
Abstract
Malaria elimination faces challenges from drug resistance, stemming from mutations within the parasite’s genetic makeup. Genetic adaptations in key erythrocyte proteins offer malaria protection in endemic regions. Emulating nature’s approach, and implementing methodologies to render indispensable host proteins inactive, holds the potential to reshape antimalarial therapy. This study delves into the functional implication of the single-span membrane protein Kell ectodomain, which shares consensus sequence with the zinc endopeptidase family, possesses extracellular enzyme activity crucial for parasite invasion into host erythrocytes. Through generating Kell-null erythrocytes from an erythroid progenitor, BEL-A, we demonstrate the indispensable nature of Kell activity in P. falciparum invasion. Additionally, thiorphan, a metallo-endopeptidase inhibitor, which specifically inhibits Kell activity, inhibited Plasmodium infection at nanomolar concentrations. Interestingly, individuals in malaria-endemic regions exhibit low Kell expression and activity, indicating a plausible Plasmodium-induced evolutionary pressure. Both thiorphan and its prodrug racecadotril, demonstrated potent antimalarial activity in vivo, highlighting Kell’s protease role in invasion and proposing thiorphan as a promising host-oriented antimalarial therapeutic.
Similar content being viewed by others


Introduction
The escalating challenge of widespread resistance to antimalarial drugs calls for a transformative shift in therapeutic strategies, particularly focusing on host proteins indispensable for parasite development. This innovative “host-directed” approach offers a promising alternative to combat drug resistance, presenting new horizons in antimalarial research1,2,3,4. Among the key targets are erythrocyte proteins, which play a crucial role in both the initiation and continuation of the parasite’s intraerythrocytic cycle, particularly during the pivotal process of merozoite invasion. Despite the recognized importance of endogenous erythrocytic proteases in parasite development, our current understanding of host-specific proteases involved in orchestrating parasite invasion remains insufficient. Expanding this knowledge is essential to identify novel, druggable effector proteases within the host cell5,6,7,8.
Merozoite invasion of erythrocytes represents a critical juncture in the parasite lifecycle, offering a highly attractive target for therapeutic intervention1,2. While extensive research has been dedicated to uncovering the molecular mechanisms underpinning this process, significant gaps persist9 especially in understanding the proteolytic pathways governed by host erythrocyte proteases. Notably, the calcium-dependent cysteine protease, Calpain 1, has emerged as the only known host protease facilitating malaria parasite egress from infected erythrocytes, underscoring the need for further investigations into other potential effector proteases10.
One such promising candidate is the Kell blood group antigen, an erythrocyte protein with functional significance in parasite invasion. The Kell antigen system is the third most immunogenic in transfusions after the ABO system, featuring over 30 antigens on a glycoprotein, and is known for its clinical significance in hemolytic transfusion reactions and hemolytic disease of the newborn11,12. Kell’s structural homology with the M13 family of zinc-dependent endopeptidases, along with its involvement in converting big endothelin-3 to its active form, points to its dual role in both physiological and pathological processes13,14,15,16.
In our study, we investigated the functional role of the Kell antigen during Plasmodium falciparum invasion through an integrative approach, employing genetic, cellular, and biochemical techniques. Our findings propose a model in which P. falciparum utilizes Kell as a co-receptor to enhance its invasion efficiency. Inhibition of Kell’s protease activity using the zinc metalloprotease inhibitor thiorphan led to a significant reduction in parasite invasion, both in vitro and in vivo. In a murine model, thiorphan administration notably decreased parasitemia and enhanced survival rates, highlighting its therapeutic potential. Ex vivo susceptibility assays further demonstrated thiorphan’s potent antimalarial activity in field isolates. Interestingly, field isolates with lower Kell expression exhibited reduced sensitivity to thiorphan, suggesting an evolutionary adaptation of host surface antigens in malaria-endemic populations. These findings provide critical insights into the role of host factors in malaria pathogenesis and potential therapeutic strategies targeting Kell.
In conclusion, this study elucidates the vital role of the Kell antigen in malaria parasite invasion, highlighting its potential as a novel target for host-directed antimalarial therapies. Our findings not only provide insight into the molecular interplay between host genetic factors and parasite survival strategies but also suggest new avenues for integrated chemotherapy-vaccine approaches to combat malaria in endemic regions.
Result
CRISPR-Cas9 mediated Kell knockout in BEL-A cell line
To rigorously establish the role of Kell in the invasion process, we employed a CRISPR-Cas9-mediated gene knockout approach targeting the Kell gene in the immortalized erythroid progenitor Bristol erythroid line adult (BEL-A) cell line17. The BEL-A cell line presents significant advantages for genetic manipulation over primary erythrocytes, which are notoriously recalcitrant to modification18.
Prior to implementing the knockout, we confirmed the expression of Kell in both undifferentiated (cytoplasmic and plasma membrane-localized) and differentiated BEL-A-derived reticulocytes (plasma membrane-localized), demonstrating its functional relevance across stages. Flow cytometry analysis revealed that Kell expression in BEL-A-derived reticulocytes mirrors that of mature erythrocytes, further validating the model system (Fig. 1A, B).

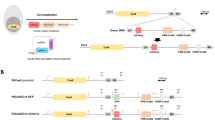
A Immunofluorescence analysis (IFA) depicting the localization of Kell (green) expression in BEL-A-derived undifferentiated erythroblasts, differentiated reticulocytes, and mature erythrocytes. The scale bar represents 5 µm, highlighting the differential expression patterns across stages of erythroid development. B Immunostaining of wild-type (WT) and Kell-null reticulocytes derived from BEL-A cells using Amnis image flow cytometry. The histogram presents the “normalized frequency” parameter (channel 1, BF, y-axis) against fluorescence “Intensity” in channel 2 (x-axis). Representative images of reticulocytes in individual fluorescence channels were captured at ×20 magnification, revealing the distinct fluorescence intensities between WT and Kellnull reticulocytes. C Schematic representation of the CRISPR-Cas9-mediated knockout strategy (created by Biorender.com) for Kell in the BEL-A cell line. The approach demonstrates the precise targeting of exons using small guide RNAs (sgRNAs) to disrupt the Kell gene locus. D Detailed schematic illustrating the strategy to disrupt the Kell genomic locus by targeting exon 1 with sgRNAs. These sgRNAs were nucleofected into BEL-A cells, and resulting clones were screened by Sanger sequencing. The Cas9 cut sites in exon 1 are indicated by vertical arrows, with the adjacent PAM sequence in red. E Chromatogram analysis of the Kell-null clone, showing the deletion of a four-nucleotide sequence (AAGG) in exon 1, in contrast to the intact genomic sequence in WT BEL-A cells. F, G Immunofluorescence analysis of Kell expression in WT and Kell-null BEL-A-derived reticulocytes. The adjacent graph illustrates the mean fluorescence intensity (MFI) of Kell (green), demonstrating a significant reduction in Kellnull reticulocytes. H Histogram showing the “normalized frequency” parameter (channel 1, BF, y-axis) versus fluorescence ‘Intensity’ in channel 2 (x-axis) of immunostained reticulocytes (WT and Kellnull), with images acquired at ×20 magnification using the Amnis imaging flow cytometer. I Immunoblot analysis of membrane fractions from WT and Kellnull erythrocytes, probed with anti-Kell antibody. GAPDH served as the loading control (to normalize the equal no. of RBCs loaded). All experiments were conducted with at least three independent biological replicates (n = 3). Statistical significance is denoted as *p < 0.05 and **p < 0.01 as calculated through the student’s t-test.
To execute the knockout strategy (Fig. 1C), we initially designed and validated multiple small guide RNA (sgRNAs) targeting Kell exon 1 in human embryonic kidney cells (HEK293) cells (Supplementary Fig. 1 and Supplementary Tables 1, 2). Among these, sgRNA1 was selected (target sequence shown in Fig. 1D) for electroporation into BEL-A cells (Supplementary Fig. 2A). Following positive selection for eGFP, individual clones were sorted, and subsequent screening via the Surveyor assay (Supplementary Fig. 2A, B) identified two successful knockout clones (23 and 31), both of which were genotyped and confirmed through Sanger sequencing (Fig. 1E and Supplementary Fig. 3A), immunofluorescence assays (IFA), and enzymatic assays, showing significantly reduced proteolytic activity in the undifferentiated stage (Supplementary Fig. 3B–D). Both clones exhibited an identical deletion in the target region, likely originating from the same transfection event.
