
Abstract
Enveloped viruses enter cells by binding to receptors present on host cell membranes, which trigger internalization and membrane fusion. For many viruses, this either directly or indirectly involves interaction with membrane-anchored carbohydrates, such as heparan sulfate, providing a potential target for a broad-spectrum antiviral approach. Based on this hypothesis, we screened a library of functionalized chitosan sulfates that mimic heparan sulfate in cellular membranes for inhibition of SARS-CoV-2 and respiratory syncytial virus (RSV) entry. An array of compounds blocking SARS-CoV-2 and RSV were identified, with the lead compound displaying broad-spectrum activity against multiple viral strains and clinical isolates. Mechanism of action studies showed the drug to block viral entry irreversibly, likely via a virucidal mechanism. Importantly, the drug was non-toxic in vivo and showed potent post-exposure therapeutic activity against both SARS-CoV-2 and RSV. Together, these results highlight the potential of functionalized carbohydrates as broad-spectrum antivirals targeting respiratory viruses.
Similar content being viewed by others


Introduction
The COVID-19 pandemic has had an unprecedented global economic and societal impact, highlighting the need for broad-spectrum antiviral agents for a rapid response to emerging and re-emerging viral infections1,2. Indeed, it is estimated that a large number of lives could have been saved if an antiviral was available early in the pandemic3. To this end, the development of antiviral drugs targeting mechanisms commonly used by viruses can provide a rapid response to new viral threats. Heparan sulfate proteoglycans (HSPGs) are a family of glycoproteins that are present on the cell surface or in the extracellular matrix (ECM) of mammalian cells and are involved in numerous key biological processes, such as cell adhesion, proliferation, differentiation, and inflammation4,5,6. They consist of a core protein that is covalently bound to unbranched, sulfated heterogeneous polysaccharides known as heparan sulfate (HS). The basic building block of HS consists of repeating disaccharide units of uronic acid (β-D-glucuronic acid or α-L-iduronic acid) linked to α-D-glucosamine by (1,4)-glycosidic bonds (Fig. 1a). This disaccharide is sulfated at different positions, mainly in the glucosamine unit.

a Schematic representation of the role of native heparan sulfate (HS) in virus attachment and infection. b HS mimics act as receptor decoys, blocking the binding of the virus to target cells and inactivating viral glycoproteins, preventing future entry into cells.
Many viruses have evolved to use the HSPGs present on the surface of host cells as an attachment site before entering the cell7,8,9. Due to the highly sulfated HS chains, HSPGs have a global negative charge that can interact electrostatically with the basic residues of viral surface glycoproteins or viral capsid proteins of non-enveloped viruses. The many individual ionic bonds that can be established between the viral protein and the polysaccharide facilitate adhesion of the virus to the cell surface. Viruses capitalize on this interaction to increase their concentration on the cell surface and improve their chances of binding a more specific entry receptor10,11,12 (Fig. 1a). In some cases, this interaction triggers a conformational change in the viral protein that enables binding to the specific cellular receptor13.
Among the viruses that use HSs as attachment points to infect cells are Severe Acute Respiratory Syndrome Coronavirus14,15 (SARS-CoV-2) and respiratory syncytial virus16 (RSV). Several studies have shown that the protein Spike (S) of SARS-CoV-2 binds HSs prior to interacting with specific cellular receptor14,15. This binding to HS has been suggested to occur cooperatively, shifting the S structure to an open conformation that facilitates binding to the viral cognate receptor, ACE215.
Likewise, the binding to cells of RSV involves an initial interaction between the viral envelope proteins G and F and HS16. These interactions induce a conformational change in the F protein resulting in the exposure of its fusion peptide, promoting fusion of the viral envelope and the cell plasma membrane and release of the nucleocapsid into the cytoplasm17. Multiple additional viruses from different viral families utilize HSs to attach to cells, including dengue virus18, human papillomaviruses19, and others20.
Considering the amount of data showing the role of HSs in facilitating viral infection, a promising antiviral strategy could be to employ soluble HS fragments to coat viral entry proteins, thereby preventing their binding to target cells (Fig. 1). However, despite major advances in the chemical synthesis of HS oligosaccharides21, the preparation of these molecules is still challenging and the scarce availability of homogeneous HS from natural sources remains a major obstacle to such antiviral therapy. Using more accessible polysaccharides that can act as HS mimics provides an attractive alternative22. In this context, sulfated polysaccharides derived from marine and natural biota, such as fucoidan23 or carrageenan24, have been shown to have promising pharmacological properties for antiviral drug development25. Heparin, a highly sulfated HS derivative, has provided promising results for the treatment of virus infections26,27. Additionally, a non-sulfated amphiphilic chitosan was shown to decrease the infectivity of SARS-COV-228, while cationic chitosan derivatives have been employed to inactivate HIV‑1 and SARS-CoV‑229. Finally, chitosan itself has been reported to exhibit antiviral activity30.
In 2018 we reported that chitosan sulfate can mimic some biological functions of HS31,32,33. Chitosan is a polysaccharide derived from chitin, an abundant polysaccharide that can be found in the exoskeleton of crustacea. It is a linear polysaccharide composed of varying amounts of (β1→4)-linked residues of glucosamine (GlcN) and N-acetyl-glucosamine (GlNAc) residues34. Chitosan contains a primary amino group and primary and secondary hydroxyl groups with different reactivity, allowing for selective O- and N-sulfonation31,35, which can alter their biological properties.
Herein, we evaluate the antiviral activity of multiple chitosan sulfate derivatives of different chain lengths having varying numbers and positions of sulfonyl groups and substitution at the C-2 amino group. Specifically, the amino group of chitosan was acylated with an oleic acid chain, since the introduction of a hydrophobic residue in sulfonated carbohydrate derivatives was shown to give rise to compounds with virucidal activity against a variety of viruses, including SARS-CoV-236 and RSV37,38. Herein, we identify a chitosan sulfate with a long hydrocarbon chain in the amino group (N-oleoyl derivative) with a broad-spectrum antiviral activity against multiple strains of SARS-CoV-2 and RSV. Cell culture and structural studies showed this compound to block entry in a non-reversible manner, likely via a virucidal mechanism. The compound was well tolerated in vivo and blocked SARS-CoV-2 and RSV replication in mice when administered both pre- and post-infection. Overall, we identify an N-oleoyl-substituted chitosan sulfate as a promising, broad-spectrum antiviral compound that overcomes production limitations confronted by natural HS sources.
Results
Synthesis of Chitosan Derivatives
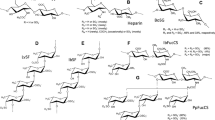
The structure of the synthesized compounds is shown in general formula I (Fig. 2). Compounds were designated by a code Mw-mX-nY-N, O-Z, where Mw is the molecular weight of the starting chitosan, X is the percentage of GlcN units with N-acetyl group, Y is the percentage of GlcN units with N-oleoyl group, O- and N- designates sulfonation on hydroxyl and amine groups, respectively, and Z the degree of sulfation. The synthesis of chitosan sulfate was carried out following previously described procedures31,35 (see “Material and Methods”).

General chemical structure of the synthesized compounds and their designated code.
Chitosan derivatives show antiviral activity against SARS-CoV-2
To evaluate whether the chitosan derivatives possessed antiviral activity in the absence of toxicity, we used a previously described pseudotyped vesicular stomatitis virus (VSV) assay that utilizes SARS-CoV-2 S protein to enter cells39. Briefly, VSV pseudotyped with the Wuhan-Hu-1 S protein (VSV-Swh) was treated with the polysaccharides at a single concentration of 1 mg/mL prior to infection of Vero cells. Viral infection was then assessed by measuring the levels of viral-expressed luciferase, followed by evaluation of toxicity using a resazurin reduction assay (i.e. Alamar Blue assay). Heparin, carrageenan, and chondroitin sulfate were included as natural source-derived sulfated polysaccharides with reported antiviral activity25. The chitosan sulfate compounds with an oleoyl group (compounds 12–19 in Fig. 3b) showed inhibitory effects on VSV-Swh entry that were comparable to or more pronounced than those of sulfate polysaccharides found in natural sources (compounds on the left side of the graph in Fig. 3b). The strongest antiviral activity was observed for compounds 12, 13, 14, 17 and 19, which achieved >92% reduction (p < 0.05 for all by two-tailed t-test on log-transformed data) at non-toxic concentrations. Of note, the beneficial effect of the oleoyl group on antiviral activity agrees with previous observations in other sulfated carbohydrate derivatives with a hydrophobic moiety36,37.

