
Abstract
Paclitaxel combination therapy is the main chemotherapy regimen for endometrial cancer (EC); however, subsequent drug resistance is a bottleneck limiting its widespread clinical application. We found that human insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) was abnormally elevated in paclitaxel-resistant EC cells and confirmed that the reduction of IGF2BP3 can effectively improve the sensitivity of EC cells to paclitaxel in vitro and in vivo. Mechanistically, elevated IGF2BP3 promotes the half-life of c-Myc by competitively inhibiting Speckle-type POZ protein (SPOP)-mediated ubiquitination and degradation of c-Myc. As a transcription factor, c-Myc can bind to the promoter of IGF2BP3, thus contributing to the increased transcription of IGF2BP3 via positive feedback and forming a signaling loop that ultimately causes the accumulation of c-Myc-induced paclitaxel resistance. Based on these findings, the application of c-Myc inhibitors (10058-F4) combined with paclitaxel helped paclitaxel-resistant EC cells regain paclitaxel sensitivity in vitro and in vivo. Together, we reveal the underlying mechanism of paclitaxel resistance in endometrial cancer cells and provide insights into treatment strategies for paclitaxel-resistant EC patients.
Similar content being viewed by others


Introduction
Endometrial cancer (EC) is an epithelial malignant tumor of the endometrium, also known as corpus uteri cancer, and is the second most common gynecological malignant tumor after cervical cancer1. In developed countries, the incidence of EC is 5.9%, and approximately 320,000 women are diagnosed with EC annually2. EC treatment includes surgery, chemotherapy, radiotherapy, targeted therapy, immunotherapy, and endocrine therapy3. Postoperative external pelvic irradiation therapy combined with chemotherapy is recommended for patients with high-risk EC, and chemotherapy alone can be used as an alternative4. For patients with stage III/IV EC who undergo surgery, if there are residual metastatic lymph node lesions, positive surgical margins (including positive vaginal margins and pelvic lateral wall involvement), or residual lesions in the pelvic cavity after surgery, individualized treatment with chemotherapy should be discussed by a multidisciplinary team4. Paclitaxel plus carboplatin is the preferred chemotherapy regimen for advanced, metastatic, and recurrent EC5. Other commonly used regimens include docetaxel plus carboplatin, doxorubicin plus cisplatin, carboplatin plus paclitaxel plus bevacizumab, liposomal doxorubicin, albumin-bound paclitaxel, and topotecan5. For patients with carcinosarcoma, paclitaxel plus carboplatin is the preferred chemotherapy5. Other chemotherapy options include paclitaxel plus ifosfamide or cisplatin plus ifosfamide5.
Although paclitaxel is one of the cornerstone drugs in the treatment of EC, some patients will gradually develop resistance to paclitaxel with the use of paclitaxel, resulting in poor efficacy of paclitaxel6. The underlying mechanisms of paclitaxel resistance include activation of the AKT signaling pathway, activation of the Notch signaling pathway, regulation of microRNA (miR-135a, miR-155, miR-34a, etc.), upregulation of P-glycoprotein, and enhancement of stemness7. Among them, the expression of c-Myc is strongly associated with the enhancement of stemness, the AKT signaling pathway, and Notch signaling pathway activation7,8,9. In addition, HMGB1 expression and release confer paclitaxel resistance in castration-resistant prostate cancer by activating and maintaining c-Myc signaling10. Therefore, c-Myc may play an important role in the development of paclitaxel resistance.
Speckle-type POZ protein (SPOP) is an E3 ubiquitin ligase adaptor protein mainly responsible for the ubiquitination and degradation of abnormally increased proteins through the proteasome11. SPOP inhibits the development of cancers and promotes chemotherapy sensitivity by downregulating a variety of oncoproteins, including the androgen receptor (AR), estrogen receptor (ER), c-Myc, and caprin1, through the ubiquitin-proteasome system (UPS)11,12,13,14. For example, wild-type SPOP promotes ubiquitination and degradation of caprin1, whereas prostate cancer-associated SPOP mutations enhance cell survival and docetaxel resistance by upregulating caprin1-dependent stress granule assembly12.
Insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3), a member of the insulin-like growth factor mRNA-binding protein family, mainly acts as an m6A reader, playing an important role in decoding m6A and performing various biological activities15. In recent years, it has been gradually found that IGF2BP3 can mediate chemotherapy resistance in a variety of cancers, such as lung cancer, breast cancer, and liver cancer16,17,18. For instance, IGF2BP3 promotes the translation of EGFR mRNA and further activates the EGFR pathway by cooperating with METTL14 to act as a reader in an m6A-dependent manner, thereby increasing the resistance of colorectal cancer cells to cetuximab19.
Herein, we demonstrated that IGF2BP3 is highly expressed in paclitaxel-resistant EC, and that decreased expression or activation of IGF2BP3 could improve the efficacy of paclitaxel. Mechanistically, we found that IGF2BP3 promoted c-Myc expression by suppressing SPOP-mediated ubiquitination and degradation of c-Myc. In turn, elevated c-Myc can directly promote the transcription of IGF2BP3 via a positive feedback mechanism, thereby forming a signaling loop that ultimately causes the accumulation of c-Myc-induced paclitaxel resistance. Functionally, IGF2BP3 promotes EC proliferation, metastasis, and invasion via the IGF2BP3/SPOP/c-Myc loop. Finally, we suggest that the combination of paclitaxel and c-Myc inhibitor 10058-F4 could improve the efficacy of paclitaxel in EC, which may provide a new direction for the treatment of EC.
Results
IGF2BP3 overexpression leads to resistance to paclitaxel in EC
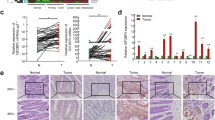
To search for target genes that promote EC resistance to paclitaxel, we subjected normal Ishikawa cells and paclitaxel-resistant Ishikawa cells to RNA-seq analysis (Supplementary Data 1). In the principal component analysis (PCA) plot, the biological replicates of the control group and the paclitaxel-resistant EC cells group were found to be consistent (Fig. 1A), indicating reliable experimental results. After this, we screened for differentially expressed genes (DEGs) between the control group and the paclitaxel-resistant EC cells group and presented them in a heatmap (Fig. 1B). Combined with GSE228106 and GSE50831 in the GEO dataset and UCEC in TCGA database, we screened out three potential genes that are highly expressed in paclitaxel-resistant EC cells, namely IGF2BP3, MSH2 and NUDT3 (Fig. 1C). The further qRT-PCR results showed that the expression of IGF2BP3 was the highest among these three (Fig. 1D), consistent with the RNA-seq analysis (Fig. 1A, B), indicating that IGF2BP3 had the strongest effect on paclitaxel resistance. After determining that the IC50 of paclitaxel was 7 nM (Fig. S1A), we overexpressed and knocked out IGF2BP3 in the EC cells. After IGF2BP3 was overexpressed in EC cells, the IC50 value of paclitaxel in Ishikawa cells and HEC-1-A cells increased to 20.11 nM and 10.01 nM (Fig. 1E), respectively, which may be attributed to the different instantaneous transfection efficiency of Ishikawa cells and HEC-1-A cells. When IGF2BP3 was completely knocked out in EC cells, the IC50 of paclitaxel was down-regulated to 5.5 nM in both Ishikawa cells and HEC-1-A cells (Fig. 1F). In paclitaxel-resistant Ishikawa cells, IGF2BP3 knockdown significantly decreased the IC50 value of paclitaxel from 225.5 nM (Fig. S1B) to 163.0 nM (Fig. S1C), suggesting that IGF2BP3 knockout could improve the sensitivity of paclitaxel. Next, we explored whether IGF2BP3 induces paclitaxel resistance in xenograft nude mouse models (Fig. 1G). Knockout of IGF2BP3 significantly inhibited xenograft tumor weight and volume, and enhanced paclitaxel-induced suppression (Fig. 1H–J). Immunohistochemistry of Ki-67 on xenograft tumors showed the lowest expression of Ki-67 in xenograft tumors with paclitaxel combined with IGF2BP3 knockout (Fig. 1K), indicating the lowest proliferation ability; thus, IGF2BP3 knockout can improve the sensitivity of paclitaxel. Together, IGF2BP3 overexpression induces the development of paclitaxel resistance in EC cells.