Clone 23, chosen for further study, was differentiated along the erythroid lineage, and the Kell knockout was confirmed in these differentiated cells using immunofluorescence assay (IFA) (Fig. 1F, G), flow cytometry (Fig. 1H) and immunoblotting (IB) (Fig. 1I). Importantly, the knockout of Kell did not alter the expression levels of other critical erythrocyte surface receptors implicated in parasite invasion, such as Band 3, Glycophorin A and B and cytoskeletal protein spectrin, and ankyrin, which are present on the inner leaflet of the plasma membrane on erythrocyte surface (permeabilized with 0.001% Triton X-100), which remained consistent with unedited BEL-A-derived reticulocytes as analyzed through IFA, Amnis imaging flow cytometer and Immunoblotting (Fig. 2A–C). Similarly, the expression of Xk, a protein that co-expresses with Kell on the cell surface, remained unchanged in the absence of Kell

A Immunofluorescence staining and quantification of protein expression levels for the specified host receptors in reticulocytes derived from unedited and Kell-null BEL-A cells. The staining demonstrates comparable expression profiles between the two cell types. B, C Flow cytometry histograms and immunoblot analyses revealed no significant alterations in the expression levels of the indicated host receptors (Spectrin, Glycophorin A, Xk, Band 3, and Glycophorin C with loading control GAPDH) between reticulocytes derived from unedited and Kell-null BEL-A cells, confirming that the Kell knockout does not impact other key receptor expressions. D, E Bar graph and representative Giemsa-stained cytospins illustrating parasite multiplication rate (PMR), calculated as the ratio of ring-stage parasites to added mature schizonts, in reticulocytes derived from WT and Kellnull BEL-A cells. The invasion was assessed 10 h post-infection by manual counting of ring-stage parasites on Giemsa-stained cytospins. Statistical significance was determined using a two-tailed student’s t-test, where *** indicates p ≤ 0.001 and “ns” denotes non-significance (p > 0.05). F Schematic representation (created by Biorender.com) illustrating Kell’s metalloprotease activity in BEL-A WT and Kell-null reticulocytes, highlighting the enzymatic role of Kell in these cell models. G Bar graph displaying enzymatic (protease) activity in reticulocytes derived from WT and Kell-null BEL-A cells. All experiments were conducted with at least three independent biological replicates (n = 3), and the data were presented as the mean ± standard deviation (SD). Statistical significance is denoted as *p < 0.05 and **p < 0.01 calculated through the student’s t-test.
To assess potential off-target effects of our sgRNA, we conducted amplification and sequencing of eight predicted off-target sites, identified using the bioinformatics tool COSMID (CRISPR Off-target Sites with Mismatches, Insertions, and Deletions; Supplementary Table 1). The selection of the eight off-target sequences for amplification and sequencing was based on predictive algorithms that prioritize sequences with the highest likelihood of off-target activity. These algorithms rank potential off-target sites based on sequence homology and Protospacer adjacent motifs (PAMs) compatibility, thus allowing us to focus on those sites most likely to be affected by off-target cleavage. Sequencing results revealed no detectable off-target mutations, supporting the precision of our CRISPR-Cas9-mediated gene editing approach.
Invasion and metalloprotease activity of Kellnull BEL-A derived reticulocyte
To assess the invasion susceptibility of genetically modified reticulocytes, P. falciparum schizonts were percoll-purified and introduced to target cells in a 96-well plate, maintaining parasitemia at ~1%. After a 10 h incubation, cytospins were prepared, Giemsa-stained, and the parasite multiplication rate (PMR)—defined as the ratio of resultant ring-stage parasites to mature schizonts added—was calculated. Results showed that Kellnull BEL-A-derived reticulocytes exhibited a substantial ~60% reduction in invasion efficiency (Fig. 2B, C). A similar invasion assay performed with another Kellnull mutant (clone 31) revealed a comparable ~60% reduction in parasitemia, thereby confirming the reproducibility of the genetic modification’s effect on parasite invasion (Supplementary Fig. 4A).
Beyond its established function as a blood group antigen, Kell exhibits zinc-dependent endopeptidase activity. This was substantiated by incubating both wild-type WT and Kellnull BEL-A-derived reticulocytes with a Kell-specific fluorogenic substrate, Glut-FAAF-AMC. The results revealed enhanced fluorescence in WT BEL-A reticulocytes, reflecting Kell’s intrinsic protease activity, whereas the Kellnull reticulocytes displayed significantly reduced or no protease activity (Fig. 2E, F).
Thiorphan, an inhibitor of Kell’s metalloprotease activity
To further investigate whether Kell’s essential role in the intricate and highly regulated process of merozoite invasion is specifically linked to its metalloprotease activity, we employed a pharmacological inhibition strategy19,20.
Initially, an in vitro enzymatic assay on intact erythrocytes was monitored using Kell-specific substrate (Glut-FAAF-AMC)13, which showed the Kell-specific protease activity on erythrocyte (Fig. 3A). The inhibitory efficacy of thiorphan on Kell’s protease activity was further evaluated which yielded a ~ 50% reduction in activity at a concentration of 500 nM (Fig. 3B). To confirm that the observed protease activity was exclusively due to Kell, Kell was purified from erythrocyte membrane lysates using zinc-dependent affinity purification (schematic in Fig. 3C). Successful purification was verified by Coomassie blue staining and immunoblotting (Supplementary Fig. 4C (right panel)). Further analysis of the protease activity from the Kell-enriched fraction provided compelling evidence that Kell is indeed responsible for the observed endopeptidase activity in erythrocytes. When purified, Kell was treated with thiorphan, a ~50% reduction in enzymatic activity was observed, consistent with the results obtained using intact erythrocytes (Fig. 3D).

A Bar graph depicting Kell protease activity on the erythrocyte surface. Erythrocytes (2 × 10⁶) were incubated with or without the substrate Glut-FAAF-AMC (10 µM), followed by the measurement of Kell’s proteolytic activity at excitation/emission wavelengths (λex/λem: 340/440 nm). Results are based on three independent biological replicates (n = 3). B Bar graph illustrating the inhibition of Kell protease activity on the erythrocyte surface in the presence of varying concentrations of Thiorphan. The data represent three independent biological replicates (n = 3). C Schematic representation (created by Biorender.com) of the zinc-dependent affinity purification process for isolating Kell from the erythrocyte surface, emphasizing the metalloprotease’s reliance on zinc for activity. D Bar graph demonstrating Thiorphan’s inhibitory effect on Kell’s enzymatic activity. The assay was performed using 500 ng of purified Kell in the presence of increasing concentrations of Thiorphan. Data represents three independent biological replicates (n = 3). E Bar graph illustrates the percentage of protease activity between wild-type (WT) and Kellnull derived reticulocytes under identical experimental conditions. Error bars represent standard deviations from the mean of three biological replicates (n = 3). Statistical significance was assessed using a two-tailed Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). F Schematic representation (created by Biorender.com) of the in vitro antimalarial activity of thiorphan in two experimental conditions in the first condition, uninfected red blood cells (RBCs) were pretreated with Thiorphan at various concentrations for 2 h at 37 °C, followed by extensive washing to remove the inhibitor. Purified schizonts were then added to the pretreated RBCs. In the second condition, schizonts were added to untreated RBCs in the presence of different concentrations of Thiorphan. After 10 h of incubation, the successful invasion was quantified by counting ring-stage parasites. G Dose-response curve showing the IC50 values of thiorphan in two experimental conditions. Data points represent the average of three biological replicates (n = 3), with 95% confidence intervals (C.I.) calculated from nonlinear regression curves using GraphPad Prism 8.0 software. H Representative Giemsa-stained images comparing control and experimental conditions (at Thiorphan IC50 concentration) for both experimental conditions. I, J Dose-response curves showing 72-h growth inhibition assays with Thiorphan on Plasmodium strains Pf3D7 and PfRKL-9 at different concentrations. Giemsa-stained images illustrate untreated parasites and those treated with Thiorphan at IC50 concentrations. All experiments were conducted with at least three independent biological replicates (n = 3), and the data were presented as the mean ± standard deviation (SD). Statistical significance is denoted as *p < 0.05 and **p < 0.01 calculated through the student’s t-test.