a Compound numbering and structures of the tested polysaccharides. The effect of the indicated compounds on viral infection and cellular viability of VSV pseudotyped with SARS-CoV-2 Swh in Vero cells (b) or RSV-AmKate2 in Vero cells (c) or A549 (d) cells. Data represents the mean and SEM of three replicates.
Chitosan derivatives show potent antiviral activity against RSV
As RSV is also known to utilize GAGs to enter cells40, we similarly evaluated the ability of these derivatives to block RSV infection. Sulfated polysaccharides from natural sources were included for comparison, although some chitosan-based compounds were omitted due to poor activity against VSV-Swh (Fig. 3c, d). Briefly, an RSV-A strain expressing the fluorescent protein mKate2 (RSV-AmKate2) was pretreated with 1 mg/mL of the compounds prior to infection of either Vero or human lung-derived A549 cells. Examination of viral-driven fluorescent protein expression showed in both cell lines a > 95% reduction of infection compared to mock treatment for all synthetic derivatives with minimal toxicity for most compounds (>75% viability using the resazurin reduction assay; Fig. 3c, d). Stronger inhibition of RSV infection was observed for natural sulfated polysaccharides than against SARS-CoV-2, highlighting the increased dependence of this virus on polysaccharides for entry. For the remainder of the paper, compound 17 was selected due to its strong inhibition of both viruses and efficient isolation process.
Compound 17 shows broad antiviral activity
We first validated the ability of compound 17 to block infection by genuine SARS-CoV-2. For this, the ability of compound 17 to block infection with SARS-CoV-2 encoding a D614G S mutation found in nearly all variants of concern (VOC) was assessed in both VeroE6-TMPRSS2 cells and A549 cells encoding the human ACE2 receptor (A549-ACE2). Ribavirin, a nucleoside analog with broad-spectrum antiviral activity41, was included as a positive control. As expected, ribavirin resulted in strong antiviral activity (>99% reduction in virus production) in both cell lines (p = 0.004 and 0.002 by two-sided t-test on log-transformed data for A549-ACE2 and VeroE6-TMPRSS2, respectively; Fig. 4a). Compound 17 led to a reduction of 82.3 ± 16.4% and 95.9 ± 1.53% reduction of virus production in A549-ACE2 and VeroE6-TMPRSS2, respectively, confirming the results obtained with VSV-Swh (p = 0.04 and 0.002 by two-sided t-test on log-transformed data for A549-ACE2 cells and VeroE6-TMPRSS2, respectively; Fig. 4a). Finally, as SARS-CoV-2 VOC encoding numerous mutations in the S gene have arisen during the evolution of the virus in the human population, we assessed the ability of compound 17 to inhibit VSV pseudotyped with different omicron variants (BA1, BA2, BA4.5). The entry of all VOC was inhibited by compound 17, with the different omicron S showing increased sensitivity relative to Wuhan-Hu-1 (range 6.8–10 fold; p < 0.05 by two-tailed t-test for all; Fig. 4b). This likely reflects an increase of positively charged amino acids in the S receptor binding domain (RBD) of omicron variants42,43,44, which can enhance binding to the negative charges present in compound 17. Importantly, no toxicity was observed at concentrations as high as 5 mg/mL (Supplementary Fig. 1a).

a Effect of compound 17 (1 mg/mL) or ribavirin (20μM) on SARS-CoV-2 production in A549-ACE2 or VeroE6-TMPRSS2 cells. Virus production was assayed by limiting dilution and the results are expressed as tissue culture infectious dose (TCID50) per mL. b The IC50 of compound 17 against VSV pseudotyped with the indicated SARS-CoV-2 S protein in VeroE6-TMPRSS2 cells. c The IC50 of compound 17 against RSV-AmKate2. d The effect of compound 17 on the production of the indicated RSV strains. Viruses were treated with 10 μg/mL of compound 17 for 1 h prior to infection of HEp-2 cells and virus production was assayed at 72 h post-infection by plaque assay. Data represent the number of plaque forming units (PFU) per mL. All assays indicate the mean and SE of 3 independent replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-tailed t-test on log-transformed values (a, d), untransformed data (b), or model parameter comparison (c).
To better understand the sensitivity of RSV to compound 17 compared to different SARS-CoV-2 variants, the IC50 for the laboratory RSV-AmKate2 strain was obtained in human A549 and HEp-2 cells. Strikingly, IC50 values for RSV-AmKate2 in both cell lines were >3 orders of magnitude lower than for any of the SARS-CoV-2 variants, highlighting the strong dependence of RSV on HS for entry (Fig. 4c and Supplementary Fig. 1b for cell viability). To examine if compound 17 was efficacious against different RSV-A strains as well as RSV-B, we tested its ability to reduce virus production of two RSV-A lab strains (RSV-AmKate2 and RSV-A2) as well as a single clinical isolate from 2016 of RSV-A and RSV-B. In all cases, compound 17 reduced virus production by >98% in all conditions (p < 0.05 by two-tailed t-test on log-transformed data; Fig. 4d). Finally, compound 17 also inhibited infection by Sindbis virus, a representative member of the unrelated Alphavirus genus, with an IC50 of 1.23 ± 0.29 μg/mL, supporting a broad-spectrum antiviral activity against a diverse array of viruses (Supplementary Fig. 2).
Mechanism of action of compound 17
To assess whether compound 17 binds directly to the SARS-CoV-2 S protein and define the binding region, microscale thermophoresis was initially attempted using either purified S or the S RBD. However, the fluorescently labeled proteins precipitated in the presence of the compound, precluding the use of this technique but suggesting an interaction (data not shown). Consequently, surface plasmon resonance (SPR) experiments were performed to quantify the binding of the compounds with either the full extracellular domain of S, the N-terminal S1 subunit alone (encompassing RBD), or the RBD alone. These proteins were covalently linked to the surface of the sensor chips and different concentrations of compound 17 were flowed. The results showed that RBD had the lowest binding affinity (KD = 87 nM), the full S had higher binding affinity (KD = 59 nM), and the S1 subunit showed the highest affinity (KD = 38 nM). The high binding affinity of the S1 subunit to polysaccharide was further supported by the fact that the chip could only be regenerated using a harsh regeneration reagent. To examine whether the binding of compound 17 to S results in S trimer destabilization, thermal shift assays were performed using a highly stable version of Wuhan-Hu-1 S (S-HexaPro)45. A concentration-dependent destabilization of the S protein was observed in the thermogram upon compound addition, most notably at concentrations >0.52 mg/mL (Fig. 5a).

a Thermostability of SWuhan Hexapro in the presence of compound 17 measured using Thermofluor assays. A stock solution of compound 17 (39 mg/mL) was diluted 1:100, 1:75, 1:50, or 1:20 in the reaction mixture. b A time-of-addition assay for RSV shows compound 17 to affect viral entry. c The effect of compound 17 is not readily reversible. Virus stocks were treated with the indicated drug concentrations during the pre-infection, infection, and post-infection steps. Virus infection was quantified by plaque assay and standardized relative to mock-treated virus stocks. All assays indicate the mean and SE of at least 3 independent replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-tailed t-test on log-transformed values.
