
Abstract
Bone-destructive diseases are caused by dysregulated osteoclastogenesis. Osteoclast differentiation is positively regulated by the ligand for receptor activator of nuclear factor kappa B (RANKL) binding to the RANK on progenitor cells. RANK then forms a multivalent interaction with an adapter molecule, tumor necrosis factor receptor-associated factor 6 (TRAF6), to transduce various downstream signals. We used affinity-based screening of a multivalent random-peptide library to identify a tetravalent peptide, WHD-tet, that binds to the RANK-binding region of TRAF6 through a multivalent interaction. CR4-WHD-tet, a cell-permeable form of WHD-tet, efficiently inhibited the RANKL-induced differentiation of bone-marrow cells to osteoclasts and osteoclastogenesis in a mouse model. CR4-WHD-tet specifically inhibited the recruitment of MAPK kinase 3 to TRAF6 without affecting other signal transducers in a late stage of differentiation, inhibiting the activation of p38-MAPK, which promotes the final stage. Thus, the interaction modulator CR4-WHD-tet fine-tunes the formation of a critical signaling complex to inhibit osteoclastogenesis.
Similar content being viewed by others

Introduction
Osteoclast-related diseases, such as chronic inflammatory arthritis and osteoporosis, are caused by disorders of osteoclastogenesis1,2. The osteoclastogenesis of myeloid progenitor cells depends on the stimulation of the receptor activator of nuclear factor kappa B (RANK) by its ligand, a receptor activator of nuclear factor kappa B ligand (RANKL)3,4,5,6,7. Activated RANK induces the recruitment and activation of a major adaptor protein, tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), to transduce various downstream signals, including the activation of nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1)8,9,10,11, to induce the expression of nuclear factor of activated T cells 1 (NFATc1), the master osteoclastogenic transcription factor12. Therefore, developing a molecule that specifically modulates the signals to inhibit osteoclastogenesis could be a promising approach to the treatment of these diseases.
Upon stimulation with RANKL, RANK interacts with TRAF6 to activate the E3 ubiquitin ligase activity of TRAF6 and then induce the formation of a K63-type polyubiquitin (K63-Ub) chain on TRAF6, which acts as a platform for the formation of a signal complex7,13,14. The complex consists of TGFβ-activated kinase 1 (TAK1)/TAK1 binding protein 2 (TAB2), the IκB kinase (IKK) complex, which comprises the catalytic subunits IKKα/β and the regulatory subunit NF-κB essential modulator (NEMO), and mitogen-activated protein kinase kinases (MKKs). TAK1 accumulates on the K63-Ub chain through TAB2 to phosphorylate and activate IKKβ, which then phosphorylates the IκBα associated with NF-κB to induce IκBα degradation and subsequent NF-κB activation. TAK1 also phosphorylates and activates MKK3/6 and MKK4/7 to induce the activation of the downstream mitogen-activated protein kinases (MAPKs), such as p38-MAPK (p38) and c-Jun N-terminal kinase (JNK), respectively, resulting in the activation of AP-115.
In an early stage of osteoclast differentiation, MKK6 rather than MKK3 activates p38 to activate AP-1, which functions cooperatively with NF-κB to strongly induce NFATc1 expression15,16,17. In a late stage of differentiation, MKK6 also activates p38, but this time to directly phosphorylate and activate the NFATc1 present in the nucleus18. In contrast, it has been shown using knockout mice that the knockout of MKK3, but not MKK6, inhibits not only osteoclast differentiation from bone marrow cells but also osteoclastogenesis in ovariectomized mice, emphasizing the essential role of MKK3 in osteoclastogenesis19. However, it remains to be clarified at which stage of differentiation MKK3 activates p38.
One of the promising targets for specifically modulating RANK signaling is the interaction between RANK and TRAF6, the most upstream event. The binding of RANKL to RANK induces the trimerization of RANK, which then interacts with three molecules of TRAF6 through a multivalent interaction between the Pro–X–Glu–X–X–(aromatic/acidic) motif of RANK and the C-terminal domain of TRAF6 (TRAF-C), which markedly increases the binding affinity based on avidity contribution20,21,22,23. Based on these data, a synthetic peptide, L-T6DP-1, with the sequence of the TRAF-C-binding region of RANK (18 amino acids)23, and a small compound (TRI4) that targets TRAF-C24 have been developed. L-T6DP-1 and TRI4 inhibit osteoclast differentiation, but their 50% inhibitory concentrations (IC50s) are around 30 μM and >200 μM, respectively, probably because of their inability to display avidity. Recently, we developed a tetravalent peptide, RANK-tet, that contains the TRAF-C-binding motif of RANK (nine amino acids) as the functional motif to form the multivalent interaction with TRAF625. A cell-permeable form of RANK-tet (CR4-RANK-tet) inhibited osteoclast differentiation with an IC50 value of 0.54 μM. However, the functional motif is not necessarily optimal for binding to TRAF6 because the motif was derived from the native sequence of RANK and then was not customized to bind to TRAF-C with high affinity.
In the present study, we screened a multivalent random-peptide library and identified a tetravalent peptide with a unique functional motif, WHD-tet, that efficiently binds to TRAF-C through a multivalent interaction. This technique is highly effective for identifying high-affinity binding motifs against target proteins that function through multivalent interactions26,27,28,29,30. Notably, the functional motif differs substantially from that of RANK-tet. CR4-WHD-tet, a cell-permeable form of WHD-tet, efficiently inhibited osteoclastogenesis with more potency than that of RANK-tet. We found that CR4-WHD-tet functions at a late stage of differentiation when a substantial amount of NFATc1 was observed in the nucleus, to specifically inhibit the recruitment of MKK3 to TRAF6 and the subsequent activation of p38, without affecting TAK1 activation, JNK activation, the recruitment of the IKK complex to TRAF6, or subsequent NF-κB activation. Therefore, CR4-WHD-tet may be a novel type of therapeutic agent that inhibits osteoclastogenesis by modulating the formation of the critical signaling complex downstream from TRAF6.
