
Abstract
Spodoptera frugiperda is a common and severely damaging agricultural pest. In-depth analysis of its population genomics and transcriptomics is crucial for providing references for pest control efforts. This study, focused on the extensive variation in the genome size of S. frugiperda, constructed its pan-genome and identified 1.37 Gb of non-reference sequences, highlighting significant genetic variation within the population. Analysis of Long Terminal Repeat (LTR) Presence/Absence Variation (PAV) suggests that LTR alterations may be one of the driving factors for genome size variation. Additionally, population gene PAV analysis revealed that variable genes are enriched in functions like acetyltransferase activity, which might be associated with detoxification, implying diverse selection pressures related to detoxification in different S. frugiperda populations. Moreover, 19 horizontal gene transfer (HGT) acquired genes were identified in the reference genome used in this study, which responded to 16 different treatments. Notably, three HGT-acquired genes (SFR02618, SFR05248, and SFR05249) co-expressed with heat shock protein family and responded under treatments with Avermectin and Cypermethrin. This may indicate their involvement in a detoxification mechanism coordinated with heat shock proteins. These results offering new insights into its genomic evolution and the potential functions of HGT-acquired genes.
Similar content being viewed by others


Introduction
The fall armyworm (Spodoptera frugiperda) is a significant agricultural pest, widely distributed across the American continent and has recently invaded Asia and Africa1. This pest poses severe threats to crucial economic crops like maize and cotton, emerging as a global challenge in agricultural production. Despite extensive research, our understanding of the genetic mechanisms underlying its adaptability, resistance to pesticides, and other key biological characteristics remains limited. However, with the advancement of high-throughput sequencing technologies, significant progress has been made in the genomic studies of S. frugiperda, with several genome sequences now published2,3,4,5. These studies systematically analyse the evolution and expression of key gene families such as detoxification related and chemosensory related genes, shedding light on the fundamental genomic characteristics of its biological traits.
Analysing the genetic patterns of populations, as opposed to individual genome assemblies, further elucidates the genetic diversity, gene functions (such as detoxification-related functions), and evolutionary history (such as pesticide resistance related) of S. frugiperda. Clark et al. utilised amplified fragment length polymorphism to assess genetic variation and found limited gene flow between S. frugiperda populations, with the majority of the genetic variability within populations6. Gui et al.‘s whole genome resequencing analysis of 137 samples revealed that S. frugiperda in China likely originated from African populations, rather than directly from the Americas. Furthermore, through these whole genome resequencing data, they also noted substantial genetic differences even among different populations within the Americas7. Their study also revealed significant differences in genome size among different individuals of S. frugiperda through genome surveys. Multiple studies have indicated that variations in transposon numbers are a primary factor driving changes in genome size8,9,10; however, whether the variation in genome size of Spodoptera frugiperda is driven by transposons requires further investigation. Additionally, Zhang et al. utilised population resequencing data to analyse genes in the S. frugiperda genome associated with resistance to pesticides and Bacillus thuringiensis (Bt), finding a high occurrence of resistance development to conventional pesticides11. Similarly, Guan et al.‘s analysis of mutational load in multiple detoxification-related gene families in S. frugiperda populations revealed an unreported 12-base insertion mutation in multiple exons of the ATP-binding cassette transporter C2 gene (ABCC2) in Brazilian populations4.
Long-term exposure to insecticides has led to many populations developing resistance to specific insecticides. For example, in lufenuron-resistant S. frugiperda, larvae exhibited significantly higher gene expression of cytochrome P450 monooxygenases compared to lufenuron-sensitive larvae12. Extensive research has been conducted on the gene response patterns of S. frugiperda under various stresses, such as toxins and insecticides. For instance, transcriptome analysis clarified the impact of azadirachtin (an insecticide) on chitin and hormone synthesis pathways, thereby modulating the growth of S. frugiperda13. Gui et al. treated S. frugiperda with 23 types of insecticides, including biopesticides, chemical pesticides, and their combinations, thoroughly analysing the response patterns of detoxification-related genes to different insecticides7.
Significant progress has been made in understanding genetic patterns and detoxification mechanisms from both genomic and transcriptomic perspectives in the aforementioned studies. However, due to the complexity of genomic variations, a single reference genome is insufficient to fully capture the genetic diversity within a species. For example, the pan-genome constructed using long-read sequencing data from 545 domestic silkworms revealed a high density of genomic variations (SNPs and indels) in the silkworm population. Specifically, 7308 new genes were identified, and 88% of the genes in the pan-genome were found to be variable genes14. The studies mentioned above have elucidated the rich genetic variation in S. frugiperda due to its long-term coevolution with plants and recent human influences. The pan-genome represents the complete set of genes within a species or population, including core genes shared by all individuals and variable genes present in some but not all. By studying the pan-genome, it is possible to uncover hidden genetic diversity, understand population-specific traits, and identify genes involved in adaptation and resistance. Thus, this study aims to construct the pan-genome of S. frugiperda to investigate genomic changes, gene PAV, and their functions. It also reveals potential patterns of horizontal gene transfer and functional insights, providing new perspectives for better understanding the evolution of S. frugiperda genome and for improving control measures against this pest.
