generated Fudamen genomes with published ancient and modern genomes listed in Supplementary Data 3 and 4.
The genetic affinity between Fudamen and previously published Shandong historical individuals
We first assessed Shandong_H_Fudamen individuals using principal components analysis (PCA) (Fig. 1C and Supplementary Data 5) by projecting ancient samples on the genetic variation of 2127 present-day individuals from Eastern Eurasia. We observed that Fudamen individuals clustered with previously published Shandong historical individuals (represented by Shandong_HE, reported by ref. 12) and Central Plain-related ancestry (represented by YR_LN and YR_LBIA). This genetic affinity was confirmed by the outgroup f3 statistics of the form f3 (Yoruba; Shandong_H_Fudamen, X), where X is one of the ancient Eurasian populations in the 1240k dataset (Supplementary Data 6 and Supplementary Fig. 2). In line with the results above, ancestry proportions estimated by unsupervised ADMIXTURE for K = 3 using LD-pruned HO dataset suggested that our newly generated Fudamen individuals were genetically similar to Shandong_HE and Central Plain-related ancestry as previous studies suggested12 (Fig. 1D and Supplementary Data 7).
The genetic impact of Western Eurasian/Central Asian-related ancestry on Fudamen
To quantitively investigate whether Shandong_H_Fudamen individuals derived from the same source population that gave rise to previously published historical era people from Shandong (represented by Shandong_HE with large sample size), we computed f4 statistics of the form f4 (Yoruba, X; Shandong_HE, each Shandong_H_Fudamen individual) (Supplementary Data 8 and Supplementary Fig. 3). To capture any marginal but detectable genetic influence from non-YR-related ancestry to Shandong_H_Fudamen individuals, we set Z-score = 2 as the cutoff to label a statistical significance. If Shandong_H_Fudamen individuals were the descendants of Xianbei (represented by AR_Xianbei_IA), we would expect positive values in f4 (Yoruba, AR_Xianbei_IA; Shandong_HE, each Fudamen individual). Instead, we observed that AR_Xianbei_IA shared equal amounts of alleles with Shandong_HE and Shandong_H_Fudamen individuals (except for Shandong_H_Fudamen individual second_8S: Shandong_HE shared more alleles with AR_Xianbei_IA compared with second_8S. Z-score = -2.184). We observed that 10 of 13 Shandong_H_Fudamen individuals shared more alleles with diverse Western Eurasians/Central Asians (such as Russia_MLBA_Krasnoyarsk and Uzbekistan_BA_Dzharkutan1) than Shandong_HE did. In f4 (Yoruba, X; Shandong_HE, Shandong_H_Fudamen group (n = 13)), we also observed that diverse Western Eurasian/Central Asian-related populations shared more alleles with the Shandong_H_Fudamen population than with Shandong_HE (Supplementary Data 9). To explore whether Shandong_HE-related ancestry adequately explained the genetic profile of these 13 Shandong_H_Fudamen individuals, we first conducted qpWave analysis by adding each population which produced significant f4 values into the outgroup set in turn (Supplementary Data 9). We found the robustness of 1-way Shandong_HE modelling for 11 of 13 Shandong_H_Fudamen individuals (Supplementary Data 10). To explore the ancestry composition of the other two Shandong_H_Fudamen individuals, i.e., second_8S and second_19-2, we used populations who shared more alleles with second_8S and second_19-2 than with Shandong_HE inferred from the f4 statistics (Supplementary Data 8) as the second source and conducted two-source admixture modelling via qpAdm method (Supplementary Data 11). We found the proposed models were feasible for second_8S and second_19-2. These 2-source models also fitted better than the one-source Shandong_HE models: second_8S and second_19-2 showed ~5% Western Eurasian/Central Asian-related component (represented by Russia_MLBA_Krasnoyarsk and Uzbekistan_BA_Dzharkutan1) and ~95% Shandong_HE-related ancestry (Fig. 2, Supplementary Data 11 and Supplementary fig. 4). To explore whether western Eurasian/Central Asian signals in second_8S and second_19-2 were attributed to the ancient DNA damage pattern, we also calculated f4 on the transversion SNPs. We observed that some western Eurasian/Central Asian-related signals remained (Supplementary Data 12). It should be noted that the overlapped transversion SNPs on four populations in the formula of all f4 statistics were less than 52,426 (relatively limited) for individual second_8S and second_19-2; not all Western Eurasian/Central Asian-related signals based on all mutations were observed on the transversion SNPs. We designated a group with all Shandong_H_Fudamen individuals except for second_8S and second_19-2 as the “Shandong_H_Fudamen” population (n = 11) hereafter. We also found that all 1-way Shandong_HE modellings (qpWave) fitted well for Shandong_H_Fudamen even when each population that shared more alleles with either Shandong_H_Fudamen or Shandong_HE into the outgroup set (Supplementary Data 13 and 14). These results also suggested that Shandong_HE-related ancestry adequately explained the genetic profile of Shandong_H_Fudamen. We also calculated admixture dates for a population pooled by second_8S and second_19-2 individuals (i.e., n = 2) by DATES software. When assuming a single pulse admixture event using Russia_MLBA_Krasnoyarsk/Kyrgyzstan_TianShanHun as Western Eurasian/Central Asian origin and Shandong_HE/YR_LN as the local ancestry, the admixture date was estimated to have occurred ~7 ± 2 generations ago (Supplementary Data 15 and Supplementary fig. 5) before the death of second_8S and second_19-2, which corresponded to the Southern and Northern Dynasties (南北朝, 420–589 AD) to the Tang dynasty (618–907 AD) in Chinese history.