Results
Multivalent peptide library screening identifies tetravalent peptides that bind to TRAF6 and efficiently inhibit osteoclastogenesis
We developed a multivalent random-peptide library to identify high-affinity binding peptides targeting proteins that function through multivalent interactions, such as Shiga toxins produced by enterohemorrhagic Escherichia coli26,27,29 and hemagglutinin (HA) of influenza A virus28, or proteins that are likely to aggregate, such as amyloid β30, a causative factor in Alzheimer’s disease. Here, we screened tetravalent peptide libraries synthesized on a cellulose membrane29,30 to identify peptides with high affinity for TRAF-C, which interacts with RANK through the region, including the RANK peptide: Arg–Gln–Met–Pro–Thr–Glu–Asp–Glu–Tyr (amino acids 341–349) (Fig. 1a). The library for the first round of screening comprised tetravalent peptides with the sequence –X–X–X–X–X–X–X–, where each X was replaced individually by a fixed amino acid, excluding Cys (Fig. 1b). Screening was based on the peptides’ binding to wild-type His-tagged TRAF-C (WT), but not to TRAF-C with an Ala substitution for Arg392 (R392A), a residue known to play an essential role in the binding of TRAF-C to RANK25. Based on the results of this initial screen (Fig. S1a), a second library was designed with a fixed Asp at position 3. After five more rounds of affinity-based screening, the binding motifs in the seventh libraries were maturated (Fig. 1c, left panel, and Fig. S1b–i). In some steps, several peptide libraries were designed in parallel to verify the diversity of the potential binding motifs. Finally, we identified 11 motifs, most of which had higher WT × ratio values than the RANK peptide (Fig. 1c right panel).

a Schematic diagram of the interaction between RANK and TRAF6. b Schematic diagram showing the structure of tetravalent peptide library synthesized on a cellulose membrane (left panel). Library for the first round of screening contained tetravalent peptides with the sequence MA–X–X–X–X–X–X–X–AU– (where X is a mixture of all amino acids except Cys and U is amino hexanoic acid as a spacer), and each X was replaced individually with a fixed amino acid, excluding Cys (Fig. S1a). Membranes were blotted with His-tagged TRAF-C (WT) or R392A-TRAF-C (R392A), and the binding to each tetravalent peptide was analyzed (Fig. S1a). The library for the second round was designed based on the motifs that showed the best binding on the first screen. Similarly, five additional rounds of affinity-based screenings were performed to maturate the binding motifs and generate the seventh libraries (Fig. S1b–i). c The library for the seventh round of screening contained peptides with the following sequences: W–D–D–X–T–D–F– (Group Ic), R–R–D–X–T–D–F– (Group Ie), R–X–D–D–T–D–F– (Group If), or X–F–D–D–T–D–F– (Group Ig), where each X was replaced individually with a fixed amino acid, other than Cys; *1 indicates the RANK peptide (R–Q–M–P–T–E–D–E –Y–). The binding to each tetravalent peptide was analyzed as described above (left panel). The TRAF-C-binding motifs are identified with the screens (right panel). WT × ratio values indicate the product of the WT-binding value and the normalized WT/R392A ratio, used to evaluate both the binding intensity and the binding specificity (right panel and Fig. S1).
We synthesized tetravalent forms of the identified motifs with clustered Arg residues at the C-terminal of each motif to increase their cell-permeability based on the previous observation that tetravalent peptides with a cluster of Arg residues are highly cell-permeable (Fig. 2a)25,28. We examined the effect of the tetravalent peptides on the RANKL-induced differentiation of bone marrow-derived monocyte/macrophage precursor cells (BMMs) to osteoclasts, which was assessed by counting the number of tartrate-resistant acid phosphatase (TRAP)-positive cells containing more than three nuclei. Among the Group I and II peptides, CR4-RRK-tet and CR4-WHD-tet, respectively, most efficiently inhibited this differentiation (Fig. 2b upper panels, Fig. S2). TRAF-C bound to these tetravalent peptides and also their CR4-lacking forms in a dose-dependent manner, but much less strongly to their monovalent forms (Fig. 2b lower panels), clearly indicating the avidity contribution based on the multivalent interaction. CR4-RRK-tet and CR4-WHD-tet inhibited differentiation in a dose-dependent manner with IC50 values of 0.16 and 0.14 μM, respectively, both of which are much lower than that of RANK-tet25 (Fig. 2c). These tetravalent peptides did not affect the proliferation of BMMs in the absence of RANKL (Fig. S3).

a Schematic diagram of the structure of the tetravalent peptides. CR4- indicates a tetravalent peptide with four Arg residues in the C-terminal region. b Tetravalent peptides inhibit osteoclastogenesis (upper panels). Bone marrow cells were treated with M-CSF for 24 h to obtain BMMs. The precursor cells were cultured with M-CSF and RANKL in the presence of the indicated concentrations of tetravalent peptides with clustered Arg residues, which did not affect the viability of the precursor cells, or with DMSO as the control. After 3 days, osteoclastogenesis was assessed by counting the number of TRAP-positive cells containing more than three nuclei (Fig. S2). Data are presented as a percentage of the control (without any peptide) (mean ± SEM, n = 6 for CR4-WHD-tet and CR4-RRK-tet, n = 3 for the others). *P < 0.05 (compared with control by Dunnett’s test). ELISA was used to measure the binding of each tetravalent or monovalent peptide (1 µg/mL) to TRAF-C (lower panels) (mean ± SEM, n = 3). c CR4-RRK-tet and CR4-WHD-tet dose-dependently inhibited osteoclastogenesis. Representative images are shown (left panels). Scale bar, 500 µm. Data are presented as a percentage of the control (without peptide) (mean ± SEM, n = 4 for 0.1 µM, n = 5 for other concentrations). *P < 0.05 (compared with control by Dunnett’s test).