Pan-cancer analysis of IGF2BP3 based on TCGA database and CPTAC database revealed that IGF2BP3 was generally highly expressed in cancers (Fig. S2). The unpaired analysis (Fig. S3A) and paired analysis (Fig. S3B) of TCGA database showed that IGF2BP3 mRNA was significantly expressed in EC. In addition, Kaplan-Meier survival analysis showed that IGF2BP3 mRNA was associated with a poorer prognosis (Fig. S3C). In addition, the ROC curve revealed that IGF2BP3 mRNA had good sensitivity and specificity for the diagnosis of EC (Fig. S3D). IGF2BP3 mRNA expression was also higher in EC at higher stages (Fig. S3E) and grades (Fig. S3F). The CPTAC database presented similar results, that is, IGF2BP3 protein expression was higher in EC (Fig. S3G) and correlated with stage (Fig. S3H), grade (Fig. S3I), age (Fig. S3J), and the Myc signaling pathway (Fig. S3K). Furthermore, we collected 68 para-carcinoma and carcinoma specimens from patients with EC for IHC, which showed that the expression of IGF2BP3 protein was dramatically increased (Fig. S3L–M) and correlated with the expression of estrogen and progesterone receptors (Fig. S3N, Table 1). Finally, we knocked out and overexpressed IGF2BP3 in EC cells (Fig. S4A), and found that IGF2BP3 promoted proliferation (Fig. S4B, C), metastasis (Fig. S4D), and invasion (Fig. S4E).

A PCA map of control and paclitaxel-resistant EC cells of RNA-seq. B RNA-seq sequencing analysis of DEGs between the control group and the paclitaxel-resistant EC cells group. C Venn analysis was performed on the highly expressed genes from the RNA-seq of paclitaxel-resistant Ishikawa cells, the highly expressed genes in paclitaxel-resistant cells from GEO228106, the highly expressed genes in paclitaxel-resistant cells from GEO50831, and the highly expressed genes in TCGA’s UCEC. D The expression of IGF2BP3, MSH2, and NUDT3 mRNA in control and paclitaxel-resistant EC cells. E The IC50 of paclitaxel in EC cells with IGF2BP3 overexpression. F The IC50 of paclitaxel in EC cells with IGF2BP3 knockout. G Regimen of paclitaxel injection in NOD-SCID nude mice (Created in BioRender. Zhang, H. (2025) https://BioRender.com/3xsuwvq, Agreement number: FY283DMA1N). H Representative photographs of xenograft tumors of NC + NS, sh-IGF2BP3 + NS, NC+ paclitaxel, and sh-IGF2BP3+ paclitaxel. I The weight of xenograft tumors of NC + NS, sh-IGF2BP3 + NS, NC+ paclitaxel, and sh-IGF2BP3+ paclitaxel. J Relative growth volume of xenograft tumors of NC + NS, sh-IGF2BP3 + NS, NC+ paclitaxel, and sh-IGF2BP3+ paclitaxel at 24 days. K Immunohistochemical staining of xenograft tumors of NC + NS, sh-IGF2BP3 + NS, NC+ paclitaxel, and sh-IGF2BP3+ paclitaxel, Scale bar, 20 μm. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
IGF2BP3 promotes transcription of c-Myc target genes by inhibiting the proteasome
To determine the mechanism underlying IGF2BP3-induced paclitaxel tolerance in EC, we subjected IGF2BP3 knockdown Ishikawa cells to RNA-seq analysis (Fig. S5A–D, Supplementary Data 2). Based on the quality control assessment demonstrated by the PCA plot (Fig. S5A), DEGs were identified between the control group and the IGF2BP3 knockdown group, which were visualized using heatmaps (Fig. S5B), volcano plots (Fig. S5C), and scatter plots (Fig. S5D). Through GESA analysis of these DEGs in the si-IGF2BP3 group, we found that the transcription of c-Myc target genes was dramatically repressed (Figs. 2A, B, and S5E), while c-Myc mRNA expression was not altered (Fig. S5F). Additionally, RIP assay confirmed that the IGF2BP3 protein does not bind to c-Myc mRNA (Fig. S5G), suggesting that IGF2BP3 does not act as an m6A reader to regulate c-Myc. Thus, IGF2BP3 may affect c-Myc at the post-translational level, and we then inhibited the proteasome with MG-132 and bortezomib, and the lysosome with chloroquine (Fig. 2C). Only when the proteasome is inhibited does c-Myc expression no longer increase with elevated IGF2BP3 levels (Fig. 2C). Concurrently, the half-life of c-Myc protein was correspondingly prolonged under the condition of IGF2BP3 overexpression (Fig. 2D–E). Moreover, the IHC results showed a positive correlation between IGF2BP3 and c-Myc in patients with EC (Fig. 2F). Thus, IGF2BP3 promotes transcription of c-Myc target genes by inhibiting the proteasome.

A Heat map of DEGs of c-Myc-targeting genes in control and IGF2BP3 knockdown EC cells. B GSEA analysis of DEGs of c-Myc-targeting genes in control and IGF2BP3 knockdown EC cells. C The effects of IGF2BP3 on c-Myc expression treated with DMSO, MG132 (20 μM), bortezomib (200 nM) and chloroquine (100 nM). D The effects of IGF2BP3 on the half-life of c-Myc detected by WB. E Statistical analysis of the effect of IGF2BP3 on the half-life of c-Myc. F Immunohistochemical staining of IGF2BP3 and c-Myc in 24 patients with endometrial cancer. Scale bar, 10 μm. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
IGF2BP3 interacts with SPOP to inhibit its ubiquitination and degradation of c-Myc
To identify the E3 ligase that mediates the upregulation of c-Myc by IGF2BP3, we enriched IGF2BP3 and performed mass spectrometry (Supplementary Data 3), which revealed that SPOP may be a potential intercalating protein of IGF2BP3 (Fig. 3A, B). Co-immunoprecipitation confirmed that IGF2BP3 could interact with SPOP (Fig. 3C), and immunofluorescence assays showed that IGF2BP3 and SPOP co-localized (Fig. 3D). Intriguingly, SPOP mutants with deletions in the MATH, BTB, or NLS domains also co-localized (Fig. 3E) and interacted (Fig. 3F) with IGF2BP3, indicating that these domains of SPOP are not responsible for the interaction between SPOP and IGF2BP3. Additionally, based on the SPOP-substrate binding properties, we screened three SPOP-binding motifs (SBC) on IGF2BP3: SBC1 (124-VTYSS-128), SBC2 (176-QRGSS-180), and SBC3 (250-PEGTS-254) (Fig. 3G). Notably, SBC3 of IGF2BP3 was indispensable for the interaction with SPOP through co-immunoprecipitation (Fig. 3H), GST pull-down (Fig. 3I), and immunofluorescence (Fig. 3J). Given that the effect of SPOP on IGF2BP3 expression was at the mRNA level (Fig. 3K) but not at the protein level (Fig. 3L), IGF2BP3 may be an upstream regulator of SPOP. As shown by qRT-PCR and Western Blot, IGF2BP3 had no effect on the expression of SPOP mRNA (Fig. S6A) and protein (Fig. S6B). Therefore, IGF2BP3 does not influence the transcription and translation of SPOP, but may affect its biological functions in ubiquitination and degradation mediated by SPOP. Co-immunoprecipitation and Western blotting showed that IGF2BP3 not only inhibited SPOP-mediated degradation of c-Myc (Fig. 3M) but also suppressed SPOP-mediated ubiquitination of c-Myc (Fig. 3N). When the expression of IGF2BP3 increased, the binding of SPOP to IGF2BP3 increased, while the binding of SPOP to c-Myc decreased (Fig. 3O, P). In addition, the level of SPOP-mediated ubiquitination of c-Myc was downregulated with an increase in IGF2BP3, whereas SPOP-mediated ubiquitination was restored with an increase in IGF2BP3-ΔSBC3 (Fig. 3Q). Thus, IGF2BP3 competitively binds SPOP to c-Myc, resulting in suppression of SPOP-mediated ubiquitination and degradation of c-Myc.