Each coloured block represents the proportion of ancestry derived from a corresponding ancestry source in the legend. The error bars represented the ±1 standard error (SE) of estimated ancestry proportions. Numbers in brackets denoted the sample size for each group. See also Supplementary Data 11 for further details.
The admixture profile of Fudamen people was also shown on the paternal Y chromosome and maternal mtDNA side (Supplementary Data 1). We observed 4 males in Fudamen displayed Western Eurasian-specific Y chromosomal haplogroup H (n = 2) and East Eurasian-specific Y chromosomal haplogroup O (n = 2). Consistent with autosomal data, individual second_8S who harboured on autosomes ~5% Western Eurasian/Central Asian-related ancestry carried Western Eurasian specific Y chromosomal haplogroup H1a2b. The dominant mtDNA lineages of Fudamen people (n = 9) were commonly found in East Eurasian populations, including G2b2, B4, D4, M9a, N9a3, and Z3a; one individual second_8S who harboured on autosomes ~5% Western Eurasian/Central Asian-related ancestry had Western Eurasian-specific mtDNA haplogroup H6a1b.
ROH patterns of Neolithic to historical era Shandong people
The runs of homozygosity (ROH) are regions of an individual’s genome where the maternal and paternal chromosomes are identical because they derive from a recent common ancestor19. Individuals with the sum of long ROH (>20 cM) greater than 50 cM indicated recent inbreeding, i.e., this individual’s parents were close relatives. A population with a low effective population size would expect an excess of short ROHs (4–8 cM). To shed light on the demographic history of ancient Shandong from the perspective of ROH patterns, we applied hapROH software to infer the ROH pattern for each previously published and newly generated ancient Shandong individual with >40,000 SNP sites on a 1240k panel, which covered the middle Neolithic Age to the historical era (Supplementary Fig. 6). Previously published Middle Neolithic Dawenkou culture-related individuals and Late Neolithic Longshan culture-related individuals displayed recent inbreeding signals measured by long ROHs. In the Iron Age and historical period, no Shandong ancients displayed inbreeding signals or excess short ROHs, suggesting that people lived in larger groups or groups with an extended mating network in the historical era.
Discussion
Yellow River basin (YR) is one of the birthplaces of Chinese civilization. Recent ancient DNA studies have revealed that in the historical era, a shared ancestry can be traced from a common origin in Hexi Corridor (the upper reaches of YR)8, Central Plain (the middle reaches of YR)9, and Shandong (the lower reaches of YR)12. Most individuals could be best modelled as the direct descendants of the Late Neolithic middle Yellow River millet farmer-related ancestry (represented by YR_LN)7, with little or no trace of other Eurasian lineages. These genomic findings emphasized the long-term genetic stability of Central Plain and the strong genetic influence from Central Plain to Shandong and Hexi Corridor (Fig. 2). Specifically, lying to the east of Central Plain, Shandong had experienced a prolonged Central Plain-related geneflow since the Neolithic. Recently published ancient genomic studies suggested that during the Neolithic Age, some Dawenkou and Longshan culture-related groups still maintained the local Shandong Hunter-gatherer-related ancestry20. However, all the published Shandong populations in the historical era (represented by Shandong_HE) were the direct descendants of Neolithic Central Plain-related ancestry, with no trace of local Shandong Hunter-gatherer-related ancestry12,20. These results suggested that the dynamic demographic history of Shandong’s population was reflected in the increase of genetic affinity with Central Plains-related ancestry over time. Our ROH inferences based on published genomes from Shandong also suggested the decrease of recent inbreeding and the increase of effective population size from the Neolithic to the historical era in Shandong.