CR4-WHD-tet inhibits RANKL-induced bone loss in mice
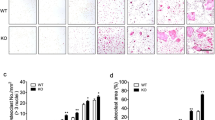
The effects of CR4-RRK-tet and CR4-WHD-tet on RANKL-induced bone loss in mice were examined. After 50 h of treatment with RANKL, a bone morphometric analysis of the femurs was performed with microcomputed tomography (μCT). Bone loss was evaluated as bone volume per tissue volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), trabecular spacing (Tb.Spac), and bone mineral content per tissue volume (BMC/TV). All the changes in these parameters induced by RANKL were efficiently inhibited by CR4-WHD-tet, but not by CR4-RRK-tet, indicating that CR4-WHD-tet inhibits osteoclastogenesis in vivo (Fig. 3). In addition, treatment with CR4-WHD-tet did not markedly affect osteoblast differentiation (Fig. S4), confirming that the attenuation of RANKL-induced bone loss by CR4-WHD-tet is due to its effect on osteoclasts rather than on osteoblasts. One possible reason why CR4-RRK-tet showed no inhibitory effect is that the relative distribution of 125I-labeled CR4-RRK-tet to bone tissues, especially to the tibia, was lower than that of CR4-WHD-tet (Fig. S5). CR4-WHD-tet, whose functional motif contains four acidic-amino acids, has 16 acidic-amino acids in total in its structure, whereas CR4-RRK-tet has eight acidic-amino acids in total. It has been shown previously that highly acidic oligopeptides, such as the l-Asp hexapeptide, can accumulate in bone31, explaining, at least in part, the preferred distribution of CR4-WHD-tet to bone tissues and its subsequent efficient inhibitory effect on RANKL-induced bone loss in vivo. Based on these observations, we used CR4-WHD-tet as the candidate compound in the following experiments.

a, b μCT analysis of the femurs of mice intraperitoneally injected with or without RANKL. CR4-WHD-tet (a) or CR4-RRK-tet (b) was coinjected with RANKL. Representative images are shown (left upper panels: axial view of the metaphyseal region; left lower panels: longitudinal view). Scale bars, 1 mm. Bone loss was evaluated as bone volume per tissue volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), trabecular spacing (Tb.Spac), and bone mineral content per tissue volume (BMC/TV) (a, b, right panels). Parameters are based on a μCT analysis of the metaphyseal region (mean ± SEM: control, n = 6; RANKL only, n = 5 or 6; RANKL plus CR4-tet, n = 6 [a] or 6–7 [b]). *P < 0.05 (by Tukey’s test). n.s., not significant.
CR4-WHD-tet functions in the late stage of differentiation to specifically inhibit the activation of p38 induced by RANKL
We examined in which stage of differentiation CR4-WHD-tet inhibited the RANKL-induced differentiation of mouse bone marrow cells into osteoclasts. CR4-WHD-tet most efficiently inhibited osteoclastogenesis in the late stage of differentiation (48–72 h), indicating that CR4-WHD-tet targets the signals induced by the third treatment with RANKL (Fig. 4a). Because NFATc1 is a master regulator of differentiation, we examined the effects of CR4-WHD-tet treatment in this late stage on the mRNA and protein expression levels of NFATc1. Although NFATc1 expression increased time-dependently at both levels, CR4-WHD-tet did not affect these levels in this stage of differentiation (Fig. 4b).

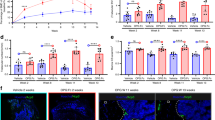
a Effect of CR4-WHD-tet treatment on RANKL-induced osteoclastogenesis in each stage of differentiation. BMMs obtained from bone marrow cells were cultured with RANKL in the presence or absence of 3 μM CR4-WHD-tet, as shown in the schematic diagram (left panel). The early, middle, and late stages of differentiation correspond to 0-24 h, 24-48 h, and 48–72 h incubation, respectively. Data are presented as a percentage of the control value (without CR4-WHD-tet) (middle panel, n = 4, mean ± SEM). *P < 0.05 (by Tukey’s test). Representative images of TRAP staining are shown (right panel). Scale bar, 500 µm. b Effect of CR4-WHD-tet treatment on the mRNA and protein expression levels of NFATc1. BMMs were cultured with RANKL in the presence or absence of 3 μM CR4-WHD-tet, as shown in the schematic diagram (left upper panel). Relative amounts of NFATc1 mRNA were determined with reverse transcription–quantitative PCR (RT–qPCR) using Gapdh as the reference gene (n = 4–8, mean ± SEM, left lower panel). NFATc1 in the lysate was analyzed with western blotting, and the intensity of each band was quantified and presented as the amount of NFATc1 relative to β-actin (n = 3, mean ± SEM, right panels). n.s., not significant (by two-sided Student’s t test). c Effect of CR4-WHD-tet treatment on the activation of signaling molecules induced by RANKL in the late stage of differentiation. BMMs were cultured with RANKL, as shown in the schematic diagram. The cells were treated with or without 3 μM CR4-WHD-tet 30 min before the third treatment with RANKL. The cell lysate was analyzed with western blotting using specific antibodies, and the intensity of each band was quantified. Activation levels of p38 (upper left panel), JNK (lower left panel), and ERK (lower right panel) were evaluated as the amount of the phosphorylated form relative to the total protein. Activation levels of NF-κB were evaluated as the amount of the phosphorylated IκB (upper middle panel) and total IκB (upper right panel) relative to β-actin (n = 3–6; mean ± SEM). *P < 0.05 (by two-sided Student’s t test). d Effect of p38 inhibitor SB203580 treatment on RANKL-induced osteoclastogenesis in the late stage of differentiation. BMMs were cultured with RANKL, as shown in the schematic diagram. The cells were treated with or without 10 μM SB203580 after the third treatment with RANKL. Data are presented as a percentage of the control value without CR4-WHD-tet (n = 3, mean ± SEM). *P < 0.05 (by two-sided Student’s t test).
