
Abstract
Immunoglobulin A nephropathy (IgA-N) is a primary glomerulonephritis that is characterized by mesangial cell proliferation and expansion. Although glomerular endothelial cells (GE) are implicated in the pathogenesis of IgA-N, their role remains poorly understood. We conduct single-cell and spatial transcriptomic analysis using human specimens with mild IgA-N compared with normal. We integrate single-cell and spatial transcriptome analyses in human samples of mild IgA-N compared to normal samples, revealing several novel findings: (1) identification of clusters of GE and their expressed gene profiles, (2) identification of novel inflammation-related molecules newly implicated in GE in mild IgA-N, (3) activation of inflammatory pathways characteristic of IgA-N. Therefore, we suggest the importance of GE in the mechanism of IgA-N, as GE initiates more active inflammatory response prior to mesangial cells in patients with mild IgA-N. These findings provide useful information for understanding the pathogenesis and choosing the best drug for IgA nephropathy.

Similar content being viewed by others


Introduction
Immunoglobulin A nephropathy (IgA-N) defined as mesangial proliferative glomerulonephritis with deposition of IgA antibodies in the renal glomerular mesangial area—is the most common primary glomerulonephritis worldwide1,2. The progression of IgA-N follows a process known as the “four-hit theory”, whereby immune complexes containing galactose-deficient IgA1 (Gd-IgA1) are first deposited in glomerular mesangial cells, which leads to inflammation and injury followed by glomerular damage2. It is thought that glomerular mesangial cells in which immune complexes containing Gd-IgA1 are deposited induce glomerular endothelial cell (GE) damage in the chronic phase of IgA-N3.
On the other hand, some studies suggest that damaged endothelial cells cause blood Gd-IgA1 immune complexes to permeate blood vessels and deposit in the mesangial area, resulting in the development and progression of IgA-N considering that endothelial cells are located in the innermost part of capillaries4,5. Additionally, studies have shown that Gd-IgA1 immune complexes enhance proinflammatory cytokines by binding to GE2,5. In fact, glomerular lesions are known to cause capillary damage in the acute phase of IgA-N, leading to hematuria4. Furthermore, in previous studies, single-cell RNA sequencing (scRNA-seq) has revealed endothelial cell damage-induced capillaritis in mice with early spontaneous IgA-N6. However, no reports have elucidated the role of vascular endothelial cells in the pathogenesis of human IgA-N, and the mechanism by which GE are involved in the pathogenesis or progression of the disease remains poorly understood. Thus, there is no definitive treatment for IgA-N, and the challenge of the disease progressing to end-stage renal disease over the long term in many patients still remains7.
ScRNA-seq, which provides transcriptomes at the single-cell level, has been used to understand cellular heterogeneity in various research studies8,9. While various scRNA-seq platforms are available, targeting tissues generally requires single-cell isolation in advance. This leads to loss of cellular spatial information. Unlike scRNA-seq, spatial transcriptome sequencing (ST-seq) uses tissue sections, allowing quantification of gene expression while preserving location information. Among the available ST-seq systems, 10 × Genomics Visium is a convenient, widely used, commercially available ST-seq platform. However, the Visium system, which captures gene expression using circular spots, each 55 μm in diameter, contains information on several to dozens of cells per spot and is therefore not compatible with gene expression analysis of only specific cells contained in aggregates comprising several cell types, such as the renal glomerulus10.
Although several studies using scRNA-seq in human IgA-N have been reported, information pertaining to GE inflammation in these studies is limited11,12,13. Furthermore, no previous reports have factored spatial location information based on ST-seq in human IgA-N. In another recently published study, we integrated data from scRNA-seq and ST-seq to analyze rat kidney14. Applying the same approach, here we successfully defined GE clusters in scRNA-seq (Fig. 1). Use of both scRNA-seq and ST-seq enabled determination of the gene expression profile of GE, one of the cell types constituting complicated renal structures such as the glomeruli, in mild IgA-N. Furthermore, activation of characteristic inflammatory responses and new inflammation-associated molecules previously not known to be involved in IgA-N were identified in GE from the mild IgA-N specimens. Especially, these results of Fig. 5 and Supplementary Data 8 indicated that in mild IgA nephropathy, various inflammatory pathways may activate in the GE prior to mesangial cells.