The ancestry composition of our newly studied Tang Dynasty people from Fudamen cemetery in the western region of Shandong could be the exception in the historical period of Shandong (Fig. 2). From an autosomal SNP perspective, Fudamen individuals were largely descended from a population represented by published historical Shandong people (Shandong_HE); 2 of 13 individuals harboured ~5% of Western Eurasian/Central Asian-related ancestry, which was previously undetected in any ancient and present-day Shandong populations. It should be noted that the limited contribution of Western Eurasian/Central Asian-related ancestry and low coverage of Fudamen genomes made it challenging to determine (1) which kinds of Western Eurasian/Central Asian-related ancestries contributed to the gene pool of Fudamen and (2) the potential sex-biased migration pattern for Western Eurasian/Central Asian-related ancestry inferred from the relative amount of X-chromosomal to autosomal genetic contribution.
Among 2 Fudamen individuals who carried ~5% Western Eurasian/Central Asian-related ancestry, individual second_19-2 did not have a tombstone; therefore, the surname of second_19-2 was unknown; another individual second_8S was identified as a biological male, and his tombstone suggested that second_8S had the surname An (安姓). His western Eurasian/Central Asian-related ancestry on autosomes indicated that the surname of An in Fudamen most likely originated from either the Parthians (安息人) or Sogdians (粟特人). Nevertheless, the admixture time of Fudamen individuals gave clues about the origin of the Western Eurasian/Central Asian-related ancestry of Fudamen people. When assuming the single pulse admixture event for Shandong_H_Fudamen_o (pooled by second_19-2 and second_8S), the estimated admixture time overlapped with the period of the Southern and Northern Dynasties (南北朝, 420–589 AD) and the Sui and Tang Dynasty (589–907 AD). The Southern and Northern Dynasties were marked by the ethnic integration between nomadic regimes and Han Chinese. The Sui and Tang Dynasties strongly developed the Silk Road and promoted cross-continental material and cultural transmission between Western Regions and ancient China. There were also a large number of Sogdians migrating to the heart place of ancient China since the Northern and Southern Dynasties21. The Prince Anqing of the Parthian Empire came to China in the Eastern Han dynasty (25–220 AD). Hu people with the surname of An who came to China after the destruction of the Parthian Empire (226 AD), for example, the Tang dynasty, should belong to the descendants of Bukhara (安国) Sogdian people11. Therefore, Fudamen individuals were most likely the descendants of the Sogdians from Bukhara (安国) who migrated to ancient China through the Silk Road trade route and rose to prominence in the military and government of ancient China. It should be noted that the actual population admixture may have been a continuous process. The initial genetic contact between West and East Eurasian might occur before the admixture time estimated by DATES software.
The large proportion of Central Plain-related ancestry in Fudamen people’s autosomes suggested that intermarriage with Han Chinese involved the Sinicization process of Hu people. Findings from the uniparental markers suggested that both Western Eurasian males and females were involved in the migration into ancient China and admixed with Eastern Eurasians, as Fudamen people carried both Western Eurasian and Eastern Eurasian-specific mtDNA and Y chromosomal haplogroups: individual second_8S carried the Western Eurasian-specific mtDNA haplogroup H which was also dominant in present-day Yaghnobis people15 who were assumed to be the only descendants of Sogdians; other Fudamen individuals carried East Asian-specific mtDNA haplogroups. Individuals second_8S and 5E carried Western Eurasian-specific Y haplogroup H, which was enriched in South Asia but not in Yaghnobis people15; other Fudamen individuals carried East Eurasian-specific Y haplogroup O. We could not infer the specific sex-bias admixture pattern of Fudamen individuals.
Interestingly, the burial customs for 2 Fudamen individuals with ~5% Western Eurasian/Central Asian-related ancestry were not significantly different from other tombs in the cemetery. Therefore, we argued that the descendants of Hu people had lived together with other residents in this area and experienced the Sinicization process from both cultural and genetic perspectives. We agreed that no Hu people-related cultural element was observed in the Tang dynasty Fudamen does not mean there was no cultural influence from Hu people to Chinese people once. Maybe the Tang dynasty was not the start of the Sinicization process for the descendants of Hu people in Fudamen, as the admixture date for Fudamen individuals suggested (the admixture date corresponded to the Southern and Northern Dynasties (南北朝, 420–589 AD) to the Tang dynasty (618–907 AD) in Chinese history).
The tombstone of M8 recorded that individual second_8S and individual second_8W were a couple and had offspring. second_8S was identified as a biological male. second_8S carried ~5% western Eurasian/Central Asian-related ancestry on autosomes and carried western Eurasian-related Y chromosomal and mtDNA haplogroup. Fortunately, we obtained the genome data for second_8N. second_8N was identified as a biological female and was genetically indistinguishable from previously published historical era Shandong people. Moreover, individual second_8S carried ~95% Central Plain-related ancestry on autosomes but western Eurasian-specific mtDNA and Y haplogroup. These genetic findings provided direct genetic evidence of the marriage between the descendants of Hu people and the Han Chinese.