A transcriptomic analysis was performed to further clarify the molecular mechanism underlying the inhibitory effects of CR4-WHD-tet. We identified Mapk11 (p38β) as a candidate gene involved in the mechanism because its mRNA expression specifically increased in the late stage of differentiation and was reduced by treatment with CR4-WHD-tet (Fig. S6a). This response was also confirmed with reverse transcription (RT)–quantitative PCR (qPCR) (Fig. S6b). Consistent with these observations, the time-dependent activation of p38 induced by the third treatment with RANKL was markedly inhibited by CR4-WHD-tet treatment (Fig. 4c). However, the RANKL-induced activation of NF-κB, which was evaluated as the time-dependent phosphorylation and subsequent degradation of IκB, was not inhibited by the CR4-WHD-tet treatment. Moreover, CR4-WHD-tet did not affect the RANKL-induced response of other MAPKs, such as JNK and extracellular signal-regulated kinase (ERK) (Fig. 4c), indicating that CR4-WHD-tet suppresses osteoclastogenesis through the specific inhibition of p38 in the late stage of differentiation. Treatment with the p38-specific inhibitor SB203580 during the late stage also inhibited osteoclastogenesis (Fig. 4d), confirming the importance of p38 in this stage. Previously, it has been shown that p38 determines the balance of apoptosis pathways. However, CR4-WHD-tet did not induce the emergence of TUNEL-positive apoptotic cells 24 h after the RANKL treatment (Fig. S7), confirming that CR4-WHD-tet inhibits osteoclastogenesis without inducing apoptosis of the precursor cells.
It has previously been shown that activated p38 localizes to the nucleus to phosphorylate and activate the NFATc1 present in the nucleus in the late stage of osteoclastogenesis18. As shown above, CR4-WHD-tet specifically inhibits RANKL-induced p38 activation in the late stage of differentiation, so we examined the effect of CR4-WHD-tet on the nuclear localization of NFATc1 and p38. After the incubation of BMMs with RANKL for 72 h, the expression of NFATc1 was markedly enhanced, and the major proportion of NFATc1 was localized in the nucleus in the absence or presence of CR4-WHD-tet that efficiently inhibited osteoclastogenesis (Fig. 5a), consistent with the observation that CR4-WHD-tet does not affect the expression of NFATc1 in this stage of differentiation (Fig. 4b). In contrast, the nuclear localization of p38, which was clearly induced by the third treatment with RANKL, was markedly inhibited by the presence of CR4-WHD-tet (Fig. 5b). Consistent with this, the increased mRNA expression of osteoclast stimulatory transmembrane protein (Ocstamp), matrix metallopeptidase 9 (Mmp9), and cathepsin K (Ctsk), all of which are marker proteins of late-stage differentiation regulated by NFATc1, was markedly reduced by CR4-WHD-tet (Fig. 5c). However, the mRNA expression of TRAP (Acp5), whose expression is even observed in mononuclear cells before the fusion process, was not reduced by CR4-WHD-tet (Figs. 2c, 4a, 5a, c). These observations confirm that the inhibitory effects of CR4-WHD-tet are mediated by the inhibition of p38 activation and the subsequent NFATc1 activation induced by RANKL in the late stage of differentiation.

a, b Effect of CR4-WHD-tet treatment on the nuclear localization of NFATc1 (a) and p38 (b). BMMs obtained from bone marrow cells were cultured with RANKL in the presence or absence of 3 μM CR4-WHD-tet, as shown in the schematic diagram (a, b, left panels). Nuclear localization was analyzed with immunocytochemistry using a specific antibody directed against NFATc1 (a, middle panel) or p38 (b, middle panel). Representative images of TRAP staining are shown (a). Scale bars, 50 µm. The percentage of nuclei with NFATc1 was measured (a, right panel; mean ± SEM, n [number of area with 5–31 nuclei] = 11–18, from 2 independent experiments). n.s., not significant (by two-sided Student’s t test). The percentage of nuclei with p38 was measured (b, right panel; mean ± SEM, n [number of area with 5–29 nuclei] = 14–19, from 3 independent experiments). ***P < 0.001 (by Tukey’s test). c Effect of CR4-WHD-tet treatment on the mRNA expression levels of genes involved in the late stage of differentiation. BMMs were cultured with RANKL in the presence or absence of 3 μM CR4-WHD-tet, as shown in the schematic diagram (upper panel). Relative amounts of each mRNA were determined with RT–qPCR using Gapdh as the reference gene with the amount of RANKL treatment only equal to 1 (n = 3–5, mean ± SEM, lower panels). *P < 0.05 (by Tukey’s test). n.s., not significant. If no expression levels were detected even after 40 cycles of amplification, the data were presented as zero.
CR4-WHD-tet specifically suppresses recruitment of MKK3 to TRAF6 to inhibit p38 activation
To clarify the mechanism by which CR4-WHD-tet inhibits p38 activation in the late stage of differentiation, we examined whether the binding of CR4-WHD-tet to TRAF6 affects the downstream signals induced by the activation of TRAF6. Streptavidin beads charged with biotinylated CR4-WHD-tet specifically precipitated TRAF6 from the cell lysates of RAW264.7 cells (Fig. 6a), in which CR4-WHD-tet also efficiently inhibits their RANKL-induced differentiation into TRAP-positive multinucleated cells (Fig. S8). In addition, CR4-WHD-tet partially inhibited the increase in the amount of RANK co-precipitated with TRAF6 by the third treatment with RANKL in bone marrow cells (Fig. S9). These observations indicate that CR4-WHD-tet interacts with TRAF6 in cells and then affects the downstream signals. The binding of RANKL to RANK causes the activation of the E3 ubiquitin ligase of TRAF6 to form K63-Ub chains on TRAF6, which acts as a platform upon which a signal complex consisting of TAK1/TAB2, the IKK complex, and MKKs is formed7,13,14. The formation of K63-Ub chains induced by the third treatment with RANKL was evaluated by the formation of smeared bands of immunoprecipitated TRAF6 (Fig. 6b) and immunoprecipitated TRAF6 labeled with HA-tagged ubiquitin with K63 only (Fig. S10). In both cases, the formation of K63-Ub chains was inhibited by CR4-WHD-tet. However, the subsequent activation of TAK1, which is induced by autophosphorylation after its recruitment to the K63-Ub chain through TAB213,14, was not affected by CR4-WHD-tet (Fig. 6c). In contrast, the recruitment of MKK3 to TRAF6, which was clearly induced by the third treatment with RANKL, was inhibited by CR4-WHD-tet (Fig. 6d and S11), whereas the amounts of MKK6 and NEMO present in the complex were not affected by the RANKL treatment in the presence or absence of CR4-WHD-tet (Fig. 6d, e). In addition, the time-dependent activation of MKK3 induced by the third treatment with RANKL was substantially and consistently inhibited by CR4-WHD-tet treatment (Fig. S12). These observations indicate that the third treatment with RANKL induced the formation of K63-Ub chains on TRAF6, which is involved in the recruitment and subsequent activation of MKK3, and that CR4-WHD-tet suppresses this process, thus inhibiting the downstream activation of p38 by MKK3 (Fig. 6f). Combined with the observation that the phosphorylation and subsequent degradation of IκB was unaffected by CR4-WHD-tet (Fig. 4c), the amount of NEMO present on the K63-Ub chain before the third treatment with RANKL may have been enough to respond to the treatment, thus activating the NF-κB pathway at this stage. Furthermore, the amounts of MKK3 and p38 were not affected by CR4-WHD-tet during the late stage of differentiation (Fig. S13), further confirming that the rapid responses of these signaling molecules, but not their protein levels, play essential roles in osteoclastogenesis.

