
Abstract
Esophageal squamous cell carcinoma (ESCC) is a deadly cancer with a poor prognosis and a high recurrence rate after chemotherapy, posing a significant clinical challenge. To elucidate the molecular basis of chemotherapy (chemo)-resistance and to develop methods to effectively eliminate chemo-resistant tumor clones, we established an ESCC organoid (ESCCO) library from 24 ESCC patients of various stages, ages, and treatments. These ESCCOs faithfully recapitulate the oncogenic mutations observed in the original ESCC tissues and manifest tumorigenic properties when xenografted. The ESCCOs respond differently to cisplatin and 5-fluorouracil, chemotherapeutic agents commonly used to treat ESCC patients, with 7 ESCCOs exhibiting potent chemo-resistance. Notably, the chemo-resistant ESCCOs show higher genes involved in antioxidant stress response pathways and more accessible chromatin at their loci than the sensitive ESCCOs. These genes can serve as valuable biomarkers to stratify chemo-resistant ESCCs in histopathological specimens. Through drug screening using the ESCCO library, we reveal that fedratinib effectively induces cell death in chemo-resistant ESCCOs. Collectively, our human ESCCO model offers novel insights into the mechanism of chemo-resistance in ESCCs, which is critical for developing effective therapeutic approaches to eradicate the recurrence of ESCCs.
Similar content being viewed by others


Introduction
Esophageal cancers occur in the mucosa of the esophagus and are one of the cancers with the poorest prognosis, ranking 7th in morbidity and 6th in mortality worldwide1. There are two histological types of esophageal cancer: squamous cell carcinoma and adenocarcinoma, and in East Asia, more than 90% of esophageal cancers are squamous cell carcinoma. In recent years, multimodality treatment combining surgical resection, chemotherapy, including cisplatin (CDDP) and 5-fluorouracil (5-FU), and radiotherapy has improved mortality rates for patients with esophageal squamous cell carcinoma (ESCC). However, treatment-resistant clones often emerge post-treatment, and the disease-free survival rate is around 55% to 63.6% within 5 years2,3. Additionally, there are no effective treatment options for those recurrent ESCCs. Therefore, there is an urgent need to develop methods to prevent the recurrence of ESCCs.
Patient-derived cancer organoids (PDOs) have been recently established as novel preclinical models for various types of cancers, including colorectal cancer4, liver cancer5, pancreatic cancer6, breast cancer7, prostate cancer8, head and neck cancer9, and oral cancer10,11, and those reproduce inter-patient and intra-tumor heterogeneity of the parental tumor tissues in vitro. PDOs have been widely used for understanding the cancer properties of individual patients and as a drug screening model to discover novel anti-cancer drugs. However, reports of ESCC-derived PDOs (ESCCOs) are quite limited. Kijima et al. successfully generated ESCCOs from 15 ESCC patients, but those have not been used for detailed analysis of genetic alterations or drug screening12. Therefore, it is important to create a larger ESCCO library, and use it to elucidate the molecular basis of chemo-resistance induction and to propose effective treatments for therapy-resistant ESCCs.
Here, we established an ESCCO library that reproduces the primary tumor tissues of 24 ESCC patients. Based on the responsiveness of ESCCOs to chemotherapeutic agents, which varied from patient to patient, we showed that basal activation of the anti-oxidative stress response is closely associated with chemo-resistance in ESCC. The results reveal that these gene signatures are useful as predictive biomarkers for chemo-resistant ESCCs in tumor specimens and that the chemo-resistant ESCCs were vulnerable to fedratinib, both of which may become new options for diagnosing and treating chemo-resistant ESCCs.
Results
Establishment of an ESCCO library
We generated a large-scale ESCCO library using resected primary ESCC tissues from 40 patients with diverse ages, cancer stages, and treatments (Supplementary Table 1). Tumor tissues from each ESCC patient were divided into small pieces that were used for analyses of histology, mutation, gene expression, and organoid culture (shown schematically in Fig. 1a). Esophageal normal epithelia (hereafter referred to as EN) specimens were obtained from the marginal regions of ESCCs without any cancerous lesions and were subjected to the same procedures as the cancer tissues. Next, we optimized culture conditions to support robust and reproducible organoid growth and found that a combination of Noggin, epidermal growth factor (EGF), R-spondin 1, and the Rock inhibitor Y-27632 was optimal for ESCCO growth. On the other hand, for the culture of EN organoids (ENOs), further addition of FGF-2, Forskolin, Prostaglandin E2, the TGF-β inhibitor A 83-01, Nicotinamide, and the GSK3 inhibitor CHIR 99021 improved the growth efficiency. Using these optimized methods, we developed 24 ESCCO lines and 40 ENO lines from different patients with extremely high success rates of 60% and 100%, respectively. Dissociated primary ESCC cells and EN cells formed organoids 100–200 μm in size within 9 days (Fig. 1b). Moreover, the organoid formation efficiency of each ESCCO line varied substantially (Supplementary Fig. 1). Furthermore, most of the ESCCO lines could be passaged for more than 3 months, but all ENO lines could only be maintained for around 30 days (Fig. 1c). TP53 mutations are the most common mutations in many types of cancers, including ESCCs, and the mutant form of p53 accumulates in the nucleus13. Consistently, the nuclear accumulation of p53 protein was confirmed in all analyzed ESCCOs and their parent tumor tissues, but not in ENOs or normal esophageal tissues (Fig. 1d). Additionally, tumor cells with p53 mutations became resistant to Nutlin-3, an MDM2 inhibitor14. Indeed, ESCCOs that showed the nuclear accumulation of p53, including ESCCO7, ESCCO11, and ESCCO12, survived even in the presence of Nutlin-3, whereas ENOs could not (Fig. 1e). Collectively, these results indicated that ESCCOs are composed exclusively of tumor cells.

a Overview of the experiment; ENOs and ESCCOs were established from a total of 24 patients. Histological, exome, transcriptome, and chromatin accessibility analyses were performed on ENOs, ESCCOs, and their parental cancer tissues. Established ESCCOs were also used for xenografts, drug response tests and to identify molecules associated with the chemotherapy resistance of ESCC. b Representative time course images of ENOs (P0) and ESCCOs (P0). Scale bars, 1 mm and inset scale bars, 200 µm. c Time in the culture of ENOs (n = 6) and ESCCOs (n = 8); the endpoint was 90 days. Data are presented as means ± standard deviation (s.d.). d Immunostaining for p53 of 1 ENO, 4 ESCCOs, and their parental cancer tissues. ESCCO12 (a TP53 deleted line, described in Fig. 3a) and ENO6 did not stain positive for p53. Scale bars, 100 µm. e Representative brightfield images of 1 ENO and 3 ESCCOs after 9 days of culture in the presence or absence of 10 μM Nutlin-3. Nutlin-3 or vehicle was added from day 2 to day 9 after seeding; images were taken on day 9. Scale bars, 1 mm and inset scale bars, 200 µm. f Analysis of niche factor requirement of ESCCOs. Each symbol represents one well from 2 separate experiments. Data are presented as means ± s.d. N: Noggin, E: EGF, R: R-spondin 1, Y: Y-27632.