A Coomassie blue staining of the enrichment of IGF2BP3. B Mass spectrometry of the enrichment of IGF2BP3. C The interaction between IGF2BP3 and SPOP was detected by co-IP. D The co-localization between IGF2BP3 and SPOP was detected by IF. Scale bar, 50 μm. E The co-localization between IGF2BP3 and SPOP-WT, SPOP-ΔMATH, SPOP-ΔBTB, or SPOP-ΔNLS. Scale bar, 50 μm. F The interaction between IGF2BP3 and SPOP-WT, SPOP-ΔMATH, SPOP-ΔBTB, or SPOP-ΔNLS was detected by co-IP. G Potential three SBCs and their deletion mutants: SBC1 (124-VTYSS-128), SBC2 (176-QRGSS-180), and SBC3 (250-PEGTS-254). H The interaction between SPOP and ΔSBC1, ΔSBC2, or ΔSBC3 was detected by co-IP. I The interaction between SPOP and IGF2BP3-WT or IGF2BP3-ΔSBC3 was detected by GST pull-down. J The co-localization between SPOP-WT or SPOP-ΔNLS, and IGF2BP3-WT or IGF2BP3-ΔSBC3 was detected by IF. Scale bar, 50 μm. K The expression of IGF2BP3 mRNA and c-Myc mRNA was affected by SPOP overexpression detected by qRT-PCR. L The effects of SPOP on IGF2BP3 expression treated with DMSO, MG132 (20 μM), bortezomib (200 nM), and chloroquine (100 nM) were detected by WB. M The effect of IGF2BP3 on SPOP-mediated degradation of c-Myc. N The effect of IGF2BP3 on SPOP-mediated ubiquitination of c-Myc. O The effect of IGF2BP3 on the interaction of SPOP and c-Myc was detected by WB. P The effect of IGF2BP3 on the interaction of SPOP and c-Myc was detected by GST-pulldown assay. Q The effects of IGF2BP3-WT and IGF2BP3-ΔSBC3 of on SPOP-mediated ubiquitination of c-Myc. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
IGF2BP3/SPOP/c-Myc forms a positive feedback loop
Considering that SPOP, as the adaptor protein of E3 ligase, mediates ubiquitination and degradation of substrate proteins, we also determined whether SPOP can mediate ubiquitination and degradation of IGF2BP3. The results showed that SPOP-WT, SPOP-ΔMATH, SPOP-ΔBTB, and SPOP-ΔNLS did not mediate the ubiquitination of IGF2BP3 (Fig. 4A, B), but mediated the degradation of IGF2BP3 (Fig. 4C–E). In addition, the half-life of IGF2BP3 did not change with SPOP overexpression (Fig. 4F, G), indicating that SPOP did not affect the half-life of IGF2BP3. After inhibiting the proteasome and lysosome, SPOP still degraded IGF2BP3 (Fig. 3L), suggesting that SPOP may downregulate IGF2BP3 at the transcriptional level, which was confirmed by qRT-PCR (Fig. 3K).

A The effect of SPOP on ubiquitination of IGF2BP3 detected by co-IP. B The effects of SPOP-WT, SPOP-ΔMATH, SPOP-ΔBTB, and SPOP-ΔNLS on ubiquitination of IGF2BP3 detected by co-IP. C The effect of SPOP on degradation of exogenous IGF2BP3. D The effect of SPOP on degradation endogenous IGF2BP3. E The effects of SPOP-WT, SPOP-ΔMATH, SPOP-ΔBTB, and SPOP-ΔNLS on degradation of IGF2BP3. F The effects of SPOP on the half-life of IGF2BP3 detected by WB. G Statistical analysis of the effect of SPOP on the half-life of IGF2BP3. H The expression of IGF2BP3 mRNA and SPOP mRNA affected by c-Myc overexpression detected by qRT-PCR. I The expression of IGF2BP3 mRNA and SPOP mRNA affected by c-Myc knockdown detected by qRT-PCR. J The expression of IGF2BP3 mRNA and SPOP mRNA affected by 10058-F4 detected by qRT-PCR. K Analysis of the effect of c-Myc on IGF2BP3 promoter by ChIP. L Mechanism of IGF2BP3 promoter plasmids with firefly luciferase and human renilla luciferase (hRluc). M Potential two E-box and their deletion mutants: E-box1 (-174CACGTG-169) and ΔE-box2 (-146CACGTG-141). N The transcriptional activity of c-Myc on IGF2BP3 promoter-WT, IGF2BP3 promoter-ΔE-box1, and IGF2BP3 promoter-ΔE-box2, and the effects of SPOP on c-Myc-mediated activities of IGF2BP3 promoter-WT, IGF2BP3 promoter-ΔE-box1, and IGF2BP3 promoter-ΔE-box2. O The interaction between SPOP and c-Myc. P The effect of SPOP on degradation of c-Myc. Q The effect of SPOP on ubiquitination of c-Myc. R The effects of SPOP-WT, SPOP-E47K, SPOP-G75R, and SPOP-ΔMATH on ubiquitination of c-Myc. S The effects of SPOP-WT, SPOP-E47K, SPOP-G75R, and SPOP-ΔMATH on degradation of c-Myc. T The effects of SPOP-WT, SPOP-E47K, SPOP-G75R, and SPOP-ΔMATH on expression of IGF2BP3 mRNA. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
c-Myc is a transcription factor for IGF2BP320. When c-Myc was overexpressed, the expression of IGF2BP3 mRNA increased (Fig. 4H), whereas when c-Myc was knocked down (Fig. 4I) or its transcriptional activity was inhibited by 10058-F421 (Fig. 4J), the expression of IGF2BP3 mRNA decreased. Through CHIP assay, we demonstrated that c-Myc can bind to the IGF2BP3 promoter, which can be weakened by SPOP (Fig. 4K). Then, we constructed a dual-luciferase reporter plasmid carrying the IGF2BP3 promoter for further confirmation using the dual-luciferase reporter assay (Fig. 4L). Considering that c-Myc promotes transcription by binding to the E-box in the promoters22, we identified two potential E-boxes in the IGF2BP3 promoter: E-box1 (-174CACGTC-169) and E-box2 (-146CACGTC-141) (Fig. 4M). Using the dual-luciferase reporter assay, we found that c-Myc binds to E-box1 to promote IGF2BP3 transcription (Fig. 4N). In addition, dual-luciferase reporter assay also confirmed that c-Myc transcription of IGF2BP3 could be reversed by SPOP (Fig. 4N). On the other hand, c-Myc is a substrate protein of SPOP13; that is, SPOP mediates the ubiquitination and degradation of c-Myc (Fig. 4O–Q). Therefore, we inferred that SPOP might downregulate IGF2BP3 mRNA expression by degrading c-Myc. To confirm this hypothesis, three loss-of-function SPOP mutants, namely SPOP-E47K, SPOP-G75R, and SPOP-ΔMATH, were constructed. Using co-immunoprecipitation and western blot assays, we confirmed that these three mutants lost the ability to mediate ubiquitination and degradation of c-Myc (Fig. 4R, S). Furthermore, only SPOP-WT downregulated IGF2BP3 mRNA (Fig. 4T), whereas the other three SPOP mutants upregulated IGF2BP3 mRNA (Fig. 4T), indicating that SPOP downregulates IGF2BP3 mRNA through ubiquitination and degradation of c-Myc.
In summary, IGF2BP3 inhibits SPOP-mediated ubiquitination and degradation of c-Myc, which in turn promotes the c-Myc-mediated transcription of IGF2BP3, thus forming a positive feedback loop consisting of IGF2BP3/SPOP/c-Myc.