a Coprecipitation assay using biotinylated CR4-WHD-tet. Lysates obtained from RAW264.7 cells were treated with streptavidin beads charged with or without biotinylated CR4-WHD-tet for 24 h at 4 °C. The beads and lysates were analyzed with western blotting. b Poly-ubiquitination of TRAF6. BMMs were cultured with RANKL, as shown in the schematic diagram. TRAF6 was immunoprecipitated and analyzed with western blotting using specific antibodies. c Effect of CR4-WHD-tet treatment on the activation of TAK1. BMMs were cultured with RANKL, as shown in the schematic diagram. The cell lysates were analyzed with western blotting using specific antibodies, and the intensity of each band was quantified. The activation level of TAK1 was evaluated as the amount of the phosphorylated form relative to the total protein (n = 6, mean ± SEM). *P < 0.05 (by two-sided Student’s t test). d, e Effect of CR4-WHD-tet treatment on the recruitment of MKK3 and MKK6 (d), or NEMO (e) to TRAF6. BMMs were cultured with RANKL, as shown in the schematic diagram. TRAF6 was immunoprecipitated and analyzed with western blotting using specific antibodies. The intensity of each band was quantified and presented as the amount relative to TRAF6 (n = 3 for MKK3 and NEMO, n = 4 for MKK6; mean ± SEM). *P < 0.05 (by Tukey’s test). n.s., not significant. The cells were treated with 3 μM CR4-WHD-tet 30 min before the third treatment with RANKL (b–e). f Schematic diagram of the effect of CR4-WHD-tet on the downstream signals from TRAF6.
Discussion
In this study, we identified WHD-tet as a novel TRAF6-binding peptide by targeting the RANK-binding region of TRAF6 using affinity-based screening of a random-peptide library. CR4-WHD-tet, a cell-permeable form of WHD-tet, efficiently inhibited RANKL-induced osteoclastogenesis in vitro and in vivo. CR4-WHD-tet specifically inhibited the recruitment of MKK3 to the K63-Ub chain on TRAF6, resulting in the inhibition of both p38 activation and its subsequent nuclear localization, which promoted the final stage of differentiation through the activation of NFATc1. Therefore, CR4-WHD-tet may be a novel type of therapeutic agent for osteoclast-related diseases that functions as an interaction modulator to tune the formation of the critical signaling complex downstream from TRAF6.
The affinity-based screening of a random-peptide library identified RRK-tet, WHD-tet, and other tetravalent peptides that bind TRAF-C with higher efficacy than RANK-tet. Interestingly, both RRK-tet and WHD-tet, but not their monovalent forms, efficiently bound TRAF-C (Fig. 2b), indicating that a multivalent interaction is strictly required for high-affinity binding to TRAF-C. This finding is consistent with the previous observation that the mushroom-shaped trimeric structure of TRAF-C interacts with three molecules of RANK through its TRAF-C-binding domain to markedly increase the binding affinity based on the avidity contribution, which represents a conformational rearrangement of the interaction22,23. Therefore, it is theoretically difficult to identify such functional motifs using regular peptide library screening or phage display screening, neither of which is based on the avidity contribution, demonstrating the superiority of this screening method.
All the TRAF-C-binding tetravalent peptides identified had the X–X–Asp–X–X–Asp/Glu–Phe motif, the sequence of which differs from the consensus TRAF6-binding motif, Pro–X–Glu–X–X– (acidic/aromatic residue), which is present not only in RANK but also in other TRAF6-interacting molecules, such as CD40 and the IRAK adaptor kinases23. In particular, Pro344 of RANK, which corresponds to the first Pro of the consensus motif, plays an essential role in TRAF6 binding through its hydrophobic interaction with Phe471 and Tyr473 in the β7-strand of TRAF623, whereas the preferential selection of Pro has not been observed in any inhibitory motifs identified. Furthermore, strong selection of C-terminal Phe was observed in all the binding motifs identified, but is not preferred in the consensus motif. In contrast, multiple (2–4) acidic-amino acids are commonly observed in the motifs identified as well as in the consensus motif. Glu–Asp–Glu (amino acids 346–348) of RANK plays an essential role in TRAF6 binding, through electrostatic interactions with Arg392 and Lys469 of TRAF623,25. This Arg392 is a target amino acid for the peptide library screening and may explain the common selection of an acidic-amino-acid cluster in both motifs. These observations indicate that the identification of the best binding motifs, which TRAF-C potentially possesses, is necessary to develop the optimal inhibitory motif, because in general the best binding motif for the intended target molecule is essentially different from the native interacting motif(s)32.
p38 has been shown to cause AP-1 activation and then to induce the expression of c-Fos and NFATc1 to promote the osteoclastogenesis induced by RANKL in the early stage of differentiation33,34, whereas in the late stage, activated p38 localizes to the nucleus to phosphorylate and activate NFATc1, enhancing cathepsin K gene expression18. Here, we found that in the late stage of differentiation, CR4-WHD-tet does not affect the expression level of NFATc1, but specifically inhibits the activation of p38 and its subsequent nuclear localization to suppress the mRNA expression of Ocstamp, Mmp9, and Ctsk, which is regulated by NFATc1 in the late stage of differentiation. These observations confirm the different roles of p38 in the various stages of differentiation and demonstrate its essential contribution to the late stage of osteoclastogenesis.