Result
S. frugiperda pan-genome construction and gene PAV analysis
After mapping the resequencing data of 137 samples to the S. frugiperda reference genome, unmapped reads were extracted and assembled into contigs. Following the removal of redundancy and contamination, a total of 1.37 Gb of non-reference sequences were obtained. Employing a variety of methods including transcript assembly from RNA-seq data, ab initio gene prediction, and genomic structure prediction based on homologous proteins, 8603 genes were identified within these non-reference sequences. The number of protein-coding genes in S. frugiperda reference genome is 20,659. By merging the reference and non-reference sequences, a comprehensive pan-genome of S. frugiperda was constructed, encompassing 1.92 Gb of sequence and 29,262 protein-coding genes. Resequencing data were re-aligned to the pan-genome for PAV calling, revealing that the gene count in the 137 resequenced samples ranged between 23,557 and 26,806 (Supplementary Data 1). The fitted curves of pan-genome and core-genome gene numbers, derived from 100 repeated samplings, indicate a continuous decrease in the number of core genes within the population of S. frugiperda with an increase in sample size. Notably, when the number of sequenced individuals is below 50, an increase in their number significantly raises the total gene count in the pan-genome (Fig. 1A). This suggests that S. frugiperda samples from different regions are subject to environmental selection on gene PAV or genetic drift during adaptation to various regional environments. Based on the PAV information of genes in the pan-genome, this study identified 17,616 core genes (present in all samples), 1200 softcore genes (present in 99% to 100% of samples), 10,291 shell genes (present in 1% to 99% of samples), and 155 cloud genes (present in <1% of individuals) (Fig. 1B).

A The fitting graph showing the total number of genes in the pan-genome and the number of core genes as the sample size varies (each sample size point is randomly drawn 100 times, followed by fitting). B The distribution of core genes, softcore genes, shell genes, and cloud genes, with a pie chart showing the proportions of different gene types. C Distribution of gene numbers in different regions of S. frugiperda, calculated based on gene PAV. D Phylogenetic tree of S. frugiperda and population structure obtained based on binary gene PAV data. E KEGG pathway enrichment analysis results for shell genes. F Pfam enrichment analysis results for shell genes.
Violin plots of gene counts from samples across different regions revealed that the gene count in S. frugiperda from Kenya was higher than that in individuals from other regions (Fig. 1C). Population structure analysis using binary PAV data of shell genes indicated significant genetic differentiation in gene population structure among four American S. frugiperda populations, with potential gene flow between Chinese and African populations (Fig. 1D). Furthermore, enrichment analysis of shell genes revealed that these genes are enriched in several gene functions, such as the L-glutamate biosynthetic process, acetyltransferase activity, and histone acetyltransferase activity, which may relate to S. frugiperda’s resistance to external factors and accelerated metabolism of pesticides (Supplementary fig. S1, Supplementary Data 2). KEGG enrichment analysis showed that these genes are enriched in the Alanine, aspartate, and glutamate metabolism pathways (Fig. 1E, Supplementary Data 3). PFAM enrichment results for shell genes showed significant enrichment in several PFAM domains related to transposons (Fig. 1F, Supplementary Data 4), including Transposase IS4 (PF13843), Pao retrotransposon peptidase (PF05380), Integrase core domain (PF00665), and DDE superfamily endonuclease (PF03184).
Characteristics of detoxification-related genes in the genome
The S. frugiperda consists of two morphologically identical but genetically distinct strains: the corn strain (C strain) and the rice strain (R strain). Each strain exhibits specific physiological traits that lead to distinct responses to biological and chemical agents15. Gui et al. collected S. frugiperda samples from populations with varying diets and environmental conditions, which may result in different selective pressures on detoxification-related genes across populations in the pan-genome constructed in this study. This study identified 74, 114, 171, 53, and 39 genes respectively for ATP-binding cassette (ABC), Carboxyl esterase (CES), Cytochrome P450 (CYP), Glutathione S-transferase (GST), and UDP-glucuronosyl transferase (UGT), which are all associated with insect detoxification, from a pan-genomic perspective (Supplementary Data 5). Among these, the ABC gene family had the highest proportion of core genes (81%), whereas the CES gene family had the lowest (54%) (Fig. 2A). There were 10, 47, 30, 23, and 6 genes located in non-reference regions for these five gene families, respectively. Gene PAV originates from structural variations, revealing that different gene families bear diverse structural variation loads within populations due to environmental influences.

Characteristics of the gene distribution in the detoxification gene family, including the distribution of five different family genes (A), exon length (B), gene length (C), number of exons (D), number of expressed genes (E), and maximum expression level in FPKM values (F). The upper bar graphs represent the number of genes, while the stacked charts below depict the distribution of core and shell genes.