Interestingly, recently published Tang Dynasty ancients from Central Plain9, three individuals from Chang’an City10 (the capital of the Tang Dynasty) and one individual from Hexi Corridor8 could also be best modelled as the mixture of Late Neolithic Central Plain-related ancestry and Western Eurasian/Central Asian-related ancestry (~2%, ~3–15% and ~30%, respectively). Therefore, the eastern-western Eurasian admixture profile might be expected during the Tang Dynasty in ancient China. Future ancient DNA studies should aim to expand the geographical and temporal range of samples to understand better the genetic influence of non-Han-related people in ancient China.
Methods
Archaeological information for Fudamen (傅大门)
The specimens for this study are from the Fudamen cemetery in Liaocheng City, Shandong Province. In 2021, the Shandong Provincial Institute of Cultural Relics and Archaeology conducted a rescue excavation, which uncovered 35 tombs. Through typological research on unearthed burial objects and epitaph records, it is reflected that most of these tombs were dated to the Tang Dynasty. 17 individuals from the Fudamen cemetery were sampled. The research protocol was approved by the Medical Ethics Committee of Xiamen University (XDYX202412K88). The Shandong Provincial Institute of Cultural Relics and Archaeology that managed the samples also approved the use of archaeological materials. The archaeological specimens are being stored at the Shandong Provincial Institute of Cultural Relics and Archaeology. All ethical regulations relevant to human research materials were followed. One individual 34 N was directed dated to the Tang dynasty (i.e., C14 radiocarbon dating). We note that other samples were not directly radiocarbon-dated. The date used in this study was based on archaeological context.
▪ 34 N: 1282–1130 cal BP
Radiocarbon dating
The preparation of bone/tooth samples involves standard acid-base-acid (ABA) procedure and collagen extraction. Firstly, bone/tooth samples are ultrasonicated in ultrapure water, dried, grinded and sieved to get the appropriately sized sample fraction (0.5–1 mm). Then samples are treated with 0.5 M hydrochloric acid (~18 h), 0.1–0.2 M sodium hydroxide (30 min–1 h), and 0.5 M hydrochloric acid (1 h). Bone/tooth collagen gelatinization is performed in pH 3 solution at 70 °C for 20 h. Gelatine solution is filtered using a cleaned filter and freeze-dried. The quality of collagen is monitored by carbon and nitrogen content in collagen, atomic C/N ratio determination and collagen yield. Samples are dated if the collagen yield is above 1% and the C:N ratio of the collagen is between 2.9 and 3.5. The samples that deviate from these ratios are deemed unsuitable for dating22. The pure collagen samples were combusted with CuO and silver in vacuo to CO2 at 900 °C for 2 h. The CO2 was purified, graphitized and measured on a compact 0.5 MeV NEC Accelerator Mass Spectrometry in the Guangzhou Institute of Geochemistry, Chinese Academy of Sciences23. All four radiocarbon dates were modelled in OxCal24 using the IntCal2025 calibration curve.
Ancient DNA extraction and sequencing
A total of 17 skeletal specimens were subjected to experimental analysis in this study. All experimental procedures were conducted in the specially designed ancient DNA cleanroom laboratory at Xiamen University26,27,28. We utilized 75% ethanol and 10% sodium hypochlorite (NaClO) to eliminate external contaminants. After surface abrasion and ultraviolet (UV) radiation treatment, we employed a drill bit to obtain inner core bone powder ranging from 130 to 230 mg. For each 100 mg of bone powder, we added 1 ml of 0.5 M EDTA and 1 μl of 20 mg/ml proteinase K, followed by incubation at 37 °C on a shaker at 300 rpm for 20 h. We used the MinElute System (Qiagen, Germany) to obtain concentrated DNA fragments. We prepared double-stranded libraries using the NEBNext® Ultra™ II DNA Library Prep Kit, paired with specially in-house designed sticky-end adaptors. We then sequenced the libraries on the DNBSEQ-T7 platform.
DNA sequence data processing
Using AdapterRemoval (version 2.3.2)29, we removed adaptors from both read pairs, trimmed bases at 5’/3’ termini with quality scores ≤20 and ambiguous bases (N) (–trimns –trimqualities --minquality 20), and collapsed forward and reverse reads (--collapse), and discarded reads shorter than 30 bp. Collapsed reads were aligned against the human reference genome hs37d5 (GRCh37 with decoy sequences) using the aln and samse modules in the Burrows-Wheeler Aligner (BWA) programme (version 0.7.17-r1188)30, with parameters ‘-l 1024’ (seeding disabled) and ‘-n 0.01’ (additional mismatches allowed). BAM files were sorted and indexed using Samtools (version 1.7)31 before being utilized to remove PCR duplicates using dedup (version 0.12.8)32. BAM data were filtered with Samtools (version 1.7)31 for a minimum Phred-scaled mapping quality score of 30.