CR4-WHD-tet inhibited the formation of K63-Ub chains induced by the third treatment with RANKL, which specifically inhibited the recruitment of MKK3, but not MKK6 or NEMO, to the K63-Ub chain and the subsequent activation of p38, even though the activation of TAK1, an upstream p38 kinase, was not affected by CR4-WHD-tet. It has been shown with RAW264.7 cells overexpressing a constitutively active form of MKK6 that MKK6 activates p38 to phosphorylate and activate the NFATc1 present in the nucleus in the late stage of differentiation18, but the contribution of MKK3 at this stage has been unknown. Our results clearly indicate the important role of MKK3 in osteoclastogenesis in the late stage of differentiation, consistent with a previous report showing that the knockout of MKK3, but not MKK6, inhibits osteoclastogenesis in vivo19.
A previously developed synthetic peptide, L-T6DP-1, containing a sequence of the TRAF-C-binding region of RANK, inhibited osteoclast differentiation with an IC50 value of 30 μM, which is 210 times higher than that of CR4-WHD-tet23. In stark contrast to CR4-WHD-tet, L-T6DP-1 inhibited NF-κB activation in RANKL-treated RAW264.7 cells23. Therefore, although these two peptides commonly target the TRAF-C region of TRAF6 including Arg392, the inhibitory mechanism of CR4-WHD-tet is quite different from that of L-T6DP-1, in that the binding of CR4-WHD-tet may induce a unique conformational rearrangement of the TRAF6 trimer. This affects the subsequent formation of K63-Ub chains on TRAF6 to inhibit the recruitment of MKK3 to TRAF6, without affecting the activation of TAK1. In addition to its higher binding affinity based on the avidity contribution, this unique binding mode of CR4-WHD-tet to TRAF6 may contribute to its highly efficient inhibitory effect on osteoclastogenesis, although the precise molecular mechanisms remain to be clarified.
The RANK/TRAF6 axis, including the downstream signaling molecules such as NF-κB and p38, is implicated in various biological processes, such as the immune system35,36 and mammary gland development37. Therefore, a series of compounds that directly target these molecules, such as inhibitors of NF-κB or p38, may have adverse side effects if they were used as therapeutic agents38. The usage of denosumab, a specific monoclonal antibody against RANKL, also potentially has the same risk as these compounds. In contrast, CR4-WHD-tet, which inhibits osteoclastogenesis by tuning the formation of the critical signaling complex downstream from TRAF6 with minimum effects on other signaling molecules, such as TAK1, JNK, and NF-κB, is potentially a novel type of therapeutic agent for osteoclast-related diseases with fewer side effects.
A possible limitation of CR4-WHD-tet may be its stability in vivo. The relatively high doses of CR4-WHD-tet required to inhibit RANKL-induced bone loss in mice may reflect this problem. We have already observed that various chemical modifications of tetravalent peptides, such as N-terminal acetylation26,30 and the usage of non-natural D-amino acid28 or artificial amino acid29 to avoid the proteolytic degradation, significantly enhance their biological activities in vivo. Thus, adding these modifications to CR4-WHD-tet would solve the stability issue. To further promote the therapeutic application of CR4-WHD-tet, it is necessary to evaluate the effect of CR4-WHD-tet on bone loss inhibition using other osteoporosis models2, such as ovariectomy and glucocorticoid-induced animal models.
Methods
Antibodies
Antibodies were obtained from the vendors and used at the indicated dilutions shown in Table S1.
Screening tetravalent peptide libraries on cellulose membrane
Tetravalent peptide libraries were synthesized on a cellulose membrane with a MultiPep spot synthesizer (CEM Corporation)28,29,30. The Met–Ala sequence and Ala at the carboxyterminus of each peptide were included to match the structure of a previously published tetravalent peptide library26. After the membrane was blocked with 5% skim milk for 1 h at room temperature, it was blotted with recombinant-histidine-tagged TRAF-C-WT or TRAF-C-R392A (10 μg/mL) for 1 h, both of which were prepared using a pET expression system25. After extensive washing, the bound TRAF-C was detected on the membrane with a rabbit anti-6×histidine-tag antibody (1:1000; Merck KGaA, Darmstadt, Germany, cat.# 70796-3), followed by horseradish peroxidase (HRP)-labeled mouse anti-rabbit IgG antibody (Cell Signaling Technology, MA, USA) and Western Lightning Plus-ECL (PerkinElmer, Inc., Washington, MA, USA). The intensity of each peptide spot was quantified as a pixel value with the ImageQuant LAS 500 CCD imager (GE Healthcare Life Sciences, Marlborough, MA, USA).
Peptides
The tetravalent peptides were synthesized with N-α-Fmoc-protected amino acids and standard BOP/HOB coupling chemistry26. To prepare the biotinylated tetravalent peptides, the terminal amino groups of the tetravalent peptides were biotinylated with biotin (Sigma-Aldrich, MO, USA) and 1-(bis[dimethylamino]methylene)-1H-benzotriazolium 3-oxide hexafluorophosphate (Peptide Institute Inc., Japan) in the last cycle of peptide synthesis. The synthesized peptides were confirmed with a mass spectrometric analysis using the autoflex II TOF/TOF system (Bruker Corp., MA, USA).
Osteoclast differentiation
Bone-marrow cells prepared from the femurs of 6-week-old male C57BL/6 mice (Shimizu Laboratory Supplies Co., Ltd., Kyoto, Japan) were cultured in a 24-well plate in α-modified Eagle’s medium (α-MEM) supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 µg/mL streptomycin25. The cells were treated with culture medium from mouse M-CSF producing Ltk- cell line, CMG14-12 cells (diluted 1:50), which were kindly provided by Dr. A. Kudo in Tokyo Institute of Technology (Yokohama, Japan), as a source of macrophage colony-stimulating factor (M-CSF) for 24 h to obtain BMMs. The obtained precursor cells were treated with the culture medium from CMG14-12 cells (diluted 1:50) and 66 ng/mL glutathione S-transferase (GST)–RANKL, which was prepared using a pGEX vector system25, in the presence of the indicated concentrations of peptide or dimethyl sulfoxide (DMSO) (final concentration 0.2%) as the control. The culture medium was changed every 24 h to the same medium containing M-CSF, GST-RANKL, and peptide. After 3 days, osteoclastogenesis was assessed by counting the number of TRAP-positive cells containing more than three nuclei. TRAP staining and nuclear staining with naphthol AS-MX phosphate (Sigma-Aldrich) and 4′,6-diamidino-2-phenylindole (DAPI; Dojindo Laboratories, Kumamoto, Japan), respectively25.