To investigate which genes are more likely to exhibit gene PAVs in S. frugiperda. The present study also investigated the exon length distribution of these detoxification genes. The average exon length for core genes was 2089 bp, and for shell genes, it was 920 bp. In the detoxification gene families, the exon lengths of shell genes were mainly distributed in the range of 0.2 ~ 1 kb, whereas those of core genes were broadly distributed between 0.6 ~ 5 kb (Fig. 2B). The total length of the genes followed a similar pattern, with core genes being longer than shell genes (Fig. 2C). Similarly, compared to core genes, shell genes in the detoxification group tended to have fewer exons (Fig. 2D). Research by Gui et al. on the transcriptome of S. frugiperda under 23 different insecticides, including flutolanil, thiamethoxam, and cyantraniliprole, revealed the response patterns of detoxification-related genes under different insecticide treatments. This study found that 91.86% of the detoxification-related genes responding to at least one insecticide were core genes (Fig. 2E). These genes also exhibited a trend where core genes were more numerous than shell genes in having the highest expression levels under insecticide treatment (Fig. 2F). However, it is crucial to consider shell detoxification genes that are highly expressed in response to insecticide treatment, as these genes may exhibit population or individual specificity. Genomic differences among individuals could lead to observable phenotypic variations.
Transposons influence the genome size of the fall armyworm
To investigate the potential impact of transposable element (TE) abundance on genome size, we quantified the number of Long Terminal Repeats (LTRs) in the pan-genome of S. frugiperda using LTRharvest, identifying a total of 721,422 LTRs (Supplementary Data 6). These LTRs mainly fall into three categories: Gypsy, Copia, and Unknown, all of which exhibit similar distribution trends across chromosomes, though with uneven distribution on individual chromosomes and higher densities on certain chromosomes (Fig. 3A). In this study, we conducted a genome survey on 32 high-depth sequenced samples (>60x coverage), with genome sizes ranging from 510 Mb to 977 Mb. Considering that LTRs are repetitive sequences, using a low threshold for LTR PAV calling could lead to erroneous results. Therefore, we performed LTR PAV calling at three coverage thresholds: greater than or equal to 0.8, greater than or equal to 0.9, and equal to 1. The results of the simple linear regression analysis with genome size revealed a significant correlation between the number of LTRs and genome size when the coverage of 0.8 indicates the presence of LTR. However, the correlation was weak (R2 was relatively small). At a coverage threshold of 1, the significance of the correlation between LTR number and genome size increased to 1.28 ×10-5, with an R2 of 0.4751 (Fig. 3B). This progressive increase in correlation coefficients may be due to false positives in LTR PAV at lower thresholds. Since LTRs are regions of significant variation in the genome, their alterations often lead to changes in genome size9,10. These findings suggest that the content of LTRs is one of the reasons for the considerable variation in genome size within the species of S. frugiperda. Interestingly, this aligns with the results of the shell gene pfam enrichment analysis mentioned earlier.

A Density distribution of LTRs across chromosomes; B Correlation between LTR PAV at different coverage levels (80%, 90%, 100%) and genome size in S. frugiperda populations; C Histogram of the length distribution of LTR retrotransposons; D Distribution of Core and Shell LTR numbers in three different types of pesticide treatments; E Distribution of Copia, Gypsy, and Unknown transposon numbers under three different pesticide treatments.
The identified transposable elements ranged in length from 100 bp to 998 bp, with the majority being <200 bp (Fig. 3C). To study the transcription patterns in LTR regions, we analyzed the expression of transposable elements >500 bp, identifying 6708 such elements. We calculated their expression under 23 different pesticide treatments (Supplementary Data 7), and the results showed that the largest number of LTRs were expressed in mixed pesticide treatments (Fig. 3D). The fewest differentially expressed LTRs were observed under biological pesticide treatments, and most of these expressible transposable elements were shell LTRs. Different families of LTRs respond to insecticides in varying numbers, with the Gypsy family showing the highest number, 1128, under mixed insecticide treatments (Fig. 3E).
Horizontal gene transfer analysis
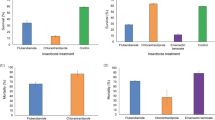
In this study, 19 HGT-acquired genes (Supplementary Data 8, Supplementary fig. S2) were identified in the SFynMstLFR reference genome (indexed in the CNSA database as CNP0000513). The coding sequence (CDS) lengths of these 19 HGT-acquired genes ranged from 82 bp to 60097 bp. They exhibited longer average lengths compared to other genes. While the GC content at the third codon position and the distribution of intron numbers were similar to other genes (Fig. 4A). The HGT-acquired genes play multiple important biological roles in insects. This study found that HGT-acquired genes in S. frugiperda are mainly enriched in domains like ubiquitin (Fig. 4B). Additionally, these genes are enriched in pathways such as transferase activity and pyridoxal phosphate binding (Fig. 4C). Insects’ detoxification processes for toxic secondary metabolites in plants or artificially applied insecticides often involve oxidation and transfer of functional groups16. Hence, the pathway enrichment results of HGT in S. frugiperda suggest that these HGT-acquired genes might play a role in detoxification. According to the PAV analysis of HGT-acquired genes in S. frugiperda population, only SFR04193 (softcore) and SFR19266 (shell gene) are variable genes. SFR19266 is absent in 9 USB_D samples, resulting in a gene frequency of only 0.3 in the USB_D population. The UniProtKB-ID of SFR04193 and SFR19266, as per their alignment with UniProt data, is YGI2_BACTU, a gene from Listeria. This gene, which is not functionally annotated, contains the Papain-like_cys_pep_sf domain and belongs to the papain-like cysteine peptidase superfamily. USA_D, a hybrid population of USA A, B, and C, predominantly inhabits cornfields. Transcriptome data analysis (PRJNA893082) found that the SFR19266 gene is significantly lowly expressed in Bt-resistance-strain vs WT (Fig. 4D). Before encountering Bt-resistance-strain, SFR19266 may have played an important biological role, such as in detoxification17. However, the artificially created Bt-resistance-strain, which lacks detoxification functionality against Bt proteins and is suppressive in expression, leads to the loss of this gene in some individuals of the USB_D population. In the fall armyworm population, the 19 HGT-acquired genes exhibit numerous start lost and missense variants (Fig. 4E, Supplementary Data 9), suggesting that environmental selection acts on these genes post-transfer.