IGF2BP3/SPOP/c-Myc loop promotes the proliferation, metastasis, and invasion of EC
First, we used siRNA to knock down IGF2BP3, knock down SPOP, and knock down IGF2BP3 plus knock down SPOP in EC cells (Fig. 5A) and found that knockdown of IGF2BP3 decreased the proliferation of EC cells, which was reversed by knockdown of SPOP (Fig. 5B, C). Similar results were obtained in the cell migration assay (Fig. 5D), cell invasion assay (Fig. 5E), and xenograft tumor model (Fig. 5F–I). Thus, IGF2BP3-mediated proliferation, metastasis, and invasion are SPOP-dependent. Next, we overexpressed IGF2BP3 or IGF2BP3-ΔSBC3, knocked down SPOP, overexpressed IGF2BP3 plus knocked down SPOP, and overexpressed IGF2BP3-ΔSBC3 plus SPOP (Fig. S7A). The results showed that the overexpression of IGF2BP3 or IGF2BP3-ΔSBC3 resulted in enhanced cell proliferation (Fig. S7B, C), migration (Fig. S7D), and invasion (Fig. S7E), and the overexpression of IGF2BP3 was more significant than that of IGF2BP3-ΔSBC3 (Fig. S7B–D). However, the proliferation, metastasis, and invasion of cells overexpressing IGF2BP3 combined with SPOP knockdown were higher than those of cells overexpressing IGF2BP3 (Fig. S7B–E). Since IGF2BP3-ΔSBC3 overexpression combined with SPOP knockdown was not dramatically different from IGF2BP3 overexpression combined with SPOP knockdown (Fig. S7B–E), IGF2BP3-mediated proliferation, metastasis, and invasion may be partially, but not completely, SPOP-dependent.

A IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown detected by WB. B The effects of IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown on proliferation detected by colony formation assay. C The effects of IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown on proliferation detected by CCK8 cell proliferation assay. D The effects of IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown on metastasis. Scale bar, 20 μm. E The effects of IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown on invasion. Scale bar, 20 μm. F Schematic representation of xenograft tumor treated by IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown. G The weight of xenograft tumor treated by IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown. H The volume of xenograft tumor treated by IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown. I The immunohistochemical staining of IGF2BP3, SPOP, and Ki-67 of IGF2BP3 knockdown, SPOP knockdown, and IGF2BP3 knockdown plus SPOP knockdown. Scale bar, 20 μm. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In addition, when IGF2BP3 was overexpressed, the viability of cells treated with paclitaxel increased, whereas when IGF2BP3-ΔSBC3 was overexpressed, there was no marked change (Fig. S7F). When IGF2BP3 or IGF2BP3-ΔSBC3 was overexpressed in IGF2BP3 knockout cells, cell viability showed the same trend (Fig. S7G). Therefore, IGF2BP3-induced paclitaxel resistance is also SPOP-dependent.
Furthermore, we knocked down SPOP, and knocked down SPOP plus knocked down c-Myc in Ishikawa and HEC-1-A cells, respectively (Fig. S8A). Compared with SPOP knockdown, the proliferation (Fig. S8B, C), migration (Fig. S8D), and invasion (Fig. S8E) of Ishikawa and HEC-1-A cells were significantly decreased when SPOP knockdown was combined with c-Myc knockdown, indicating that c-Myc could reverse the inhibitory effect of SPOP on EC cells. In other words, the function of SPOP is c-Myc-dependent. Third, we overexpressed IGF2BP3, overexpressed IGF2BP3 plus c-Myc knockdown, and overexpressed IGF2BP3 plus treatment with 10058-F4 in EC cells (Fig. S9A). Overexpression of IGF2BP3 enhanced the proliferation (Fig. S9B, C), metastasis (Fig. S9D), and invasion (Fig. S9E) of EC cells, which were reversed by c-Myc knockdown or c-Myc transcriptional inhibitor 10058-F4 (Fig. S9B–E), indicating that the oncogenic role of IGF2BP3 was c-Myc dependent. Moreover, c-Myc was overexpressed in EC cells, leading to increased proliferation, metastasis, and invasion, which were also reversed by IGF2BP3 knockdown (Fig. S9B–E), suggesting that the oncogenic role of c-Myc was partly dependent on IGF2BP3. Thus, the oncogenic effects of IGF2BP3 and c-Myc are interdependent and form a loop.
Moreover, given that SPOP exerts its tumor-suppressive effect by promoting the degradation of c-Myc by binding to the SBC of c-Myc, we constructed a c-Myc mutant with deleted SBC (Fig. 6A), whose function was not affected by SPOP. Through WB and co-immunoprecipitation, we confirmed that SPOP did not mediate the ubiquitination and degradation of c-Myc-ΔSBC (Fig. 6B–D). Then, we overexpressed SPOP plus c-Myc-ΔSBC and overexpressed SPOP plus overexpressed c-Myc-ΔSBC plus knockout IGF2BP3 in Ishikawa and HEC-1-A cells (Fig. 6E). The proliferation (Fig. 6F, G), metastasis (Fig. 6H), invasion (Fig. 6I), and xenograft tumor model (Fig. 6J–N) of the SPOP overexpression plus c-Myc-ΔSBC overexpression group could all be reversed by IGF2BP3 knockout, indicating that SPOP-mediated degradation of c-Myc is dependent on IGF2BP3.

A Two SBCs in c-Myc and the deletion mutant: 185-VCSTS-189 plus 261-PTTSS-265. B The effect of SPOP on degradation of c-Myc-ΔSBC. C The interaction between SPOP and c-Myc-WT or c-Myc-ΔSBC. D The effect of SPOP on ubiquitination of c-Myc-WT and c-Myc-ΔSBC. E SPOP overexpression plus c-Myc-ΔSBC overexpression and SPOP overexpression plus c-Myc-ΔSBC overexpression plus IGF2BP3 knockout detected by WB. F, G The effects of SPOP overexpression plus c-Myc-ΔSBC overexpression and SPOP overexpression plus c-Myc-ΔSBC overexpression plus IGF2BP3 knockout on proliferation. H The effects of SPOP overexpression plus c-Myc-ΔSBC overexpression and SPOP overexpression plus c-Myc-ΔSBC overexpression plus IGF2BP3 knockout on metastasis. Scale bar, 20 μm. I The effects of SPOP overexpression plus c-Myc-ΔSBC overexpression, and SPOP overexpression plus c-Myc-ΔSBC overexpression plus IGF2BP3 knockout on invasion. Scale bar, 20 μm. J–N Xenograft tumor model of SPOP overexpression plus c-Myc-ΔSBC overexpression and SPOP overexpression plus c-Myc-ΔSBC overexpression plus IGF2BP3 knockout. Scale bar, 20 μm. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In summary, IGF2BP3/SPOP/c-Myc forms a positive feedback loop to promote the proliferation, metastasis, and invasion of EC, thereby ensuring that IGF2BP3 exerts its function through c-Myc in EC.