Data quality control
We used a variety of methods to assess the ancient DNA’s authenticity. First, we used mapDamage (version 2.2.1)33 to compute the postmortem damage pattern. We determined if each library had >10% C-to-T misincorporations at 5’ termini and >10% G-to-A misincorporations at 3’ termini, as is expected for double-stranded libraries. Second, we estimated the contamination rate. To calculate the mtDNA contamination rate, (1) we used the schmutzi.pl module in schmutzi software34 and the contaminant database developed for Eurasian samples from share/schmutzi/alleleFreqMT/eurasian/freqs; and (2) we followed the pipeline posted in https://github.com/mnievesc/Ancient_mtDNA_Pipeline/blob/master/MT_DataAnalysis_Pipeline.sh to conduct contamMix35-related analysis. The X chromosomal contamination rate was calculated for males using the contamination module in ANGSD software (version 0.910)36. We used the HapMap resources for CHB (Han Chinese in Beijing) provided by ANGSD software to define X chromosomal polymorphic sites. We focused our investigation on the non-recombining part of the X chromosome (X:5,000,000–154,900,000). The library was treated as contaminated when the mtDNA contamination was greater than 5%, except when nuclear DNA contamination was assessed by the X chromosome <5% for males.
Biological sex determination
An individual with one X chromosome and one Y chromosome was designated as “male,” whereas an individual with two X chromosomes was designated as “female.” Rx and Ry statistics37,38 were computed to estimate the biological sex of the samples based on shotgun data.
Genotyping
We used the trimBam module in BamUtil (version 1.0.15)39 to mask 9 bp from both ends to reduce the bias caused by ancient DNA deamination. We randomly picked one high-quality base (–q 30 and –Q 30) as a pseudo haploid using pileupcaller (https://github.com/stschiff/sequenceTools).
Estimation of genetic relatedness
We first applied PLINK software (version 1.9)40 to exclude non-polymorphic and low-frequency variants (--maf 0.01). We then applied the default parameters of the READ software41 to estimate the biological relatedness between each pair of Fudamen individuals.
Uniparental haplogroup assignment
We produced mitochondrial consensus sequences of quality ≥20 using the log2fasta tool in Schmutzi software34. Y-chromosomal and mitochondrial haplogroups were assigned by Yleaf (version 2.2)42 with “-r 1 -q 30 -b 90” option and Haplogrep (version 2) software43, respectively.
We here described the manual check for the Y chromosomal haplogroup assignment for 5 males:
-
(1)
Fudamen sample 34N could be assigned as O2a2b1a1a3 based on the mutation O2a2b1a1a3-CTS7634:17578411T- > C. This sample also had upstream derived mutations for haplogroup O2a2b1a1: O2a2b1a1-CTS5128:16028396T- > C, O2a2b1a1-F342:16472742A- > G, O2a2b1a1-F373:16880085C- > T. This sample, however, showed inconsistent mutations at haplogroup O2a2b1a1a3a with a derived allele at O2a2b1a1a3a-F3455:23540900T- > C, O2a2b1a1a3a-F574:21306283T- > C, O2a2b1a1a3a-F2631:17897541C- > T, but ancestral alleles at O2a2b1a1a3a-F1401:8766750C- > G, O2a2b1a1a3a-F317:15981403A- > G, O2a2b1a1a3a-Y16696:15964719C- > T, O2a2b1a1a3a-F2000:15825642A- > G, O2a2b1a1a3a-F3431:23353724G- > A, O2a2b1a1a3a-F375:16898689C- > G, O2a2b1a1a3a-F1870:14945490C- > T. This sample also showed ancestral alleles at O2a2b1a1a3b-PH3555:17962646C- > T and O2a2b1a1a3b-PH3164:17552442G- > A.
-
(2)
Fudamen sample 5E could be assigned as H1a2b1 based on the mutation H1a2b1-Z14308:28683664A- > G. This sample also had upstream derived mutations for haplogroup H1a2b: H1a2b-Y2586:21265071T- > C, H1a2b-Z14277:14040666G- > A, H1a2b-Z14258:2697625G- > A, H1a2b-Z14269:8068519C- > G, H1a2b-Y2579:8143732C- > A, H1a2b-Z14306:24417307A- > G, H1a2b-Z14266:7396848G- > A, H1a2b-Y2583:14004420T- > G, H1a2b-Z14299:23041312C- > A, H1a2b-Z14290:21299587G- > A, H1a2b-Z14297:22800803T- > C, H1a2b-Z14279:14886830A- > G, H1a2b-FGC3219:16210460C- > A, H1a2b-Z14301:23302776A- > G, H1a2b-Z14286:17483823G- > T, H1a2b-Z14283:16289044G- > A. This sample, however, showed inconsistent mutations at haplogroup H1a2b1b with a derived allele at H1a2b1b-Z34827:22579488G- > A, H1a2b1b-Z34790:17080968C- > T, H1a2b1b-Z34831:22766386G- > A, H1a2b1b-Z34738:7038591G- > A but ancestral alleles at other sites defined H1a2b1b. We caution the observed derived alleles might be caused by ancient DNA damage.