We next evaluated whether compound 17 affected S structure directly using negative stain electron microscopy of both S-Hexapro and the SD614G. In the absence of the compound, particles of both S proteins were evenly distributed across the grid and exhibited the characteristic triangular shape for the intact Spike trimer (Supplementary Fig. 3). However, the addition of compound 17 resulted in strong alterations of both S variants, reducing particle numbers and leading to the emergence of aggregates and smaller particles (Supplementary Fig. 3). Together, these results demonstrate a direct effect of the compound on S integrity.
To examine if compound 17 affected RSV entry, we performed a time-of-addition assay. For this, the drug was present throughout the experiment (pre-, during, and post-infection; pre+post), pre- and during infection (pre), or only post-infection (post; Fig. 5b). Virus infection was quantified at 24 h when the fluorescent signal is readily observed from the initial round of infection. An antiviral effect was observed only when the compound was present during the infection process (pre+post or pre), confirming a direct role in viral entry (Fig. 5b) and excluding a post-entry mechanism. As compound 17 was found to disrupt SARS-CoV-2 structure, we evaluated whether the anti-RSV activity was also a result of virucidal activity (e.g. non-reversible). For this, concentrated RSV stocks were preincubated with compound 17 at a concentration resulting in strong antiviral activity (1 μg/mL) and then diluted 1000-fold to infect cells at a concentration at which the compound no longer shows antiviral activity (1 ng/mL; Fig. 5c). Following infection, cells were washed to remove both unbound virus and compound and incubated for seven days for plaques to develop. Despite infection occurring at a concentration lacking antiviral activity (1 ng/mL), a strong reduction in virus production was observed when the virus was pre-treated with drug concentrations exhibiting antiviral activity (1 μg/mL), similar to that observed for cells infected with RSV in the presence of 1 μg/mL compound 17 (Fig. 5c). In contrast, low concentrations of the drug did not affect virus infection (1 ng/mL; Fig. 5c). Hence, the effect of the drug on RSV infectivity is not readily reversible, potentially as a result of protein misfolding of the glycoproteins as observed for SARS-CoV-2 S.
Preliminary toxicological assessment of compound 17
To assess the tolerability of compound 17 in vivo, a dose range finding experiment was done to determine both the maximum tolerated concentration and the dose without observed adverse effects (NOAEL). As both viruses affect the airways, the intranasal route of administration was chosen. An initial single-dose escalation study with concentrations ranging from 1–100 mg/kg was performed with mice receiving 25 µL per nostril of compound 17 in saline solution. No behavioral alterations, changes with toxicological relevance, or macroscopic alterations in organs were observed even at the highest dose (See Supplementary Information section 7.1). Next, the effect of four consecutive daily intranasal administrations of three doses (45, 60, and 100 mg/kg) was evaluated. Again, no adverse effects were observed (see Supplementary Information section 7.2). Hence, compound 17 shows good tolerability, with multiple doses of up to 100 mg/kg causing no adverse observable effects.
Biodistribution of nasally administered compound 17 in mice
Having established the safety profile of the drug in vivo, we next examined its biodistribution to define the kinetics of accumulation and clearance across different tissues. To enable visualization of the drug in mice using single photon emission computerized tomography (SPECT) whole-body imaging, a 4-hydroxyphenylmethyl residue was attached to a small proportion (4%) of amino groups of the chitosan by reductive amination of compound 17 with 4-hydroxybenzaldehyde and the resulting product was labeled with iodine-131 (131I) using iodination beads (see Supplementary Information, section 8, and Supplementary Fig. 4). In order to determine the residence time of the labeled polymer in the nostril under anesthetic conditions, the labeled polymer was administered in the left nostril (10 µL; 5.7 ± 0.7 MBq; 14.0 ± 0.8 mg/Kg) with the animals under anesthesia. A sequence of static 8-min SPECT images was then obtained over ~2 h, followed by computerized tomography (CT) images for anatomical reference. SPECT images revealed that intranasal administration resulted in ~90% of the dose deposited in the nostril, while the remaining dose was deposited in the throat or swallowed by the animal immediately after or during administration (Fig. 6a). Interestingly, the concentration of radioactivity in the nostril remained constant over the whole study, thus confirming minimal translocation of the labeled polymer in this time window (Fig. 6b).

a Representative SPECT/CT images (coronal views) obtained at different times after intranasal administration of the 131I-labeled compound 17 with the animal under anesthesia. b Percentage of administered dose remaining at the nostril at different times post-administration relative to the amount of radioactivity found immediately after administration for each mouse. c Representative SPECT/CT whole body images (coronal views) obtained at different times after intranasal administration of the 131I-labeled compound 17 with the animal recovering from anesthesia after administration and in between imaging sessions. For A and C images are maximum intensity projections (MIP) and have been co-registered with a 3D-rendered image of the CT. Scale bars, expressed as standard uptake value (SUV), have been adjusted for proper visualization of the presence of radioactivity in the different regions.
To define the long-term biodistribution of the labeled polymer at the whole-body level, mice were similarly treated and allowed to recover from anesthesia immediately after administration and between imaging sessions. SPECT image analyzes revealed the accumulation of the compound at the administration site (nostril and throat) shortly following administration (30 min), as well as the gastric tract (Fig. 6c). At 4 h post-administration, radioactivity was observed in the stomach, abdominal region, and the administration site, while at 24 h radioactivity was mainly localized at the administration site (nostril and the throat; Fig. 6c).
To obtain a quantitative measurement of compound accumulation in organs, the labeled compound was administered to mice (n = 20) as above, after which mice were allowed to recover from anesthesia. Mice were then sacrificed at different time points (0.5, 3, 6, 24 and 48 h post-administration; n = 4 per group) and radioactivity was measured in different organs (heart, lungs, spleen, muscle, stomach, large intestine, small intestine, liver, kidneys, and brain) and body fluids (blood and urine; Supplementary Fig. 5). The analysis confirmed that a large fraction of the radioactivity is eliminated through the gastrointestinal tract, with the concentration of radioactivity peaking in the stomach at 0.5 h post-administration (7.6 ± 4.7% of the injected radioactivity dose [ID]) and in the small (8.3 ± 4.9% ID) and large intestines at 3 h post-administration (13.0 ± 3.5% ID; Supplementary Fig. 5). Other organs showed low levels of radioactivity except for the urine, suggesting progressive deiodination of the polymer46. The lungs, where virus replication occurs, showed a low concentration of radioactivity (0.11 ± 0.09, 0.14 ± 0.11 and 0.08 ± 0.04%ID at 0.5, 3, and 6 h post-administration), with almost negligible signal at 24 h (~0.007%ID). Overall, >95% of the radioactivity was eliminated from the body at 48 h post administration. In summary, imaging and biodistribution studies are consistent with the passage of most of the labeled compound 17 through the esophagus to the gastrointestinal tract and almost complete elimination after 48 h.
Compound 17 shows antiviral and anti-inflammatory activity in vivo
We next evaluated whether compound 17 was active in vivo. For this, k18-hACE2 mice were treated intranasally with vehicle alone or compound 17 at either 24 mg/kg or 12 mg/kg of compound 17 three hours before intranasal challenge with SARS-CoV-2. Mice were then treated once daily for two additional days with vehicle or compound 17. Viral load in the lungs was determined on day 3 post-infection (Fig. 7a). Strikingly, no virus was detected in any of the lungs of mice treated with the higher dose of compound 17 prior to infection (reduction in viral load >6-log reduction; p = 0.0004 by Mann–Whitney test), while the lower dose reduced viral load by >5 logs (p = 0.001 by Mann-Whitney test; Fig. 7a). To assess the ability of compound 17 to act post-infection, one group of mice received 12 mg/kg of the drug starting at 24 h post-infection. Even when administered at this later time point, viral loads were reduced >3-logs (p = 0.001 by Mann–Whitney test; Fig. 7a). As SARS-CoV-2 pathogenesis is known to be exacerbated by cytokine induction, the levels of IL-1β, IL-6, and tumor necrosis factor-alpha (TNF-α) were analyzed in the lungs of these mice. As expected, SARS-CoV-2 infection resulted in significant induction of all three cytokines (see Vehicle-treated mice vs. uninfected mice; Fig. 7b). All treatments with compound 17, even when applied one day post-infection, resulted in a significant reduction of cytokine induction (Fig. 7b). In particular, TNF-α induction was completely abolished in all treatment conditions.