a Scheme and (b) flow diagram for integrated analysis of scRNA-seq and Visium. scRNA-seq single-cell RNA sequencing.
Results
Diagnosis of IgA-N
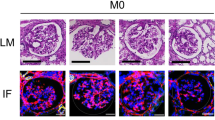
All patients had never been diagnosed with IgA-N or normal glomeruli by renal biopsy prior to nephrectomy for renal cancer. We performed pathological diagnosis using FFPE and immunofluorescence using frozen sections on non-tumorous regions surrounding the tumor in the resected kidney. The samples were diagnosed as IgA nephropathy or normal. Fig. 1b depicts the flow of specimens who underwent tissue diagnosis for IgA-N in this study. The final analysis included 9 specimens with normal tissue and 6 specimens with IgA-N. All histopathological images diagnosed with IgA-N showed IgA deposition in the mesangial area and mesangial cell proliferation and mesangial matrix expansion, characteristic features of IgA-N (Supplementary Fig. 1a–d). Additionally, the IgA-N lesions were of grade I or II according to the Japanese histologic classification (JHC), indicating mild IgA-N15,16 (Table 1). There were no significant differences between normal and IgA-N in antihypertensive drugs use and angiotensin II receptor blockers use (Table 1).
Cell identification using scRNA-seq
ScRNA-seq was performed using human kidney samples from 3 specimens with IgA-N and 3 normal subjects with normal tissue (Fig. 1b), yielding a total of 45,488 cells with a median of 2375 genes per cell and a mean read count per cell of ~51,260 (post-normalization mean reads per cell) (Supplementary Data 1). Unsupervised clustering analysis yielded 32 cell clusters, with the following 13 cell types identified by the expression of known renal cell marker genes: GE, Cluster #18; mesangial cell (mesa), Clusters #19 and #24; podocyte (podo), Cluster #20; proximal tubule (PT), Clusters #1, #3, #8, #9, and #26; distal tubule (DT), Clusters #0, #10, and #22; distal convoluted tubule (DCT), Clusters #5 and #21; collecting duct (CD), Clusters #4, #6, #7, #25, #29, and #31; endothelial cell (Endo), Clusters #2, #13, #15, #17, and #27; smooth muscle cell (SMC), Clusters #11 and #12; B cell (B), Cluster #30; T cell (T), Cluster #16; monocyte (Mono), Clusters #14 and #23; and NK cell (NK), Cluster #28 (Fig. 2a, b, Supplementary Fig. 1, Supplementary Data 2). All clusters defined as GE were detected from both IgA-N and normal specimens (Fig. 2c).

a UMAP plots of major renal cell populations identified by unsupervised clustering and annotation with marker genes; (b) Violin plots showing cluster-specific expression of genes representative of each cluster of known marker genes in major renal cells. Based on the marker genes, the following cells were identified: glomerular endothelium (GE), Cluster #18; mesangial cell (mesa), Clusters #19 and #24; podocyte (podo), Cluster #20; proximal tubule (PT), Clusters #1, #3, #8, #9, and #26; distal tubule (DT), Clusters #0, #10, and #22; distal convoluted tubule (DCT), Clusters #5 and #21; collecting duct (CD), Clusters #4, #6, #7, #25, #29, and #31; endothelial cell (Endo), Clusters #2, #13, #15, #17, and #27; smooth muscle cell (SMC), Clusters #11 and #12; B cell (B), Cluster #30; T cell (T), Cluster #16; monocyte (mono), Clusters #14 and #23; and NK cell (NK), Cluster #28; (c) Count ratio of cells from IgA-N specimens versus cells from normal specimens for each annotated cell type; and (d) Violin plot showing gene expression in each cluster of IL13RA2, a new GE marker gene. IgA immunoglobulin A, scRNA-seq single-cell RNA sequencing, UMPA uniform manifold approximation and projection.
Further, both EH domain containing 3 (EHD3)17 and sclerostin (SOST)18, known GE markers, were expressed in the GE clusters of interest. The expression of interleukin 13 receptor subunit alpha 2 (IL13RA2), a molecule previously not known GE marker, was also high in GE clusters (Fig. 2d). Analysis of several publicly human renal scRNA-seq datasets reveals consistent IL13RA2 expression in GE (Supplementary Fig. 6a–e.). Next, immunostaining of SOST and IL13RA2 using EHD3 as the index demonstrated a specific staining pattern of the GE in both IgA-N and normal specimens (Fig. 3a, b).