The different characteristics of ESCCOs were confirmed by different dependencies of their growth on culture components. For example, the growth of ESCCO2, ESCCO3, and ESCCO6 was substantially reduced in the absence of Noggin, whereas the growth of ESCCO7 and ESCCO11 was hardly affected by Noggin removal. Furthermore, the growth of ESCCO6 was also heavily dependent on R-spondin 1. Removal of EGF significantly reduced the growth of ESCCO6 and ESCCO12 compared with other ESCCOs. Y-27632 is required for growth of all analyzed ESCCOs (Fig. 1f).
Tumorigenic capacity of ESCCOs in vivo
To analyze whether the established ESCCOs retain the capacity of tumorigenesis with the characteristics of the parental tumors in vivo, ESCCOs were subcutaneously transplanted into the backs of immunodeficient NOD/Scid/IL2Rγnull (NOG) mice. Implanted ESCCO9, ESCCO15, and ESCCO17 showed tumor outgrowth in NOG mice, although the size of the tumor tissue formed varied (Fig. 2a, b). Hematoxylin & eosin staining confirmed the stratified and keratinized features of parental ESCCs, especially when ESCCO9 and ESCCO15 were transplanted (Fig. 2c). In addition, immunostaining for human p53 protein showed that its nuclear accumulation was observed (Fig. 2c), recapitulating the original ESCC phenotypes with genetic abnormalities in a PDO xenograft model.

a Macroscopic image of xenografts derived from ESCCO15 transplanted subcutaneously into the back of a NOG mouse. b Summary of results for xenografts; the indicated number of mice were subcutaneously injected with ESCCOs containing approximately 1 × 106 cells in the back, and tumor cells were successfully engrafted in all recipients. Tumor volumes were calculated at 1 month after the transplantation. c H&E and p53 staining of xenograft sections at 1 month after the transplantation. ESCCO9, ESCCO15, and ESCCO17 were transplanted as in b. Scale bars in lower magnification images, 500 µm, Scale bars in higher magnification images, 100 µm.
Genomic and transcriptome features of ESCCOs
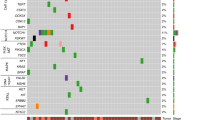
To determine whether the ESCCOs retain the mutational profiles of the parental ESCC tissues, we performed whole-exome sequencing (WES) from 11 different pairs of ESCCOs, ESCC tissues, and ENOs. The ESCC-specific mutations were then extracted by excluding ENO-derived germline mutations. The TP53 mutation, the most frequently detected mutation in ESCCs15, was detected in all ESCCOs and ESCC tissues analyzed (Fig. 3a). Most of the other frequent somatic mutations in the parental ESCC tissues were retained in ESCCOs (Fig. 3a), and the mutation profile of each ESCCO was unique for each patient like a fingerprint. These results demonstrate that ESCCOs accurately reproduce the diversity of genetic mutations among ESCC patients (Fig. 3a and Supplementary Table 2).

a Summary of somatic mutations identified in ESCCOs and their parental cancer tissues. Types of mutations are indicated at the bottom. b Summary of cancer driver mutations classified as Tier 1 COSMIC Cancer Gene Census (CGC) genes identified in ESCCOs and their parental cancer tissues. Types of mutations are indicated at the bottom. c Hierarchical cluster dendrogram based on the ward.D2 method showing transcriptome differences between ESCCOs and ENOs. The height of the branches indicates the dissimilarity between clusters. d GSEA of transcriptional profiles in ESCCOs and ENOs. The top 10 ranked GO terms of the BP (C2 (CP) gene sets (MSigDB)) significantly enriched in either ESCCOs (Red) or ENOs (blue) (NOM p-value < 0.05) are shown. e GSEA plots showing the most enriched gene sets in ESCCOs (upper) and ENOs (lower). NES: normalized enrichment score, NOM p-value: nominal p-value.
We next assessed Tier 1 oncogenic mutations, the most clinically significant variants listed by the COSMIC Cancer Gene Census (CGC) (Fig. 3b). Several variants of Tier 1 oncogenic mutations especially related to the oncogenic pathway in ESCCs15, which include Hippo pathway genes (FAT1, FAT4), histone-modifying genes (KDM family genes, BAP1, EP300), NOTCH pathway genes (NOTCH1, NCOR2), RTK-RAS pathway genes (ERBB4, MET, FGFR1), cell cycle-related genes (CDKN2A, ATM, CHEK2), PI3K pathway genes (PI3KCA, PTEN, PI3KCB) and Nuclear factor erythroid 2-related factor 2 (NRF2) pathway genes (NFE2L2) were identified. Importantly, most of those mutations were commonly identified both in ESCCOs and in the corresponding ESCC tissues (Fig. 3b and Supplementary Table 2), indicating that ESCCOs reproduce the oncogenic machinery of their parental ESCCs. In this context, ESCCO3, ESCCO15, ESCCO18, and ESCCO34, had more diverse Tier 1 oncogenic mutations than the parental ESCC tissues, implying the possibility that the organoid culture process induces new mutations or selects rare clonal variants in some ESCCOs.
We next performed transcriptome analysis of 15 ESCCOs and 5 ENOs. Hierarchical clustering based on the transcriptome analysis revealed that ENOs and ESCCOs were separated into distinct clusters (Fig. 3c). We then analyzed the biological processes overrepresented either in ESCCOs or ENOs by comparing their transcriptomes using Gene Set Enrichment Analysis (GSEA)16. As a result, most of the top-ranked overrepresented biological processes in ESCCOs were related to DNA replication, indicating that progression of the cell cycle is the most obvious feature in ESCCOs compared to ENOs (Fig. 3d, e). On the other hand, the top-ranked biological processes in ENOs were “Formation of the cornified envelope” and “Keratinization”, suggesting that normal epithelial differentiation is suppressed in ESCCOs (Fig. 3d, e). Therefore, ESCCOs are less differentiated with higher proliferative ability than ENOs.
The mechanism of chemotherapy resistance in ESCCOs
We tested the susceptibility of each ESCCO line to CDDP + 5-FU (hereafter abbreviated as CF), commonly used agents in ESCC chemotherapy. Notably, the sensitivity against those agents was highly variable among ESCCO lines, and they were subdivided into a chemo-sensitive group (ESCCO12, 18, 19, 20, 25) and a chemo-resistant group (ESCCO7, 11, 15, 17, 29, 34, 37) (Fig. 4a, b and Supplementary Table 1). We analyzed the CF sensitivity of organoids derived from patients who received chemotherapy and those who did not. In brief, of the 14 ESCCOs established from patients who had received chemotherapy, nine were sensitive (64.3%), and five were resistant (35.7%). Of the 10 ESCCOs established from patients without chemotherapy, eight were sensitive (80%), and 2 were resistant (20%). Of the total 24 ESCCO strains, 17 were sensitive (70.8%), and seven were resistant (29.2%) (Supplementary Fig. 2). Thus, a slightly higher number of ESCCOs from patients who received chemotherapy during clinical treatment showed chemotherapy drug resistance, but this did not indicate a clear causal relationship. Further studies are needed to increase the number of ESCCOs further.


