
Abstract
Climate change is threatening agricultural production across the globe. Germplasm collections provide an opportunity to explore where variation exists with important crop species. Genome environment association (GEA) is a standard approach for investigating the genetic basis of adaptation to natural environments. While these analyses provide insight into local adaptation, they have not been widely adopted in breeding or conservation programs. This may be attributable to the difficulty in identifying the best individuals for transplantation/relocation in conservation efforts or identification of the best parents in breeding programs. To explore the potential utility in future breeding programs, we used the cereal crop - barley (Hordeum vulgare L.) due to its wide adaptability to different environments and agroecologies, ranging from marginal and low input fields to high-productive farms. Here, we conduct environmental genomic selection (EGS) on 753 landrace barley accessions using a mini-core of 31 landrace accessions and a de-novo core of 100 as the training populations. Since local adaptation to the environment is polygenic, a whole-genome approach is likely to be more accurate for selection. Here we show how an integrative approach coupling environmental genomic selection and species distribution modelling can help identify key parents for adaptation to specific environmental variables.
Similar content being viewed by others

Introduction
Climate resilience requires adaptation to changing climatic regimes, including warmer day and nighttime temperatures and more sporadic but potentially more intense rainfall events. For grassland and forest environments, this could cause major changes in species composition. In some cases, human efforts may be necessary to identify and relocate individuals or genotypes well adapted for these new environments. Agriculture faces a similar issue, in terms of a realignment of where crops are produced, and which genetic backgrounds will be successful in new growth environments. Adapting to a changing environment is a major concern for natural and managed plant populations, this includes forests, orchards, and field crops1,2. There is an expanding scope of research exploring methods for accelerating plant breeding for a changing climate3,4. Many have emphasized an understanding of climate resilience targets to help define interventions that will be the most useful for specific geographies and species5,6,7,8.
Early efforts to identify the genetic basis of environmental adaptation examined allele frequency differences among populations9. Refinements to these approaches have sought to account for the variance among related individuals and populations relative to environmental variables10,11 and to identify sharp changes in allele frequency in species with an essentially continuous geographic range12. Collectively the approaches have been identified as landscape genomics13,14,15, which uses evolutionary relationships among wild relatives of crops and landraces to identify potentially adaptive loci7,16,17. A popular approach in landscape genomics involves genome-environment association (GEA) analysis, which seeks to identify relationships between allele frequencies or marker allele dosages and environmental variables that characterize the climate where each accession originates. Some early GEA studies made use of populations sampled from natural environments18,19. Another popular source of samples for GEA has been germplasm collections where collection localities are represented by individual (often inbred) accessions and no population-level information is available. GEA studies often focus on identifying loci underlying local adaptation20,21,22. The implicit assumption is that environmental variables such as temperature can be used as proxies for the selective pressures that have shaped the differences in allele frequencies among populations23,24. By correlating genetic information with environmental data that is most relevant to an important abiotic stress in a crop (e.g., temperature for heat stress), GEA analyses provide a means to select individuals from germplasm collections that may confer tolerance to those stresses.
There are many frameworks for GEA analysis25,26, however, in crops popular approaches involve the use of linear mixed-models, a well-established tool for other genomics-enabled plant breeding as well as quantitative genetic methods such as genome-wide association studies (GWAS) and genomic selection (GS). In many GEA analyses, the standard GWAS model is modified to use explicit bioclimatic data instead of trait phenotypic values as the response variable, in what is known as environmental GWAS (E-GWAS)27. While standard GWAS has become an important method for understanding the genetic architecture of complex traits in crops, it is less useful as a plant breeding selection tool, since only a fraction of heritable polygenic variation may be detected28,29. Instead, many breeding programs now use GS, which estimates the effect of genome-wide markers simultaneously and predicts the breeding value or total genetic merit of individuals30,31. Similarly, while E-GWAS can advance our understanding of the genetic architecture underlying local adaptation and potentially identify loci associated with tolerance to abiotic stresses, the polygenic nature of local adaptation32, suggests that E-GWAS alone may not capture the full extent of genetic variation needed to identify the genetic basis of local adaptation to be advantageous for applications such as plant breeding. There is a similar conceptual and practical advance to be made by moving from GEA and E-GWAS to environmental genome-wide selection to estimate the whole-genome adaptive value of an individual to bioclimatic conditions. Making use of environmental variables can extend genomic prediction approaches in meaningful ways that apply to many different species and scenarios.
Crop germplasm collections are a readily accessible resource for landscape genomic studies. However, they often include large numbers of accessions and phenotypic characterization at even a single location may be prohibitive or not possible due to differences in traits such as days to flowering33,34. This issue motivated the development of the “core” collection concept, which tries to maximize the amount of genetic diversity in the smallest number of accessions35. The development of core and mini-core collections allows a sufficiently diverse subset of a germplasm collection to be evaluated phenotypically for many traits of interest across different environments, with a cost of missing low-frequency beneficial genetic variation. For example, the entire U.S. Department of Agriculture (USDA) barley germplasm collection numbers 33,176 accessions; however, the development of core (n = 2417) and mini-core (n = 186) collections from this germplasm has allowed practical molecular marker genotyping and phenotyping of different traits36. Barley landraces are a useful system for exploring this approach because of prior studies of environmental adaptation and the large number of cloned genes associated with adaptation to broad production environments.