Binding between TRAF-C and each peptide
Each peptide (1 µg/mL) was applied to a well of a typical 96-well ELISA plate (Thermo Fisher Scientific, MA, USA) and incubated overnight for 24 h. After the plate was blocked, it was incubated with various concentrations of His–TRAF-C or His–TRAF-C-R392A for 1 h at room temperature. The bound His–TRAF-C or His–TRAF-C-R392A was detected with a mouse monoclonal anti-His-tag antibody (clone: 9C11, FujiFilm Wako Pure Chemical Corporation, Osaka, Japan) and an HRP-conjugated horse anti-mouse IgG secondary antibody (Cell Signaling Technology, MA, USA).
RANKL-induced bone loss
Seven-week-old female C57BL/6 mice were intraperitoneally injected with 3 mg/kg GST-RANKL in the presence of CR4-RRK-tet (130 mg/kg) or CR4-WHD-tet (150 mg/kg). After 50 h, all the mice were sacrificed, and the femurs from both legs were collected39. Computed tomography (CT) images of the femurs were acquired with a cone-beam X-ray μCT system (ScanXmate-RB090SS150; Comscantecno Co., Ltd, Yokohama, Japan). Three-dimensional microstructural image data were reconstructed. A 1 mm thick region located 0.1 mm from the epiphyseal growth plate was reconstructed for the axial view, and the structural indices were calculated with the TRI/3D-BON software (Ratoc Systems Inc., Osaka, Japan). A bone morphometric analysis was performed as described previously40. The analysis was performed on both legs of each mouse. All animal experiments were approved by the Animal Ethics Committee of Doshisha University before their commencement and were performed in accordance with the approved protocols. We have complied with all relevant ethical regulations for animal use.
Quantitative PCR
After the BMMs were harvested, their total RNA was extracted with Sepasol®-RNA I Super G (Nacalai Tesque, Inc., Kyoto, Japan), and transcribed into cDNA with ReverTra Ace® qPCR RT Master Mix (Toyobo, Osaka, Japan), according to the manufacturers’ protocols. PCR was performed for 40 cycles with the obtained cDNA as a template and specific primers (Table S2). The mRNA levels of each gene were quantified with qPCR using Thunderbird® Next SYBR® qPCR Mix (Toyobo) and the same primers. The data were analyzed with relative quantification based on the ΔΔCt method, using Gapdh as the reference gene.
Western blotting
BMMs cultured in a six-well plate (Iwaki, Shizuoka, Japan) were treated as indicated and then lysed in lysis buffer containing 1% NP-40, 0.2% sodium dodecyl sulfate (SDS), 10% glycerol, 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, and Protease Inhibitor Cocktail (Nacalai Tesque). The lysates (10 μg protein) were separated with SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After the membrane was blocked with 5% skim milk, it was immunoblotted with the indicated primary antibodies, followed by HRP-labeled goat anti-rabbit IgG or horse anti-mouse IgG antibody (Cell Signaling Technology). The membrane was finally visualized with Western Lightning Plus-ECL (PerkinElmer), and analyzed with an ImageQuant LAS 500 CCD imager (GE Healthcare Sciences). β-Actin was used as the internal control.
Immunocytochemical analysis
BMMs cultured on a 35 mm glass-bottom dish (Iwaki) were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then incubated with a specific antibody directed against NFATc1 or p38, and then with an Alexa-546-labeled secondary antibody (Thermo Fisher Scientific). The nuclei were stained with 1 μg/mL DAPI. All the fluorescent images were analyzed with an LSM710 Laser Scanning Confocal Microscope (Carl Zeiss Co., Ltd, Oberkochen, Germany).
Coprecipitation assay using biotinylated peptide
RAW264.7 cells (ATCC, VA, USA) cultured in a 10 cm culture dish (Corning, NY, USA) were lysed in lysis buffer (20 mM Tris-HCl [pH 7.0], 150 mM NaCl, 10% glycerol, 1% NP-40, 10 mM EDTA, 1 mM EGTA, 10 mM NaF, 10 mM NaPPi, 200 µM sodium orthovanadate (V), and Protease Inhibitor Cocktail (Nacalai Tesque)). For coprecipitation, the lysates were treated with streptavidin beads (Sigma-Aldrich) charged with or without biotinylated CR4-WHD-tet for 24 h at 4 °C. After extensive washing, the beads were analyzed with western blotting, as described above.
Immunoprecipitation
BMMs cultured in a 10 cm culture dish (Corning) were treated as indicated, and then lysed with lysis buffer, as described above. The lysates (100 μg protein) were pretreated with protein G Sepharose beads (Cytiva, Tokyo, Japan). After centrifugation, the lysates were incubated with an anti-TRAF6 antibody at 4 °C. After 1 h, the lysates were treated with protein G Sepharose beads for 1 h at 4 °C. After extensive washing, the beads were analyzed with western blotting using specific antibodies, as described above.
Statistics and reproducibility
Significant differences between the two groups were analyzed with an unpaired two-sided Student’s t test. Significant differences between each group and the control group were analyzed with a one-way analysis of variance (ANOVA) followed by Dunnett’s test. Multiple comparisons of differences among every group were analyzed with one-way ANOVA followed by Tukey’s range test. All statistical analyses were performed with the IBM SPSS Statistics software (ver. 27.0.0.0).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
References
Al-Bari, A. A. & Al Mamun, A. Current advances in regulation of bone homeostasis. FASEB Bioadv. 2, 668–679 (2020).
Song, S., Guo, Y., Yang, Y. & Fu, D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharm. Ther. 237, 108168 (2022).
Dougall, W. C. et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 13, 2412–2424 (1999).
Kong, Y. Y. et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323 (1999).
Takayanagi, H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 7, 292–304 (2007).