a Viral load in the lungs of SARS-CoV-2 infected mice, treated daily with either vehicle alone or with compound 17 at the indicated dose starting either 3 h prior to virus challenge (Pre-infection) or 24 h post viral infection (Post-infection; n = 8 per group). b Relative mRNA levels of the indicated cytokine in the lungs of mice from (a). c The number of RSV genome copies per gram of lung tissue for mice infected with RSV-A2 and treated daily with Vehicle or 12 mg/kg of compound 17 starting 24 h post-infection (n = 5 per group). *p < 0.05, **p < 0.01, ***p < 0.001 by Mann-Whitney test.
To assess if compound 17 affected immune activation directly or whether the reduction in cytokine secretion was due to its effect on viral replication, we tested the influence of compound 17 on cytokine secretion in a lipopolysaccharide (LPS)-induced lung inflammation model47 (see Supplementary Information, section 9). Briefly, mice were treated with vehicle or compound 17 (12 mg/kg) daily for three days starting three hours post challenge with either LPS or vehicle. The expression of cytokines involved in the Th1/Th2 response was then determined on day four (GM-CSF, IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-12p70, IL-18 and TNF-α). Compound 17 did not affect the activation of most cytokines analyzed regardless of whether mice were challenged with LPS (Supplementary Fig. 6) except for increased IL-6 and TNF-α secretion in the absence or presence of LPS challenge, respectively (p < 0.05 by Mann-Whitney test with Bonferroni multiple test correction for both). Hence, compound 17 shows a strong and specific reduction of both SARS-CoV-2 replication and cytokine secretion in the murine model of infection.
We next assessed whether compound 17 showed antiviral activity against RSV infection in vivo. As treatment was efficacious when applied 24 h post-infection in SARS-CoV-2 infected mice, a similar protocol was evaluated. Specifically, mice were challenged with RSV-A2 intranasally, and either mock-treated or treated with 12 mg/kg of compound 17 daily starting 24 h after viral infection. Viral load in the lungs was then assessed on day 4 following infection by qPCR. A statistically significant, four-fold reduction in viral load was observed in the lungs of compound 17 treated mice compared to vehicle-treated mice (p = 0.00794 by Mann–Whitney test; Fig. 7c). Of note, due to the relatively poor replication of RSV in murine lungs, post-infection treatment does not lead to strong reductions in viral titers by qPCR even with the use of clinically approved monoclonal antibody therapy48,49,50, and hence the 4-fold reduction observed for compound 17 is likely clinically relevant.
Finally, since HS and chitosan sulfates are known to affect coagulation51,52, we assessed whether compound 17 treatment altered these processes in vivo (see Supplemental Information, section 9). For this, mice were treated i.n. for four days with compound 17 or vehicle, and coagulation was assessed on day five using prothrombin time (PT) and activated partial thromboplastin time (APTT). PT was unaffected by compound 17 treatment (7.58 ± 1.60 s and 7.17 ± 0.28 s for vehicle and compound 17, respectively) while APTT was increased (19.03 ± 1.96 s and 23.27 ± 2.1 s for vehicle and compound 17, respectively; p < 0.01 by Mann–Whitney test; Supplementary Fig. 7). These patterns are consistent with the effect of widely utilized anticoagulants on clotting, such as unfractionated heparin52, and should be taken into account for any antiviral therapy based on such compounds.
Discussion
Despite the tremendous global burden imposed by viral infection, few effective prevention or treatment options are currently available. Vaccination remains the most effective means of reducing the burden of viral infection, yet vaccines are only available for a few viruses and can be inefficient in the most affected population groups, such as the immune-compromised, elderly, or newborns53,54. Biologicals, such as monoclonal antibodies, have also been employed to treat viral infections55,56 but their implementation on a large scale remains limited due to high production and administration costs. Moreover, the highly specific nature of antibody-antigen binding can frequently be selected for variants that bypass neutralization56, limiting their long-term utility. Antiviral compounds, on the other hand, have proven to be useful for the treatment of viral infections for the few viruses for which they are available57, although treatment with multiple compounds is frequently required to overcome issues with drug resistance. In general, such therapies can be less costly to produce and implement.
Effective antivirals can in principle interfere with all steps of the viral replication cycle. However, targeting the initial viral entry steps can increase efficiency by blocking the virus prior to its logarithmic replication phase and concomitantly reduce toxicity by bypassing the need to enter the host cell. Moreover, viral entry inhibitors can have broad-spectrum antiviral activity as multiple viruses can utilize common receptors and/or host factors to enter cells58. An example of such host attachment factors are HSPGs, which are utilized by multiple viruses to bind host cells9,40,44 via electrostatic interactions between positively charged amino acid residues of viral surface proteins and the negatively charged sulfate groups of HSPGs. In the current work, we screened a diverse panel of sulfated chitosans and found multiple compounds to show antiviral activity with low or no toxicity (Fig. 2c–e). In particular, compounds with sulfonyl groups on the oxygen atoms and a small percentage of a hydrophobic oleoyl residue on the amino group of the glucosamine unit showed strong antiviral activity. Overall, the number and diversity of derivatives showing antiviral activity highlights the broad applicability of antiviral approaches based on mimics of cellular HSPGs and the potential for combinatorial therapy should antiviral resistance become problematic.
From our initial screening, we selected compound 17 for further study. Aside from the strong antiviral activity of this compound, it is obtained from readily available commercial chitosan. Evaluation of compound 17’s antiviral activity showed its ability to block the entry of multiple SARS-CoV-2 and RSV strains into host cells, highlighting its broad-spectrum antiviral activity. Mechanism of action studies revealed compound 17 to bind to the S protein of SARS-CoV-2, in particular the S1 subunit containing the RBD, and to disrupt S trimers (Fig. 5a). A similar virucidal type activity was observed for RSV, where dilution of the compound to concentrations without antiviral effect could not recover infectivity (Fig. 5b). RSV encodes two viral glycoproteins that are involved in entry, the fusion (F) protein and the attachment glycoprotein (G), both of which have been reported to bind heparan sulfate in vitro16. However, as only the F protein is required for entry in cell culture16 compound 17 is likely to directly interact with it.
Unfortunately, we were unable to obtain a high-resolution structure of compound 17 bound to two different SARS-CoV-2 constructs due to the ability of the compound to disrupt S trimers. Hence, detailed structural insights into the binding interactions could not be directly defined. However, the detrimental effect of the polysaccharide on the protein could be due to the presence of both the negatively charged sulfate groups and the hydrophobic oleoyl residue. Indeed, a fully sulfated tetrasaccharide containing a hydrophobic cholesterol (PG545) with potent antiviral activity against SARS-CoV-2 has been shown to induce destabilization of the S1-RBD structure, with the side chain of the lipophilic steroid being essential for its activity36. What makes RSV over 1000-fold more sensitive to compound 17 compared to SARS-CoV-2 entry remains to be investigated. This could be the result of multiple factors, such as the number of binding sites per viral glycoprotein, the inherent lower stability of the protein, or the slower rate of infection of RSV versus SARS-CoV-2, which allows for a larger window of time for the antiviral to act. Additional experiments are required to better understand these mechanisms.