Barley (Hordeum vulgare L.) is cultivated in an extremely wide geographic and ecological range (from the equator to the inside the Arctic Circle) and has become known as an excellent model for studying and responding to climate change due to its ability to adapt to multiple stresses37,38. In barley, there have been efforts to use GEA and E-GWAS to identify genetic differentiation among wild populations39 and to find loci associated with abiotic stress tolerance20,40. Phenotypic recurrent selection and even marker-assisted backcrossing often do not effectively, nor efficiently, transfer quantitative traits into breeding germplasm41. However, exotic germplasm has been used to explore quantitative (polygenic) traits for centuries, especially in small grains42. The use of GS provides a theoretical basis for greatly increasing the efficiency of using exotic (unadapted) germplasm as a donor parent to introgress polygenic traits40. When exploring this strategy in germplasm collections, mini-core collections containing a large proportion of total genetic variation have been shown to be very useful as the initial training population43. Considering the successful application of GS for phenotypic traits, we argue that its use for environmental traits will boost the efficiency in the selection of germplasm better adapted to climate change. Barley provides an exemplar source to test environmental genomic selection (EGS) as there are many robust landrace collections20, and several well-characterized core collections36. Previous research has generally shown decreases in yield associated with increases in temperature in barley growing regions44,45. As a result of these predictions, various adaptation strategies have been suggested, but they have presented limited contributions to breeding populations37. Further, while GS has become the norm in breeding programs, it has yet to become common in the utilization of germplasm collections, with respect to the use of landscape genomic techniques46. Here we propose to explore the use of EGS to identify the best potential parental accessions from the collection of landraces that is maintained by the USDA; thus, improving the speed with which breeding for climate adaptation can occur.
Results and discussion
Population structure
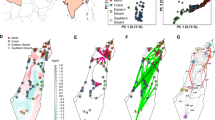
The first goal was to recapitulate the previous analysis20, where four populations had been identified. These relationships were established using 5800 single nucleotide polymorphisms—SNPs—in Lei et al.20 and the dataset was divided into central European, Asian, coastal Mediterranean, and East African populations. We identified a similar population structure to that which has been previously identified20 (using 3175 SNPs) finding five populations; namely East African (population 1, n = 89), Levant/Mediterranean (population 2, n = 205), North African/Mediterranean (population 3, n = 117), Northern Europe (population 4, n = 95) and Asia (population 5, n = 278) (Figs. 1 and S1). A comparison of these clusters between the previous study and this study can be found in Supplementary Data 2. These population clusters recapitulate historic cultivation history and match well with previous studies.

A Hierarchical clustering of landrace barley (N = 784) samples from the Lei et al.20 dataset based on 3175 SNPs following an LD prune at 0.2. B PCA with samples colored by populations identified in HCPC. C Relationship between hierarchical clustering of samples and geographic location.
Environmental genomic prediction
In this study, we explore the utility of core and mini-core collections for GS by using them as the training population for GS calculations. Core and mini-core collections have been previously published for Barley36. We used 31 lines from Munoz-Amatriain et al. which had the designation of landrace and overlapped with the Lei et al. publication. Additionally, a de-novo core collection (n = 100) was selected to represent the 784 lines from Lei et al. using the corehunter software47. Prediction accuracy for the mini-core (n = 31) and the de-novo core (n = 100) was assessed (Fig. 2). Using 10-fold cross-validation with 50 iterations we assessed four models; rrBLUP (ridge-regression best linear unbiased prediction), Gaussian Kernel, Exponential Kernel, and BayesCπ (Bayesian method of estimated GEBV that has a short computation time) models. The rrBLUP method was selected as the optimal model for GS because it had the highest prediction accuracy. We found good predictive accuracy for the entire collection irrespective of the core size used as the training population (Fig. 2) with some variables having higher predictive accuracy than others (e.g., bio1—mean annual temperature, bio3—Isothermality, bio4—Temperature seasonality, bio6—Minimum temperature of the coldest month, bio11—Mean temperature of the coldest quarter, bio14—precipitation of the driest month, and bio17—Precipitation of the driest quarter). Exploring the established core collection (n = 31) versus the de-novo core collection (n = 100), we found differences in prediction accuracy with the larger de-novo core had higher predictive accuracies, especially for bio12—mean annual precipitation, bio13—precipitation in the wettest month, bio16—precipitation of the wettest quarter and bio18—Precipitation of the warmest quarter (Figs. 2 and S2). Genomic estimated adaptive values (GEAVs) were assessed for each line (Supplementary Data 3–6) and population (Fig. 3A, B). For some variables, there were wider distributions of GEAV values (e.g., bio4—temperature seasonality Fig. 3), which indicates that for some environmental stressors, landrace accessions have more potential to adapt, but there is not a straightforward interpretation in all cases. Different variables had higher predictive accuracy in different populations (Figs. 2 and S2). This shows that if a core is developed, prediction accuracies should be high enough for GEAVs to be useful. The distinct evolutionary histories of the populations may mean that to fully take advantage of GEAV, inferring the population structure may provide a more accurate assessment of which accessions may be the best line. For example, East Africa (population 1) has poor GEAV values for bio 4 (temperature seasonality), but Asia (population 5) has the highest mean GEAVs for this climate variable (Fig. 3).

Cross-validation of genomic prediction. Four genomic prediction methods (RR-BLUP, G-BLUP with a Gaussian Kernel, G-BLUP with an exponential kernel, and BayesCπ) were evaluated using 6 cross-validation schemes. Prediction accuracy (r(PGE, y)) was estimated using 10-fold cross-validation with 50 replicates. The line shows the mean value across all runs and replicates, and the blue ribbon shows the mean value ± SD.