a, b Representative brightfield images (a) and cell viability data (b) of chemo-sensitive ESCCOs (ESCCO12, 18, 19, 20, 25) and chemo-resistant ESCCOs (ESCCO7, 11, 15, 17, 29, 34, 37) cultured in the presence or absence of chemotherapeutic drugs (cisplatin, 5-FU, or a combination of cisplatin and 5-FU (CF)). After seeding ESCC cells at day 0, chemotherapeutic drugs were added from day 5 to day 10, then removed and cultured for an additional 5 days (day 15). Cell viability was measured on day 15. Scale bars in a, 100 µm. Each symbol in b represents the average relative viability of one experiment. Data are presented as means ± s.d. c Paired plots showing changes in relative viability at P0 and P1 ESCCOs after treatment with CF. Cell viability assays were performed as in b. Each symbol represents the relative viability of an individual ESCCO (Red: chemo-resistant ESCCOs, Blue: chemo-sensitive ESCCOs) (n = 18). Statistical significance was determined by paired Student’s t-test (p = 0.8307). d Paired plot showing changes in relative viability against CF in CF treatment-naïve (CF tx: none) and previously CF-exposed ESCCOs (CF tx: 1). Cell viability assays were performed as in b. Each symbol represents an individual chemo-resistant ESCCO (ESCCO11, 15, 17, 34) (n = 8). Statistical significance was determined by paired Student’s t-test (p = 0.5186). e GSEA of transcriptional profiles in chemo-resistant ESCCOs and chemo-sensitive ESCCOs. The top 10 ranked GO terms of the BP (C2 (CP) gene sets (MSigDB)) significantly enriched in either chemo-resistant ESCCOs (Red) or chemo-sensitive ESCCOs (blue) (NOM p-value < 0.05) are shown. f GSEA plots showing the most enriched gene sets in chemo-resistant ESCCOs and chemo-sensitive ESCCOs. NES: normalized enrichment score, NOM p-value: nominal p-value. g Single-cell transcriptome analysis of chemo-resistant ESCCOs (Red: ESCCO15, 17, 34) and chemo-sensitive ESCCOs (Blue: ESCCO18, 20, 25). UMAP representation depicting the clustering results of each ESCCO. Feature plots depicting the expression of representative NRF2 pathway target genes (ALDH3A1, SPP1, UCHL1, TXNRD1).
We further validated whether they show a “heritable” chemo-resistant nature to daughter cells or whether chemo-resistant ESCCOs contain a more significant number of chemo-resistant subclones than chemo-sensitive ESCCOs (Fig. 4c, d). If the latter scenario is correct, CF treatment should induce the selective expansion of chemo-resistant subclones and thus increase resistance by passaging. We found that the resistance/sensitivity of each ESCCO to CF did not significantly change after passaging (Fig. 4c), which suggests that CF resistance/sensitivity is determined by the original ESCCO traits in the genome and/or epigenome before the CF treatment and is inherited by daughter cells. We next treated CF-resistant ESCCOs with CF and then amplified viable cells to re-evaluate the resistance to CF (Fig. 4d). As a result, the CF treatment did not increase their resistance to CF (Fig. 4d), denying the selective expansion of CF-resistant subclones. We recently reported similar features of chemo-resistance in human tongue cancer organoids11.
To analyze the overrepresented pathways in chemo-resistant ESCCOs compared with chemo-sensitive ESCCOs, we performed GSEA and found that the “NRF2 pathway” ranked at the top. In contrast, CF-sensitive ESCCOs highly expressed IFN signal target genes. Thus, we concluded that CF-resistant ESCCs are characterized by the activated NRF2 pathway and a repressed IFN signal pathway (Fig. 4e, f). NRF2 is a transcription factor that plays an important role in cellular defense against toxicity and oxidative stress17. In this context, CDDP not only inhibits cell division by cross-linking the genome but also exhibits cytotoxicity by inducing higher intracellular reactive oxygen species (ROS) levels from mitochondria18. Indeed, gain-of-function mutations in NRF2 are found in 22% of advanced ESCCs, and those mutations confer chemoradiotherapy resistance to those ESCCs19.
To examine the difference in NRF2 pathway activation between chemo-resistant and chemo-sensitive ESCCOs at the single-cell level, we performed single-cell RNA sequencing (scRNA-seq) of three chemo-resistant (ESCCO15, 17, 34) and three chemo-sensitive ESCCOs (ESCCO18, 20, 25). Using the Seurat package, which is widely used to classify cells into different subtypes (clusters) based on their gene expression patterns, ESCCO cells consisting of each organoid line were subdivided into 4 to 9 clusters. To visualize cells in which the NRF2 pathway is activated, we created feature plots of representative NRF2 target genes, i.e. ALDH3A1, SPP1, UCHL1, and TXNRD1. Of note, NRF2 target gene-expressing cells did not form unique clusters in chemotherapy-resistant ESCCOs but rather were distributed across clusters at substantially higher levels than in chemo-sensitive ESCCOs (Fig. 4g). In line with Fig. 4d, these results suggested the absence of specific chemo-resistant subclones in the chemo-resistant ESCCOs. In contrast, the distribution of cell cycle-related genes (G2/M phase genes: MKI67 and CCNB1, G1/S phase genes: MCM2, MCM3, Supplementary Fig. 3) was not uniform and tended to be strongly expressed in certain clusters. Furthermore, there was no obvious reduction in proliferating cells in the chemo-resistant ESCCOs. Therefore, these results implied that the feature of chemo-resistance in ESCCs does not depend on cell cycle status.
Epigenomic features of chemotherapy resistance in ESCCOs
WES analysis of six chemo-resistant ESCCO lines showed that only three have NFE2L2 mutations (Fig. 3b and Supplementary Table 1). These results indicated that chemo-resistant ESCCs without NFE2L2 mutations exist. Notably, the expression of NRF2 target genes involved in antioxidant stress response was significantly increased in chemo-resistant ESCCOs with or without NFE2L2 mutation, compared to chemo-sensitive ESCCOs (Fig. 4e, f and Supplementary Fig. 4). KEAP1 and CUL3 are negative regulators of NRF2 activity, and their loss-of-function mutations are associated with aberrant NRF2 stabilization20,21,22. However, those mutations were not detected in the ESCCO lines without NFE2L2 mutation. In addition, the expression levels of NFE2L2 and CUL3 genes were comparable between chemo-sensitive and -resistant ESCCOs, and the expression of KEAP1 was unexpectedly increased in chemo-resistant ESCCOs (Supplementary Fig. 4), indicating that copy number alterations in these genes are also unlikely to be involved in the upregulation of NRF2 target genes in chemo-resistant ESCCOs. Collectively, these results showed that chemo-resistant ESCCOs cannot be stratified solely by NFE2L2 gene mutations or by gene mutations of KEAP1 and CUL3, genes related to the NRF2 pathway.