Furthermore, this property of thiorphan was also assessed in BEL-A WT and Kellnull derived reticulocyte model with a Kell-specific synthetic fluorophore in presence and absence of thiorphan respectively13. Results showed enhanced fluorescence intensity attributed to Kell’s protease activity with BEL-A reticulocyte. However, BEL-A-derived Kellnull reticulocyte model showed significant reduced/no protease activity, emphasizing Kell’s role in intrinsic proteolytic processes (Fig. 3E). Furthermore, while thiorphan exhibited an inhibitory effect on BEL-A-derived WT reticulocyte, no such significant inhibition was observed with Kellnull reticulocyte model. Moreover, in vitro invasion assay with BEL-A-derived WT erythrocyte model (IC50: 503 nM; determined from an invasion assay with individual erythrocytes) showed 50% invasion inhibition. However, in Kellnull erythrocyte model, which inherently showed a 60% reduction in parasitemia, the addition of thiorphan at its IC50 did not lead to a further reduction (Supplementary Fig. 6), implying that the thiorphan-mediated invasion defect is specifically attributed to Kell.
To further quantify the interaction between thiorphan and Kell, we employed microscale thermophoresis (MST), which detects interaction-induced shifts in the thermophoretic mobility of proteins. This analysis revealed a strong binding affinity, with a dissociation constant (Kd) of 33 μM between thiorphan and Kell (Supplementary Fig. 5A). Complementary to these findings, a cellular thermal shift assay (CETSA) confirmed significant enhancement in Kell’s thermostability upon thiorphan binding, with a calculated binding energy of −5.83 kcal/mol, indicating strong engagement with Kell’s catalytic site (Supplementary Fig. 5B, C).
Host-specific efficacy of thiorphan against Kell metalloprotease
To further elucidate the antimalarial activity of thiorphan, we conducted an in vitro invasion assay using purified P. falciparum (Pf3D7) schizonts exposed to different concentrations of thiorphan, yielding an IC50 of approximately 621.6 nM. Given that Kell is the primary host target of thiorphan, an additional assay was performed to confirm that the observed inhibition was specifically due to Kell inhibition. In this parallel experiment, erythrocytes were pretreated with varying concentrations of thiorphan, followed by thorough washing to remove any unbound drug (Fig. 3F (Schematic representation)). When the pretreated erythrocytes were subsequently exposed to Pf3D7 schizonts, the invasion was reduced by ~50% at a concentration of 507.3 nM, consistent with the IC50 obtained from direct parasite exposure. These results reinforce that thiorphan’s inhibitory effect on parasite invasion is mediated through its interaction with the host protein Kell (Fig. 3G, H).
Moreover, to assess the impact of thiorphan on the entire intraerythrocytic developmental cycle (IDC), a 72-h growth inhibition assay was performed using both chloroquine-sensitive (Pf3D7) and chloroquine-resistant (PfRKL-9) strains. The results demonstrated 50% growth inhibition at concentrations of 579.9 nM for Pf3D7 and 655.5 nM for PfRKL-9, indicating potent antimalarial activity of thiorphan irrespective of the parasite strain (Fig. 3I, J). Notably, no morphological abnormalities were observed in parasites treated with thiorphan during the IDC, suggesting that its primary mode of action targets the invasion stage without affecting the later stages of parasite development.
Collectively, these data underscore Kell’s crucial role in facilitating protease-dependent Plasmodium falciparum invasion of erythrocytes21. Nevertheless, while these findings offer significant insights, further mechanistic studies are warranted to delineate the exact nature of Kell’s interaction with plasmodium proteins and its broader implications in the merozoite invasion pathway. Advanced experimental approaches will be required to fully elucidate this interaction and its potential as a therapeutic target.
In vivo antimalarial efficacy of thiorphan and its prodrug racecadotril
Murine Kell shares ~74% sequence homology with its human counterpart22, and our in vitro protease assays with murine erythrocytes subjected to varying concentrations of thiorphan, revealed a concentration-dependent reduction in Kell’s activity (Fig. 4A, B). It is noticeable that the IC50 for the rodent cell enzymatic assay is lower than that for the human cell. The observed lower IC50 in rodent

A Schematic representation (created by Biorender.com) of the in vitro Kell enzymatic activity with the mice erythrocytes. B Bar graph illustrating the enzymatic activity of the Kell antigen in murine erythrocytes under varying concentrations of thiorphan, highlighting the dose-dependent inhibition of Kell-mediated proteolytic activity. C Line graph depicting the antimalarial efficacy of thiorphan (2.5 mg/kg) in reducing parasite density in a murine model infected with Plasmodium berghei, demonstrating its potential therapeutic effects as compared to untreated (no. of mice in each group = 3). D Representative Giemsa-stained microscopic images displaying the morphological impact of Thiorphan treatment on P. berghei-infected erythrocytes in mice, emphasizing parasite clearance post-treatment. E Survival curve comparing Thiorphan-treated versus untreated mice, showcasing improved survival rates in the treated group. F Dose-response graph showing the effects of different concentrations of Racecadotril (5, 25, 125 mg/kg) administered via intraperitoneal injection on parasite density in P. berghei-infected mice, indicating a concentration-dependent reduction in parasitemia. G Representative microscopic images of Giemsa-stained blood smears from mice treated with Racecadotril, illustrating the extent of parasitic reduction at its different concentrations. H Survival plot comparing the longevity of Racecadotril-treated and untreated mice, with significant survival benefits observed in the treated cohort (*p < 0.05).
cell assays compared to human cells may be attributed to several factors. One critical difference is the size of red blood cells (RBCs), with mouse RBCs being significantly smaller than human RBCs. The reduced size impacts the overall surface area-to-volume ratio, potentially affecting drug uptake and the interaction of the parasite with the host cell membrane. These variations can influence drug binding affinity, catalytic efficiency, and overall sensitivity, leading to a lower IC50 in rodent cells.
In the animal study, both the thiorphan and racecadotril treatment was performed together according to IAEC guidelines for racecadotril treatment, each group contained five mice, while for thiorphan treatment, each group contained three mice. This sample size was chosen to ensure reliable and reproducible results, while minimizing unnecessary animal use. The inclusion of the number of mice per group allowed for consistent comparisons across experimental conditions, supporting robust data generation while adhering to best practices in animal research.
To evaluate the in vivo antimalarial efficacy of thiorphan, we administered the compound to P. berghei-infected mice. Strikingly, by the second day of treatment, we observed a significant fivefold reduction in parasitemia (Fig. 4C), as evidenced by representative Giemsa-stained blood smears (Fig. 4D). However, by the third day, parasite resurgence was evident, likely due to the suboptimal attainment of thiorphan’s minimum inhibitory concentration (MIC) in the bloodstream across the dosing regimen. To address this concern, a more comprehensive assessment of pharmacokinetics analysis of thiorphan, with a particular focus on its clearance, is necessary. Despite this, thiorphan-treated mice exhibited improved survival rates compared to the untreated control group (Fig. 4E).
In parallel, we tested racecadotril, a clinically approved anti-secretory enkephalinase inhibitor used to treat diarrhea, which acts as a prodrug of thiorphan. Racecadotril’s distinctive characteristics—high therapeutic index, exceptional safety profile, inability to cross the blood-brain barrier, and rapid conversion to thiorphan—render it a promising candidate for antimalarial activity. Administering varying oral doses of Racecadotril to P. berghei-infected mice resulted in a 50–60% reduction in parasitemia by the third day (Fig. 4F), as shown by Giemsa-stained smears (Fig. 4G), along with an improved survival rate relative to the control group (Fig. 4H). Notably, Racecadotril treatment was well-tolerated, with no significant adverse effects observed in the mice. While we observed significant effects in the mouse model, we cannot conclusively exclude the involvement of alternative mechanisms, including the potential inhibition of Kell’s ET-3 activity, in contributing to the observed reduction in parasitemia following treatment.
Ex vivo antimalarial efficacy of thiorphan
In malaria-endemic regions, host-parasite evolutionary interactions often drive mutations in erythrocytic proteins, providing critical insights into the natural selection of host factors essential for parasite survival. To further our understanding, we assessed Kell expression levels on erythrocytes from individuals residing in the malaria-endemic regions of Tripura (sample was collected from individuals infected with the malaria parasite), India. IFA (Fig. 5A, B) and Flow cytometry analysis (Fig. 5C, D) revealed a notably lower expression of Kell on erythrocytes from individuals in endemic regions compared to non-endemic areas (Delhi, India). This suggests that Plasmodium-induced selective pressure may drive alterations in Kell expression profiles.