GEAV values are grouped and colored by hierarchical clustering within the dataset (k = 5) A for training set n = 31 and B for training set n = 100.
When exploring the overlaps in accessions in the top 5% of GEAVs for temperature-related climate variables (bio1, 4, 6, and 11) specialists were identified in non-overlapping regions (bio1 (n = 15), bio11 (n = 1), and bio4 (n = 40) (Fig. S3). Specifically, we detected an overlap among accessions (n = 15) with high GEAVs for bio1—mean annual temperature, bio 6—minimum temperature of the coldest month, and bio11—mean temperature in the coldest quarter, but poor overlap with bio4—temperature seasonality (Fig. S3). Similarly exploring the lines that had high GEAVs for precipitation namely bio14 and bio17, a complete overlap in the accessions was seen in the top 5% of GEAV values), with 32/40 coming from the Levant/Mediterranean population (population 2), 1/40 from North African/Mediterranean (population 3), 3/40 from Northern Europe (population 4) and 4/40 lines from East Asia (population 5).
Environmental variation and GEAV association
The populations defined by genetic assignment differ in terms of which environmental variables are most strongly associated with population-level variance. When looking at all the populations together, we observed that distinct variables were more associated with each population (Figs. 4A–H and S4–S9). For example, bio4—temperature seasonality was associated with the Asian population (population 5) (Fig. S4E), while bio17—Precipitation of the Driest Quarter was more associated with the Levant/Mediterranean population (population 2) (Fig. S4B). GEAV patterns can be explored by plotting values for environmental variables at each line’s geographic origin. For example, for bio3—isothermality more southern latitudes and more specifically lines from population 1 (East African population) have high GEAVs for this trait (Figs. 3A, B and 4C). In contrast, when breeding for temperature seasonality—bio4 (Fig. 4D) lines in more northern latitudes and more specifically population 5 have high GEAVs for this trait (Fig. 3A, B).

A Relationship between environmental variables and lines associated with each population. B Geographic distribution of GEAV values for Bio 1—Annual mean temperature. C Geographic distribution of GEAV values for Bio 3—Isothermality. D Geographic distribution of GEAV values for Bio 4—Temperature seasonality. E Geographic distribution of GEAV values for Bio 6—Min temperature of the coldest month. F Geographic distribution of GEAV values for Bio 11—Mean temperature of the coldest month. G Geographic distribution of GEAV values for Bio 14—Precipitation of the driest month. H Geographic distribution of GEAV values for Bio 17—Precipitation of the driest quarter.
Leveraging population distribution models
The geographic coordinates from each sample within each population were used to create population distribution models (PDMs), like previous work in common bean48 and created a new response factor (suitability score for each sampling location). The different environmental characteristics of each PDM may be driving genetic architecture and population divergence and thus could be explored for favorable alleles in parent selection for breeding for potential future environments. There were clear differences in optimal areas for each population identified (Figs. 5 and S6–S10). For example, the East African population (Figs. 5B and S5) showed a narrower range than populations from Northern Europe (Figs. 5E and S8) and Asia (Figs. 5F and S9). Sources of local adaptation can be found in each of the populations and suitability for different environmental variables (Figs. S5–S9). Each pixel in the suitability model shows the habitat conditions of the geography which greatly impacts the ability of a population to grow in that region, typically if a suitability value is above 0.2 plants will be able to grow with agricultural intervention. We further explored the GEAV values for species distribution models (SDM) suitability scores (Fig. S10). Prediction accuracies were highest for the specific population the PDMs originated from, except for population 4 (Fig. S10A). All the accessions with all the GEAVs for specific environmental variables and for each subpopulation are in Supplementary Data 7–18, this allows breeders with different priorities to choose which accessions match their environment (PDM) and environmental variable of choice. Additionally, PDMs can be utilized to identify key parental lines for breeding programs aimed at adapting to specific environmental conditions. By intersecting distribution ranges with lines that fall within these ranges (Fig. 5B–F; Supplementary Data 9–13 (core n = 31) and Supplementary Data 13–17 (core n = 100)), breeders can select genetic backgrounds that are optimal for a target environment (Supplementary Data 9–18).

A Overlaps in species distribution for all five populations. B Population 1—East African population-specific species distribution with range-specific lines for bio1. C Population 2—Levant/Mediterranean population-specific species distribution with range-specific lines for bio1. D Population 3—North African/Mediterranean population-specific species distribution with range-specific lines for bio1. E Population 4—Northern Europe population-specific species distribution with range-specific lines for bio1. F Population 5—Asian population-specific species distribution with range-specific lines for bio1.
Chromosomal patterns of adaptation across populations
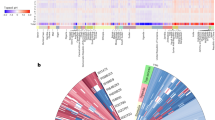
We examined chromosomal patterns across the five populations. When exploring individual marker effects on each chromosome there were distinctly different patterns across the populations (Figs. 6, S11 and S12). For example, the marker effect pattern on chromosome one in the Levant/Mediterranean population differs from other populations, while the East African population shows a different pattern on chromosome 2 (Fig. 6). This shows that despite many of the response factors being highly quantitative there are patterns that are genetic background specific that can lead to more local adaptation. Despite these different patterns there were often common SNPs that had consistently large effects for specific traits (Fig. S13; Supplementary Data 1 and 5). While these markers had larger predicted effects on the phenotype (e.g., sometimes being related to a change in temperature of ~0.1 °C) these SNPs did not always follow the geographic distributions of the populations, often the SNPs would have patterns where the beneficial allele would be present in many populations (Fig. S15). Marker effects were not distributed evenly across chromosomes or among populations (Figs. S11 and S12). There seem to be many private alleles within populations that have an adaptive effect (Fig. S12). These variants are not evenly distributed by trait, suggesting that some populations apparently have more tolerance than others for specific environmental stressors.