A Distributions of sequence lengths, GC content at the third codon position (GC3), and the number of introns in HGT and other genes. B Most common PFAM protein domains identified within the candidate HGT-acquired genes. Enrichment p-values (p ≤ 0.05) for selected functions are shown. C Gene ontology functions enriched in the putative HGT set. Enrichment p-values (p ≤ 0.05) for selected functions are displayed. D Frequency of gene SFR19266 differing in populations (top) and its expression level between resistant strains and WT (wild type) (bottom), ** indicates significant difference in differential expression (q < 0.01). E Waterfall plot of the of HGT-acquired genes within populations.
Expression patterns of HGT-acquired genes under various stress conditions
In this study, we collected 123 RNA-seq samples from 8 different BioProjects (Supplementary Data 10). These samples encompassed a range of 23 sample groups, including virus infection, pesticide treatment, and feeding on various food sources. A significant proportion of these samples involved stress treatments on S. frugiperda, such as Microplitis manila parasitized, various pesticide treatments, virus infection, and feeding on insect-resistant plants. Differential expression was identified across 19 of these groups. The reference genome of S. frugiperda comprises 20,659 genes, of which 9807 exhibited differential expression in these 19 sample groups compared to the control (Fig. 5A, Supplementary Data 11). There were 285 genes differentially expressed under 19 treatments. Specifically, during responses to pesticide and resistance-strain, the numbers of DEGs increased to 5320 and 5102, respectively, with 1765 and 1738 unique DEGs in each treatment (Fig. 5B). This pattern may be because plant resistance to insects is related to secondary metabolites. The diversity of secondary metabolites in plants is considered a crucial adaptive determinant for the interactions between plants and other organisms in the natural environment and the diversity of secondary metabolites results in an increased variety of genes involved in the interactions between insects and plants. Similarly, the diversity in artificially applied pesticides, including a wide range of biological and chemical pesticides, has also contributed. Long-term natural and anthropogenic selection pressures have shaped the unique genomic composition and gene expression system of S. frugiperda.

A Expression patterns of genes differentially expressed (q-value < 0.05, |log2FoldChange | >1) across all treatments. B Venn diagram showing the number of DEGs across different treatment groups. C Expression patterns of differentially expressed HGT-acquired genes under various treatments. D Network diagram of DEGs co-expressed with HGT-acquired genes. Treatments A, B, C, and D correspond to different treatment groups: A: Parasitized; B: Pesticide; C: Resistance-strain; D: Virus-infected.
Differing from the overall genomic expression pattern, 17 out of 19 HGT-acquired genes showed differential expression in at least one stress condition (Fig. 5C). This revealed the significant role of horizontal gene transfer events in the environmental adaptation of S. frugiperda. The highest number of differentially expressed HGT-acquired genes was observed under HdIV infection and feeding on different corn varieties (14 genes). There were no differentially expressed HGT-acquired genes under Cypermethrin and Nora-virus-infected treatments. To further elucidate the biological functions undertaken by the HGT-acquired genes, this study conducted a co-expression analysis with other DEGs (Fig. 5D). 10 HGT-acquired genes were co-expressed with other DEGs. Functional enrichment analysis of co-expressed genes in the four major categories (Pesticide, Resistance-strain, Virus-infected, and Parasitized) revealed that the pfams of genes co-expressed with SFR02618, SFR05248, and SFR05249 under pesticide treatment were enriched in the HSP20 family, potentially related to drug resistance.