Therefore, we next investigated the non-genetic features of chemo-resistant ESCCO using ATAC-seq. DEseq2 analysis of ATAC-seq data, revealed that compared with chemo-sensitive ESCCOs, chemo-resistant ESCCOs significantly overrepresented 4,112 chromatin regions and underrepresented 1232 chromatin regions (Fig. 5a). Motif analysis showed that NRF2, p53, and AP1 binding motifs were significantly enriched in chemo-resistance ESCCOs (Fig. 5b, right), whereas such enrichment was not observed in chemo-sensitive ESCCOs (Fig. 5b, left). Notably, similar patterns of TF binding motif enrichment in ATAC-seq peaks were observed when MCF7 human breast cancer cells were exposed to menadione, a potent inducer of intracellular ROS23. These results suggested that chemo-resistant ESCCOs exhibit a “poised antioxidant state” even before chemotherapeutic agent exposure. Supporting these findings, in chemo-resistant ESCCOs, genomic loci of putative NRF2 target genes, including SPP1, UCHL1, and ABCC2, were under constitutively open chromatin states (Fig. 5c–e), whereas the genomic loci of ALDH3A1 and TXNRD1 were not (Supplementary Fig. 5a, b). KEAP1 is a direct binding partner and regulator of NRF2. Hypermethylation of the promoter region of the KEAP1 gene locus reduces KEAP1 gene expression and eventually activates NRF2 in lung and kidney cancers24,25. However, unlike those cancers, chemo-resistant ESCCOs showed neither decreased KEAP1 gene expression (Supplementary Fig. 4) nor suppression of chromatin accessibility in the promoter region of the KEAP1 gene (Supplementary Fig. 5c).

a Scatter plot showing the differential chromatin accessibility between chemo-resistant ESCCOs (ESCCO7, 15, 17, 29, 34, 37) compared with chemo-sensitive ESCCOs (ESCCO12, 18, 19, 20, 25, 35), with a fold change cutoff of 2-fold and a significance cutoff of p < 0.05 (in red). b Analysis of open chromatin regions lost or gained in chemo-resistant ESCCOs compared with chemo-sensitive ESCCOs. c–e Genome browser tracks of ATAC-seq signals at NRF2 target gene loci, e.g., SPP1 (c), UCHL1 (d), ABCC2 (e). The average ATAC-seq signal of chemo-resistant ESCCOs (red) or chemo-sensitive ESCCOs (blue), and the signal of individual ESCCOs are shown.
Identification of gene biomarkers stratifying chemo-resistant ESCCs
As NAC has little tumor regression effect in approximately 28% of ESCC patients26, the identification of biomarkers that can predict the efficacy of CF-based chemotherapy is important for early and appropriate ESCC treatment.
To select candidate biomarker genes to identify chemo-resistant ESCCs, we analyzed the correlation between the expression level of each gene in each ESCCO line (Fig. 3c) and their area under the dose-response curve (AUC) values of CDDP (CDDP-AUC, Fig. 6a, b). As a result, 117 genes showed a positive correlation with the AUC value (r > 0.5, mean TPM > 50, Fig. 6c). When 57 of those genes that correlated with the AUC significantly (p < 0.05) were subjected to GO analysis, GO terms related to “xenobiotic metabolic process” and “cellular response to oxidative stress” were ranked as the most overrepresented GO terms (Fig. 6d). Of note, 13 of the 57 genes were NRF2 pathway target genes, and some representative examples are ALDH3A1 (r = 0.84, p = 0.0023, Fig. 6e), ABCC1 (r = 0.76, p = 0.010), SRXN1 (r = 0.69, p = 0.028), UGT1A7 (r = 0.68, p = 0.032), CBR1 (r = 0.66, p = 0.037), KEAP1 (r = 0.65, p = 0.041), and UGT1A1 (r = 0.63, p = 0.050).

a Dose-response curves of ESCCOs after the treatment with CDDP. After the seeding of ESCC cells at day 0, CDDP was added from day 5 to day 10. Cell viability was measured on day 10. Data are presented as means of technical triplicates. One or two experiments were performed for each ESCCO. b Heat map showing AUC values for each ESCCO treated with CDDP. c The rank of Spearman correlation coefficients (r) for genes whose expression levels are positively correlated with the dose-response curve AUC values of CDDP in ESCCOs. Only genes with an r > 0.5 and mean TPM > 50 were plotted. A total of 117 genes were identified as positively correlated genes. d Gene ontology analysis of CDDP resistance-associated genes (r > 0.5, p-value < 0.05, and mean TPM > 50). Bar chart showing the top 10 GO terms for biological process, ranked by -Log10 p-value. e Scatter plot, with the line of best fit, showing the correlation between dose-response curve AUC values of CDDP [x-axis] and ALDH3A1 mRNA expression level (TPM) [y-axis] in each ESCCO line. Correlation coefficients (r) and p-values are also shown. f Detection of ALDH3A1, SPP1, and TXNRD1 mRNA by RNAscope in pathological specimens from patients #15, 17, 18, and 20. ESCCOs derived from patients #15 and 17 were chemo-resistant, and ESCCOs derived from patients #18 and 20 were chemo-sensitive. g Quantification of the area of the mRNA molecule spot relative to the area of the tumor region using QuPath software. n = 5 technical replicates for each sample. Data are presented as means are (%) ± s.d. Statistical significance was determined by a two-tailed Mann–Whitney test (ALDH3A1: p < 0.0001; SPP1: p < 0.0001; TXNRD1: p < 0.0001).
Based on those results, we used RNAscope to quantify the expression levels of those NRF2 target genes (ALDH3A1, TXNRD1, and SPP1) in pathological specimens of primary tumors from ESCC patients who had been evaluated for CF reactivity using established organoids. As expected, many mRNA molecule dots were detected in the pathological specimens of patients #15 and #17, whose ESCCOs were CF-resistant. On the other hand, mRNA dots were undetectable in specimens of patients #18 and #20, whose ESCCOs were CF-sensitive (Fig. 6f). We confirmed that those results were significantly different between the two groups (Fig. 6g), which demonstrates that ESCCOs are useful for extracting biomarker genes that can predict chemo-resistant ESCCs.
Identifying second-line drugs in chemo-resistant ESCCs using the ESCCO library
A major current clinical challenge is that there are few treatment options for CF-resistant ESCCs. Thus, we tested whether NRF2 itself could be a molecular target for removing chemo-resistant ESCC cells. To this end, we generated NFE2L2 knockdown (KD) chemo-resistant ESCCOs by lentivirally transducing a shRNA that targets NFE2L2 and evaluated their CF responsiveness. The expression of NFE2L2 and representative NRF2 target genes was significantly reduced in all NFE2L2 KD lines tested (ESCCO15, 17, 34), indicating that the KD effect of NFE2L2 was sufficient (Supplementary Fig. 6a). However, only ESCCO15 showed an enhanced CF sensitivity (Supplementary Fig. 6b). That result suggests that the NRF2 pathway is commonly activated in chemo-resistant ESCCOs, but the reliance on this pathway to acquire chemotherapy resistance varies from case to case in ESCCs.