The x-axis represents marker positions for each marker from a 9K Illumina Infinium iSelect Custom Genotyping BeadChip, this means that marker spacing is uneven across each chromosome, thus positions are in slightly different places on each chromosome. The y-axis represents each individual line in each subpopulation, for East African (Population 1, n = 89), Levant/Mediterranean (Population 2, n = 205), North African/Mediterranean (Population 3, n = 117), Northern Europe (Population 4, n = 95), and Asia (Population 5, n = 278).
Adapting to climate change
Using a large collection of publicly available landraces of barley that contained georeferences and were genotyped with a common SNP panel, we explored which individuals may be the best parents for use in breeding programs targeting environmental stress adaptation. Genomic prediction was conducted using a subset of the barley core collection that was represented in the georeferenced accessions as the training population. Further, to try and better understand the potential for different climate niches, population-specific distribution models were created to explore if parental material from population-specific niches would be more useful. In general, we found specific accessions that had high GEAVs across a wide range of variables (temperature, precipitation, climate niche), indicating there are some parents that have much higher potential. There were often population-specific patterns of variation, indicating that there is potential for combining different genetic architectures to create more resilient plants, with individuals with high GEAVs likely to have different polygenic adaptations. The approach explored here is broadly applicable to a range of problems, including selection of trees for plantations, conservation efforts (e.g., using genomic prediction instead of genetic offsets), and other crops. With the decreasing cost of generating sequencing and improvements in the ability to get geographically relevant information, the analytical approach here can be applied to many systems. It is also important to understand the distribution of GEAVs as recent work has shown that there is increased genetic load at range edges49 this has implications for where individuals for future breeding work should be selected from.
Exploration of previously identified candidate genes
In previous work, known genes were identified with large impacts on local adaptation20,50. There were often allelic series identified across populations where in different environments different alleles were favored50. When exploring the GEAVs specific genes had a disproportionate impact on the total variance explained. Although some of these genes were previously identified through GEA analysis, they were at the extreme ends of the distribution for marker effects (Supplementary Data 6; Fig. S14) and explained a small amount of variance. Using bio 1 as a test case we explored the annotation among the markers with the 10 largest marker effects. Among these, there was no clear annotation associated with heat tolerance. However, this is not unexpected, previous work20,39 did not identify a large effect QTL for bio 1, the approach here (genomic prediction) is designed to identify combined marker effects that accumulate to create polygenic adaptation. In essence, since there are large differences in GEAVs the approach is working as intended despite not finding large effect markers. This suggests that climate change adaptation will be largely polygenic and that individual genes will not be strong enough for these environmental responses. This suggests that a marker-assisted backcrossing approach to create resilience will be less successful than a genomic prediction approach. Using the model where we treated candidate genes from previous GEA analysis as fixed effects the model fit was improved, but there was no improvement in prediction accuracy using either core collection when fixing cold tolerance (Fig. S15A) and flowering time (Fig. S15B) related genes. These candidate genes had different impacts on prediction accuracy for different traits, which is expected based on the relationship between a particular gene and the environmental response of interest. Unlike previous work in rice51 and wheat52, but like previous simulation work53, we found that using fixed effects did not improve prediction accuracy (Fig. S15). This could be due to the highly quantitative nature of the abiotic stress tolerance, or it could be due to overfitting the model by including fixed effects that are not related to the specific abiotic stress. In either case, it seems like the best approach will likely be to use a genomic prediction method rather than a marker-assisted backcrossing method to enhance adaptation to climate change.
Maximizing the efficiency of selection to incorporate exotic germplasm for adaptation to climate change
Plant breeding takes a long time, depending on the plant system it can take between 8 and 20 years to release a new cultivar54. Breeding programs have long appreciated the utility of collecting and storing genetically diverse accessions of crops and their wild relatives to maintain the genetic variation essential to breeding progress55,56. Germplasm collections of the wild relatives of crops have had great value to agriculture by providing, for example, new alleles for disease resistance or crop quality57,58; however, they have been underutilized with respect to climate change15,59,60. While many studies have examined approaches to best exploit germplasm collections in plant breeding programs43,61, optimizing the selection of accessions remains a challenge. Currently, breeding programs are not releasing cultivars fast enough to keep up with predictions for climate change62. This implies that the evolutionary relationships between populations can perhaps provide insight into adaptation in modern cultivars to future climate change, due to understanding how both genetic background and loci of large effect interact in populations that were the founders of proximate modern cultivars. Targeted decisions can greatly increase the speed of breeding. New developments in high-throughput phenomics63, high-throughput genotyping64, and speed breeding65 may provide ways to rapidly introduce novel abiotic stress tolerance genes. Further, combining EGS with other metrics of local adaptation, for example, home field advantage66, i.e., combining locally adapted with environmentally adapted material, maybe a pathway to more rapidly develop cultivars for specific geographies. EGS is an extension of GEA analysis to make diverse collections more available to plant breeding programs. While classic GEA analysis provides important information about specific alleles that are putatively adaptive, it does not provide direct information about parental line performance or value as a parent7. Traditional GS has been employed to decrease cycle time67, extending this approach by exploiting local adaptation should enhance breeding for climate change. EGS should provide better parental selection because instead of focusing on loci of large effect that may be in an unadapted genetic background, it can incorporate more quantitative information. Also, this approach still allows for a mechanistic understanding while pushing forward populations within active programs, in effect, it allows for both a retrospective and prospective exploration at the same time.