A Immunofluorescence assay (IFA) images depict a significant reduction in surface expression of the Kell antigen on erythrocytes isolated from malaria-infected individuals residing in malaria-endemic regions (Tripura, India) compared to those from a non-endemic region (Delhi, India). B The mean fluorescence intensity (MFI) analysis of Kell expression, derived from IFA results, highlights notable regional variations in antigenic presentation between erythrocytes from both malaria-endemic and non-endemic areas. C The histogram illustrates flow cytometry analysis of Kell expression in erythrocyte samples, corroborated by microscopy assessment, further emphasizing the differences in antigen levels across the studied populations. D Flow cytometry results reveal a statistically significant reduction in Kell surface expression on clinical isolates from malaria-endemic regions compared to non-endemic regions (*p < 0.05). Data were acquired using a BD FACS™ machine and analyzed via FlowJo software. The y-axis represents relative Kell expression, comparing Kell expression in erythrocytes from Delhi residents with samples from a malaria-endemic region, ensuring an accurate assessment of regional differences in Kell expression as a potential factor in disease susceptibility. E Ex vivo antimalarial efficacy of Thiorphan on clinical isolates of Plasmodium from the endemic region (Tripura) is demonstrated, with IC50 values depicted. These are contrasted with IC50 values obtained from a laboratory strain of the parasite (Pf3D7). F MFI analysis of Kell’s metalloendopeptidase activity in erythrocytes from both endemic and non-endemic regions. G A positive correlation is observed between donor erythrocytes with reduced Kell expression (X-axis) and diminished Kell-mediated protease activity (Y-axis), as reflected by a Pearson’s correlation coefficient of 0.3685. H Donors with lower Kell expression exhibited a positive correlation with the enhanced ex vivo antimalarial efficacy of thiorphan, with a Pearson’s correlation coefficient of 0.4170. I A correlation graph highlights a significant positive association between reduced Kell-mediated enzymatic activity and the ex vivo antimalarial efficacy of thiorphan, with a Pearson’s correlation coefficient of 0.4114. The linear regression line is depicted in brown, with a 95% confidence interval band (dashed line).
Ex vivo antimalarial assays further revealed that thiorphan exhibited enhanced efficacy against field strains of the malaria parasite compared to its in vitro performance against Pf3D7 (Fig. 5E). This increased sensitivity may stem from the diminished Kell expression on erythrocytes from malaria-endemic populations. Furthermore, we observed a reduced level of protease activity in erythrocytes from these individuals relative to those from non-endemic regions (Fig. 5F). A correlation analysis demonstrated that individuals with low Kell expression not only exhibited reduced protease activity but also showed heightened sensitivity to thiorphan (Fig. 5F–H).
Our data suggest that lower Kell expression on erythrocytes may confer a protective phenotype, rendering them less susceptible to parasite invasion. This supports the hypothesis that populations exposed to malaria have evolved resistance mechanisms through the selective pressure toward reduced Kell expression and activity. However, the samples collected were a relatively small number and were not controlled for age, ethnicity, other blood groups, etc.
Discussion
Malaria, instigated by parasites of the Plasmodium genus, remains a significant global health burden. Current pharmacological interventions have predominantly targeted the intrinsic pathways of these parasites; however, the alarming proliferation of drug-resistant strains necessitates the exploration of novel therapeutic strategies. The identification of host molecular components that Plasmodium manipulates for its advantage can illuminate the intricacies of host-pathogen interactions and unveil potential host-directed targets for innovative drug development3,4.
The present study elucidates the pivotal function of the Kell antigen, a zinc-dependent endopeptidase23, in the invasion of P. falciparum, thereby contributing to the evolving paradigm of host-directed antimalarial therapy.
The Kell blood group system, encompassing over 35 antigens, is encoded by the KEL gene on chromosome 715,24. It plays a crucial role in erythrocyte physiology and immunity. The Kell antigen is a zinc-dependent endopeptidase that cleaves endothelin-3, influencing erythropoiesis and vasoregulation14,16. Polymorphisms within the KEL gene lead to variations in Kell antigen expression, with notable clinical implications, including hemolytic disease of the newborn (HDN) and transfusion reactions25. The KEL1 and KEL2 alleles encode the K1 (Kell) and K2 (Cellano) antigens, respectively13,25,26.
Our findings highlight Kell as a novel and indispensable factor co-opted by the malaria parasite to facilitate erythrocyte invasion, a critical event in malaria pathogenesis. This research signifies a substantial advancement in our comprehension of host-parasite interactions, particularly emphasizing the role of erythrocyte surface proteins in facilitating malaria invasion.
Utilizing CRISPR-Cas9 gene editing, we successfully generated a Kell knockout (KO) in the BEL-A erythroid cell line18, which resulted in a significant reduction in parasite invasion, validating Kell’s critical role in this process. Furthermore, the expression of other surface receptors, such as Band 3, Glycophorin A and C, as well as the cytoskeletal proteins spectrin and ankyrin, which do not form a direct interaction with Kell but rather interact through Band 327,28, remained unaffected compared to unedited BEL-A-derived reticulocytes.
The incomplete inhibition of merozoite invasion suggests that the parasite may exploit additional host factors or redundant invasion pathways when Kell is absent or inhibited. To further validate the role of Kell’s proteolytic activity in invasion, we utilized thiorphan, a potent metalloprotease inhibitor. Thiorphan-mediated inhibition of Kell significantly impaired parasite invasion, yet no additional reduction was observed in Kell-deficient erythrocytes. This finding underscores a Kell-dependent mechanism underlying thiorphan’s antimalarial efficacy.
Supporting this, biochemical assays—including enzymatic inhibition and target engagement analyses—confirmed thiorphan’s specific binding to Kell’s catalytic pocket, reinforcing its potential as a targeted inhibitor against malaria parasite invasion.
Furthermore, inhibition of Kell protease with thiorphan did not completely block the invasion process. This redundancy likely represents an evolutionary adaptation by the parasite, ensuring successful erythrocyte entry even when a single invasion receptor is targeted under selective pressure29. Our findings are consistent with those of Soubes et al., who reported that the absence of Kell does not fully block invasion but rather supports the parasite’s ability to utilize alternative invasion routes30.
The ability to disrupt and complement phenotypes through genetic manipulation is a fundamental approach in cell biology and genetics. However, applying such techniques directly to red blood cells is challenging due to their anucleate nature. The development of in vitro culture systems for nucleated erythroid precursors, which can be genetically modified before differentiating into enucleated reticulocytes, has opened new avenues for exploring erythrocyte biology and host-parasite interactions17,31.
We further confirmed the specificity of Kell’s proteolytic activity in invasion through thiorphan, a potent metalloprotease inhibitor32,33. Moreover, we extended our findings to an in vivo context utilizing a murine model of malaria, where thiorphan administration markedly reduced parasitemia and enhanced survival rates. Despite the resurgence of parasites following treatment cessation, the results indicate promising therapeutic potential. Notably, we also demonstrated the antimalarial efficacy of racecadotril, a clinical prodrug of thiorphan, which displayed enhanced bioavailability and in vivo potency34,35,36. This repurposing of an already-approved drug highlights the translational potential of our approach, potentially expediting clinical applications37,38.
Despite the promising nature of these findings, several limitations merit consideration. The murine model, although prevalent in malaria research, does not fully encapsulate the complexities of human red blood cell biology, potentially limiting the translatability of these results. Differences in red blood cell size, surface properties, and drug pharmacokinetics may account for the potency observed for thiorphan in murine versus human erythrocytes. Furthermore, while thiorphan and racecadotril exhibit robust antimalarial effects, the incomplete clearance of parasites and subsequent resurgence in parasitemia post-treatment underscore the challenges in achieving sustained therapeutic efficacy. Future investigations should prioritize the optimization of dosing regimens, the exploration of combination therapies, and the assessment of potential resistance development. A promising avenue for future research includes the employment of Kell knockout mice to comprehensively evaluate the impact of Kell inhibition on malaria infection. Such models would facilitate a direct assessment of how the absence of Kell influences parasite invasion and host-parasite interactions in vivo, providing invaluable insights into Kell’s role in malaria pathogenesis. Additionally, utilizing Kell knockout mice could address safety concerns and off-target effects associated with Kell inhibition, thereby offering a more physiological context for therapeutic interventions.
Furthermore, this study illustrates the ability of BEL-A-derived reticulocytes to support intracellular development and reinvasion, paving the way for potential continuous growth assays over multiple cycles. However, the current high costs associated with producing sufficient reticulocytes for large-scale experiments remain a significant obstacle. Future efforts should focus on optimizing reticulocyte production, curtailing culture expenses, and enhancing long-term storage conditions to facilitate such assays.