Toxicological assessment of compound 17 showed it to be non-toxic following multiple administrations near the limit of its solubility (100 mg/mL) when administered intranasally. This dose was considered as the NOAEL. Biodistribution and radio imaging of 131I-labeled compound 17 also revealed that most of the compound is eliminated from the body at 48 h post-administration, indicating that the risk of systemic toxicity is negligible. Administration of lower doses than the NOAEL of 100 mg/mL was sufficient to result in a profound reduction of both viral titers and inflammatory responses following challenge with SARS-CoV-2, even when treatment was initiated one-day post-challenge. As a cytokine storm induced by SARS-CoV-2 is known to play an important role in pathogenesis59, these results highlight the potential of compound 17 to limit viral-induced pathology. In this context, it has been shown that heparin, which is a highly sulfated form of heparan sulfate, can modulate immune responses in COVID-19 patients60 by reducing the expression of pro-inflammatory cytokines61. Interestingly, we found that compound 17 did not reduce cytokine expression in an LPS-induced model of inflammation, suggesting the reduction of inflammatory cytokine in the context of SARS-CoV-2 infection is likely due to effects on viral replication. It is important to note that we did not include a direct comparison with an approved antiviral for SARS-CoV-2 in the current study to comply with the 3 R principle (Replacement, Reduction, and Refinement) as we have previously evaluated the effect of remdesivir in our group under the same settings47. A stronger antiviral activity was observed in the current study with compound 17 compared to remdesivir (>6-log reduction versus 2-log reduction for remdesivir), which was also accompanied by a more significant reduction in cytokine production (see Fig. 7 versus Fig. 5 in47).
Antiviral activity was also observed in a mouse model of RSV infection, albeit at a more modest (4-fold) yet statistically significant level. However, it has been shown that post-infection treatment of mice or rats infected with RSV with even clinically approved monoclonal antibodies does not lead to strong reductions in viral replication when assayed by qPCR48,49,50, as was done in our work. This is likely because RSV does not replicate efficiently in murine infection models, providing a limited window for observing a reduction in viral loads following the initial infection.
In sum, we demonstrate that a synthetic chitosan derivative is a promising antiviral, with broad-spectrum antiviral activity against different SARS-CoV-2 and RSV strains as well as Sindbis virus, is well tolerated in vivo, and is effective at reducing both viral replication and inflammation even 24 h post-infection. The high safety profile of compound 17 suggests it could be utilized prophylactically, potentially as a simple nasal spray. However, additional studies are needed to better define the effect of compound 17 on coagulation in light of the observed increase in APTT, which indicates a delay in the intrinsic coagulation pathway.
Materials and methods
Ethics and biosafety
All work with BSL2 pathogens was approved by the biosafety committees of both I2SysBio and the University of Valencia. All work with genetically modified organisms (GMOs) was approved by the relevant Spanish GMO committee. Work with SARS-CoV-2 in vitro was carried out at the Biosafety Level 3 (BSL-3) Facility of the Fundación para Fomento de Investigación Sanitaria y Biomédica (FISABIO) in Valencia, Spain, following approval by the relevant committees. Work with SARS-CoV-2 in vivo was carried out at the BSL-3 Facility of Centro de Investigación en Sanidad Animal (CISA, INA-CSIC) in Valdeolmos, Madrid, Spain. Animal experimentation for RSV was approved by the Valencian government (code GVRTE/2022/3642381), by the Division of Animal Protection of the Comunidad de Madrid (PROEX 115.5-21) for SARS-CoV-2, and by the Division of Animal Protection of the Comunidad de Madrid (PRO-AE-SS-207) for biodistribution studies. We have complied with all relevant ethical regulations for animal use.
Synthesis and characterization of polysaccharides
The synthesis of chitosan sulfate was carried out using chitosan with different molecular weights and degrees of acetylation (Mw-mX) as the starting material following the schematic in Supplementary Fig. 8. Chemical O-sulfation and N- sulfonation reactions were carried out under acidic or basic conditions, respectively. N-Oleoyl chitosan derivatives were prepared by N-acylation of chitosan using oleic acid in the presence of 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (step 2), to yield compounds Mw-mX-nY. Sulfonation reactions on hydroxyl groups were carried out under homogeneous conditions in formic acid, using chlorosulfonic acid as reagent33. Under these conditions, starting chitosan Mw-mX led to O-sulfonated products Mw-mX-O-Z (step 5). Likewise, the compounds bearing the oleoyl group furnished O-sulfonated derivatives Mw-mX-nY-O-Z (step 6). In neutral or mildly basic conditions, amine is the most reactive group, and thus chemoselective N-sulfonation can be performed using SO3·pyridine complex in the presence of a base62. Under these conditions, N-sulfonation of starting chitosan Mw-mX or N-oleoyl derivative Mw-mX-nY led to N-sulfonated products Mw-mX-N-Z and Mw-mX-nY-N-Z, respectively (steps 1 and 3, respectively). The subsequent O-sulfonation afforded products with sulfonyl groups on the oxygen and nitrogen atoms, Mw-mX-O,N-Z and Mw-mX-nY-O,N-Z (steps 4 and 7). The target products were characterized by NMR and IR spectroscopy and the degree of sulfation was determined by elemental analysis. Further details for the experimental procedures and compound characterization of the polysaccharides can be found in the Supplementary Information, sections 1–5, and NMR and ATR-FTIR Spectra in section 10.
Cells, culture conditions, and reagents
HEK293T (CRL-3216), Vero (CCL-81), BHK-21 (CCL-10), and HEp-2 (CCL-23) cells were obtained from ATCC. VeroE6-TMPRSS2 were purchased from the JCRB Cell Bank (catalog JCRB1819), A549-ACE2-TMPRSS2 were purchased from InVivoGen (catalog a549-hace2tpsa), A549-ACE2 were a kind gift of Dr. Oliver Schwartz (Institut Pasteur), and VeroE6 cells were a king gift of Dr. Sonia Zúñiga (the Spanish National Centre for Biotechnology, CNB-CSIC). All cells were cultured in DMEM containing pen-strep with either 10% or 2% FBS for maintenance or infection, respectively. When required for maintain the transgene, the appropriate antibiotic was included. All cells were validated to be mycoplasma-free at regular intervals. Resazurin (Sigma–Aldrich, R7017), MTT (Sigma–Aldrich, 475989), ribavirin (Thermo Scientific, Catalog number 460480010), and neutral red (Sigma–Aldrich, Catalog number N2889-100mL) were purchased from commercial sources.
Virus strains, production, and titration
Pseudotyped vesicular stomatitis virus lacking its native glycoprotein encoding both GFP and firefly luciferase (VSVΔG) was produced by transfection of HEK293T cells using calcium phosphate with the indicated plasmids for 24 h, followed by infection with VSVΔG pseudotyped with the VSV G protein for 45 min, addition of media containing a neutralizing VSV G targeting antibody, and collection of supernatants containing pseudotyped viruses after 16 h. Pseudotyped VSV was concentrated by high-speed centrifugation (50,000 × g for 4 h) and resuspended in DMEM. Viral titer was obtained by infecting the relevant cell type for 16 h in a 96-well plate and quantification of GFP-expressing cells on a live-cell microscope (Incucyte SX5, Sartorius). Plasmids encoding the RSV-A (strain A2 encoding the F gene of Line 19) expressing the mKate2 fluorescent protein were obtained from BEI resources (catalog NR-36460 and related plasmids) and produced from plasmids as previously described63. RSV-A2 was obtained from ATCC (VR-1540) and clinical strains of RSV-A and RSV-B were obtained from EVAG (011V-01970 and 011V-01970, respectively). All RSV strains were amplified in HEp-2 cells at low multiplicity of infection (MOI) of 0.001-0.01. Virus titration was carried out by counting fluorescent cells (RSV-AmKate2) on the Incucyte SX5 live cell microscope and/or by plaque assay on HEp-2 cells using an overlay containing 0.4% Agarose and 2.5% FBS for 7 days. The SARS-CoV-2 isolate utilized for in vitro antiviral activity was kindly provided by Sonia Zúñiga, Isabel Sola and Luis Enjuanes from the Spanish National Centre for Biotechnology (CNB-CSIC) and harbored the following mutations relative to the Wuhan-Hu-1 reference strain (GenBank MN908947): C3037 > T, resulting in a silent mutation, C14408 > T in nsp12, and A23403 > G (D614G in S protein). This virus was amplified in VeroE6-TMPRSS2 cells and tittered in the same cells by limiting dilution as previously described39. Sindbis virus expressing eGFP was recovered from the pTE3'2J-GFP plasmid (a kind gift from Dr. Carla Salah, Institut Pasteur, France) in BHK-21 cells as previously described64. For animal experiments SARS-CoV-2 isolate hCoV-19/Spain/SP-VHIR.02, D614G(S) was propagated and titrated in VeroE6 cells65.