-
(3)
Fudamen sample second_19-1 could be assigned as H1a2b based on the mutation H1a2b-Y2584:14387228T- > C, H1a2b-Z14268:7957243C- > A, H1a2b-Z14297:22800803T- > C, H1a2b-Z14267:7553253A- > G. This sample also had upstream-derived mutations for haplogroup H1a: H1a-M2712:6853560G- > A, H1a-M3069:23149791G- > T, H1a-M2764:8845931A- > G. This sample also showed ancestral alleles at sites defined as H1a1 and H1a2a and the subclades of H1a1 and H1a2a. This sample showed ancestral alleles at H1a2b1a-Z14316:6901411C, H1a2b1b-Z34816:21360775C, H1a2b1c-Z34851:9820626T.
-
(4)
Fudamen sample second_38-2 could be assigned as O2a2b1a2a1d1 based on the mutation O2a2b1a2a1d1-F2903:18939070A- > G. This sample also had upstream-derived mutations for haplogroup O2a2b1a2a1-F174:8869263G- > A, O2a2b1a2a1-F323:16193325T- > G. This sample also showed ancestral alleles at O2a2b1a2a1d1a-Y30488:21689545A.
-
(5)
Fudamen sample second_8S could be assigned as H1a2b based on the mutation H1a2b-Y2590:23808189T- > G, H1a2b-Z14290:21299587G- > A, H1a2b-Y2579:8143732C- > A, H1a2b-Z14268:7957243C- > A. This sample also had upstream-derived mutations for haplogroup H1a: H1a-M69:21894058T- > C, H1a-M2817:14667241G- > A, H1a-M3036:21944665C- > T, H1a-M2812:14572769C- > T. This sample also showed ancestral alleles at sites defined as H1a1 and H1a2a and the subclades of H1a1 and H1a2a. This sample showed ancestral alleles at H1a2b1a-Z14323:8749969G, H1a2b1b-Z34769:14531658C, H1a2b1c-Z34859:17486729G.
Data merging
We merged Fudamen samples with published genome-wide SNP data of present-day and ancient Eurasians using the mergeit function in EIGENSOFT software44. The information on co-analysed ancient and modern genomes is listed in Supplementary Data 3 and 4. Two datasets were used in our analysis: (1) We merged Fudamen data with the 1240k dataset curated by The Allen Ancient DNA Resource (AADR)45, which covered ancient DNA data with maximum SNP sites (1,135,618). (2) We merged Fudamen data with the Human Origin (HO) dataset curated by The Allen Ancient DNA Resource (AADR)45, which covered ancient DNA data with overlapped SNP sites between 1240k and HO SNP panel (593,120), as well as present-day Eurasians genotyped on Human Origin SNP chip.
Principal components analysis (PCA)
We ran PCA on the HO dataset using the smartpca (version 16000) algorithm in EIGENSOFT software44 with the options “lsqproject: YES” and “numoutlieriter:0”, and “shrinkmode: YES”. We employed the “lsqproject: YES” option to project all ancient genomes onto the PC spaces calculated with the modern genomes.
Unsupervised ADMIXTURE
We pruned the strong linkage disequilibrium using PLINK software (version 1.9)40 with option “--indep-pairwise 200 25 0.4” on the merge Human Origin dataset. We run the unsupervised ADMIXTURE using ADMIXTURE software v1.3.046 with default parameters and set K ranging from 2 to 12.
F statistics
We applied qp3pop (version 651) from the ADMIXTOOLS software44 with the option “inbred:YES” To calculate outgroup f3 in the form of f3 (A, B; Yoruba). We used qpDstat (version 980) from the ADMIXTOOLS software44 with the option “f4mode:YES” to calculate f4 statistics in the form of f4 (Yoruba, A; B, C). Standard error (Std.err) was calculated with 5 cM block jackknifing as implemented in the ADMIXTOOLS software44.
Admixture modelling using qpAdm
We modelled our genomes as an admixture of two source populations and estimated the proportions of ancestry using qpAdm (version 1000) in ADMIXTOOLS software44, with settings “details:YES” and “allsnps:YES”. We chose following populations as the base outgroups: Mbuti.DG, Loschbour.DG, CHG, Onge.DG, Anatolia_N_published, Russia_EHG, Iran_GanjDareh_N, Shandong_EN (pooled by Boshan, Bianbian, Xiaogao, and Xiaojingshan), Mongolia_N_East, Japan_Jomon,Kazakhstan_Eneolithic_Botai, Taiwan_Hanben, YR_MN. All the conditions listed below should be satisfied: (1) Coefficient >0 and Coefficient ± Std.err >0; (2) p-value > 0.01; (3) p-value for the nested model <0.05.