Paclitaxel combined with 10058-F4 improves the efficacy in EC models
To explore the clinical value of the IGF2BP3/SPOP/c-Myc loop in the treatment of EC, we overexpressed IGF2BP3 in EC cells to mimic paclitaxel-resistant EC patients with elevated IGF2BP3 levels. The EC cells were then treated with paclitaxel, paclitaxel plus c-Myc knockdown, and paclitaxel plus 10058-F4. Compared with paclitaxel alone, the weight and volume of tumors were smaller than those of paclitaxel plus c-Myc knockdown and paclitaxel plus 10058-F4, whereas there was no significant difference between paclitaxel plus c-Myc knockdown and paclitaxel plus 10058-F4 (Fig. 7A–D), indicating that the knockdown or transcriptional inhibition of c-Myc could enhance paclitaxel sensitivity. Thus, the combination of 10058-F4 and paclitaxel is expected to improve the efficacy of paclitaxel in patients with paclitaxel-resistant EC. Venn analysis of c-Myc-targeted hallmark genes in GSEA, upregulated genes in RNA-Seq of paclitaxel-resistant Ishikawa cells (Fig. 1B), downregulated genes in RNA-Seq of IGF2BP3 knockdown Ishikawa cells (Fig. S5B), and UCEC in TCGA database showed that CCNA2, CCT2, BUB3, PSMD1, and NCBP1 may play a role in IGF2BP3/SPOP/c-Myc loop-induced paclitaxel resistance (Fig. 7E). Considering that only CCNA2 and CCT2 have been confirmed as target genes of c-Myc-mediated transcription in previous studies23,24, we selected CCNA2 and CCT2 as the downstream indicators of IGF2BP3/SPOP/c-Myc loop-mediated paclitaxel resistance. Immunohistochemistry showed that when IGF2BP3 was overexpressed, the expression of CCNA2 and CCT2 was elevated, which could be reversed by c-Myc knockdown or 10058-F4 treatment, suggesting that CCNA2 and CCT2 are downstream indicators of IGF2BP3/SPOP/c-Myc loop-mediated paclitaxel resistance (Fig. 7F).

A Schematic representation of xenograft tumor of IGF2BP3 overexpression, IGF2BP3 overexpression treated by paclitaxel, IGF2BP3 overexpression plus c-Myc knockdown treated by paclitaxel, and IGF2BP3 overexpression treated by paclitaxel and 10058-F4. B The weight of xenograft tumor of IGF2BP3 overexpression, IGF2BP3 overexpression treated by paclitaxel, IGF2BP3 overexpression plus c-Myc knockdown treated by paclitaxel, and IGF2BP3 overexpression treated by paclitaxel and 10058-F4. C The volume of xenograft tumor of IGF2BP3 overexpression, IGF2BP3 overexpression treated by paclitaxel, IGF2BP3 overexpression plus c-Myc knockdown treated by paclitaxel, and IGF2BP3 overexpression treated by paclitaxel and 10058-F4. D The immunohistochemical staining of IGF2BP3, c-Myc, and Ki-67 of IGF2BP3 overexpression, IGF2BP3 overexpression treated by paclitaxel, IGF2BP3 overexpression plus c-Myc knockdown treated by paclitaxel and IGF2BP3 overexpression treated by paclitaxel and 10058-F4. Scale bar, 20 μm. E Venn analysis based on c-Myc-targeted genes in GSEA, up-regulated genes in RNA-seq of paclitaxel-resistant Ishikawa cells, and down-regulated genes in RNA-seq of IGF2BP3 knockdown Ishikawa cells. F The immunohistochemical staining of CCNA2 and CCT2 of of IGF2BP3 overexpression, IGF2BP3 overexpression treated by paclitaxel, IGF2BP3 overexpression plus c-Myc knockdown treated by paclitaxel and IGF2BP3 overexpression treated by paclitaxel and 10058-F4. Scale bar, 20 μm. G Molecular mechanism of IGF2BP3/SPOP/c-Myc loop mediated paclitaxel resistance (Created in BioRender. Zhang, H. (2025) https://BioRender.com/bjcmb9j, Agreement number: DY282PE8YM). Scale bar, 20 μm. Data are shown as mean ± SD (n ≥ 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
In conclusion, under physiological conditions, SPOP promoted ubiquitination and degradation of c-Myc, thereby inhibiting the development of EC and ensuring the efficacy of paclitaxel for EC (Fig. 7E). When paclitaxel resistance occurs, the expression of IGF2BP3 is upregulated, resulting in the accumulation of c-Myc through the IGF2BP3/SPOP/c-Myc loop, which leads to the development of paclitaxel resistance in patients with EC (Fig. 7E). Using this underlying mechanism, 10058-F4 can improve the efficacy of paclitaxel in EC patients by blocking the transcriptional activity of c-Myc, thereby inhibiting the IGF2BP3/SPOP/c-Myc loop (Fig. 7E).
In this study, we found that IGF2BP3 interacts with SPOP, but the interaction site of SPOP is not in the conventional SPOP domains (MATH, BTB, and NLS), which is very similar to that of G3BP1, another SPOP upstream repressor25. Interestingly, the interaction between c-Myc and SPOP occurs in the MATH domain, whereas IGF2BP3 inhibits SPOP through competitive inhibition13. Therefore, we hypothesized that IGF2BP3 is more likely to bind to the BACK domain of SPOP, which is primarily responsible for SPOP oligomerization11, thereby affecting SPOP oligomerization and resulting in the reduced binding of c-Myc to SPOP, thus exhibiting the illusion of competitive inhibition. To verify this hypothesis, we performed a molecular docking analysis of SPOP and IGF2BP3 and found that E251 in the SBC of IGF2BP3 may bind to H345 and H347 in the BACK domain of SPOP (Fig. S10A). However, co-immunoprecipitation indicated that IGF2BP3 could interact with BACK domain-missing SPOP, and immunofluorescence indicated that IGF2BP3 co-localized with BACK domain-missing SPOP, indicating that the interaction sites of IGF2BP3 and SPOP were not in the BACK domain but in the region outside the traditional domains (Fig. S9B, C). In addition, SPOP binds to SBC3 (250-PETTS-254) on IGF2BP3, which is located in the hnRNP-K homology domain of IGF2BP3 and is responsible for m6A read and further translation26. However, our results confirmed that IGF2BP3 did not affect SPOP mRNA expression. Therefore, whether there is an association between IGF2BP3 and SPOP mRNA, and the specific relationship between them, needs to be further explored.
IGF2BP3 is the only member of the IGF2BP family, along with two other members, IGF2BP1 and IGF2BP227. Moreover, similar SBC sequences were present in the amino acid sequences of IGF2BP1 and IGF2BP2, which were 251-PEGCS-254 and 248-PEGTS-252, respectively. Given that IGF2BP1 and IGF2BP2 are m6A readers of c-Myc mRNA, there is a significant positive correlation between IGF2BP1, IGF2BP2, and c-Myc28,29. In addition, RNA-seq analysis showed that IGF2BP1 and IGF2BP2 levels were both dramatically increased in paclitaxel-resistant Ishikawa cells (Fig. 1B). Therefore, it is possible that both IGF2BP1 and IGF2BP2 can further increase c-Myc expression by inhibiting SPOP-mediated c-Myc degradation, thereby jointly promoting paclitaxel resistance, which needs to be confirmed in further studies.
Paclitaxel is a widely used chemotherapeutic drug, which is not only used in endometrial cancer but also in ovarian, cervical, esophageal, and breast cancers30. Given that our current study was limited to EC, future studies could focus on whether IGF2BP3 has a similar role in other cancers, and if so, the improved efficacy of paclitaxel revealed in this study could be more generalizable. In addition, there are many other paclitaxel analogs, including paclitaxel liposomes, albumin-bound paclitaxel, and docetaxel7. Their mechanisms are similar, all of which are M-phase cyclin-specific drugs that can promote the polymerization of microtubules into stable microtubules and inhibit their depolymerization, thereby significantly reducing the number of tubules and destroying the microtubule network structure31. Therefore, it is worth exploring whether the IGF2BP3/SPOP/c-Myc loop has the same role as these paclitaxel analogs. The strategy to improve the sensitivity of paclitaxel in this study was to combine 10058-F4 with paclitaxel, but the research stage of 10058-F4 is only in the preclinical stage, so there is still a long way to go before clinical application32. IDP-121 and OTX-2002 are the latest drugs targeting c-Myc, but they are only in phase I/II clinical research, and their targets are not only in cancer33,34. Thus, IDP-121 and OTX-2002 may be clinically relevant in combination with paclitaxel.
Methods
Cell culture
Ishikawa cells were purchased from Wuhan Pricella Biotechnology Co., Ltd with the catalog number CL-0283. HEC-1-A (catalog number HTB-112), HEK-293T (catalog number CRL-3216), and HeLa (catalog number CCL-2) cells were purchased from American Type Culture Collection. All cells were cultured in DMEM (Zhongqiaoxinzhou, ZQ-100, China) with 10% FBS and (Standard Quality, FBSST-01033, China) and 1% penicillin-streptomycin solution (MeilunBio, MA0110, China) at 37 °C and 5% CO2.