Antiviral and cell viability assays
For the primary screening, viruses were pre-incubated with the indicated compounds at a concentration of 1 mg/mL for 1 h prior to infection of the indicated cell line in 96-well plate format, using ~1000 FFU of virus per well. The virus/drug mixture was then added to cells for 16 h or 24 h for VSV-S or RSV-AmKate2, respectively. Virus infection was assessed by quantification of virus-expressed luciferase using the Thermo Scientific™ Pierce™ Firefly Luciferase Glow Assay Kit (VSV pseudotypes) or examination of red fluorescence using a live cell microscope (Incucyte SX5, Sartorius) for RSV-AmKate2. For the time of addition studies, 1000 FFU RSV-AmKate2 was either pre-treated (pre and pre+post conditions) or mock-treated (post) with 1 μg/mL compound 17 for 1 h, after which cells were infected for 2 h with the mixture. Subsequently, cells were washed, and either media alone (pre) or compound 17 at 1 μg/mL added (pre+post and post). Following 24 h, virus infection was quantified by measuring mKate2 expression. The dose resulting in 50% reduction of viral infection was performed as in the primary screening using a range of 5-fold compound dilutions. Virus infection was assessed via the examination of viral expressed fluorescent proteins at 16 h, 12 h, and 24 h for VSV, Sindbis, and RSV-AmKate2, respectively, and the relative infection was standardized to that of mock-treated cells. The IC50 was then calculated using the DRC package in R using the LL3 model, as previously described39. For the virucidal assay, 104 PFU of RSV-A2 were incubated with PBS or 1 µg/mL compound 17 for 1 h at 37 °C in 0.5 mL, subjected to 3 serial 10-fold dilutions, and used to infect HEp-2 cells for 2 h. Finally, the inoculum was removed, and cells were overlayed with 0.6% agar for 7 days to allow for plaque to develop, followed by fixation with 10% formaldehyde and staining of plaques with 0.05% neutral red in water. In parallel, to ensure antiviral activity was not due to the residual amount of compound 17, 100 PFU of virus were similarly incubated with 1 µg/mL, 1 ng/mL, or dilutant alone and then used to similarly infect cells. For the SARS-CoV-2 virus production assay, 105 tissue culture infectious dose units (TCID50) of the virus were incubated with 1 mg/mL compounds 17, remdesivir (20 μM) or dilutant for 1 h, after which the mix was used to infect VeroE6-TMPRSS2 cells or A549-ACE2 cells for 1 h. Subsequently, the inoculum was removed and fresh media with compound or dilutant was added. After 24 h, the supernatant was collected and tittered via limiting dilution on VeroE6-TMPRSS2 cells. For assessment of virus production by different RSV strains, 5000 PFU of each RSV strain were incubated with dilutant or 10 µg/mL of compound 17 for 1 h at RT and then used to infect HEp-2 cells for 2 h. Cells were then washed and incubated with or without the drug in infection media for 72 h. Viral titer was then obtained in the supernatant by plaque assay on HEp-2 cells. Experiments were performed in triplicate. For viability assessment, cells were treated with different compound concentrations for 24 h, followed by addition of resazurin (44 μM final concentration) or MTT (0.5 mg/mL final concentration) until visible changes in color were observed (~2 h). Subsequently, the relative reduction of these compounds was read using fluorescence (resazurin; excitation of 535 nm and emission of 595 nm) or absorbance (MTT; 570 nM) on a Tecan Spark plate reader and results standardized to those of untreated cells.
Biophysical and structural analyzes
Detailed methods for biophysical and structural analyzes can be found in the Supplementary Information, section 6.
In vivo assays
Detailed methods for the toxicological, biodistribution, inflammation, and coagulation assays can be found in sections 7–9 of the Supplementary Information. To assess antiviral activity against SARS-CoV-2, 6-week-old hemizygous K18-hACE2 transgenic female mice were used (n = 8 per group)65. At day 0, animals were anesthetized under isoflurane and inoculated intranasally with 35 µL of DMEM containing 5 × 104 PFU of SARS-CoV-2 isolate hCoV-19/Spain/SP-VHIR.02, D614G(S)65. On days 0–3, mice were similarly anesthetized and treated with PBS or either 24 or 12 mg/kg of compound 17 and anesthetized and humanly sacrificed at 3 dpi. For prophylactic efficacy assays, animals were inoculated with compound 17 starting 3 h before infection. In the case of post-exposure therapeutic assays, animals were inoculated with compound 17 starting on day 1 post-infection. The right lungs of the mice were harvested, homogenized in TriReagent (Thermo Fisher Scientific) using a TissueLyser II and RNA was extracted with RiboPure RNA Purification Kit (Thermo Fisher Scientific). Viral load was determined by one-step quantitative RT-PCR as described66 using a QuantStudio5 Real-time PCR system (Applied Biosystems). Data are expressed as PFU equivalents/g of tissue by comparison with previously titrated samples. For cytokine expression analyzes, cDNA was synthesized with Biotools High Retrotrasncriptase Starter Kit with Oligo dT (Biotools). The relative quantification of proinflammatory cytokines induction over uninfected control animals was performed with specific PrimeTime Std qPCR Assays (Integrated DNA Technologies) for IL-1β, IL-6, TNFα, and GAPDH (as housekeeping gene) as described47.
To assess antiviral activity against RSV-A2, 10 female Balb/C (6–7 weeks of age; Charles River Laboratories, France, strain code 028) were lightly anesthetized with isoflurane and infected with 5 × 105 PFU of highly purified RSV-A2 in 50μL of PBS by intranasal administration (day 0)67. On days 1–3, mice were similarly anesthetized and treated with either PBS (n = 5) or 12 mg/kg of compound 17 (n = 5) diluted in PBS in a volume of 35 μl (6.9 mg/mL). Finally, on day 4, mice were sacrificed and lungs were removed. The left lung was weighed, resuspended in 0.5 mL cold PBS, and homogenized on Precellys® homogenizer using ceramic beads (1.4/2.8 mm, catalog 431-0170) for 1 cycle of 10 s at 4500 rpm. The supernatant was clarified and stored at −70 °C. For qPCR analysis, RNA from 55 μL of the homogenized lung supernatant was extracted using the NZY total RNA isolation kit (MB13402) and eluted in 40 μl. cDNA was then produced with M-MLV reverse transcriptase (Promega) containing 8uL of RNA and both oligo-dT18 and RSV-specific primer CAATGAACTAGGATATCAAGAC in a final volume of 25 μL. To define the number of genome copies per lung, qPCR was performed using the PowerUp SYBR Green Master Mix (ThermoFisher Scientific) with primers binding to the RSV G gene (CGGCAAACCACAAAGTCACA and TTCTTGATCTGGCTTGTTGCA) and an annealing temperature of 60 °C. The relative copy of RSV genomes was obtained relative to a standard curve of the RSV-AmKate2 antigenomic plasmid.
Statistical analysis
All statistical analysis was performed using the R software (version 4.3.3.).