One of the most important implications of generating PDOs is that they can be used to evaluate the effects of various drugs on an individual patient’s tumor. In this context, our ESCCO library contains several chemo-resistant lines, thereby providing a novel and useful drug screening platform to identify second-line drugs for chemo-resistant ESCCs. Considering the need for prompt clinical use, we evaluated 27 kinase inhibitors as candidate drugs that are already in clinical use and have known maximum plasma concentrations (Cmax) (Supplementary Table 3). Four chemo-resistant ESCCOs, including ESCCO15, ESCCO17, ESCCO29, and ESCCO34, were cultured in the presence of candidate kinase inhibitors at their Cmax concentration, and their cytotoxicity was compared to CF. Of note, fedratinib, a jak2-selective inhibitor, reduced survival more effectively than CF in all 4 chemo-resistant ESCCO lines. Recent studies showed that fedratinib treatment is safe and effective in patients with myelofibrosis27, and this drug could be an effective treatment option for head and neck squamous cell carcinoma (HNSCC) subgroups28. As ESCCs are squamous cell carcinomas, these results strongly support the potential of fedratinib as a treatment option for chemo-resistant ESCCs. Based on the qRT-PCR results, fedratinib did not alter the expression of NRF2 target genes in chemo-resistant ESCCOs, which suggests that the decreased survival rate of chemo-resistant ESCCOs by fedratinib treatment is not due to direct inhibition of the NRF2 pathway (Supplementary Fig. 7).
Discussion
In this study, we established tumor organoids capable of long-term passage from 24 ESCC patients of various genders and ages, cancer stages, and neoadjuvant therapies (Supplementary Table 1). Those ESCCOs were characterized using histology, mutation profiling, gene expression profiling at both the bulk and single-cell levels, epigenetic profiling, and responses to anticancer drugs. ESCCOs retained most of the oncogenic mutations of the parental tumor cells (Fig. 3a, b and Supplementary Table 2) and tumorigenicity in vivo (Fig. 2). In addition, ESCCOs and ENOs showed significantly different gene expression profiles (Fig. 3c). Thus, the ESCCOs faithfully recapitulated the features of the parental ESCCs.
The biggest challenge in the clinical treatment of ESCCs is the emergence of chemo-resistant clones and the resulting cancer recurrence. Therefore, it is first necessary to clarify the mechanism(s) by which minimal residual tumors occur after chemotherapy. In this regard, we identified chemo-resistant ESCCOs in our ESCCO library, and the chemo-resistant ESCCOs commonly showed increased chromatin accessibility to and expression of NRF2 target genes. A previous study reported gain-of-function mutations of NRF2 in 22% of patients with advanced ESCCs, and those mutations caused their chemoradiotherapy resistance19. However, we could not distinguish chemo-resistant from chemo-sensitive ESCCOs by the presence of NFE2L2 mutations. Instead, our data suggest that the “latent” open chromatin state of genomic loci of various NRF2 target genes may be at least partially involved in acquiring and inheriting chemotherapy resistance traits in ESCCOs. In this context, the results from NFE2L2 KD experiments implied that in addition to the NRF2 pathway, other pathways may be responsible for the induction of chemoresistance in some ESCCOs, which remains an important topic for future study.
At present, the reasons for the aberrant NRF2 pathway activation in “NFE2L2 unmutated” chemo-resistant ESCCOs remain unclear. These ESCCOs showed neither decreased KEAP1 gene expression nor suppression of chromatin accessibility. In addition, no changes were observed in the expression of NFE2L2 and CUL3, critical genes for NRF2 target gene expression, between chemo-resistant and chemo-sensitive ESCCOs (Supplementary Fig. 4). In this regard, our ESCCO library may provide an insight into previously unappreciated mutation-independent mechanisms of aberrant NRF2 pathway activation and resulting chemo-resistance observed in ESCCs.
Unlike artificial models created by exposing cancer cell lines to chemotherapy drugs for a long period29, chemo-resistant ESCCOs reflect the characteristics of the original patient tumors. Since we have established multiple chemo-resistant ESCCO lines with various oncogenic mutations, they should be very useful in identifying effective drugs for various types of chemotherapy-resistant cases. Taking advantage of this, we screened existing drugs in clinical use for the chemo-resistant ESCCOs. As a result, we found that fedratinib showed a higher growth-inhibiting effect than CF at the plasma concentration level. Additionally, multiple kinase inhibitors (regorafenib, cabozantinib malate, pexidartinib, and erdafitinib) showed similar levels of growth inhibition as CF for all chemo-resistant ESCCOs, suggesting that they may also be effective as second-line agents for recurrent ESCCs. Fedratinib might become an effective treatment option for chemo-resistant ESCCs, but its effect is independent of NRF2 pathway inhibition (Supplementary Fig. 7). Regarding this point, fedratinib also has off-target effects, such as inhibiting the activity of BRD4. The expression and function of BRD4 are dysregulated in various human cancers30, and many reports have shown that BRD4 inhibition reduces cancer cell proliferation both in vitro and in vivo31,32,33. Moreover, BRD4-targeting BETi are currently in clinical trials for multiple cancers30. Therefore, the inhibition of BRD4 function may be one of the molecular mechanisms by which fedratinib suppresses the survival of chemo-resistant ESCCOs. In any case, we would like to emphasize that our unique ESCCO library can be used as a powerful tool for screening new drugs for ESCCs, especially to discover candidate drugs that can eradicate chemo-resistant ESCCs.
Identifying biomarkers that can predict the efficacy of chemotherapy is valuable in clinical treatment. In patients with ESCCs, it would avoid ineffective CF therapy and improve the patient’s quality of life. Using pathological specimens of patients’ primary tumors, we succeeded in identifying NRF2 target genes (ALDH3A1, SPP1, and TXNRD1) as biomarkers for chemo-resistant ESCCs, which will be an important guide in determining the direction of ESCC treatment.
Given that our ESCCO library could be an important resource for discovering more effective new drug targets, biomarkers, and drug screening platforms for chemo-resistant ESCCs, prospectively establishing and analyzing ESCCOs from many patients with various types of ESCCs will accelerate the development of personalized medicine.
Methods
Human tissues
Human tissues were obtained from patients diagnosed with ESCC at the Tokyo Medical and Dental University Hospital and at the Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital. Normal and cancer tissues were obtained from surgically resected specimens. This study was approved by the Medical Research Institute of the Tokyo Medical and Dental University and the Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital. Informed consent was obtained from all patients. All ethical regulations relevant to human research participants were followed.