Construction and transfection of siRNA, shRNA, and plasmid
siRNA was constructed by Tsingke Biotechnology (Hangzhou, China). The shRNA and wild-type plasmids were constructed using Miaolin Biology (Wuhan, China). The mutant plasmids were constructed using the ClonExpress Ultra One Step Cloning Kit V2 (Vazyme, C116, Nanjing, China) and KOD-Plus Mutagenesis Kit (TOYOBO, SMK-101, Tokyo, Japan). The siRNA, shRNA, and plasmids were transfected according to Lipo6000™ (Beyotime, Shanghai, China). The oligonucleotide sequences of siRNA and shRNA and the primers for the mutant plasmids are shown in Table S1.
Antibodies and reagents
The following antibodies were used: IGF2BP3 (1:2000 for WB, 1:100 for IHC, Proteintech, 14642-1-AP, Wuhan, China), SPOP (1:5000 for WB, Proteintech, 16750-1-AP, Wuhan, China), c-Myc (1:2000 for WB, 1:50 for IHC, Proteintech, 10828-1-AP, Wuhan, China), GAPDH (Proteintech, 60004-1-Ig, Wuhan, China), Ki-67 (1:500 for IHC, Proteintech, 27309-1-AP, Wuhan, China), CCNA2 (1:50 for IHC, Abclonal, A19036, Wuhan, China), CCT2 (1:50 for IHC, Abclonal, A4700, Wuhan, China), Goat Anti-Rabbit (1:200 for IF, Proteintech, SA00013-4, Wuhan, China), FLAG (1:10000 for WB, MBL, M185-7, Tokyo, Japan), Myc (1:5000 for WB, MBL, M192-7, Tokyo, Japan), HA (1:10000 for WB, MBL, M180-7, Tokyo, Japan), Anti-Rabbit (1:5000 for WB, 1:500 for IHC, Abclonal, AS038, Wuhan, China), anti-mouse (1:50000 for WB, Proteintech, SA00001-1, Wuhan, China). The following reagents were used: anti-FLAG M2 agarose beads (Smart-Lifesciences, SA042100, Changzhou, CHINA), MG-132 (Selleckchem, S2619, Houston, USA), bortezomib (Selleckchem, S1013, Houston, USA), chloroquine (Selleckchem, S6999, Houston, TX, USA), CHX (MCE, HY-B0713, Shanghai, China), paclitaxel (Meilunbio, MB1178, Liaoning, China), and 10058-F4 (MCE, HY-12702, Shanghai, China).
Co-immunoprecipitation (co-IP)
After transfection for 48 h, the cells were treated with MG132 for 6 h and then lysed in Weak RIPA lysis buffer (Meilunbio, MA0153, China). Next, anti-Flag M2 agarose beads were added and incubated for 6 h at 4 °C. The beads were then washed several times with Weak RIPA lysis buffer. The bound proteins were added to the SDS-PAGE solution to boil for western blot. All co-IP assays were conducted in at least three independent assays.
Western blot (WB)
Proteins were transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK) using cell lysates and co-immunoprecipitated after SDS-PAGE. After the membrane was blocked in 5% skim milk for 2 h, the primary antibody was incubated at 4 °C overnight, and then the secondary antibody was incubated at 4 °C for 2 h. The target proteins were visualized using an ECL chemiluminescence system (Kermey, M0101, China). All western blots were conducted in at least three independent assays and are presented as representative images. The original WB images are presented in Fig. S11.
Real-time quantitative polymerase chain reaction (qRT-PCR)
First, total RNA was extracted using the RNA isolator Total RNA Extraction Reagent (Vazyme, R401-01, Nanjing, China) according to the manufacturer’s instructions. cDNA synthesis was performed according to the instructions of HiScript® III All-in-one RT SuperMix (Vazyme, R333, Nanjing, China). Finally, PCR amplification was performed using ChamQ SYBR qPCR Master Mix (R331; Vazyme, Nanjing, China) according to the manufacturer’s instructions. All quantifications were normalized to GAPDH. All qRT-PCR assays were conducted in at least four independent assays. Primers used for qRT-PCR are listed in Table S1.
GST pull-down assay
After fixing GST and GST-SPOP on BeyoGold™ GST-tag Purification Resin (Beyotime, P2262, China), they were cleaned 3 times with pull-down buffer (Beyotime, P2262, China). The beads were incubated with the protein lysate overnight and washed three times with binding buffer (Beyotime, P2262, China). The binding protein was separated by SDS-PAGE and boiled for western blotting. All GST pull-down assays were conducted in at least three independent assays.
Immunohistochemistry (IHC)
Paraffin sections were repaired with ethylenediaminetetraacetic acid (EDTA) antigen repair buffer (Beyotime, P0085, China), inactivated endogenous peroxidase with 3% hydrogen peroxide, blocked with 10% donkey serum (Biosharp, BL1051A, China), and incubated with primary antibody at 4 °C overnight. After incubation with the secondary antibody (Goat Anti-Rabbit antibody) for 1 h, the paraffin sections were stained with DAB assay kit (Solarbio, G1212, Beijing, China), and stained with hematoxylin (Solarbio, G4070, Beijing, China). Immunohistochemical scores were scored in two ways: one was evaluated by pathologists, and the other was analyzed and evaluated using ImageJ. The IHC assays of patient samples were approved by the Medical Ethics Committee of Ningbo University (Certificate No. NBU-2024-315). The study has received informed consent from these patients, and all ethical regulations relevant to human research participants were followed. The original IHC images are presented in Fig. S12, and the clinical information of patients is presented in Supplementary Data 4.
Immunofluorescence (IF)
HeLa cells were seeded onto chamber slides and fixed using a 4% paraformaldehyde solution under ambient conditions for 30 min. Following three PBS washes, cellular membranes were subjected to permeabilization with 0.1% Triton X-100 dissolved in PBS for 15 min. Subsequently, the cells underwent sequential treatments: 1. Blocking non-specific binding sites with PBS containing 0.5% BSA for 1 h; 2. Primary antibody incubation in PBS at 4°C overnight. After removing unbound antibodies through PBS rinses, the samples were exposed to fluorophore-conjugated secondary antibodies and counterstained with DAPI at room temperature for 60 min to visualize nuclei. The following antibodies were used for IF: FLAG (1:100, Proteintech, 20543-1-AP/66008-4-Ig, Wuhan, China), Myc (1:100, Proteintech, 60003-2-Ig/80845-1-RR, Wuhan, China), HA (1:100, Proteintech, 51064-2-AP/81290-1-RR, Wuhan, China), anti-rabbit (1:200, Abclonal, AS038, Wuhan, China), and anti-mouse (1:200, Abclonal, AS033, Wuhan, China). Nuclei were stained with DAPI (Solarbio, C0065, Beijing, China). Image acquisition was performed under a confocal laser scanning microscope (LEICA TCS SP8). All IF assays were conducted in at least three independent assays.
Chromatin immunoprecipitation (ChIP) assay
For the ChIP assay, we strictly followed the protocol provided with the BeyoChIP™ Enzymatic ChIP Assay Kit (Beyotime, P2083S, China). Briefly, cells cultured in 10 cm dishes were cross-linked with 1% formaldehyde, and the chromatin was fragmented by ultrasonic treatment. The chromatin was then incubated with FLAG antibody (Proteintech, 66008-4-Ig, China) overnight at 4 °C, with Mouse IgG (Proteintech, B900620, China) serving as a negative control. Protein A/G magnetic beads (Beyotime, P2083S, China) were added and incubated at 4 °C for 2 h. The DNA-bead complexes were washed sequentially with low salt immune complex wash buffer (Beyotime, P2083S, China), high salt immune complex wash buffer (Beyotime, P2083S, China), LiCl immune complex wash buffer (Beyotime, P2083S, China), and TE buffer (Beyotime, P2083S, China). The DNA was eluted using Elution Buffer (Beyotime, P2083S, China), followed by treatment with 5 M NaCl, 0.5 M EDTA + 1 M Tris (pH 6.5) + 20 mg/ml proteinase K to reverse the cross-links. The DNA was purified using a DNA purification kit (Beyotime, D0033, China) and subjected to PCR and real-time PCR to evaluate the enrichment level of IGF2BP3 promoter DNA in the samples. All CHIP assays were conducted in at least four independent assays.