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Wang, W.-H., Thitithanyanont, A., Urbina, A. N. & Wang, S.-F. Emerging and re-emerging diseases. Pathogens 10, 827 (2021).
Delardas, O., Kechagias, K. S., Pontikos, P. N. & Giannos, P. Socio-economic impacts and challenges of the coronavirus pandemic (COVID-19): an updated review. Sustainability 14, 9699 (2022).
Han, A. X. et al. Estimating the potential impact and diagnostic requirements for SARS-CoV-2 test-and-treat programs. Nat. Commun. 14, 7981 (2023).
Xu, D. & Esko, J. D. Demystifying heparan sulfate–protein interactions. Annu. Rev. Biochem. 83, 129–157 (2014).
Li, J.-P. & Kusche-Gullberg, M. Chapter Six - Heparan Sulfate: Biosynthesis, Structure, and Function. in Proc. International Review of Cell and Molecular Biology (ed. Jeon, K. W. B. T.-I. R. of C. and M. B.) 215–273 (Academic Press, 2016).
Bedini, E., Corsaro, M. M., Fernández-Mayoralas, A. & Iadonisi, A. Chondroitin, dermatan, heparan, and keratan sulfate: structure and functions BT - extracellular sugar-based biopolymers matrices. in (eds. Cohen, E. & Merzendorfer, H.) 187–233 (Springer International Publishing, Cham, 2019). https://doi.org/10.1007/978-3-030-12919-4_5.
Kamhi, E., Joo, E. J., Dordick, J. S. & Linhardt, R. J. Glycosaminoglycans in infectious disease. Biol. Rev. 88, 928–943 (2013).
Bauer, S., Zhang, F. & Linhardt, R. J. Implications of glycosaminoglycans on viral zoonotic diseases. Diseases 9, 85 (2021).
Cagno, V., Tseligka, E. D., Jones, S. T. & Tapparel, C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses 11, 596 (2019).
Zhang, Q. et al. Host heparan sulfate promotes ACE2 super-cluster assembly and enhances SARS-CoV-2-associated syncytium formation. Nat. Commun. 14, 5777 (2023).
Zhang, Q. et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 6, 80 (2020).
Kearns, F. L. et al. Spike-heparan sulfate interactions in SARS-CoV-2 infection. Curr. Opin. Struct. Biol. 76, 102439 (2022).
Alotaibi, F. S. et al. Interactions of proteins with heparan sulfate. Essays Biochem. EBC20230093 https://doi.org/10.1042/EBC20230093 (2024).
Liu, L. et al. Heparan sulfate proteoglycans as attachment factor for SARS-CoV-2. ACS Cent. Sci. 7, 1009–1018 (2021).
Clausen, T. M. et al. SARS-CoV-2 Infection depends on cellular heparan sulfate and ACE2. Cell 183, 1043–1057.e15 (2020).
Battles, M. B. & McLellan, J. S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 17, 233–245 (2019).
Feldman, S. A., Audet, S. & Beeler, J. A. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74, 6442–6447 (2000).
Chen, Y. et al. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3, 866–871 (1997).
Giroglou, T., Florin, L., Schäfer, F., Streeck, R. E. & Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75, 1565–1570 (2001).
Salvador, B. et al. Filoviruses utilize glycosaminoglycans for their attachment to target cells. J. Virol. 87, 3295–3304 (2013).
Zong, C. et al. Heparan sulfate microarray reveals that heparan sulfate–protein binding exhibits different ligand requirements. J. Am. Chem. Soc. 139, 9534–9543 (2017).
Lu, W., Yang, Z., Chen, J., Wang, D. & Zhang, Y. Recent advances in antiviral activities and potential mechanisms of sulfated polysaccharides. Carbohydr. Polym. 272, 118526 (2021).
Pradhan, B. et al. A state-of-the-art review on fucoidan as an antiviral agent to combat viral infections. Carbohydr. Polym. 291, 119551 (2022).
Figueroa, J. M. et al. Efficacy of a nasal spray containing iota-carrageenan in the postexposure prophylaxis of covid-19 in hospital personnel dedicated to patients care with covid-19 disease. Int. J. Gen. Med. 14, 6277–6286 (2021).
Ray, B. et al. Antiviral strategies using natural source-derived sulfated polysaccharides in the light of the covid-19 pandemic and major human pathogenic viruses. Viruses 14, 35 (2022).
Conzelmann, C. et al. Inhaled and systemic heparin as a repurposed direct antiviral drug for prevention and treatment of COVID-19. Clin. Med. 20, e218 LP–e218221 (2020).
Vitiello, A. & Ferrara, F. Low molecular weight heparin, anti-inflammatory/immunoregulatory and antiviral effects, a short update. Cardiovasc. Drugs Ther. 37, 277–281 (2023).
Pyrć, K. et al. SARS-CoV-2 inhibition using a mucoadhesive, amphiphilic chitosan that may serve as an anti-viral nasal spray. Sci. Rep. 11, 20012 (2021).
Cele, Z. E. D. et al. Cationic chitosan derivatives for the inactivation of HIV-1 and SARS-CoV-2 enveloped viruses. ACS Omega 8, 31714–31724 (2023).
Jaber, N. et al. A review of the antiviral activity of Chitosan, including patented applications and its potential use against COVID-19. J. Appl. Microbiol. 132, 41–58 (2022).
Revuelta, J. et al. Heparanized chitosans: towards the third generation of chitinous biomaterials. Mater. Horiz. 8, 2596–2614 (2021).
Revuelta, J. et al. Unraveling the structural landscape of chitosan-based heparan sulfate mimics binding to growth factors: deciphering structural determinants for optimal activity. ACS Appl. Mater. Interfaces 12, 25534–25545 (2020).
Doncel-Pérez, E. et al. Synthesis, physicochemical characterization and biological evaluation of chitosan sulfate as heparan sulfate mimics. Carbohydr. Polym. 191, 225–233 (2018).
Junceda-Mena, I., García-Junceda, E. & Revuelta, J. From the problem to the solution: chitosan valorization cycle. Carbohydr. Polym. 309, 120674 https://doi.org/10.1016/j.carbpol.2023.120674 (2023).
Carvalho, L. C. R., Queda, F., Santos, C. V. A. & Marques, M. M. B. Selective modification of chitin and chitosan: en route to tailored oligosaccharides. Chem. Asian J. 11, 3468–3481 (2016).
Guimond, S. E. et al. Synthetic heparan sulfate mimetic pixatimod (PG545) potently inhibits SARS-CoV-2 by disrupting the spike–ACE2 interaction. ACS Cent. Sci. 8, 527–545 (2022).
Jones, S. T. et al. Modified cyclodextrins as broad-spectrum antivirals. Sci. Adv. 6, eaax9318 (2024).
Heida, R. et al. Development of an inhalable antiviral powder formulation against respiratory syncytial virus. J. Control. Release 357, 264–273 (2023).
Gargantilla, M. et al. C-2 Thiophenyl tryptophan trimers inhibit cellular entry of SARS-CoV-2 through interaction with the viral spike (S) Protein. J. Med. Chem. 66, (2023).
Hallak, L. K., Kwilas, S. A. & Peeples, M. E. Interaction between respiratory syncytial virus and glycosaminoglycans, including heparan sulfate bt - glycovirology protocols. in (ed. Sugrue, R. J.) 15–34. https://doi.org/10.1007/978-1-59745-393-6_2 (Humana Press, Totowa, NJ, 2007).
Beaucourt, S. & Vignuzzi, M. Ribavirin: a drug active against many viruses with multiple effects on virus replication and propagation. Molecular basis of ribavirin resistance. Curr. Opin. Virol. 8, 10–15 (2014).
Nie, C. et al. Charge matters: mutations in omicron variant favor binding to cells. ChemBioChem 23, e202100681 (2022).