Discussion
The S. frugiperda is a common and severely harmful agricultural pest. The short breeding cycle and rapid intraspecific genomic variation of agricultural pests significantly impact their ability to quickly adapt to various environments, including developing resistance to pesticides18. This study conducted a survey analysis of the genomes of 32 high-depth sequenced S. frugiperda and found that the size of their genomes ranged from 510 Mb to 977 Mb. Given the considerable variation in genome size, it is essential to further investigate differences in genomic components, gene count, and gene function catalogues within these genomes. Currently, large-scale genome resequencing efforts for pan-genome studies are limited in agricultural pests (such as Heliconius butterflies)19, while similar studies in plants and mammals have uncovered numerous functional genes20,21. To comprehensively study this rich genomic variation, this research constructed the first pan-genome of S. frugiperda, which includes 1.37 Gb of non-reference sequences. These sequences are 2.5 times the size of the reference genome (550 Mb), highlighting the extensive intraspecific genomic variation within S. frugiperda. Previous studies have shown that the genetic diversity within Spodoptera frugiperda populations is greater than that between populations6,7,22. In this study, the phylogenetic tree constructed based on PAV analysis places the four populations from the USA at opposite ends, indicating a significant level of genetic differentiation within the US populations. Based on the pan-genome, an intriguing question arises: What genomic components drive the variation in genome size? Analysis of the non-reference sequences revealed that 7% of these sequences are transposable elements. Transposable elements are often variable parts of the genome, and multiple studies have shown that changes in transposable elements can affect genome size23. In plants, for example, LTRs are an important type that influences genome size24. So, do LTRs in insect genomes also drive changes in genome size? This study, based on rigorous LTR PAV calling (with 100% read coverage alignment), correlated LTR PAVs with genome size and found a correlation coefficient of 0.4751, with a p-value of 1.28 e-5. These findings suggest that LTR variation is one of the significant drivers of changes in the genome size of S. frugiperda.
The construction of a pan-genome not only aids in understanding the driving factors behind genome size variation but also facilitates a comprehensive understanding of protein-coding genes in S. frugiperda. In the reference genome, the number of protein-coding genes identified is 20,659, while an additional 8603 protein-coding genes have been identified in the non-reference segment. The diversity in genome size is also reflected in the variability in the number of protein-coding genes within the genome, particularly in S. frugiperda from the Kenya region, where the gene count is notably higher than in other regions. This provides clues for studying the impact of regional environmental factors on the genomic evolution of agricultural pests. Moreover, the pan-genome analysis identified shell genes in the population that may arise through various pathways, such as genes present in ancestors being lost in some individuals or the creation of new genes in some individuals due to gene duplication. The enrichment of these variable genes in pathways like ‘drug metabolism—cytochrome P450’ and ‘drug metabolism—other enzymes’ suggests the influence of exogenous chemicals (such as plant secondary metabolites or insecticides) on the variability of insect-encoded protein gene numbers. It is noteworthy that the detoxification-related genes in insects co-evolve with the diversity of plant secondary metabolites25, Insects living in different geographical environments encounter different plants, leading to different natural selection pressures on their detoxification-related genes. Thus, the 116 detoxification-related genes identified in the non-reference segment of this study represent an expansion of the detoxification gene pool of S. frugiperda. Although many studies have found that point mutations or indels contribute to insecticide resistance in insects26, the impact of gene PAV should not be overlooked. This can help us to more comprehensively understand the development of insect disease resistance.
Genes from other organisms such as fungi, bacteria, plants, and viruses are commonly integrated into the insect genome through HGT, impacting the insects’ detoxification capabilities and defence mechanisms27. In this study, 19 HGT genes were identified in the SFynMstLFR version genome, a number higher than that observed in orders outside Lepidoptera28. This may be associated with the presence of PiggyBac elements, a unique type of transposon found in Lepidoptera29. Pfam enrichment analysis of shell genes revealed an accumulation in the Transposase IS4. Transposases can recognize the ITRs at both ends of a transposon. If a protein-coding gene is present between two ITRs, it may also be integrated into the insect30. This phenomenon may represent one of the potential mechanisms underlying HGT in S. frugiperda.
The phenomenon of HGT from the genomic level of microorganisms to the genome of insects, followed by the acquisition of transcriptional activity, often accompanies the emergence of new functions. These functions play a significant role in aspects such as insect reproduction and response to toxic substances31,32. This study investigates the HGT-acquired genes in S. frugiperda using various types of RNA-seq data. Compared to control, 17 out of 19 HGT-acquired genes in S. frugiperda showed differential expression under multiple treatments. In organisms, genes with closely related expression patterns often have similar functions, and they may be upstream or downstream genes in a metabolic or signalling pathway. Consequently, this study further infers the potential functions of HGT-acquired genes in S. frugiperda genome using co-expression analysis. Results revealed that under pesticide treatments, the pfams of DEGs co-expressed with the HGT-acquired genes (SFR02618, SFR05248, and SFR05249) were enriched in the HSP20. Previous research has identified the involvement of the insect HSP gene family in resistance to pesticide like pyrethroids33. The identification of differential expression of HGT-acquired genes SFR02618, SFR05248, and SFR05249 under pesticide treatment and their co-expression with other HSP genes provides new clues for discovering novel mechanisms of pesticide resistance.
In summary, this study includes a series of analyses, including pan-genome construction, based on the population resequencing data of S. frugiperda. The possible driving factors for the significant changes of the genomic size variation in S. frugiperda have been identified. Combined with the identification of HGT-acquired genes and expression analysis under various treatments, the study offers conjectures on the patterns and potential functions of HGT in S. frugiperda. These findings will contribute new insights into further understanding the genomic evolutionary patterns of S. frugiperda, an important agricultural insect, and offer novel strategies for controlling S. frugiperda.