RNA Immunoprecipitation (RIP) Assay
After washing the treated cells with PBS, digest the cells with trypsin (Beyotime, C0201, China) and count the cells to obtain 1.5 million cells. Add 300 μL of Lysis Buffer (Beyotime, P1801S, China) (containing 1 mM DTT, 100 U/ml RNase Inhibitor, and 0.2 mM PMSF). After incubating on ice for 15 min, centrifuge at 14,000 × g for 10 min at 4 °C. Take 30 μL of the supernatant as Input. Incubate IgG (Proteintech, B900620, China) and IGF2BP3 (Proteintech, 14642-1-AP, Wuhan, China) antibodies with Protein A/G Agarose (Beyotime, P1801S, China) according to the instructions. Add the remaining lysate to the Protein A/G Agarose and incubate overnight, then wash four times with NT2 Wash Buffer (Beyotime, P1801S, China). Add 100 μL of Elution Buffer (Beyotime, P1801S, China) and incubate at 55 °C for 30 min. Purify the RNA using the RNAeasy™ Animal RNA Extraction Kit (Spin Column) (Beyotime, R2077, China) according to the instructions and perform qPCR detection. All RIP assays were conducted in at least four independent assays.
Dual-luciferase reporter assays
Ishikawa and HEC-1-A cells were seeded in 96-well plates at 10,000 cells/well. The IGF2BP3-WT, IGF2BP3-ΔE-box1, and IGF2BP3-ΔE-box2 reporter plasmids were co-transfected with c-Myc or control. After 48 h, relative luciferase activity was measured using the Duo-Lite Luciferase Assay System (Vazyme, DD1205, Nanjing, China) according to the manufacturer’s protocol. All dual-luciferase reporter assays were conducted in at least three independent assays.
Cell counting kit-8 (CCK8) assay
Treated Ishikawa and HEC-1-A cells were seeded in 96-well plates at a density of 1500 cells/well. Within 0–5 days, 10 μl of Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan) was added to each well at the same time every day. After incubation for 2 h, OD values were measured at 450 nm using a microplate absorbance instrument (Bio-Rad, US). Each assay was repeated thrice.
Colony formation assay
Treated Ishikawa and HEC-1-A cells were inoculated into a 6-well plate at a density of 1500 cells/well. After ~2 weeks of culture, the cells were fixed in 4% paraformaldehyde for 30 min and then stained with crystal violet solution (Solarbio, G4070, Beijing, China) for 10 min. After capturing cell colony images with smartphones, ImageJ software was used for the analysis. Each assay was repeated thrice.
Cell migration and invasion assays
For the migration assay, 30,000 treated Ishikawa and HEC-1-A cells were inoculated with 200 μl medium without FBS in the upper chamber of Transwell chambers (8.0 μm, 3342, Corning, USA), and 500 μl medium containing 10% FBS was added to the lower chambers. After 48 h, the cells were fixed in 4% paraformaldehyde for 30 min and stained with crystal violet solution (Solarbio, G4070, Beijing, China) for 10 min. Finally, the upper chamber was cleaned, and the number of cells on the lower surface of the chamber was counted. Each assay was repeated thrice. For the invasion assay, migration inserts (Costar) coated with matrix gel/fibronectin (BD Biosciences, USA) were placed in the upper chamber of the Transwell chamber prior to cell inoculation, and the subsequent steps were the same as those for the migration assay. Each experiment was repeated four times.
Stable cell line establishment
The psPAX2, pMD2.G, and target plasmid/shRNA were transfected into HEK293T cells in a 4:3:1 ratio, and the cell supernatant was collected and filtered through a 0.45 μm Sterile Syringe Filter (Biosharp, BS-PVDF33-45-S, China). EC cells were infected with the filtered lentivirus suspension, and stable cell lines were screened using 10 μg/ml puromycin dihydrochloride (Beyotime, ST551, Shanghai, China).
Paclitaxel resistant cell lines establishment
After treating EC cells with 0 nM, 2.5 nM, 5 nM, 10 nM, 15 nM, 20 nM, 30 nM, 40 nM, 60 nM, 80 nM paclitaxel for 48 h, the viability of EC cells was detected by CCK8 assay, and the IC50 of paclitaxel was calculated to be 7 nM. Ishikawa cells were treated with 1 nM paclitaxel for 48 h, cleaned twice with PBS, replaced with paclitaxel-free medium, and allowed to return to normal shape and normal growth speed. Then, the concentration of paclitaxel was successively increased to 3 nM, 7 nM, 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, and 70 nM, and the above steps were repeated until the cells returned to their normal shape and normal growth speed, and the establishment of paclitaxel-resistant cell lines was completed.
Xenograft tumor models
NOD-SCID nude mice aged 4–6 weeks (strain no. T001492) (weighing 15–25 g) were obtained from Jiangsu GemPharmatech Co. Ltd (Nanjing, China) for xenograft tumor models. The treated stable cell lines were transplanted subcutaneously into the right side of mice. The tumor volume was measured every 4 days, and when the tumor volume reached 50 mm3, the abdominal injection of paclitaxel or intravenous injection of 10058-F4 was administered every 2 days for five consecutive injections. When the tumors had grown to a suitable size, the mice were euthanized, and the tumors were imaged and weighed. All animal experiments were conducted in accordance with guidelines approved by the Animal Protection Committee of Ningbo University, and we have complied with all relevant ethical regulations for animal use. All assays used at least four nude mice. Tumor volume = (length × width2) × 1/2.
Bioinformatic analysis
The expression of mRNA (FPKM) and clinicopathological features were derived from GSE228106 and GSE50831 in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and UCEC in the TCGA database (https://portal.gdc.cancer.gov/). Protein expression and clinicopathological features were derived from the UCEC in the CPTAC database (https://cptac-data-portal.georgetown.edu/). FPKM is converted to TPM [Log2 (FPKM + 1)]. The “BiocManager,” “heatmap,” “limma,” “ggplot2,” “survival,” and “survminer” packages in R software (version 4.2.1) were used for analysis.
Statistics and reproducibility
Statistical data were analyzed using Student’s t-test and expressed as the mean value ± standard deviation of at least triplicate measurements in three separate assays. The calculations were performed using GraphPad Prism version 8. The images were analyzed and quantified using Image J. The numerical source data is presented in Supplementary Data 5. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq datasets discussed in this publication are accessible through GEO series accession number GSE228106 and GSE50831 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). The mRNA expression datasets discussed in this publication are accessible through TCGA (https://portal.gdc.cancer.gov/) series accession UCEC. The protein expression datasets discussed in this publication are accessible through CPTAC series accession UCEC. All other source data and the uncropped western blots are available from the corresponding author Xiaofeng Jin.
Change history
04 July 2025
A Correction to this paper has been published: https://doi.org/10.1038/s42003-025-08406-z
References
Makker, V. et al. Endometrial cancer. Nat. Rev. Dis. Prim. 7, 88 (2021).
Banz-Jansen, C., Helweg, L. P. & Kaltschmidt, B. Endometrial cancer stem cells: where do we stand and where should we go? Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23063412 (2022).
Colombo, N. et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 27, 16–41 (2016).
van den Heerik, A., Horeweg, N., de Boer, S. M., Bosse, T. & Creutzberg, C. L. Adjuvant therapy for endometrial cancer in the era of molecular classification: radiotherapy, chemoradiation and novel targets for therapy. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 31, 594–604 (2021).