Zhang, Z., Zhang, J. & Wang, J. Surface charge changes in spike RBD mutations of SARS-CoV-2 and its variant strains alter the virus evasiveness via HSPGs: a review and mechanistic hypothesis. Front. Public Health 10, 952916 (2022).
Gelbach, A. L. et al. Interactions between heparin and SARS-CoV-2 spike glycoprotein RBD from omicron and other variants. Front. Mol. Biosci. 9, https://doi.org/10.3389/fmolb.2022.912887 (2022).
Hsieh, C.-L. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020).
Petrov, S. A., Yusubov, M. S., Beloglazkina, E. K. & Nenajdenko, V. G. Synthesis of radioiodinated compounds. Classical approaches and achievements of recent years. Int. J. Mol. Sci. 23, 13789 (2022).
Botella-Asunción, P. et al. AG5 is a potent non-steroidal anti-inflammatory and immune regulator that preserves innate immunity. Biomed. Pharmacother. 169, 115882 (2023).
Asunción, M. et al. Comparative effects of two neutralizing anti-respiratory syncytial virus (RSV) monoclonal antibodies in the rsv murine model: time versus potency. Antimicrob. Agents Chemother. 49, 4700–4707 (2005).
Bannister, R. et al. Use of a highly sensitive strand-specific quantitative PCR to identify abortive replication in the mouse model of respiratory syncytial virus disease. Virol. J. 7, 250 (2010).
Caidi, H., Harcourt, J. L., Tripp, R. A., Anderson, L. J. & Haynes, L. M. Combination therapy using monoclonal antibodies against respiratory syncytial virus (RSV) G glycoprotein protects from RSV disease in BALB/c Mice. PLoS ONE 7, e51485 (2012).
Liu, J. & Pedersen, L. C. Anticoagulant heparan sulfate: Structural specificity and biosynthesis. Appl. Microbiol. Biotech. 74, 263–272 (2007).
Yang, J. et al. Preparation, characterization and in vitro anticoagulant activity of highly sulfated chitosan. Int. J. Biol. Macromol. 52, (2013).
Lee, A. R. Y. Bin et al. Efficacy of covid-19 vaccines in immunocompromised patients: systematic review and meta-analysis. BMJ 376, e068632 (2022).
Weinberger, B. Vaccines for the elderly: current use and future challenges. Immun. Aging 15, 3 (2018).
Tam, E. H., Peng, Y., Cheah, M. X. Y., Yan, C. & Xiao, T. Neutralizing antibodies to block viral entry and for identification of entry inhibitors. Antivir. Res. 224, 105834 (2024).
Pantaleo, G., Correia, B., Fenwick, C., Joo, V. S. & Perez, L. Antibodies to combat viral infections: development strategies and progress. Nat. Rev. Drug Discov. 21, 676–696 (2022).
Tompa, D. R., Immanuel, A., Srikanth, S. & Kadhirvel, S. Trends and strategies to combat viral infections: a review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 172, 524–541 (2021).
Zhou, Y. & Simmons, G. Development of novel entry inhibitors targeting emerging viruses. Expert Rev. Anti Infect. Ther. 10, 1129–1138 (2012).
Cron, R. Q., Goyal, G. & Chatham, G. G. Cytokine Storm Syndrome. Annu. Rev. Med. 74, 321–337 (2023).
Braz-de-Melo, H. A. et al. The use of the anticoagulant heparin and corticosteroid dexamethasone as prominent treatments for COVID-19. Front. Med. 8, 615333 (2021).
Shi, C. et al. The potential of low molecular weight heparin to mitigate cytokine storm in severe COVID-19 patients: a retrospective cohort study. Clin. Transl. Sci. 13, 1087–1095 (2020).
Holme, K. R. & Perlin, A. S. Chitosan N-sulfate. A water-soluble polyelectrolyte. Carbohydr. Res. 302, 7–12 (1997).
Latorre, V. & Geller, R. Identification of cytoplasmic chaperone networks relevant for respiratory syncytial virus replication. Front. Microbiol. 13, 880394 (2022).
Saleh, M. C. et al. Antiviral immunity in Drosophila requires systemic RNA interference spread. Nature 458, 346–350 (2009).
Casasnovas, J. M. et al. Nanobodies protecting from lethal SARS-CoV-2 infection target receptor binding epitopes preserved in virus variants other than omicron. Front. Immunol. 13, 863831 (2022).
Rodríguez-Pulido, M. et al. Non-coding RNAs derived from the foot-and-mouth disease virus genome trigger broad antiviral activity against coronaviruses. Front. Immunol. 14, 1166725 (2023).
Ramos, M., Lao, Y., Eguiluz, C., Del Val, M. & Martínez, I. Urokinase receptor-deficient mice mount an innate immune response to and clarify respiratory viruses as efficiently as wild-type mice. Virulence 6, 710–715 (2015).
Acknowledgements
We thank Elba Barberà Ferragud and Sebastian Velandia-Alvarez for supporting the RSV animal experiments. We thank Isidoro Martinez Gonzalez (ISC-III, Majadahonda) for helping with the RSV-A2 purification. This work acknowledges the use of ICTS-ReDIB, supported by the Ministry of Science, Innovation and Universities (MICIU) at biomaGUNE. We thank Raquel Palao (ICTP-CSIC) for the GPC analysis. Funding for this project was provided by grants SGL2021-03-009, SGL2103053, and SGL2021-03-052 from European Union NextGenerationEU/PRTR through the CSIC Global Health Platform established by EU Council Regulation 2020/2094, to RG, to MAM-A, J. R., A. F.-M., J.L.L, and the IBV-Covid19-Pipeline acknowledge the financial support provided by CSIC (grants CSIC-COV19-013, CSIC-COV19-082) and the MICIN (grants PID2019-105337RB-C21, PID2020-120322RB-C21, PID2020-116880GB-I00). RG also acknowledges funding from the Generalitat Valenciana (GVA, GVCOVID19-2020-05). MAM-A acknowledges funding from the European Union’s Horizon Europe research and innovation program under grant agreement number 101137506 (NAVIPP).
Author information
Authors and Affiliations
Consortia
Contributions
E.T. and S.I. performed the synthesis of polysaccharides. J.R. and A.F-M. contributed to the polysaccharide design, synthesis and characterization, and supervised the toxicological assessment. R.G. designed, supervised, and analyzed all antiviral assays with both RSV and SARS-CoV-2, except for the in vivo evaluation against SARS-CoV-2, which was supervised by M.A.M-A. and performed by E.C-P. and A.-B.B. R.G., L.R., A.I.A-A., and C.F.G., performed and analyzed all in vitro antiviral assays. A.G.A. contributed the k18-hACE2 mice. A.F.N performed the thermal shift assays and helped to make the samples for in vitro functional assays. M.L.L.R. performed the electron microscopy experiments of negative stained samples. Some members of the IBV-Covid19-Pipeline produced the RBD, S1 subunit and spike proteins used in surface plasmon resonance, thermal shift and negative stain electron microscopy assays and other members of the Pipeline supervised the work. J.L.L. supervised the work, analyzed the data and helped to write a few parts of the manuscript. J.L., A.K., O.M. and M.G.M. performed radiolabeling and biodistribution experiments. R.G., J.R., A.F.-M., M.A.M.-A., and the IBV-Covid19-Pipeline obtained funding. R.G., J.R., and A.F.-M. wrote the manuscript. A.F.-M. put forward the hypothesis.
Corresponding authors
Ethics declarations
Competing interests
R.G., J.R., A. F-M., C.F.G., M.A.M.-A. are inventors of a patent application pertaining to the compounds utilized in this work (European Patent Application EP4227324A1).
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Shitao Li and Mengtan Xing. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Revuelta, J., Rusu, L., Frances-Gomez, C. et al. Synthetic heparan sulfate mimics based on chitosan derivatives show broad-spectrum antiviral activity. Commun Biol 8, 360 (2025). https://doi.org/10.1038/s42003-025-07763-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07763-z