Updating your training population in the local breeding program
Historically heat and drought stress have been very difficult to phenotype68. Recent advances in controlled environment agriculture and phenomics have increased the measurement precision69, which combined with the advent of artificial intelligence in breeding70 are expected to advance breeding for climate change. These two phenotypes are projected to be some of the most important traits under climate change. The difficulty of phenotyping these traits makes them excellent candidates for GEA and EGS, however, once parents are selected and incorporated into breeding programs the initial training population will no longer be the most appropriate71. The GEAV approach can be explored for any climate response (Fig. 4; Supplementary Data 3 and 4). For example, while drought is a problem in North America, in northern Europe waterlogging/flooding is likely to be a larger problem. Here for the precipitation-related climate variables examined (bio14 and bio17), we can see that the highest GEAVs for these traits occur in regions that have high precipitation, even in dry parts of the year (Fig. 4G, H). Given the difficulty in phenotyping abiotic stress, ensuring that continued progress can be made in the next cycle, it is important to update the training population. The first step in doing this will be making sure that your elite parents have been phenotyped for abiotic stress tolerance in the normal way it is assessed in the breeding program. Further, it will be important to test multiple training populations to optimize resource allocation for a specific breeding program. A major next step will be to understand if you can continue to select for abiotic adaptation after more than one generation of crossing or if this method is best suited for parental selection.
Caveats
It is important to note that in this study there was limited marker coverage, which may impact the overall GEAVs. For example, the IPK germplasm collection has been genotyped with GBS having much higher marker resolution72. While we speculate on the best way to incorporate this exotic germplasm into breeding programs it will be important to conduct both simulation and empirical studies to make sure the rate of gain per cycle is like marker-assisted selection. The GEAV calculation is based on historic mean values for climate variables from 1970 to 200073. Having more accurate climatic data with or making use of the entire time-series would lead to better results.
Conclusion
Barley is grown from the tropics to the Arctic Circle. Despite this large range, there are clear populations which differ greatly in their predicted distributions. These different populations have different genetic values (GEAVs) for breeding for climate change. Large-scale genotyping of landrace material followed by genetic characterization with EGS can identify promising parents and reduce the time required for the breeding process. Here we have used publicly available data (genotypes, with georeferences for accessions and worldwide climate data) and identify landraces that have shown polygenic adaptation to climate niches and specific environmental variables and likely host beneficial alleles for introgression when breeding for target environments.
Methods
Core collection/genotype
The USDA barley core collection comprises 2417 accessions36. Based on 9K Illumina Infinium iSelect Custom Genotyping BeadChip74, a set of 1860 non-redundant samples were retained, identified as the iCore. These accessions originated from 94 countries and included 815 landraces. From the iCore collection, Muñoz-Amatriaín et al.36 further developed a mini-core collection comprising 186 accessions, 31 of which were landrace samples, which are represented here as the n = 31 core. Genotypes reported here derive from automated genotype calling implemented in the software Alchemy75. SNP calls with posterior probability >0.95 were retained, while calls below the threshold were treated as missing data42. The VCF file used for analysis here was reported in Lei et al.20 using SNP physical positions in the Morex_v2 assembly76. Lei et al.20 selected 803 landrace accessions from the iCore and following quality filtering and the exclusion of accessions lacking distinct locality information, they identified a final set of 784 georeferenced landrace accessions, which was selected as the genetic material in this study.
De-novo core collection methods
The genotypic data from the Lei et al.20 was provided in Supplemental_dataset_1.vcf. This VCF was converted into a genlight object using the “vcfR”77 and “adegenet” packages78. A distance matrix was calculated using the “poppr” package79. Hierarchical clustering was performed on the distance matrix using the Ward method, and the dataset was clone-corrected to account for potential duplicates. To show this method would be applicable to species where no core was developed, we also developed a de-novo core of the 784 lines. This was generated using the “corehunter” package47 where 100 lines (core n = 100) were sampled from the precomputed distance matrix.