Abu-Rustum, N. et al. Uterine neoplasms, version 1.2023, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. JNCCN 21, 181–209 (2023).
Mekhail, T. M. & Markman, M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 3, 755–766 (2002).
Hashemi, M. et al. Paclitaxel and docetaxel resistance in prostate cancer: molecular mechanisms and possible therapeutic strategies. Biomed. Pharmacother. Biomed. Pharmacother. 160, 114392 (2023).
Cerquetti, L. et al. C-MYC modulation induces responsiveness to paclitaxel in adrenocortical cancer cell lines. Int. J. Oncol. 46, 2231–2240 (2015).
Supino, R., Favini, E., Cuccuru, G., Zunino, F. & Scovassi, A. I. in Signal Transduction Pathways, Pt C: Cell Signaling in Health and Disease Vol. 1095 Annals of the New York Academy of Sciences (ed M. Diederich) 175–181 (2007).
Lei, X. et al. HMGB1 release promotes paclitaxel resistance in castration-resistant prostate cancer cells via activating c-Myc expression. Cell. Signal. 72, https://doi.org/10.1016/j.cellsig.2020.109631 (2020).
Zhang, H., Jin, X. & Huang, H. Deregulation of SPOP in Cancer. Cancer Res 83, 489–499 (2023).
Shi, Q. et al. Prostate Cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol. Cancer 18, https://doi.org/10.1186/s12943-019-1096-x (2019).
Geng, C. et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 36, 4767–4777 (2017).
Ge, Y. et al. Endometrial cancer (EC) derived G3BP1 overexpression and mutant promote EC tumorigenesis and metastasis via SPOP/ERα axis. Cell Commun. Signal. 21, https://doi.org/10.1186/s12964-023-01342-7 (2023).
Wang, P.-F. et al. The oncogenic functions of insulin-like growth factor 2 mRNA-binding protein 3 in human carcinomas. Curr. Pharmacol. Des. 26, 3939–3954 (2020).
Stangl, A. et al. Molecular features and race-associated outcomes of SPOP-mutant metastatic castration-resistant prostate cancer. Prostate 83, 524–533 (2023).
Li, M. et al. An isocorydine derivative (d-ICD) inhibits drug resistance by downregulating IGF2BP3 expression in hepatocellular carcinoma. Oncotarget 6, 25149–25160 (2015).
Zhou, S., Sheng, L., Zhang, L., Zhang, J. & Wang, L. METTL3/IGF2BP3-regulated m6A modification of HYOU1 confers doxorubicin resistance in breast cancer. Biochim. Et. Biophys. Acta Gen. Subj. 1868, 130542 (2024).
Chen, L. J. et al. IGF2BP3 promotes the progression of colorectal cancer and mediates cetuximab resistance by stabilizing EGFR mRNA in an m(6)A-dependent manner. Cell Death Dis. 14, 581 (2023).
Du, M. et al. MYC-activated RNA N6-methyladenosine reader IGF2BP3 promotes cell proliferation and metastasis in nasopharyngeal carcinoma. Cell Death Discov. 8, 53 (2022).
Tan, A. W. et al. c-Myc Drives inflammation of the maternal-fetal interface, and neonatal lung remodeling induced by intra-amniotic inflammation. Front. Cell Dev. Biol. 11, 1245747 (2023).
Madden, S. K., de Araujo, A. D., Gerhardt, M., Fairlie, D. P. & Mason, J. M. Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 20, https://doi.org/10.1186/s12943-020-01291-6 (2021).
Wang, Z. et al. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell 33, 706–720.e709 (2018).
Ghozlan, H., Showalter, A., Lee, E., Zhu, X. & Khaled, A. R. Chaperonin-containing TCP1 complex (CCT) promotes breast cancer growth through correlations with key cell cycle regulators. Front. Oncol. 11, https://doi.org/10.3389/fonc.2021.663877 (2021).
Mukhopadhyay, C. et al. G3BP1 inhibits Cul3(SPOP) to amplify AR signaling and promote prostate cancer. Nat. Commun. 12, 6662 (2021).
Liu, X. et al. Targeting IGF2BP3 in Cancer. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24119423 (2023).
Bell, J. L. et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression?. Cell. Mol. Life Sci. CMLS 70, 2657–2675 (2013).
Zhu, Q. et al. MNX1-AS1 promotes phase separation of IGF2BP1 to drive c-Myc-mediated cell-cycle progression and proliferation in lung cancer. Cancer Res. 82, 4340–4358 (2022).
Hu, C. et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int. J. Biol. Sci. 18, 507–521 (2022).
Ying, N. et al. Nano delivery system for paclitaxel: recent advances in cancer theranostics. Colloids Surf. B Biointerfaces 228, 113419 (2023).
Aapro, M. The scientific rationale for developing taxoids. Anti Cancer Drugs 7, 33–36 (1996).
Zhang, M., Fan, H. Y. & Li, S. C. Inhibition of c-Myc by 10058-F4 induces growth arrest and chemosensitivity in pancreatic ductal adenocarcinoma. Biomed. Pharmacother. Biomed. Pharmacother. 73, 123–128 (2015).
Calvis, C. et al. IDP-121, a first-in-class staple peptide targeting c-MYC. Cancer Res. 81, 3 (2021).
Rodriguez-Rivera, I. I. et al. A phase 1/2 open-label study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of OTX-2002 as a single agent and in combination with standard of care in patients with hepatocellular carcinoma and other solid tumor types known for association with the MYC oncogene (MYCHELANGELO I). J. Clin. Oncol. 41, TPS627–TPS627 (2023).
Acknowledgements
Thanks for the technical support of confocal laser scanning microscope (LEICA TCS SP8) by the Core Facilities (Disen Mei, meidisen@nbu.edu.cn; Danhong Wan, wandanhong@nbu.edu.cn), Health Science Center, Ningbo University. We thank Mengjie Yang (yangmengjie@nbu.edu.cn) and Chao Dong (dongchao@nbu.edu.cn) from the Laboratory Animal Center of Ningbo University for their technical support. We thanked Shumin Ni of The First Affiliated Hospital of Ningbo University for technical assistance. This work was supported by the National Natural Science Foundation of Ningbo (No.2024J037), the Youth Science and technology innovation leader of Ningbo (No.2023QL052), the National Natural Science Foundation of Zhejiang (No.LY24C050001, No.LTGY24H16004), the National Natural Science Foundation of China (No.32270821), the K.C. Wong Magna Fund in Ningbo University, Student Research and Innovation Program (2024SRIP2502, 2024SRIP1917 and 2024SRIP1925), Ningbo University 2025 graduate research innovation project (IF2025058, IF2025055) and Natural Science Fund of Ningbo (2022J219).
Author information
Authors and Affiliations
Contributions
Investigation: Y.G., L.K., F.Z., Z.Z. G.C. and S.W. Analysis: H.W., Y.W., and J.Z. Bioinformatics: L.H., J.M., S.W. and J.X. Design and Supervision: P.L., M.Y. and X.J. Funding acquisition: X.J., M.Y., M.Z., Y.G., L.H., S.W. and J.S. Manuscript Preparation (original draft and review): Y.G., L.K., Y.L., Z.Y. and X.J. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All animal assays were approved by the Animal Ethics and Welfare Committee (AEWC) of Ningbo University (Certificate No. AEWC-NBU20210057). The IHC assays of patient samples were approved by the Medical Ethics Committee of Ningbo University (Certificate No. NBU-2024-315). The study has received informed consent from these patients and all ethical regulations relevant to human research participants were followed.
Consent for publication
All authors have consented to submit this article for publication.
Peer review
Peer review information
Communications Biology thanks Xiangbing Meng and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Myron Evans II and Kaliya Georgieva. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, Y., Kong, L., Li, Y. et al. The role of IGF2BP3/SPOP/c-Myc loop in paclitaxel resistance of endometrial cancer. Commun Biol 8, 624 (2025). https://doi.org/10.1038/s42003-025-08065-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08065-0