Estimating the admixture dates
We applied the DATES algorithm (version: 753)47 under a single pulse admixture model to date the admixture for a single ancient genome. We used the following parameters: “binsize: 0.001”, “maxdis: 1”, “runmode: 1”, “mincunt: 1”, and “lovaifit: 0.45”. We assumed 29 years per generation44.
Runs of homozygosity
ROH refers to segments of the genome where the two chromosomes in an individual are identical to each other owing to recent common ancestry. Therefore, the presence of long ROH segments strongly suggests that an individual’s parents are related. We applied the hapROH method using the Python library hapROH19 with default parameters. The method was developed to identify ROH from low-coverage genotype data typical of ancient DNA and is still robust enough to identify ROH for individuals with coverage down to 0.5×. We reported the total sum of ROH longer than 4, 8, 12 and 20 cM and visualized the results using Datagraph.
Statistics and reproducibility
Modern Human contamination rates were estimated <1% based on ContamMix and ANGSD software. Detailed information of the statistical analyses carried out as described in the ‘Methods’ section. All analyses can be reproduced by accessing the associated data linked in the Data availability statement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The BAM files reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA009378). All other data are available from the corresponding author on reasonable request.
Code availability
No new code and sequence analysis methods were developed. Details on the analysis settings are provided in ‘Methods’ section.
Abbreviations
- SNPs:
-
single nucleotide polymorphisms
- HO:
-
Human Origin
- PCA:
-
principal components analysis
- N:
-
Neolithic; EN, Early Neolithic
- MN:
-
Middle Neolithic
- LN:
-
Late Neolithic
- BA:
-
Bronze Age
- IA:
-
Iron Age
- HE:
-
Historical era
- YR:
-
Yellow River
- ANA:
-
ancient Northeast Asian
- cal BP:
-
calibrated years before present
- ROH:
-
Runs of homozygosity
- cM:
-
centiMorgans.
References
Xiang, D. Tang Dynasty Chang’an and Western Regions Civilization (Hebei Education Press, 2001).
Kumar, V. et al. Bronze and Iron Age population movements underlie Xinjiang population history. Science 376, 62–69 (2022).
Zhang, F. et al. The genomic origins of the Bronze Age Tarim Basin mummies. Nature 599, 256–261 (2021).
Mao, Y. Hu people from the Western Regions in the Yellow River Basin during the Northern Dynasties to the Sui and Tang Dynasties. Root Explorer 2, 35–41 (2006).
Ma, Y. Examination of Central Asians coming to China in the Late Eastern Han Dynasty. J. Xinjiang Univ. (Philos. Soc. Sci. Ed.) 2, 18–28 (1984).
Fu, Y. On the integration and complementarity between the Hu and Han ethnic groups in the Tang Dynasty. J. Shandong Univ. (Philos. Soc. Sci. Ed.) 3, 55–64 (1992).
Ning, C. et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat. Commun. 11, 2700 (2020).
Xiong, J. et al. Inferring the demographic history of Hexi Corridor over the past two millennia from ancient genomes. Sci. Bull. 69, 606–611 (2024).
Ma, H. et al. Ancient genomes shed light on the long-term genetic stability in the Central Plain of China. Science Bulletin. https://doi.org/10.1016/j.scib.2024.07.024 (2024).
Lv, M. et al. Ancient genomes from the Tang Dynasty capital reveal the genetic legacy of trans-Eurasian communication at the eastern end of Silk Road. BMC Biol. 22, 267 (2024).
Fan, W. An surname Hu people in the Tang Dynasty. J. Inn. Mong. Univ. (Human. Soc. Sci. Ed.) 2, 55–62 (1998).
Shen, Q. et al. Ancient genomes illuminate the demographic history of Shandong over the past two millennia. J. Genet. Genomics. https://doi.org/10.1016/j.jgg.2024.07.008 (2024).
Narasimhan, V. M. The formation of human populations in South and Central Asia. Science 365, eaat7487 (2019).
Damgaard, P. et al. 137 ancient human genomes from across the Eurasian steppes. Nature 557, 369–374 (2018).
Cilli, E. et al. The genetic legacy of the Yaghnobis: A witness of an ancient Eurasian ancestry in the historically reshuffled Central Asian gene pool. Am. J. Phys. Anthropol. 168, 717–728 (2019).
Du, P. et al. Ancient genome of the Chinese Emperor Wu of Northern Zhou. Curr. Biol. 34, 1587–1595.e5 (2024).
Wang, C.-C. et al. Genomic insights into the formation of human populations in East Asia. Nature 591, 413–419 (2021).
Han, X. The settlement and sinicization of Central Asians in Chang’an during the Tang Dynasty. Ethn. Stud. 3, 63–72+109 (2000). (in Chinese).