Establishment of ESCCO cultures
Normal esophageal epithelial tissues were washed with phosphate-buffered saline (PBS), after which 0.5 mL of 0.8% trypsin was injected under the epithelium. After 20 min of incubation at 37 °C, the epithelium was peeled away, and the tissue was further incubated with 0.05% trypsin/EDTA (Nacalai) at 37 °C for 15 min. Digested epithelium suspensions were mechanically dissociated into single cells by pipetting and were then passed through a 70-μm nylon mesh. Additionally, 10% fetal calf serum (FCS)/PBS was added to the strained suspensions before centrifugation at 440 × g for 5 min. The cell pellets were resuspended in 5 mL Advanced DMEM/F-12 medium (Thermo Fisher Scientific) containing Primocin (Invivogen), 1 mM HEPES (Nacalai), and L-alanyl-L-glutamine (Nacalai) (AdDF+++) and were then centrifuged again at 440 × g for 5 min, after which the cells were resuspended in Matrigel (Corning). ESCC tissues were washed with PBS, cut into 2 mm3 pieces, and incubated in 1500 PU/mL dispase I (Godo Shusei) at 37 °C for 50 min, after which they were incubated in 0.05% trypsin/EDTA for 15 min. The digested suspensions were mechanically dissociated into single cells by pipetting and were then passed through a 70-μm nylon mesh. Additionally, 10% FCS/PBS was added to the strained cell suspensions and centrifuged at 440 × g for 5 min. The cell pellets were resuspended in 5 mL AdDF+++, centrifuged again at 440 × g for 5 min, and then resuspended in Matrigel (Corning).
For organoid cultures, a 25 μL drop of each cell/Matrigel mixture was seeded into 48-well plates. After solidification, 250 μL organoid medium (AdDF+++, 1× B27 (Invitrogen), 1.25 mM N-acetylcysteine (Sigma-Aldrich), 10% RspoI-culture supernatant of the 293T-HA-RspoI-Fc cell line (provided by Calvin Kuo of Stanford University), 100 ng/mL Noggin (Miltenyi), 50 ng/mL EGF (Peprotech), 10 µM Y-27632 (Nacalai) and 50 μg/mL Primocin was added to each well. For ENO cultures, the following growth factors and compounds were also added to the culture medium: A 83-01 (Nacalai), FGF2 (Miltenyi), Nicotinamide (Sigma), Prostaglandin E2 (Tocris), Forskolin (Tocris) and CHIR 99021 (Chemscene). Cells were cultured at 37 °C with 5% CO2, and the medium was changed every 3–4 days. For passage, organoids were harvested and incubated with 0.05% trypsin/EDTA at 37 °C for 12 min, then were mechanically dissociated into smaller pieces by pipetting and washed with 10% FCS/PBS. Following centrifugation at 440 × g for 5 min, the cell pellets were resuspended in AdDF+++ and centrifuged again at 440 × g for 5 min, after which they were resuspended in Matrigel. Organoids were passed every 7–12 days.
For cryopreservation, organoids were mechanically dissociated into single cells in 0.05% trypsin/EDTA. After washing with AdDF+++, the cell pellets were suspended in CryoScarless DMSO-Free (Bio Verde) and frozen at –80 °C.
Microscopic images were obtained using a BZ-X700 fluorescence microscope (Keyence).
Histology and immunostaining
Murine and human tissues were fixed overnight in 10% Formalin (Nacalai) and then embedded in paraffin. Organoids were recovered by dissolving the Matrigel with Cell Recovery Solution (Corning) and were then fixed for 4 h in 4% paraformaldehyde and embedded in iPGell (GenoStaff). They were then further fixed overnight in 10% Formalin (Nacalai) and embedded in paraffin. Five μm paraffin sections were cut and then stained with H&E using a standard protocol. For immunohistochemistry, sections were incubated with anti-p53 antibody (sc47698, 1:100; Santa Cruz Biotechnology) overnight at 4 °C. The sections were then incubated with anti-mouse IgG-Biotin Ab (405303, 1:100; BioLegend) for 30 min at 37 °C followed by incubation with streptavidin HRP (1:250; Zymed) for 30 min at 37 °C. A peroxidase stain DAB kit (Nacalai) was used to detect HRP. Microscopic images were obtained using a BZ-X700 fluorescence microscope (Keyence).
Mice
Six-week-old female NOD/Shi-scid IL-2Rγ-null (NOG) mice were obtained from CLEA Japan. All experiments using mice were approved by the Institutional Animal Care Committee of the Tokyo Medical and Dental University. All experiments were performed in accordance with relevant guidelines and regulations.
Xenotransplantation of organoids
Organoids containing approximately 1 × 106 cells were collected with Cell Recovery Solution (Corning), then resuspended in 55 μL 50% Matrigel/50% culture medium. The Matrigel-organoid suspensions were subcutaneously injected in NOG mice (female, 8 weeks old) using a 0.3 mL 31 G syringe (Becton Dickinson). At 1 month after transplantation, the mice were euthanized by cervical dislocation, and their subcutaneous tumors were excised. Tumor volumes were calculated 1 month after the transplantation using the formula V = π/6 × L (length) × W (width) × H (height).
Whole exome sequencing (WES) and somatic mutation analysis
Genomic DNA was purified from ESCC tissues, ESCCOs, and ENOs using a Gentra Puregene Tissue Kit (Qiagen). Library preparation, sequencing, mapping, and variant calls were performed by Macrogen Inc. (Japan) and RIKEN Center for Integrative Medical Sciences (IMS) (Japan). In brief, paired-end libraries were prepared using the SureSelect Human All Exon kit V6 (Agilent) or Twist Exome 2.0 according to the manufacturer’s recommendations and were sequenced using an Illumina NovaSeq 6000. After sequencing, raw data were converted into Fastq files by Illumina package bcl2fastq (v2.20.0). Sequence reads were aligned to the UCSC hg38 genome using BWA software (Burrows-Wheeler Alignment Tool) (v0.7.17).
For the analysis of cancer-specific mutations, the somatic mutations were called using GATK (v.4.2.0.0) and Mutect2 according to the GATK Best Practice guidelines. Germline variants were excluded using each patient’s normal tissues. The called somatic mutations were added to gene annotations using Annovar. We selected variants in protein-coding regions and splice sites. Variants identified in healthy individuals with more than 1% allelic frequency using gnomAD (https://gnomad.broadinstitute.org) and ToMMo (https://jmorp.megabank.tohoku.ac.jp) databases were excluded. Finally, the selected variants were confirmed using the Integrative Genomics Viewer (IGV). To select cancer-related variants, we further selected the variants listed in the Cancer Gene Census Tier134) of the COSMIC database (v99).
Bulk RNA sequencing and data analysis
Total RNA was isolated from ESCCOs and ENOs using an RNeasy mini kit (Qiagen). RNA-seq library preparation, sequencing, mapping, and gene expression analyses were performed by Macrogen Inc. (Japan) and the RIKEN Center for Integrative Medical Sciences (IMS) (Japan). Only high-quality RNA preparations with RIN values greater than 7.0 were used for RNA library construction. The RNA sequencing library was independently prepared by Macrogen Inc. (Japan) using an Illumina TruSeq Stranded mRNA Sample Prep Kit (Illumina) with 1 µg total RNA for each sample. Other RNA sequencing libraries were independently prepared by RIKEN IMS (Japan) using an Ultra II RNA Library Prep Kit for Illumina (New England Biolabs) with 100 ng total RNA for each sample. The libraries were submitted to an Illumina NovaSeq 6000 (Illumina), and the paired-end (2 × 100 bp) sequencing was performed by Macrogen Inc. Other libraries were also submitted to Illumina NextSeq 2000 (Illumina), and paired-end (2 × 50 bp) sequencing was performed at RIKEN IMS.