Ono, T. & Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell Biol. 149, 325–341 (2018).
Sun, Y. et al. Recent advances in osteoclast biological behavior. Front Cell Dev. Biol. 9, 788680 (2021).
Deng, L. et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 (2000).
Wang, C. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 (2001).
Kobayashi, N. et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. Embo J. 20, 1271–1280 (2001).
Mizukami, J. et al. Receptor activator of NF-κB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol. Cell Biol. 22, 992–1000 (2002).
Takayanagi, H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3, 889–901 (2002).
Park, J. H., Lee, N. K. & Lee, S. Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 40, 706–713 (2017).
Yamamoto, M., Gohda, J., Akiyama, T. & Inoue, J. I. TNF receptor-associated factor 6 (TRAF6) plays crucial roles in multiple biological systems through polyubiquitination-mediated NF-κB activation. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 97, 145–160 (2021).
Jianwei, W. et al. The role of TAK1 in RANKL-induced osteoclastogenesis. Calcif. Tissue Int. 111, 1–12 (2022).
Huang, H. et al. Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-κB transactivation by RANKL. Cell Death Differ. 13, 1879–1891 (2006).
Lin, J., Lee, D., Choi, Y. & Lee, S. Y. The scaffold protein RACK1 mediates the RANKL-dependent activation of p38 MAPK in osteoclast precursors. Sci. Signal 8, ra54 (2015).
Matsumoto, M. et al. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 279, 45969–45979 (2004).
Boyle, D. L. et al. Differential roles of MAPK kinases MKK3 and MKK6 in osteoclastogenesis and bone loss. PLoS One 9, e84818 (2014).
Ye, H. & Wu, H. Thermodynamic characterization of the interaction between TRAF2 and tumor necrosis factor receptor peptides by isothermal titration calorimetry. Proc. Natl Acad. Sci. USA 97, 8961–8966 (2000).
Darnay, B. G., Ni, J., Moore, P. A. & Aggarwal, B. B. Activation of NF-κB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-κB-inducing kinase: Identification of a novel TRAF6 interaction motif. J. Biol. Chem. 274, 7724–7731 (1999).
Chung, J. Y., Park, Y. C., Ye, H. & Wu, H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci. 115, 679–688 (2002).
Ye, H. et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447 (2002).
Moriya, J. et al. Structure-based development of a protein-protein interaction inhibitor targeting tumor necrosis factor receptor-associated factor 6. J. Med. Chem. 58, 5674–5683 (2015).
Anzai, M. et al. A tetravalent peptide that binds to the RANK-binding region of TRAF6 via a multivalent interaction efficiently inhibits osteoclast differentiation. Biochem. Biophys. Res Commun. 636, 178–183 (2022).
Nishikawa, K. et al. A multivalent peptide-library approach identifies a novel Shiga toxin-inhibitor that induces aberrant cellular transport of the toxin. FASEB J. 20, 2597–2599 (2006).
Nishikawa, K. Recent progress of Shiga toxin neutralizer for treatment of infections by Shiga toxin-producing Escherichia coli. Arch. Immunol. Ther. Exp. 59, 239–247 (2011).
Omi, J. et al. The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection. Nat. Commun. 11, 162 (2020).
Watanabe-Takahashi, M. et al. Identification of a peptide motif that potently inhibits two functionally distinct subunits of Shiga toxin. Commun. Biol. 4, 538 (2021).
Sato, W. et al. A tailored tetravalent peptide displays dual functions to inhibit amyloid β production and aggregation. Commun. Biol. 6, 383 (2023).
Ishizaki, J., Waki, Y., Takahashi-Nishioka, T., Yokogawa, K. & Miyamoto, K. Selective drug delivery to bone using acidic oligopeptides. J. Bone Min. Metab. 27, 1–8 (2009).
Nishikawa, K. et al. A peptide library approach identifies a specific inhibitor for the ZAP-70 protein tyrosine kinase. Mol. Cell 6, 969–974 (2000).
Matsumoto, M., Sudo, T., Saito, T., Osada, H. & Tsujimoto, M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-κB ligand (RANKL). J. Biol. Chem. 275, 31155–31161 (2000).
Huang, H. et al. Induction of c-Fos and NFATc1 during RANKL-stimulated osteoclast differentiation is mediated by the p38 signaling pathway. Biochem. Biophys. Res Commun. 351, 99–105 (2006).
Walsh, M. C. & Choi, Y. Biology of the TRANCE axis. Cytokine Growth Factor Rev. 14, 251–263 (2003).
Walsh, M. C. et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 24, 33–63 (2006).
Fata, J. E. et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 103, 41–50 (2000).
Sipos, W., Pietschmann, P. & Rauner, M. Strategies for novel therapeutic approaches targeting cytokines and signaling pathways of osteoclasto- and osteoblastogenesis in the fight against immune-mediated bone and joint diseases. Curr. Med. Chem. 15, 127–136 (2008).
Tomimori, Y. et al. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J. Bone Min. Res. 24, 1194–1205 (2009).
Shinohara, M. et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132, 794–806 (2008).
Acknowledgements
This work was supported by grants from the Japan Society for the Promotion of Science (JSPS; KAKENHI grant number JP22J11001), the Japan Science and Technology (JST; SPRING grant number JPMJSP2129), and the Joint Research Project of the Institute of Medical Science, The University of Tokyo.
Author information
Authors and Affiliations
Contributions
M.A., M.W.-T., H.K., Y.M., A.I., and Y.O. performed the experiments, and analyzed and interpreted the data. M.W.-T. and H.K. prepared the recombinant protein and performed the peptide library screening experiments. H.S. provided the antibody resources. T.W. and Kei N. analyzed and interpreted the data. J.I. and K.N. supervised the project. M.A. and K.N. interpreted the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ruili Yang, Mariana Osako and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Dr Martina Rauner, Dr Ophelia Bu. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Anzai, M., Watanabe-Takahashi, M., Kawabata, H. et al. Clustered peptide regulating the multivalent interaction between RANK and TRAF6 inhibits osteoclastogenesis by fine-tuning signals. Commun Biol 8, 643 (2025). https://doi.org/10.1038/s42003-025-08047-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08047-2