Additionally, our study unveiled a fascinating genetic adaptation within populations from malaria-endemic regions of India, where reduced Kell expression on erythrocytes correlates with a putative protective mechanism against malaria. Previous studies have illuminated the evolutionary conflict between host and parasite, exemplified by the prevalent single nucleotide polymorphism (SNP) in the Duffy antigen receptor for chemokines (DARC) gene among African populations. This mutation disrupts Duffy antigen expression, resulting in the Duffy-null phenotype, which confers resistance to Plasmodium vivax infection. However, recent reports indicate that this protection is not absolute, with increasing incidences of P. vivax infections documented in Duffy-negative individuals, thereby challenging the efficacy of this protective mechanism39,40,41. Furthermore, a noteworthy illustration of adaptive evolution in host genetic factors is the intricate relationship between the sickle cell trait (HbS) and glucose-6-phosphate dehydrogenase (G6PD) deficiency mutations in malaria-endemic regions42,43. This dynamic exemplifies the profound influence of natural selection, driven by the selective pressures exerted by infectious diseases. Both genetic mutations confer a significant survival advantage against Plasmodium infection, particularly P. falciparum, thereby shaping their prevalence in populations inhabiting malaria-prone environments. Moving forward, future research involving studying the role of Kell polymorphism to compare Kell expression across diverse environmental settings and investigate how varying intensities of malaria transmission shape these genetic adaptations. This will help unravel the role of environmental and evolutionary forces in modulating Kell expression and its connection to malaria susceptibility. However, further analysis involving larger sample sizes will be required to substantiate this relationship conclusively.
In summary, this study presents compelling evidence that the Kell blood group antigen (metalloprotease) plays a critical role in facilitating Plasmodium invasion, thereby unlocking new avenues for host-directed therapies. However, although these findings provide valuable insights, additional mechanistic research is needed to clarify the precise nature of Kell’s interaction with Plasmodium proteins and its broader role in the merozoite invasion process. More sophisticated experimental methods will be necessary to fully understand this interaction and assess its potential as a therapeutic target. The utilization of thiorphan and its prodrug racecadotril, specifically targeting Kell’s proteolytic activity, underscores the therapeutic potential of inhibiting host proteases. Future investigations should aim to optimize dosing regimens and explore combination therapies to augment antimalarial efficacy. Furthermore, deeper exploration into the broader implications of host genetic adaptations and their capacity to influence parasite invasion strategies could unveil new therapeutic targets, thereby advancing the ongoing battle against drug-resistant malaria.
Materials and methods
BEL-A cell culture
The BEL-A cells were cultured according to the methodology originally described earlier18. In the expansion phase, cells were maintained in a serum-free expansion medium (StemSpan SFEM; Stem Cell Technologies) supplemented with specific components, including 50 ng/ml human recombinant stem cell factor (hSCF), 3 IU/ml Erythropoietin, 10−6 M dexamethasone (Sigma), and 1 μg/ml doxycycline (Takara Bio). Culture media was regularly changed every 48 h and maintained at 37 °C in a 5% CO2 incubator.
sgRNA design and cloning
Four sgRNAs were designed for Kell gene using CHOP-CHOP (https://doi.org/10.1093/nar/gkz365) against Exon 1, 2, and 6. Each sgRNA was cloned into the Cas9 backbone vector with tGFP (pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988)), as previously described44. sgRNA and primer sequences are mentioned in Tables S1 and S2, respectively.
Surveyor assay
Each sgRNA was transfected into HEK293T (obtained from ATCC, mycoplasma-free) cells using Lipofectamine 3000 (Thermo Fisher Scientific) as per the manufacturer’s protocol. After 48 h of transfection, cells were harvested, genomic DNA was isolated, and amplified using genotyping primers. A surveyor assay using T7 endonuclease I (NewEngland Biolbs (NEB)) was performed to estimate the editing efficiency of sgRNAs, as described earlier45.
Electroporation in BEL-A cells
To facilitate the knockout of Kell in BEL-A cells, electroporation was conducted with pCas9-sgRNA-Kell at specified parameters: 1100 V, 30 ms, and three pulses using the Neon electroporation system (Invitrogen). Six hours post-transfection, cells were washed and resuspended in an expansion medium. After 48 h, GFP-positive cells were subjected to single-cell sorting in a 96-well plate and cultured until confluency was achieved. Gene editing was confirmed by surveyor assays and Sanger sequencing.
Erythroid differentiation of BEL-A cells
The BEL-A WT, and edited BEL-A cells were differentiated into erythroid cells by culturing with a differentiation medium of IMDM (Sigma-Aldrich) supplemented with 3% human AB serum (Sigma-Aldrich), 2% fetal bovine serum (FBS) (Thermo Fisher Scientific), 1X antibiotic antimycotic solution (Sigma-Aldrich), 200 µg/mL Holo-transferrin (Sigma-Aldrich), 10 µg/mL recombinant human insulin (Sigma-Aldrich), 3 IU/mL heparin (STEMCELLTechnologies), 3 IU/mL erythropoietin (Peprotech), 1 µg/mL doxycycline (Sigma-Aldrich), 10 ng/mL stem cell factor (Immunotools), 1 ng/mL Interleukin-3 (IL-3) (Immunotools). The cells were seeded at 2 × 105 cells/mL density at day 0, followed by 3.5 × 105 cells/mL on day 2. The cells were further seeded at 5 × 105 cells/mL on day 4, and doxycycline was removed from the medium. From day 6 onwards, 1 × 106 cells/mL density was maintained, and the concentration of holo-transferrin was increased to 500 µg/mL. On day 6, SCF and IL-3 were also removed from the medium, and the cell density was maintained at 1 × 106 henceforth until Day 1446. At the end of erythroid differentiation, reticulocytes were isolated by centrifuging at 848 rcf for 5 min. These reticulocytes is used further for invasion assay and enzymatic assay.
Characterization of Kell-KO in BEL-A-derived reticulocytes
Antibody used
Antibodies used were as follows: Kell (Invitrogen: PA5-47221); Xk (Invitrogen: PA5-40782); Band 3 (Sigma: B-9277); (Glycophorin A (Sigma: G-7900); Glycophorin C (Sigma:G7775); Band 3 (Sigma: B-9277); Spectrin α II (Santa Cruz: sc-48382); Ankyrin (Santa Cruz: sc-12719); Goat anti-Mouse IgG (H + L) Secondary Antibody, HRP (Invitrogen: 62-6520); Goat anti-Rabbit IgG (H + L) Secondary Antibody, HRP (Invitrogen: 31460); Donkey anti-Goat IgG (H + L) Secondary Antibody, HRP (Invitrogen: A15999); Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 (A-11032); Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 (A-11001); HRP-conjugated GAPDH Mouse mAb (AC035); Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 (A-11008); secondary anti-goat IgG conjugated with Alexa fluor-488 (Invitrogen; A-11055)
Immunofluorescence assay
To examine the expression of Kell in BEL-A WT and Kellnull erythrocyte. A thin smear of RBCs was fixed with chilled methanol for 30 min, blocked with 3% BSA (Sigma), and incubated with an anti-Kell primary antibody (Invitrogen: PA5-47221) at a 1:200 dilution for 1 h and secondary anti-goat IgG conjugated with Alexa fluor-488 (Invitrogen; A-11055). Microscopic analysis was performed using a fluorescence microscope (Olympus) with a 60X objective lens. For fluorescence imaging, we utilized an Olympus microscope equipped with a halogen light source in conjunction with a 60X objective lens for all images used in our quantitation. The light source settings were kept consistent throughout the experiments to ensure uniform illumination. The same set of excitation and emission filters were employed across all images, specifically optimized for the detection of [Alexa-488 (FITC channel).] Additionally, the detection module was standardized [specific gain: 4.48x or exposure time: 74.94 ms] across all experimental conditions. These settings were optimized during initial calibrations and applied consistently to avoid any variations in signal intensity that could affect quantitation. All images used for analysis were captured under identical settings, ensuring reproducibility and accuracy in the data obtained. During analysis of the images, no alteration in intensity was applied.