The reads were aligned to the reference human genome (hg38) with STAR (ver.2.7.5a) software. The reads aligned to rRNA and transfer RNA (tRNA) regions were removed. The mapped reads were assigned to genes (Ensembl database annotation version GRCh38.105) using FeatureCounts from the Bioconductor package Rsubread (ver.2.0.1). Normalization and differential expression analysis were performed using DESeq2 (ver.1.26.0). A hierarchical clustering dendrogram based on gene expression profiles was generated using an R hierarchical clustering algorithm (ward. D2) by average linkage with the Pearson distance.
GSEA
GSEA16 was performed using GSEA v2.0.13 software (Broad Institute, http://www.broadinstitute.org/gsea/msigdb/index.jsp). The number of permutations was set to 1000. Low-expressed genes, rRNAs, and mtRNAs were excluded in GSEA datasets.
Single-cell RNA-sequencing and data analysis
Cell suspensions were dropletted with a Chromium Controller (10x Genomics) using a Chromium Single Cell 3’ Reagent Kit, after which cDNA libraries were prepared. Sequencing was performed using NovaSeq6000 R1:28, R2:90 (Illumina) according to the manufacturer’s instructions. The reads from scRNA-seq were processed with Cell Ranger v6.1.2. scRNA-seq data analysis was performed using the R package Seurat v4.3.0. Cells were selected for analysis based on the following criteria: 200 < nFeature < 3000, percent.mt < 20%. Dimensional reduction was performed using PCA (Principal component analysis). UMAP (Uniform Manifold Approximation and Projection) plots were then generated using PC1 ~ PC20. These single-cell profiles were clustered by shared nearest neighbor (SNN) modularity optimization (resolution = 0.5) and projected on the UMAP plot.
Drug response assay
To assess the susceptibility of ESCCOs and ENO to Nutlin-3 (Fig. 1e), dissociated single organoid cells were resuspended in Matrigel at 2000 cells/10 μL and were seeded into 96-well plates (10 μL per well). Forty-eight hr after plating, 10 μM Nutlin-3 (Enzo) was added and cultured for 7 days. For the chemotherapy response assay shown in Fig. 4b, dissociated single ESCCO cells were seeded as detailed above at day 0, and 11 μM CDDP (Fujifilm Wako Chemicals) and/or 2.46 μM 5-FU (Sigma-Aldrich), were added from day 5 to day 10, after which drugs were removed and cultured for an additional 5 days (day 15). Cell viability was measured by CellTiter-Glo (Promega) on day 15 of the culture. For the CDDP dose-response curves analysis shown in Fig. 6a, dissociated single ESCCO cells were seeded, and then various concentrations of CDDP were added from day 5 to day 10 after seeding. Viable cells were quantified at day 10. For CF response assays with repeated passaging (Fig. 4c), CF was added from day 5 to day 10, then removed and cultured for an additional 5 days. Viable cells were quantified as detailed above. To assess the CF susceptibility of ESCC cells obtained from previously chemo-exposed ESCCOs (Fig. 4d), chemo-resistant ESCCOs were exposed to CDDP from day 5 to day 10, then removed and cultured for an additional 5 days. ESCC cells were prepared from these re-established ESCCOs, and CF susceptibility was compared to untreated ESCCOs, similar to Fig. 4d.
ATAC-seq
ATAC-seq library preparation, sequencing, mapping, and gene expression analyses were performed by DNAFORM (Japan). Briefly, cells were lysed, and a transposition reaction was performed with Tn5 Transposase (Illumina) at 37 °C for 30 min. The reaction liquid was purified with a DNAFORM Smart Extract kit. Five cycles of PCR with custom Nextera PCR primers35 were then conducted using NEBNext Q5 Hot Start HiFi PCR Master Mix (New England Biolabs). The additional number of PCR cycles was determined by qPCR of the partly amplified products. The PCR products were purified with Agencourt AMPure XP beads (Beckman), using double size selection (left ratio: 1.8x, right ratio: 0.65x) according to the manufacturerʼs protocol. Paired-end sequencing was performed using an Illumina HiSeq sequencer. Mapping and peak calls were conducted by the ENCODE ATAC-seq pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline). Reads were mapped to the mm10 reference sequence using Bowtie2 (ver. 2.3.4.3), and duplicate reads were removed with Picard (ver. 2.20.7) and samtools (ver. 1.9). Peak calling was performed using MACS2 (ver. 2.2.4) with default parameters. After the removal of blacklist regions, the consistency of peaks was tested by Irreproducible Discovery Rate (IDR) using IDR (ver. 2.0.4.2). Peak annotations were conducted by HOMER (ver. 4.9.1) with default settings. The list of differential accessible peaks was obtained using DEseq2 (Ver. 1.20.0). Known motifs and de novo consensus motifs in the central 200 bp of obtained peaks were searched by HOMER with default settings.
RNAscope
RNAscope in-situ hybridization (ISH) of FFPE samples was performed using RNAscope™ 2.5 HD Reagent Kit-Brown (DAB-HRP) from Advanced Cell Diagnostics (ACD). Sample pre-treatment and RNAscope were performed according to the manufacturer’s instructions (ACD). RNAscope probes, Hs-ALDH3A1 (Cat. No. 516111), Hs-SPP1 (Cat. No. 420101), and Hs-TXNRD1 (Cat. No. 494461) were purchased from ACD. Microscopic images were obtained using a BZ-X700 fluorescence microscope (Keyence). The area of the mRNA molecule spot relative to the area of the tumor region was quantified using QuPath software (QuPath 0.5.0, https://qupath.github.io/).
Drug screening
For the kinase inhibitor response assay shown in Fig. 7, dissociated single ESCCO cells were seeded as detailed above at day 0, and each drug (Table S3) with Cmax concentrations was added from day 1 to day 4. Cell viability was measured by CellTiter-Glo (Promega) on day 4 of the culture.

Scatter plots showing the relative survival of each chemo-resistant ESCCO (ESCCO15, 17, 29, 34) when cultured in the presence of 27 kinase inhibitors that are already in clinical use and have known Cmax concentrations. Two independent experiments were performed, and the relative survival of each drug against CF is expressed as a log2 fold change. Fedratinib reduced survival more effectively than CF in all 4 chemo-resistant ESCCO lines.