Population structure and environmental genomic selection
The above dataset was examined for population structure using “SNPRelate80.” For the Principal Component Analysis (PCA) and Hierarchical Clustering of Principal Components (HCPC), only bi-allelic SNPs further filtered for linkage disequilibrium (0.2) (3175 SNPs) were used. The genomic prediction was performed using 6068 SNPs to predict bioclimatic and biophysical variables to generate a GEAV for each accession for a given trait (conceptualized as the genetic value for a specific environmental context) (Supplementary Data 3,and 4). In previous work, these have been characterized as GEAVs46. Four genomic prediction methods were examined: (i) RR-BLUP, (ii) G-BLUP with an exponential kernel, (iii) G-BLUP with a Gaussian kernel, and (iv) BayesCπ. R packages “rrBLUP”81 and “hibayes”82 were used for the analysis (Fig. 2). The training population (core n = 31) consisted of 31 georeferenced accessions, representing the overlap between the mini-core collection identified by Munoz-Amatriain et al.36 and the 784 landrace samples used in Lei et al.20. The remaining 753 landraces from the Lei et al.20 datasets were used as the validation set. Prediction accuracy was based on Pearson correlation (r(PGE, y)) between the predicted genotypic effects and the observed environmental variable with 10-fold cross-validation. Environmental traits which were ascribed to a prediction accuracy over 50% for most methods were examined in more depth (Fig. 2). Having a prediction accuracy of greater than 50% has been empirically shown to be a lower threshold for high prediction accuracy83,84,85,86. For the de-novo core (core n = 100) prediction accuracy was examined similarly with the remaining 684 landrace lines used as the validation set (Fig. 2). When setting previously identified phenotypically validated genes of interest as fixed effects (n = 22) in the prediction and GS models, highly correlated SNPs were removed due to multicollinearity using the R package “caret87.” This resulted in 14 SNPs which were set as fixed effects. Prediction accuracy for the rrBLUP model with these fixed effect SNPs was calculated for both cores (n = 31 and n = 100) (Fig. S15) as well as GS and GEAVs (Supplementary Data 7 and 8). Rank changes across the environmental variables were also examined (Fig. S16).
Environmental data
Occurrence data from Munoz-Amatriain et al.36 were separated into populations based on the genetic assignment analysis (see above). This led to 89 individuals in population 1, 205 individuals in population 2, 117 individuals in population 3, 95 in population 4, and 278 in population 5. These occurrence points were used to query the WorldClim 2.1 climate data (all 19 bioclim variables for temperature and precipitation, Table S1). Data were downloaded at the highest available spatial resolution of 30 s (~1 km2) (https://www.worldclim.org73). These bioclimatic data were used to create SDM using the software Maxent (Version 3.4.4—88) in RStudio (Version 2022.2.0.443—89). Map overlays were created using the “raster,” “rworldmap,” “ggplot2,” “sf” and “mapdata” packages in RStudio. Suitability maps were overlaid for the present day (1970–2000), with a suitability cutoff score of 0.2. Acceptable suitability is defined as 0.2 for cultivated regions90 and 0.4 for natural areas91. Model quality was explored using the area under the curve (AUC) and the standard deviation of the AUC across replicates (SDAUC). A good model requires an AUC ≥ 0.7 and an SDAUC < 0.15. The final SDM suitability value was then used as a response factor for EGS.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Code availability
All code available at—https://github.com/ahmccormick/Barley_EGS/tree/main?tab=readme-ov-file.
References
Moore, F. C. & Lobell, D. B. Adaptation potential of European agriculture in response to climate change. Nat. Clim. Change 4, 610–614 (2014).
Snowdon, R. J., Wittkop, B., Chen, T. W. & Stahl, A. Crop adaptation to climate change as a consequence of long-term breeding. Theor. Appl. Genet. 134, 1613–1623 (2021).
Araus, J. L. & Kefauver, S. C. Breeding to adapt agriculture to climate change: affordable phenotyping solutions. Curr. Opin. Plant Biol. 45, 237–247 (2018).
Cowling, W. A., Li, L., Siddique, K. H. M., Banks, R. G. & Kinghorn, B. P. Modeling crop breeding for global food security during climate change. Food Energy Secur. 8, e00157 (2019).
Bandillo, N. B. et al. Dissecting the genetic basis of local adaptation in soybean. Sci. Rep. 7, 17195 (2017).
Fumia, N. et al. Wild relatives of potato may bolster its adaptation to new niches under future climate scenarios. Food Energy Secur. 11, e360 (2022).
Gao, L., Kantar, M. B., Moxley, D., Ortiz-Barrientos, D. & Rieseberg, L. H. Crop adaptation to climate change: an evolutionary perspective. Mol. Plant 16, 1518–1546 (2023).
Pironon, S. et al. Potential adaptive strategies for 29 sub-Saharan crops under future climate change. Nat. Clim. Change 9, 758–763 (2019).
Lewontin, R. C. & Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74, 175–195 (1973).
Coop, G., Witonsky, D., Di Rienzo, A. & Pritchard, J. K. Using environmental correlations to identify loci underlying local adaptation. Genetics 185, 1411–1423 (2010).
Günther, T. & Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 195, 205–220 (2013).
Yang, W. Y., Novembre, J., Eskin, E. & Halperin, E. A model-based approach for analysis of spatial structure in genetic data. Nat. Genet. 44, 725–731 (2012).
Bragg, J. G., Supple, M. A., Andrew, R. L. & Borevitz, J. O. Genomic variation across landscapes: insights and applications. New Phytol. 207, 953–967 (2015).
Rellstab, C., Gugerli, F., Eckert, A. J., Hancock, A. M. & Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 24, 4348–4370 (2015).
Langridge, P. & Waugh, R. Harnessing the potential of germplasm collections. Nat. Genet. 51, 200–201 (2019).
Lasky, J. R., Josephs, E. B. & Morris, G. P. Genotype–environment associations to reveal the molecular basis of environmental adaptation. Plant Cell 35, 125–138 (2023).
Lasky, J. R. et al. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 1, e1400218 (2015).
Eckert, A. J. et al. Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics 185, 969–982 (2010).
Fang, Z. et al. Megabase-scale inversion polymorphism in the wild ancestor of maize. Genetics 191, 883–894 (2012).