Flow cytometry
Expression of RBC surface proteins was measured in unedited and Kellnull BEL-A derived reticulocytes by flow cytometry. For flow cytometry, 1 × 105 cells from each of the group was resuspended in PBS + 1% BSA, blocked with 5% BSA for 30 min, and were labeled with primary antibody for 1 h at RT. Cells were washed in 1X PBS, and then incubated for 1 h at RT with appropriate Alexa fluor-conjugated secondary antibody, followed by washing and data acquisition in Amnis image flow cytometer. Staining was performed for different antigens (Band 3, Kell, Xk, Glycoporin A, Glycophorin C, Spectrin, and Ankyrin). For staining of spectrin and ankyrin cells were permeabilized with Triton X-100 (0.001% in 1X PBS) for 5 min, followed by respective primary and secondary antibody staining. The dilutions for the primary and secondary antibodies were prepared according to the manufacturer’s specifications provided in the datasheets.
Immunoblotting
To assess the expression of Kell and other surface markers, in unedited and Kellnull BEL-A derived reticulocyte equal amount of lysate fraction (total reticulocyte) was resolved on a 12% SDS-PAGE gel and blotted to a 0.45-µm nitrocellulose membrane (Bio-Rad) using a semi-dry transfer apparatus. Immunoblotting was performed using a goat-raised anti-Kell primary antibody (Invitrogen; 1:2000) and HRP-conjugated anti-goat secondary antibodies (Invitrogen; 1:10,000). The signal was analyzed using the ChemiDoc Gel Imaging System and Image Lab Software (Bio-Rad). To analyze the expression of other surface proteins in unedited and Kellnull BEL-A derived reticulocyte lysate fraction immunoblotting was performed with Band 3 (1:5000 dilution); Xk (1:500 dilution), Glycophorin A (1:5000), Glycophorin C (1:5000), Spectrin (1:1000), Ankyrin (1:1000) as per the manufacture protocols. For loading control (in terms of the number of reticulocytes), an equal number of reticulocytes as of the test sample was immunoblotted and probed with GAPDH.
Off-target analysis
For off-target analysis, off-target sites were predicted using the COSMID tool, which identifies potential genomic regions with sequence homology to the designed sgRNA47. Based on the predicted off-target sites, specific primers were designed flanking each potential off-target region. Genomic DNA was extracted from the edited cells, and PCR was performed using the designed primers to amplify the off-target regions. The PCR products were then purified and subjected to Sanger sequencing to detect any unintended mutations. Sequence alignments were performed against the reference genome to identify mismatches, insertions, or deletions at the predicted off-target sites, providing insights into the specificity of the genome editing.
Parasite culturing
In vitro Culture of P. falciparum 3D7 strain was cultured using O+ human erythrocytes, under a mixed gas environment (5% O2, 5% CO2, and 90% N2) as described previously48. The culture media was composed of RPMI 1640 (Invitrogen, Carlsbad, CA, United States), supplemented with 50 mg/L hypoxanthine (Sigma-Aldrich, St. Louis, MO, United States), 2 g/L sodium bicarbonate (Sigma-Aldrich, St. Louis, MO, United States), and 5 g/L Albumax I (Gibco, Grand Island, NY, United States). Human erythrocytes (O+), as well as the serum of healthy volunteers, were procured from the blood bank and used only for research purposes. Synchronous development of the erythrocytic stages of a human malaria parasite, P. falciparum, in culture was accomplished by suspending cultured parasites in 5% D-sorbitol and subsequent reintroduction into culture49.
Invasion assay in BEL-A-derived reticulocytes
The invasion assay was conducted using reticulocytes derived from both unedited and Kellnull-derived reticulocytes. Pf3D7 parasites were enriched at the 44–46 h schizont stage with 95% purity through Percoll gradient centrifugation (GE Healthcare). Purified schizonts were incubated with the respective reticulocytes in a 100 µL culture volume per well in a 96-well plate, with initial parasitemia and hematocrit levels set at 1 and 2%, respectively. After 10 h of incubation, slides were prepared by smearing the cells after cytocentrifuge. Fixed with methanol (Sigma-Aldrich, USA) and stained with Giemsa (Sigma-Aldrich, USA). For quantification of invasion, cells were counted per cytospin. A total of 5000 cells were counted in each of the biological replicates (n = 3).
To assess the effect of thiorphan, the invasion of reticulocytes derived from WT and Kellnull BEL-A knockout cells was evaluated in the presence of thiorphan at its IC50 concentration. For quantification of invasion, cells were counted per cytospin. A total of 5000 cells were counted in each of the biological replicates (n = 3). For flow cytometry, cells were washed in PBS, stained with ethidium bromide (10 µg/mL in PBS; Sigma-Aldrich) for 30 min at room temperature in the dark, and washed three times in PBS. In total, 1 × 105 cells from each well were acquired using channel 4 of an Amnis image flow cytometer.
Protease activity
In BEL-A-derived reticulocytes
To determine the ability of BEL-A-derived reticulocytes with the Kell phenotype to cleave a synthetic fluorogenic peptide substrate, an enzymatic assay was performed. About 10 µM of a synthetic fluorogenic peptide (Glut-FAAF-AMC) was mixed with 2 × 106 BEL-A WT and Kellnull reticulocyte, respectively in a 100 µL of reaction buffer (50 mM HEPES, pH 7, and 0.15 M NaCl)13. Following 1 h incubation at 37 °C, fluorescence intensity was measured using a microplate reader (Thermo Fisher Scientific; λex : 340 nm and λem: 440 nm).
In erythrocytes
To determine the ability of erythrocyte with the Kell phenotype to cleave a synthetic fluorogenic peptide substrate, an enzymatic assay was performed as described above with 2 × 106 erythrocyte. To determine the thiorphan sensitivity on Kell protease activity, enzymatic assay was performed with different concentration of thiorphan and fluorescence intensity was measured as described above.
Zinc-dependent affinity purification of Kell from erythrocytes
To ascertain that the surface protease activity observed on erythrocytes is solely due to Kell. We next aimed to purify Kell from ghost erythrocytes, for which membrane fraction was prepared by repeatedly washing RBCs in chilled hypotonic solution (5 mM HEPES). Membrane proteins were extracted from RBC ghost (lysis buffer:50 mM HEPES, pH 7.2; 1% Triton X-100 (Sigma); 1% IGEPAL CA-30 (Sigma); and, 1X PIC (Roche complete tablets)) Following centrifugation (9425 rcf), detergent-sensitive fraction was incubated with zinc resin (G-bioscience) pre-equilibrated with the lysis buffer as per the manufacture protocol. The resin was subsequently pelleted (94 rcf), washed, and eluted with 2 CV of elution buffer (50 mM HEPES, pH 7.2, 150 mM NaCl, and 250 mM Imidazole) followed by Immunoblot analysis using Kell-specific antibody as described above.
Protease activity with the purified fraction of Kell
To ascertain the Kell-specific inhibitory role of thiorphan on its protease activity, protease activity was performed with a purified fraction of Kell (500 ng) in the absence and presence of a range of concentrations of thiorphan as mentioned above in the same buffer condition. % Enzymatic activity was calculated by using the formula: 100× (λemm of Untreated sample – blank]-[λemm – blank]/ [λemm – blank])
Antimalarial activity of thiorphan
Parasite invasion assays were performed using synchronized late-stage schizont parasites that were isolated through percoll gradient centrifugation. For Erythrocyte invasion of Pf3D7 in the presence of thiorphan purified schizonts were incubated with RBCs to maintain the 1% parasitemia and 2% hematocrit with different concentrations of thiorphan in a total volume of 100 µL per well of a 96-well microtiter plate. Wells with DMSO served as controls. Ten hours post-incubation, smears were prepared and newly invaded ring-stage parasites were quantified by Giemsa staining.
To evaluate the host-mediated effect, RBCs were pretreated with thiorphan at concentrations ranging from 100 nM to 1.6 µM for 2 h, followed by washing with 1X iRPMI to remove any remaining drug before conducting the invasion assay. Purified schizonts were then added to the thiorphan pretreated RBCs in a 100 µL reaction volume per well, with conditions maintained at 2% hematocrit and 1% parasitemia. After 10 h of incubation, blood smears were prepared, and newly invaded ring-stage parasites were counted using Giemsa staining. A total of 3000 cells were counted in each of the experiments in three biological replicates (n = 3).
The results were expressed as the percent inhibition compared to the untreated controls, calculated with the following formula: 100× ([OD of Untreated sample – blank]-[OD– blank]/ [OD – blank]) and represented as a sigmoidal curve generated through GraphPad Prism 8.0 software.