Lentiviral transduction
To generate lentiviral particles, 293 T cells were seeded in a 10 cm dish one day before transfection. Cells were co-transfected with a Trans-lentiviral packaging mix (Horizon Discovery) and GIPZ Lentiviral Human NFE2L2 shRNA plasmid (Horizon Discovery), using Lipofectamine LTX (ThermoFisher). Six hours later, the cell culture medium was replaced with 10% FCS DMEM. After 48 h, the culture supernatant containing lentivirus particles was collected and mixed with polyethylene glycol (PEG). The virus particles were concentrated by centrifugation (7000 rpm, 4 °C, 20 min), and the pellet was resuspended to AdDF+++.
For lentivirus infection, 10,000 dissociated ESCCO cells mixed with 25 μL Matrigel and 5 μL lentivirus solution were seeded into 48-well plates. After solidification, 300 μL ESCCO medium containing 1.25 μL TransDux Virus Transduction Reagent (System Biosciences) was added. Cells were cultured at 37 °C with 5% CO2, and the medium was changed every 3–4 days. Cell passaging was performed as described above.
qRT-PCR
Total RNA was extracted using an RNeasy mini kit (Qiagen). First-strand complementary DNA was synthesized from the total RNA using FastGene Scriptase II (Fast Gene). Real-time PCR was performed using SYBR Green (Roche) and a LightCycler 480 machine, and RNA levels were calculated using the ΔCT method, normalized to GAPDH expression. Details of the primers used are provided in Supplementary Table 4.
Statistics and reproducibility
Statistical analyses were conducted using GraphPad Prism v.7. Paired Student’s t-test, a two-tailed Mann–Whitney test, and Welch’s t-test were used to evaluate the statistical significance between the two groups. A p-value < 0.05 is considered to be significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq datasets have been deposited in the Gene Expression Omnibus with the accession number GSE269447. The raw FASTQ files in the WES study will be provided for scientific research upon request due to human patient privacy concerns. Source data can be obtained in Supplementary data 1.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Ando, N. et al. A randomized trial comparing postoperative adjuvant chemotherapy with cisplatin and 5-fluorouracil versus preoperative chemotherapy for localized advanced squamous cell carcinoma of the thoracic esophagus (JCOG9907). Ann. Surg. Oncol. 19, 68–74 (2012).
Yang, H. et al. Long-term efficacy of neoadjuvant chemoradiotherapy plus surgery for the treatment of locally advanced esophageal squamous cell carcinoma: The NEOCRTEC5010 Randomized Clinical Trial. JAMA Surg. 156, 721–729 (2021).
van de Wetering, M. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Broutier, L. et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435 (2017).
Seino, T. et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467 (2018).
Sachs, N. et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386 (2018).
Gao, D. et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014).
Millen, R. et al. Patient-derived head and neck cancer organoids allow treatment stratification and serve as a tool for biomarker validation and identification. Med. 4, 290–310 (2023).
Driehuis, E. et al. Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer Discov. 9, 852–871 (2019).
Sase, M. et al. Comparative analysis of tongue cancer organoids among patients identifies the heritable nature of minimal residual disease. Dev. Cell. 60, 396–413.e6 (2025).
Kijima, T. et al. Three-dimensional organoids reveal therapy resistance of esophageal and oropharyngeal squamous cell carcinoma cells. Cell Mol. Gastroenterol. Hepatol. 7, 73–91 (2018).
Ando, K. et al. Discrimination of p53 immunohistochemistry-positive tumors by its staining pattern in gastric cancer. Cancer Med. 4, 75–83 (2015).
Matano, M. et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262 (2015).
Li, M., Zhang, Z., Wang, Q., Yi, Y. & Li, B. Integrated cohort of esophageal squamous cell cancer reveals genomic features underlying clinical characteristics. Nat. Commun. 13, 5268 (2022).
Subramanian, A., Kuehn, H., Gould, J., Tamayo, P. & Mesirov, J. P. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 23, 3251–3253 (2007).
He, F., Ru, X. & Wen, T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 21, 4777 (2020).
Santos, N. A. et al. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Arch. Toxicol. 81, 495–504 (2007).
Shibata, T. et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 13, 864–873 (2011).
Singh, A. et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med 3, e420 (2006).
Nioi, P. & Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 362, 816–821 (2007).
Ooi, A. et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 73, 2044–2051 (2013).
Levings, D. C., Lacher, S. E., Palacios-Moreno, J. & Slattery, M. Transcriptional reprogramming by oxidative stress occurs within a predefined chromatin accessibility landscape. Free Radic. Biol. Med. 171, 319–331 (2021).
Wang, R. et al. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res Commun. 373, 151–154 (2008).
Fabrizio, F. P. et al. Keap1/Nrf2 pathway in kidney cancer: frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 8, 11187–11198 (2017).
Matsuda, S. et al. Distribution of residual disease and recurrence patterns in pathological responders after neoadjuvant chemotherapy for esophageal squamous cell carcinoma. Ann. Surg. 276, 298–304 (2022).
Harrison, C. N. et al. Safety and efficacy of fedratinib, a selective oral inhibitor of Janus kinase-2 (JAK2), in patients with myelofibrosis and low pretreatment platelet counts. Br. J. Haematol. 198, 317–327 (2022).
Gu, Z. et al. Pharmacogenomic landscape of head and neck squamous cell carcinoma informs precision oncology therapy. Sci. Transl. Med. 14, eabo5987 (2022).
Sharma, S. V., Haber, D. A. & Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 10, 241–253 (2010).
Hu, J., Pan, D., Li, G., Chen, K. & Hu, X. Regulation of programmed cell death by Brd4. Cell Death Dis. 13, 1059 (2022).
Zuber, J. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 (2011).
Sui, S. et al. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 10, 331 (2019).
Sakamaki, J. I. et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 66, 517–532.e9 (2017).
Sondka, Z. et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Acknowledgements
We thank H. Kamioka for secretarial support, A. Suzuki, S. Haruki (Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital) for tissue acquisition, and Prof. M. Yamamoto (Tohoku Medical Megabank Organization, Tohoku University) for thoughtful discussion. This work was supported by a JSPS Grant-in-Aid for Challenging Research (Pioneering) under Grant Number JP21K18259 (T.S.), a Takeda Science Foundation Research Grant of the Princess Takamatsu Cancer Research Fund under Grant Number 20-25220 (T.S.), the G-7 Scholarship Foundation (T.S.), the Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP23ama121015.
Author information
Authors and Affiliations
Contributions
S.N., T.S., E.O., and M.K. contributed equally to this work. T.S. conceived the study, S.N., T.S., E.O., M.K., and H.Y. performed experiments and analyzed data. S.N., T.S., and T.O. wrote the manuscript. F.M. and T.S. performed bioinformatic analysis. Kouhei Yamamoto, Kazuya Yamaguchi, Y.N., A.M., and Y.K. provided esophageal tissue samples, advice, and discussion. T.O. supervised the overall project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Weiren Huang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nakagawa, S., Sato, T., Ohashi, E. et al. An organoid library of human esophageal squamous cell carcinomas (ESCCs) uncovers the chemotherapy-resistant ESCC features. Commun Biol 8, 507 (2025). https://doi.org/10.1038/s42003-025-07869-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07869-4