Lei, L. et al. Environmental association identifies candidates for tolerance to low temperature and drought. G3 Genes Genomes Genet. 9, 3423–3438 (2019).
Neyhart, J. L., Kantar, M. B., Zalapa, J. & Vorsa, N. Genomic-environmental associations in wild cranberry (Vaccinium macrocarpon Ait.). G3 Genes Genomes Genet. 12, jkac203 (2022).
Wang, D. R., Kantar, M. B., Murugaiyan, V. & Neyhart, J. Where the wild things are: genetic associations of environmental adaptation in the Oryza rufipogon species complex. G3 Genes Genomes Genet. 13, jkad128 (2023).
Tiffin, P. & Ross-Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 29, 673–680 (2014).
Lotterhos, K. E. The paradox of adaptive trait clines with nonclinal patterns in the underlying genes. Proc. Natl. Acad. Sci. USA 120, e2220313120 (2023).
Lotterhos, K. E. & Whitlock, M. C. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 24, 1031–1046 (2015).
Lind, B. M. & Lotterhos, K. E. The accuracy of predicting maladaptation to new environments with genomic data. Mol. Ecol. Resour. 25, e14008 (2025).
Pyhäjärvi, T., Hufford, M. B., Mezmouk, S. & Ross-Ibarra, J. Complex patterns of local adaptation in teosinte. Genome Biol. Evol. 5, 1594–1609 (2013).
Battlay, P. et al. Large haploblocks underlie rapid adaptation in the invasive weed Ambrosia artemisiifolia. Nat. Commun. 14, 1717 (2023).
Li, X. et al. An integrated framework reinstating the environmental dimension for GWAS and genomic selection in crops. Mol. Plant 14, 874–887 (2021).
Lorenz, A. J. et al. Genomic selection in plant breeding. 77–123, https://doi.org/10.1016/B978-0-12-385531-2.00002-5 (2011).
Meuwissen, T. H. E., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001).
Yeaman, S. Evolution of polygenic traits under global vs local adaptation. Genetics 220, iyab134 (2022).
Frankel, O. H. & Soulé, M. E. Conservation and Evolution. (Cambridge University Press, 1981).
Skinner, D. Z., Bauchan, G. R., Auricht, G. & Hughes, S. A method for the efficient management and utilization of large germplasm collections. Crop Sci. 39, 1237–1242 (1999).
Frankel, O. & Brown, A. Plant genetic resources today: a critical appraisal. In Crop Genetic Resources: Conservation and Evaluation. Ch. 21, 249–257 (George Allan and Unwin, London, 1984).
Muñoz-Amatriaín, M. et al. The USDA Barley Core Collection: genetic diversity, population structure, and potential for genome-wide association studies. PLoS ONE 9, e94688 (2014).
Dawson, I. K. et al. Barley: a translational model for adaptation to climate change. New Phytol. 206, 913–931 (2015).
Newton, A. C. et al. Crops that feed the world 4. Barley: a resilient crop? Strengths and weaknesses in the context of food security. Food Secur. 3, 141–178 (2011).
Fang, Z. et al. Two genomic regions contribute disproportionately to geographic differentiation in wild barley. G3 Genes Genomes Genet. 4, 1193–1203 (2014).
Russell, J. et al. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024–1030 (2016).
Bernardo, R. Genomewide predictions for backcrossing a quantitative trait from an exotic to an adapted line. Crop Sci. 56, 1067–1075 (2016).
Poets, A. M., Fang, Z., Clegg, M. T. & Morrell, P. L. Barley landraces are characterized by geographically heterogeneous genomic origins. Genome Biol. 16, 173 (2015).
Yu, X. et al. Genomic prediction contributing to a promising global strategy to turbocharge gene banks. Nat. Plants 2, 16150 (2016).
Cammarano, D. et al. The impact of climate change on barley yield in the Mediterranean basin. Eur. J. Agron. 106, 1–11 (2019).
Xie, W. et al. Decreases in global beer supply due to extreme drought and heat. Nat. Plants 4, 964–973 (2018).
Cortés, A. J., López-Hernández, F. & Blair, M. W. Genome–environment associations, an innovative tool for studying heritable evolutionary adaptation in orphan crops and wild relatives. Front. Genet. 13, 910386 (2022).
Thachuk, C. et al. Core Hunter: an algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 10, 243 (2009).
Ramirez-Villegas, J. et al. A gap analysis modelling framework to prioritize collecting for ex situ conservation of crop landraces. Divers. Distrib. 26, 730–742 (2020).
Fiscus, C. J., Aguirre-Liguori, J. A., Gaut, G. R. & Gaut B. S. Climate, population size, and dispersal influences mutational load across the landscape in Vitis arizonica. bioRxiv (2024).
Hemshrot, A. et al. Development of a multiparent population for genetic mapping and allele discovery in six-row barley. Genetics 213, 595–613 (2019).
Spindel, J. E. et al. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 116, 395–408 (2016).
Sarinelli, J. M. et al. Training population selection and use of fixed effects to optimize genomic predictions in a historical USA winter wheat panel. Theor. Appl. Genet. 132, 1247–1261 (2019).
Rice, B. & Lipka, A. E. Evaluation of RR-BLUP genomic selection models that incorporate peak genome-wide association study signals in maize and sorghum. Plant Genome 12, 180052 (2019).
Bernardo, R. Breeding for Quantitative Traits in Plants Vol. 1, 369 (Stemma Press, 2002).