Growth inhibition assay
Two different strains of P. falciparum, RKL-9 (chloroquine-resistant) and 3D7 (chloroquine-sensitive), were used for the chemosensitivity tests. For this, the compounds were dissolved in DMSO and then diluted with medium to achieve the required concentrations (final DMSO concentration <1%, which is non-toxic to the parasite). The drugs were placed in 96-well flat-bottom microplates in duplicate at different concentrations (10, 5, 2.5, 1.25, 0.625, 0.312, and 0.156 µM). Sorbitol synchronized cultures with 0.8-1% parasitemia and 2% hematocrit were aliquoted into the plates and incubated for 72 h in a final volume of 100 µL/well. Parasite growth was determined through Giemsa analysis in a blinded manner. IC50 values were determined via nonlinear regression analysis using GraphPad Prism 8.0 software. The results were expressed as the percent inhibition compared to the untreated controls, calculated with the following formula: 100× ([OD of Untreated sample – blank]-[OD– blank]/ [OD – blank]) in independent biological repeats (n = 3). As blank, uninfected RBCs were used. IC50, which is the dose required to cause 50% inhibition of parasite viability, was determined by extrapolation.
Cellular thermal shift assay (CETSA) to evaluate the interaction of Kell with thiorphan
To investigate the interaction of thiorphan with Kell, a CETSA was performed as mentioned previously50. This involved resuspending 200 µL washed RBCs in 300 µL of 1X PBS and treating them with thiorphan (10 µM). After 2 h of treatment, ghost membrane fraction lysate was prepared. Detergent-sensitive fractions from both thiorphan-treated and untreated samples were equally aliquoted, and incubated at different temperatures (40, 60, and 80 °C) for 5 min, followed by centrifugation at 9425 rcf for 20 min, the supernatant fraction was collected and loaded on 12% SDS gel. Immunoblotting was then performed using the Kell antibody, as mentioned above.
In vivo analysis of antimalarial activity with thiorphan and racecadotril
Animal studies were performed in accordance with guidelines of the Institutional Animal Ethics Committee (IEAC) of Jawaharlal Nehru University (JNU), Delhi and the Committee for Control and Supervision of Experiments on Animals (CPCSEA).
In this study, we evaluated the antimalarial efficacy of thiorphan using a murine infection model. Six mice were randomly divided into two groups: a treatment group and a control group, with three mice in each (n = 3). The treatment group received thiorphan at a dosage of 2.5 mg/kg, administered through the intraperitoneal route daily for three consecutive days, while the control group received an equivalent volume of the vehicle.
To evaluate the antimalarial efficacy of Racecadotril (prodrug of thiorphan), P. berghei ANKA infected mice were divided into four groups: three treatment groups and one control group with five mice in each group (n = 5) and given oral doses of 5, 25 mg, and 125 mg/kg of body weight, respectively, for 11 consecutive days, with dosages administered twice daily. The efficacy of thiorphan and Racecadotril treatment was assessed by comparing parasitemia levels between the treatment and control groups. To assess the impact on parasitemia in the infected mice, smears were made, stained with Giemsa, and subsequently analyzed under a light microscope.
Ex vivo antimalarial activity
Fresh blood samples were collected from malaria-infected patients following ethical approval and informed consent. Whole blood was centrifuged at 848 rcf for 10 min to separate plasma. The RBC pellet was washed twice with RPMI 1640 medium (without serum), and the hematocrit was adjusted to 2% without adding any fresh erythrocytes.
Drug preparation and assay setup
To assess the antimalarial potency of thiorphan in clinical isolates ex vivo antimalarial activity was monitored. Briefly, 100 µL of infected RBC suspension (containing 0.5–1% parasitemia) in 96-well plates containing different concentrations of thiorphan respectively was added to each well, resulting in a final hematocrit of 2%. Control wells with drug-free medium were also included. Plates were incubated at 37 °C in a gas mixture (5% CO₂, 5% O₂, and 90% N₂) for 72 h. Furthermore, along with it, the 3D7 strain was also tested against thiorphan with the same range of concentration to determine the IC50 and to compare it with the field strain.
Smears were prepared for each sample, stained with Giemsa, and assessed through blinded manual counting of Giemsa-stained smears to determine the IC50 of thiorphan.
FACS analysis for the expression of Kell on erythrocyte surface from field isolates (Tripura region, India)
Blood samples from active malaria patients were obtained with the consent of patients and in the case of minors from their parents/guardians as approved by the Institutional Ethics Committee of Agartala, Govt. Medical College, Agartala [F.4-(6-13) AGMC/Medical Education/IEC Approval/2022). The patients’ details are presented in Supplementary Table S3.
To evaluate the Kell expression on erythrocytes from the isolates of the Tripura region, India, erythrocytes were washed, centrifuged at 848 rcf, and resuspended in 1X PBS. 2 × 107 RBCs from each sample were fixed with 2.5% Glutaraldehyde for 30 min at RT, followed by blocking in 3% BSA for 30 min. Subsequently, cells were probed with primary Kell antibody (2.5 µg of the Kell antibody per million cells in 1X PBS) and secondary Alexa fluor-488-conjugated antibody (1:200 dilution in 1X PBS) followed by washing with 1X PBS three times. The sample was then run in BD FACSTM and analyzed using FlowJo V10 software. Briefly, for MFI calculation, first, the erythrocyte populations were selected based on forward scatter (FSC) and side scatter (SSC) parameters to ensure appropriate gating and identification of the desired cell population. FSC was used to assess cell size, while SSC measured granularity or internal complexity. Once the target population was accurately gated, the median fluorescence intensity (MFI) of the cells was calculated. This MFI represents the level of the specific marker expression in the selected cell populations, enabling a comparative analysis between samples from malaria-endemic and non-endemic regions. Simultaneously with microscopic analysis, thin smears of immunostained erythrocytes were prepared on glass slides, mounted with DAPI-antifade, and examined under a fluorescence microscope. The light source settings were maintained consistently across all experiments to ensure uniform illumination.
Statistics and reproducibility
Statistical analyses were performed using Student’s t-test in GraphPad Prism 8.0 software. Data were presented as the mean ± standard deviation (SD) of at least three independent biological replicates (n = 3), unless otherwise indicated. A p value of ≤0.05 was considered statistically significant, with significance levels indicated as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. Parametric Student’s t-test (two-tailed) was used to examine the significance between the two groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Cowman, A. F. & Crabb, B. S. Invasion of red blood cells by malaria parasites. Cell 124, 755–766 (2006).
Foth, B. J. et al. Quantitative time-course profiling of parasite and host cell proteins in the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteom. 10, M110.006411 (2011).
Adderley, J. & Grau, G. E. Host-directed therapies for malaria: possible applications and lessons from other indications. Curr. Opin. Microbiol. 71, 102228 (2023).
Glennon, E. K. K., Dankwa, S., Smith, J. D. & Kaushansky, A. Opportunities for host-targeted therapies for malaria. Trends Parasitol. 34, 843–860 (2018).
Burns, A. L. et al. Targeting malaria parasite invasion of red blood cells as an antimalarial strategy. FEMS Microbiol. Rev. 43, 223–238 (2019).
Wright, G. J. & Rayner, J. C. Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLoS Pathog. 10, e1003943 (2014).
Cowman, A. F., Berry, D. & Baum, J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 198, 961–971 (2012).
Cowman, A. F., Tonkin, C. J., Tham, W. H. & Duraisingh, M. T. The molecular basis of erythrocyte invasion by malaria parasites. Cell Host Microbe 22, 232–245 (2017).
Blackman, M. J. Proteases in host cell invasion by the malaria parasite. Cell. Microbiol. 6, 893–903 (2004).
Chandramohanadas, R. et al. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science 324, 794–797 (2009).
Mattaloni, S. M. et al. Clinical significance of an alloantibody against the Kell blood group glycoprotein. Transfus. Med. Hemotherapy 44, 53–57 (2017).
Moise, K. J. Fetal anemia due to non-Rhesus-D red-cell alloimmunization. Semin. Fetal Neonatal. Med. 13, 207–214 (2008).
Clapéron, A. et al. The Kell protein of the common K2 phenotype is a catalytically active metalloprotease, whereas the rare Kell K1 antigen is inactive: identification of novel substrates for the Kell protein. J. Biol. Chem. 280, 21272–21283 (2005).
Lee, S. et al. Proteolytic processing of big endothelin-3 by the Kell blood group protein. Blood 94, 1440–1450 (1999).
Lee, S., Russo, D. & Redman, C. M. The Kell blood group system: Kell and XK membrane proteins. Semin. Hematol. 37, 113–121 (2000).