Methods
Genomic and transcriptomic data acquisition
This study acquired whole-genome resequencing data of 137 S. frugiperda individuals from America, Africa, and China7. These data were converted to fastq format using fastq-dump and subsequently processed with fastp to remove low-quality and adaptor sequences34. The cleaned data were then mapped to the genome of a female Spodoptera frugiperda from Yunnan, China, assembled by Gui et al. (the genome file was retrieved from the FAW genome database at https://159.226.67.243:8080/fawmine), using BWA35. Additionally, we obtained transcriptome data from 72 samples used in Gui et al.‘s study7, which include control samples and samples treated with 23 insecticides (biopesticides, chemical pesticides, and mixed pesticides). Furthermore, RNA-seq data from 152 samples across 12 bioprojects in NCBI were used: PRJNA514995, PRJNA602034, PRJNA64563613, PRJNA72959836, PRJNA760971, PRJNA81890037, PRJNA861097, PRJNA87768938, PRJNA884429, PRJNA88609639, PRJNA89308240, and PRJNA910105 (Supplementary Data 10).
Pan-genome construction
We used an iterative mapping and assembly approach to construct the S. frugiperda pan-genome. Bam files generated as aforementioned were processed using the fastq function of samtools (-f12, -f 68 -F8, and -f 132 -F 8) to extract unmapped reads41. Unmapped reads were assembled using MaSuRCA42 individually, discarding contigs <500 bp. Each sample’s contigs were aligned against the NT (NCBI non-redundant nucleotide) database using blastn to remove contigs from archaea, viruses, bacteria, fungi, and viridiplantae. Additionally, contigs from each sample were annotated using Kraken2 against the kraken2-microbial database43, with sequences identified as of microbial origin being removed. All sample contigs were merged, and redundancies were eliminated using cd-hit (default parameters) to yield the final non-reference sequences44. These non-reference sequences file was merged with the reference genome sequences file to form S. frugiperda pan-genome.
Pan-genome annotation
Non-reference contigs of S. frugiperda were annotated for repetitive sequences using RepeatMasker (http://repeatmasker.org) based on RepBase (v17.01, http://www.girinst.org/repbase). Additionally, a de novo repeat sequence library was constructed using RepeatModeler45, followed by another round of annotation with RepeatMasker. Tandem Repeats in non-reference contigs were annotated using Tandem Repeats Finder46. RNA-seq data were mapped to non-reference contigs using hisat247, and unmapped reads were extracted with samtools. Trinity was used for assembling unmapped reads, followed by redundancy removal with cd-hit-est48. Finally, gene structures in non-reference contigs were predicted using maker2, integrating transcriptomes assembled from RNA-seq, protein sequences from the reference genome, and structures predicted de novo by august (trained on the reference genome)49.
Gene PAV analysis
The resequencing data of 163 individuals were mapped to S. frugiperda pan-genome using BWA. A gene was considered present if at least 2 reads covered 20% of its exonic regions; otherwise, it was deemed absent. This analysis was conducted using SGSGeneLoss v0.1 (minCov = 2, lostCutoff = 0.2). Presence and absence were denoted as ‘1’ and ‘0’, respectively. Based on this binary gene PAV data, a maximum-likelihood phylogenetic tree of S. frugiperda was constructed using iqtree (1000 bootstraps). Population structure analysis was performed using STRUCTURE on the binary gene PAV data.
Horizontal gene transfer analysis
This study conducted a horizontal gene transfer analysis on genes located on the chromosomes of S. frugiperda and in contigs >100 kb. The identification of HGT-acquired genes in this research was guided by the study of Li et al.28. Initially, the protein sequences of S. frugiperda were compared with the Refseq database (last accessed May 5, 2023) using the blastp function of DIAMOND (e-value < e-5). Based on the NCBI Taxonomy database, taxonomic information for the species of each sequence that matched those in the Refseq database was obtained. The sequences identified through blastp were categorised into three groups: insects (RECIPIENT), other metazoans (GROUP), and non-metazoan (OUTGROUP). Based on this taxonomic information and the blastp results, three values were calculated: bbhO (the bitscore of the best match in the OUTGROUP from the blastp results), bbhG (the bitscore of the best match belonging to the GROUP but not to the RECIPIENT), and maxB (the bitscore of self-comparison). Using these values, the Alien Index was calculated: AI = (bbhO / maxB) - (bbhG / maxB). Additionally, the percentage of species from OUTGROUP lineage (outg_pct) in the list of the top 1,000 hits that have different taxonomic species names was calculated. Genes that met the criteria of AI value > 0 and outg_pct ≥ 65% proceeded to further analysis.
Using MAFFT (with the “–auto” option), multiple sequence alignments were conducted for each gene’s top 1000 homologous sequences in the Refseq database50. The ambiguous alignment regions were trimmed using trimAl (with the “-automated1” option)51. Subsequently, IQ-TREE was used to construct the maximum likelihood trees52, with the best-fitting model of amino acid evolution and 1000 ultrafast bootstrapping replicates. The phylogenetic trees were visualised with ggtree R package (Yu et al., 2017) to identify putative HGT-acquired genes.