Ringbauer, H., Novembre, J. & Steinrücken, M. Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nat. Commun. 12, 5425 (2021).
Du, P. et al. Genomic dynamics of the Lower Yellow River Valley since the Early Neolithic. Curr. Biol. 34, 3996–4006.e11 (2024).
Rong, X. Medieval China and Foreign Civilizations (SDX Joint Publishing Company, 2014).
Van Klinken, G. J. Bone collagen quality indicators for palaeodietary and radiocarbon measurements. J. Archaeol. Sci. 26, 687–695 (1999).
Zhu, S. et al. The compact AMS facility at Guangzhou Institute of Geochemistry, Chinese Academy of Sciences. Nucl. Instrum. Methods Phys. Res. Sect. B: Beam Interact. Mater. At. 361, 72–75 (2015).
Ramsey, C. B. & Lee, S. Recent and Planned Developments of the Program OxCal. Radiocarbon 55, 720–730 (2013).
Reimer, P. J. et al. The IntCal20 Northern Hemisphere radiocarbon age calibration curve (0–55 cal kBP). Radiocarbon 62, 725–757 (2020).
Knapp, M., Clarke, A. C., Horsburgh, K. A. & Matisoo-Smith, E. A. Setting the stage—building and working in an ancient DNA laboratory. Ann. Anat. - Anat. Anz. 194, 3–6 (2012).
Llamas, B. et al. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR: Sci. Technol. Archaeol. Res. 3, 1–14 (2017).
Zhu, K. et al. Protocol for a comprehensive pipeline to study ancient human genomes. STAR Protoc. 5, 102985 (2024).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Peltzer, A. et al. EAGER: efficient ancient genome reconstruction. Genome Biol. 17, 60 (2016).
Ginolhac, A., Rasmussen, M., Gilbert, M. T. P., Willerslev, E. & Orlando, L. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27, 2153–2155 (2011).
Renaud, G., Slon, V., Duggan, A. T. & Kelso, J. Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 16, 224 (2015).
Fu, Q. et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 23, 553–559 (2013).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinforma. 15, 356 (2014).
Skoglund, P., Storå, J., Götherström, A. & Jakobsson, M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 40, 4477–4482 (2013).
Mittnik, A., Wang, C.-C., Svoboda, J. & Krause, J. A molecular approach to the sexing of the triple burial at the upper paleolithic site of Dolní Věstonice. PLoS ONE 11, e0163019 (2016).
Jun, G., Wing, M. K., Abecasis, G. R. & Kang, H. M. An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res. 25, 918–925 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Alaçamlı, E. et al. READv2: advanced and user-friendly detection of biological relatedness in archaeogenomics. Genome Biol. 25, 216 (2024).
Ralf, A., Montiel González, D., Zhong, K. & Kayser, M. Yleaf: software for human Y-chromosomal haplogroup inference from next-generation sequencing data. Mol. Biol. Evol. 35, 1291–1294 (2018).
Weissensteiner, H. et al. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63 (2016).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Mallick, S. et al. The Allen Ancient DNA Resource (AADR) a curated compendium of ancient human genomes. Sci. Data 11, 182 (2024).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Chintalapati, M., Patterson, N. & Moorjani, P. The spatiotemporal patterns of major human admixture events during the European Holocene. eLife 11, e77625 (2022).
Acknowledgements
The work was funded by the National Natural Science Foundation of China (T2425014 and 32270667), the Natural Science Foundation of Fujian Province of China (2023J06013), the Major Project of the National Social Science Foundation of China granted to Chuan-Chao Wang (21&ZD285), the National Key Research and Development Program of China (2023YFC3303701-02), Open Research Fund of State Key Laboratory of Genetic Engineering at Fudan University (SKLGE-2310), and Open Research Fund of Forensic Genetics Key Laboratory of the Ministry of Public Security (2023FGKFKT07). We sincerely thank the editors and reviewers for their contributions and suggestions. S.F. and Z.X. from the Information and Network Center of Xiamen University are acknowledged for their help with high-performance computing.
Author information
Authors and Affiliations
Contributions
C.-C.W. conceived and supervised the project. W.L. and B.S. provided the materials and resources. J.L., Y.W., and Y.Z. performed the archaeological data analysis. H.M., H.H., L.Q., and H.C. performed the wet laboratory work. R.W. performed the genetic data analysis and prepared the figures. R.W. wrote the manuscript. C.-C.W. edited the manuscript. All authors contributed to the article and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Guanglin He and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Zuzana Hofmanova and Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, R., Liu, W., Wu, Y. et al. East and West admixture in eastern China of Tang Dynasty inferred from ancient human genomes. Commun Biol 8, 219 (2025). https://doi.org/10.1038/s42003-025-07665-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07665-0