Sha, Q., Redman, C. M. & Lee, S. Endothelin-3-converting enzyme activity of the KEL1 and KEL6 phenotypes of the kell blood group system. J. Biol. Chem. 281, 7180–7182 (2006).
Trakarnsanga, K. et al. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 8, 14750 (2017).
Satchwell, T. J. et al. Genetic manipulation of cell line derived reticulocytes enables dissection of host malaria invasion requirements. Nat. Commun. 10, 3806 (2019).
Dean, L. Blood Groups and Red Cell Antigens (NCBI, 2005).
Mohandas, N. & Narla, A. Blood group antigens in health and disease. Curr. Opin. Hematol. 12, 135–140 (2005).
Yeoh, S. et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131, 1072–1083 (2007).
Lee, S., Russo, D. C. W., Pu, J., Ho, M. & Redman, C. M. The mouse Kell blood group gene (Kel): cDNA sequence, genomic organization, expression, and enzymatic function. Immunogenetics 52, 53–62 (2000).
Lee, S., Debnath, A. K. & Redman, C. M. Active amino acids of the Kell blood group protein and model of the ectodomain based on the structure of neutral endopeptidase 24.11. Blood 102, 3028–3034 (2003).
Denomme, G. A. Kell and Kx blood group systems. Immunohematology 31, 14–19 (2015).
Arnoni, C. P. et al. An easy and efficient strategy for KEL genotyping in a multiethnic population. Rev. Bras. Hematol. Hemoter. 35, 99–102 (2013).
Bibi, A. et al. Genotyping of KELL blood group in multiethnic populations of Quetta, Pakistan. Health Biotechnol. Biopharm. 7, 66–81 (2023).
Bennett, V. & Stenbuck, P. J. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature 280, 468–473 (1979).
Lux, S. E. Anatomy of the red cell membrane skeleton: Unanswered questions. Blood 127, 187–199 (2016).
Lim, C., Dankwa, S., Paul, A. S. & Duraisingh, M. T. Host cell tropism and adaptation of blood-stage malaria parasites: challenges for malaria elimination. Cold Spring Harb. Perspect. Med 7, a025494 (2017).
Soubes, S. C., Reid, M. E., Kaneko, O. & Miller, L. H. Search for the sialic acid-independent receptor on red blood cells for invasion by plasmodium falciparum. Vox Sang. 76, 107–114 (1999).
Daniels, D. E. et al. Comparing the two leading erythroid lines BEL-A and HUDEP-2. Haematologica 105, e389–e394 (2020).
Medja, F. et al. Thiorphan, a neutral endopeptidase inhibitor used for diarrhoea, is neuroprotective in newborn mice. Brain 129, 3209–3223 (2006).
Stanović, S. et al. Thiorphan, an inhibitor of neutral endopeptidase/enkephalinase (CD10/CALLA) enhances cell proliferation in bone marrow cultures of patients with acute leukemia in remission. Haematologia 30, 1–10 (2000).
Singh, N. & Narayan, S. Racecadotril: a novel antidiarrheal. Med. J. Armed Forces India 64, 361–362 (2008).
Primi, M. P., Bueno, L., Baumer, P., Berard, H. & Lecomte, J. M. Racecadotril demonstrates intestinal antisecretory activity in vivo. Aliment. Pharmacol. Ther. 13, 3–7 (1999).
Eberlin, M., Mück, T. & Michel, M. C. A comprehensive review of the pharmacodynamics, pharmacokinetics, and clinical effects of the neutral endopeptidase inhibitor racecadotril. Front. Pharmacol. 3, 93 (2012).
Chaccour, C. & Rabinovich, N. R. Advancing the repurposing of ivermectin for malaria. Lancet 393, 1480–1481 (2019).
de Carvalho, L. P., Kreidenweiss, A. & Held, J. Drug repurposing: a review of old and new antibiotics for the treatment of malaria: Identifying antibiotics with a fast onset of antiplasmodial action. Molecules 26, 2304 (2021).
Bouyssou, I. et al. Unveiling P. vivax invasion pathways in Duffy-negative individuals. Cell Host Microbe 31, 2080–2092.e5 (2023).
Bouyssou, I. et al. The Darc side of vivax malaria in Africa: unveiling invasion pathways into Duffy-negative erythroblasts. Blood 142, 1074 (2023).
Dechavanne, C. et al. Duffy antigen is expressed during erythropoiesis in Duffy-negative individuals. Cell Host Microbe 31, 2093–2106.e7 (2023).
Okafor, I., Okoroiwu, H. & Ekechi, C. Hemoglobin S and glucose-6-phosphate dehydrogenase deficiency coinheritance in AS and SS individuals in malaria-endemic region: a study in Calabar, Nigeria. J. Glob. Infect. Dis. 11, 118–122 (2019).
Mohanty, S. S., Parihar, S., Huda, R. K., Toteja, G. S. & Sharma, A. K. Prevalence of sickle cell anemia, β-thalassemia and glucose-6-phosphate dehydrogenase deficiency among the tribal population residing in the Aravali hills of Sirohi region of Rajasthan state. Clin. Epidemiol. Glob. Health 13, 100916 (2022).
Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. & Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272–W276 (2016).
Bhargava, N. et al. Development of an efficient single-cell cloning and expansion strategy for genome edited induced pluripotent stem cells. Mol. Biol. Rep. 49, 7887–7898 (2022).
Gupta, P. et al. Development of pathophysiologically relevant models of sickle cell disease and β-thalassemia for therapeutic studies. Nat. Commun. 15, 1794 (2024).
Cromer, M. K. et al. Comparative analysis of CRISPR off-target discovery tools following ex vivo editing of CD34+ hematopoietic stem and progenitor cells. Mol. Ther. 31, 1074–1087 (2023).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
Lambros, C. & Vanderberg, J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 (1979).
Chakrabarti, M. et al. Interaction of Plasmodium falciparum apicortin with α- and β-tubulin is critical for parasite growth and survival. Sci. Rep. 11, 4688 (2021).
Acknowledgements
The BEL-A2 cell line was generated by Professor Jan Frayne, Professor David Anstee, and Dr. Kongtana Trakarnsanga, with funding from the Welcome Trust (grant numbers 087430/Z/08 and 102610), NHS Blood and Transplant and Department of Health (England). We sincerely thank the Central Instrumentation Facility (CIF) of the Special Centre for Molecular Medicine (SCMM), Jawaharlal Nehru University (JNU), New Delhi. This work is supported by funding from the Indian Council of Medical Research, Government of India (No. NER/84/2022-ECD-I) and Department of Biotechnology, Government of India (No. IC-12044(11)/10/2021-ICD-DBT) to S.S. The National Bioscience Award from the Department of Biotechnology, Government of India, to S.S. is also acknowledged. This work is also supported by the Department of Biotechnology, Government of India, and Council for Scientific and Industrial Research (CSIR) grant (MLP2010) to S.R. G.K. acknowledges CSIR for financial aid. P.G. is a recipient of a Senior Research Fellowship from the Department of Biotechnology. Schematic representations in figures were created with Biorender.com. Model Rural Health Research Unit (MRHRU), Tripura is acknowledged for logistic support. We acknowledge the Department of Health Research (DHR), New Delhi, for permitting us to collect samples and conduct part of the study at the Multi-Disciplinary Research Unit (MRU), Agartala Government Medical College.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.S. and S.R.; Data curation: S.S., G.K., P.G., S.G.G, P.T., and V.S.; Formal analysis: S.S., S.G., and S.R.; Funding acquisition: S.S.; Investigation: S.S., S.R., G.K., P.G., S.G.G., P.T., and V.S.; Methodology: S.S., S.G., G.K., P.G., S.G.G., S.B., S.A., P.T., V.S., B.G., I.P.B., and V.R.A.; Project administration: S.S. and S.R.; Resources: S.S.; Supervision: S.S. and S.R.; Validation: S.S., S.R., N.M., and J.B.; Visualization: S.S. and S.R.; Writing—Original draft: S.S. and G.K.; Writing—review and editing: S.S., S.R., G.K., R.J., P.G., S.G.G., S.G., J.B., and N.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Dr. Nishith Gupta and Dr. Ophelia Bu. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kumari, G., Gupta, P., Goswami, S.G. et al. CRISPR/Cas9-engineering of Kell null erythrocytes to unveil host targeted irresistible antimalarial. Commun Biol 8, 730 (2025). https://doi.org/10.1038/s42003-025-07968-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07968-2