Retrotransposon analysis
LTR retrotransposons were identified using the default parameters of LTRharvest (v.1.5.10)53. For 32 samples with a sequencing depth >60x, a genome survey based on K-mer was conducted using the GCE software54. LTR calling was performed on these 32 samples using HUPAN55. Considering false positives, LTRs with coverage greater than or equal to 0.8, greater than or equal to 0.9, and equal to 1 were considered existent, and LTR PAV (Presence-Absence Variation) statistics were conducted. The results of these three statistics were subjected to a univariate linear regression analysis with genome size.
Gene expression analysis
Fastp was used for quality control of the transcriptome data, and the quality-controlled transcriptome data were mapped to the genome using hisat2, with the genome sourced from Gui et al.7. Reads count were calculated using featureCounts56, and differential expression analysis was conducted using deseq257, and | log2 fold change | ≥ 1 and FDR < 0.05 were used as a cut-off for significant differential expression. Heatmaps were created using the R package ComplexHeatmap58. The Pearson correlation coefficients were calculated for HGT-acquired genes and differentially expressed genes (DEGs). Gene pairs with an absolute value of Pearson correlation coefficient >0.85 and a q-value <0.01 were considered co-expressed. The co-expression results are visualized using Cytoscape (Shannon et al., 2022).
Data availability
The data are presented in the manuscript and the supporting materials. The resequencing data were acquired from CNSA with accession CNP0001020. The RNA-seq data were acquired from NCBI (PRJNA514995, PRJNA602034, PRJNA645636, PRJNA729598, PRJNA760971, PRJNA818900, PRJNA861097, PRJNA877689, PRJNA884429, PRJNA886096, PRJNA893082, and PRJNA910105) and CNSA (CNP0001020). Pan-Genome data sequences can be downloaded from this website (https://doi.org/10.6084/m9.figshare.25106663.v1, https://doi.org/10.6084/m9.figshare.25106837.v1, https://doi.org/10.6084/m9.figshare.25106852.v1).
References
Tay, W. T., Meagher, R. L., Czepak, C. & Groot, A. T. Spodoptera frugiperda: Ecology, Evolution, and Management Options of an Invasive Species. Annu. Rev. Entomol. 68, 299–317 (2023).
Kakumani, P. K., Malhotra, P., Mukherjee, S. K. & Bhatnagar, R. K. A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics 104, 134–143 (2014).
Gouin, A. et al. Two genomes of highly polyphagous lepidopteran pests (Spodoptera frugiperda, Noctuidae) with different host-plant ranges. Sci. Rep. 7, 11816 (2017).
Guan, F. et al. Whole-genome sequencing to detect mutations associated with resistance to insecticides and Bt proteins in Spodoptera frugiperda. Insect Sci. 28, 627–638 (2021).
Xiao, H. et al. The genetic adaptations of fall armyworm Spodoptera frugiperda facilitated its rapid global dispersal and invasion. Mol. Ecol. Resour. 20, 1050–1068 (2020).
Clark, P. L. et al. Population variation of the fall armyworm, Spodoptera frugiperda, in the Western Hemisphere. J. Insect Sci. 7, 5 (2007).
Gui, F. et al. Genomic and transcriptomic analysis unveils population evolution and development of pesticide resistance in fall armyworm Spodoptera frugiperda. Protein Cell 13, 513–531 (2022).
Bennetzen, J. L. Mechanisms and rates of genome expansion and contraction in flowering plants. Genetica 115, 29–36 (2002).
Zhang, Q. J. et al. The Chromosome-Level Reference Genome of Tea Tree Unveils Recent Bursts of Non-autonomous LTR Retrotransposons in Driving Genome Size Evolution. Mol. Plant 13, 935–938 (2020).
Ye, X. et al. Genomic signatures associated with maintenance of genome stability and venom turnover in two parasitoid wasps. Nat. Commun. 13, 6417 (2022).
Zhang, L. et al. Genetic structure and insecticide resistance characteristics of fall armyworm populations invading China. Mol. Ecol. Resour. 20, 1682–1696 (2020).
Nascimento, A. R. B., do, Fresia, P., Cônsoli, F. L. & Omoto, C. Comparative transcriptome analysis of lufenuron-resistant and susceptible strains of Spodoptera frugiperda (Lepidoptera: Noctuidae). BMC Genomics 16, 985 (2015).
Shu, B. et al. Identification of azadirachtin responsive genes in Spodoptera frugiperda larvae based on RNA-seq. Pestic. Biochem. Physiol. 172, 104745 (2021).
Tong, X. et al. High-resolution silkworm pan-genome provides genetic insights into artificial selection and ecological adaptation. Nat. Commun. 13, 5619 (2022).
Adamczyk, J. J. Jr., Holloway, J. W., Leonard, B. R. & Graves, J. B. Susceptibility of fall armyworm collected from different plant hosts to selected insecticides and transgenic Bt cotton. J. Cotton Sci. 1, 21–28 (1997).
Hernandez, E. P. et al. Glutathione S-transferases play a role in the detoxification of flumethrin and chlorpyrifos in Haemaphysalis longicornis. Parasit. Vectors 11, 460 (2018).
Ozhelvaci, F. & Steczkiewicz, K. Identification and classification of papain-like cysteine proteinases. J. Biol. Chem. 299, 104801 (2023).