Brown, A. H. D., Frankel, O. H. & Marshall R. D. The Case for Core Collections 136–156 (Cambridge University Press, 1989).
Milner, S. G. et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 51, 319–326 (2019). Available from: https://www.nature.com/articles/s41588-018-0266-x.
Dempewolf, H. et al. Adapting agriculture to climate change: a global initiative to collect, conserve, and use crop wild relatives. Agroecol. Sustain. Food Syst. 38, 369–377 (2014).
Dempewolf, H. et al. Past and future use of wild relatives in crop breeding. Crop Sci. 57, 1070–1082 (2017).
Henry, R. J. Genomics strategies for germplasm characterization and the development of climate resilient crops. Front. Plant Sci. 5, 68 (2014).
Hübner, S. & Kantar, M. B. Tapping diversity from the wild: from sampling to implementation. Front. Plant Sci. 12, 626565 (2021).
Stenberg, J. A. & Ortiz, R. Focused Identification of Germplasm Strategy (FIGS): polishing a rough diamond. Curr. Opin. Insect Sci. 45, 1–6 (2021).
Henry, R. J. Innovations in plant genetics adapting agriculture to climate change. Curr. Opin. Plant Biol. 56, 168–173 (2020).
Gill, T. et al. A comprehensive review of high throughput phenotyping and machine learning for plant stress phenotyping. Phenomics 2, 156–183 (2022).
Jackson, S. A., Iwata, A., Lee, S., Schmutz, J. & Shoemaker, R. Sequencing crop genomes: approaches and applications. New Phytol. 191, 915–925 (2011).
Wanga, M. A., Shimelis, H., Mashilo, J. & Laing, M. D. Opportunities and challenges of speed breeding: a review. Plant Breed. 140, 185–194 (2021).
Ewing, P. M., Runck, B. C., Kono, T. Y. J. & Kantar, M. B. The home field advantage of modern plant breeding. PLoS ONE 14, e0227079 (2019).
Bernardo, R. & Yu, J. Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 47, 1082–1090 (2007).
Tuberosa, R. Phenotyping for drought tolerance of crops in the genomics era. Front. Physiol. 3, 347 (2012).
Fumia, N. et al. Exploration of high-throughput data for heat tolerance selection in Capsicum annuum. Plant Phenome J. 6, e20071 (2023).
Hayes, B. J. et al. Advancing artificial intelligence to help feed the world. Nat. Biotechnol. 41, 1188–1189 (2023).
Neyhart, J. L., Tiede, T., Lorenz, A. J. & Smith, K. P. Evaluating methods of updating training data in long-term genomewide selection. G3 Genes Genomes Genet. 7, 1499–1510 (2017).
Jiang, Y., Weise, S., Graner, A. & Reif, J. C. Using genome-wide predictions to assess the phenotypic variation of a barley (Hordeum sp.) Gene Bank collection for important agronomic traits and passport information. Front. Plant Sci. 11, 11 (2021).
Fick, S. E. & Hijmans, R. J. WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315 (2017).
Comadran, J. et al. Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat. Genet. 44, 1388–1392 (2012).
Wright, M. H. et al. ALCHEMY: a reliable method for automated SNP genotype calling for small batch sizes and highly homozygous populations. Bioinformatics 26, 2952–2960 (2010).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Endelman, J. B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 4, 250–255 (2011).
Yin, L., Zhang, H., Li, X., Zhao, S., & Liu X. hibayes: an R package to fit individual-level, summary-level and single-step Bayesian regression models for genomic prediction and genome-wide association studies. BioRxiv (2022).
Desta, Z. A. & Ortiz, R. Genomic selection: genome-wide prediction in plant improvement. Trends Plant Sci. 19, 592–601 (2014).
Crossa, J. et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 112, 48–60 (2014).
Schrauf, M. F., de Los Campos, G. & Munilla, S. Comparing genomic prediction models by means of cross validation. Front. Plant Sci. 12, 734512 (2021).
Heslot, N., Yang, H. P., Sorrells, M. E. & Jannink, J. L. Genomic selection in plant breeding: a comparison of models. Crop Sci. 52, 146–160 (2012).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Phillips, S. J. & Miroslav Dudík, R. E. S. Maxent software for modeling species niches and distributions (Version 3.4.1). Available from: http://biodiversityinformatics.amnh.org/open_source/maxent/ (2017).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2023).
Evans, J. M., Fletcher, R. J. & Alavalapati, J. Using species distribution models to identify suitable areas for biofuel feedstock production. GCB Bioenergy 2, 63–78 (2010).
Radomski, T. et al. Finding what you don’t know: testing SDM methods for poorly known species. Divers. Distrib. 28, 1769–1780 (2022).
Author information
Authors and Affiliations
Contributions
A.H.M.: conceptualization, data analysis, and manuscript preparation; Q.C.: data analysis and manuscript preparation; S.N.: data analysis and manuscript preparation; P.L.M.:data analysis and manuscript preparation; S.H.: data analysis and manuscript preparation; J.L.N.: conceptualization and manuscript preparation; M.B.K.: conceptualization, data analysis, and manuscript preparation; all authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Paulo Izquierdo and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Jorge Duitama and Michele Repetto.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Halpin-McCormick, A., Campbell, Q., Negrão, S. et al. Environmental genomic selection to leverage polygenic local adaptation in barley landraces. Commun Biol 8, 618 (2025). https://doi.org/10.1038/s42003-025-08045-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08045-4