Cohen, Z. P., Schoville, S. D. & Hawthorne, D. J. The role of structural variants in pest adaptation and genome evolution of the Colorado potato beetle, Leptinotarsa decemlineata (Say). Mol. Ecol. 32, 1425–1440 (2023).
Ruggieri, A. A. et al. A butterfly pan-genome reveals that a large amount of structural variation underlies the evolution of chromatin accessibility. Genome Res. 32, 1862–1875 (2022).
Zhou, Y. et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 606, 527–534 (2022).
Li, M. et al. De Novo Assembly of 20 Chicken Genomes Reveals the Undetectable Phenomenon for Thousands of Core Genes on Microchromosomes and Subtelomeric Regions. Mol. Biol. Evol. 39, msac066 (2022).
Cabusas, J. V. B., Latina, R. A. & Caoili, B. L. Genetic diversity and population structure of the fall armyworm, Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae) in the Philippines. J. Appl. Entomol. 148, 983–995 (2024).
Lee, S.-I. & Kim, N.-S. Transposable Elements and Genome Size Variations in Plants. Genomics Inf. 12, 87–97 (2014).
Zedek, F., Šmerda, J., Šmarda, P. & Bureš, P. Correlated evolution of LTR retrotransposons and genome size in the genus eleocharis. BMC Plant Biol. 10, 265 (2010).
Heidel-Fischer, H. M. & Vogel, H. Molecular mechanisms of insect adaptation to plant secondary compounds. Curr. Opin. Insect Sci. 8, 8–14 (2015).
Kim, J. et al. Novel diamide resistance-linked mutation in Korean Spodoptera exigua and a LAMP assay based on a mutation-associated intronic InDel. J. Pest Sci. (2004). 94, 1017–1029 (2021).
Boto, L. Horizontal gene transfer in the acquisition of novel traits by metazoans. Proc. R. Soc. B Biol. Sci. 281, 20132450 (2014).
Li, Y. et al. HGT is widespread in insects and contributes to male courtship in lepidopterans. Cell 185, 2975–2987.e10 (2022).
Elick, T. A., Bauser, C. A. & Fraser, M. J. Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica 98, 33–41 (1996).
Häcker, I. et al. Improved piggyBac Transformation with Capped Transposase mRNA in Pest Insects. International Journal Molecular Sciences 24, 15155 (2023). vol.
Jorgenson, M. A., Chen, Y., Yahashiri, A., Popham, D. L. & Weiss, D. S. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Mol. Microbiol. 93, 113–128 (2014).
Nikoh, N. et al. Bacterial Genes in the Aphid Genome: Absence of Functional Gene Transfer from Buchnera to Its Host. PLOS Genet. 6, e1000827 (2010).
Si, F.-L. et al. HSP superfamily of genes in the malaria vector Anopheles sinensis: diversity, phylogenetics and association with pyrethroid resistance. Malar. J. 18, 132 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Malook, S., Liu, X.-F., Liu, W., Qi, J. & Zhou, S. The race goes on: A fall armyworm-resistant maize inbred line influences insect oral secretion elicitation activity and nullifies herbivore suppression of plant defense. bioRxiv https://doi.org/10.1101/2021.05.17.444416 (2021).
Gulinuer, A., Xing, B. & Yang, L. Host Transcriptome Analysis of Spodoptera frugiperda Larvae Parasitized by Microplitis manilae. Insects vol. 14 (2023).
Shu, H. et al. The transcriptomic profile of Spodoptera frugiperda differs in response to a novel insecticide, cyproflanilide, compared to chlorantraniliprole and avermectin. BMC Genomics 24, 3 (2023).
Zheng, R. et al. Comparative transcriptome analysis reveals key candidate genes mediating ovarian development in Spodoptera frugiperda fed on two host plants. Front. Physiol. 13, (2022).
Jin, M. et al. Downregulation of a transcription factor associated with resistance to Bt toxin Vip3Aa in the invasive fall armyworm. Proc. Natl. Acad. Sci. 120, e2306932120 (2023).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013).
Lu, J. & Salzberg, S. L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome 8, 124 (2020).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. Usa. 117, 9451–9457 (2020).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Grabherr, M. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. 29, 644–652 (2011).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 1–14 (2011).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, (2008).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. archivePrefix 1308, 2012 (2020).
Duan, Z. et al. HUPAN: a pan-genome analysis pipeline for human genomes. Genome Biol. 20, 149 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gu, Z. Complex heatmap visualization. iMeta 1, e43 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32100355, 32100352, 31871964) and the National Key R&D Program of China (2021YFD1400700).
Author information
Authors and Affiliations
Contributions
YXH, XDK, and YS conceived and designed the experiments. YS, HYR, BSS, JML, JJL and XW contributed to paper writing. YXH, XDK, LNX and YS contributed to the data analysis. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, YX., Rao, HY., Su, BS. et al. The pan-genome of Spodoptera frugiperda provides new insights into genome evolution and horizontal gene transfer. Commun Biol 8, 407 (2025). https://doi.org/10.1038/s42003-025-07707-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07707-7