a Double-stained images of EHD3 (green) and SOST (red), (b) Fluorescence intensity of SOST, (c) Double-stained images of EHD3 (green) and IL13RA2 (red), and (d) Fluorescence intensity of IL13RA2. b, d The ratio of fluorescence area for each antigen against EHD3. The box plot extends from the 25th to the 75th percentile, with a horizontal line marking the median, while the whiskers represent the range. Scale = 40 µm.
Validation of spatial distribution of GE
In addition to the same specimens as used for scRNA-seq, kidney samples from 3 specimens with IgA-N and 6 normal subjects with normal tissues were used for the Visium analysis (Fig. 1b), yielding a total of 22,782 spots with a median of 4918 genes detected from each spot (Supplementary Data 3) and a mean read count per spot of ~57,646 (post-normalization mean reads per spot). For spots on each section, the two renal regions of the cortex and medulla and the glomeruli were identified by experts based on pathological information (Fig. 4a, Supplementary Fig. 3, 4). Expression of representative glomerular constituent cell markers (podo; NPHS1 adhesion molecule, nephrin (NPHS1), mesa; platelet derived growth factor receptor alpha (PDGFRA), GE; EHD3) was confirmed in the glomerular clusters (Fig. 4b).

Spots detected from histological images of normal specimens and IgA-N specimens. a Detected spots classified by a pathologist into the following four regions according to their histological features: cortex, medulla, glomeruli (Glom), and other. b Dot plots showing the expression of glomerular constituent cell marker genes (podo; NPHS1, mesa; PDGFRA, GE; EHD3) in each region. (c) UMAP plots of the clusters used for integrated analysis with Visium: GE, Cluster #18; and endothelial cells (Endo) (Endo1, Cluster #2; Endo2, Cluster #13; Endo3, Cluster #15; Endo4, Cluster #17; and Endo5, Cluster #27). Gray dots indicate cells not used for the integrated analysis with Visium. Predictive score of each cell type (scRNA-seq data; X-axis) against each histological annotation (Visium data; Y-axis) of (d) normal specimens and (e) IgA-N specimens. The dot color represents the scaled predictive score, and the dot size represents the proportion of spots with a predictive score of >0 in each histological annotation. IgA immunoglobulin A, scRNA-seq single-cell RNA sequencing UMPA uniform manifold approximation and projection.
An integrated analysis of scRNA-seq and Visium was performed to confirm whether the GE population estimated in scRNA-seq was located in the glomerular area. scRNA-seq data of the endothelial cell population (Endo1, Cluster #2; Endo2, Cluster #13; Endo3, Cluster #15; Endo4, Cluster #17; and Endo5, Cluster #27) and putative GE population (Cluster #18) (Fig. 4c) were integrated with the Visium data separately for IgA-N specimens and normal specimens. We get prediction scores for each spot of Visium for each clusters of scRNA-seq data. The predictive scores for the GE clusters in the glomerular area were high for both IgA-N and normal specimens (Fig. 4d, e, Supplementary Fig. 5a, b). The Endo-2, Endo-3, and Endo-4 exhibited high prediction scores in the cortex, medulla, and arterial endothelium/vasa recta, respectively. The spatial localization of Endo-1 and Endo-5 within the vascular endothelium could not be definitively determined (Fig. 4d, e, Supplementary Fig. 5a, b).
Differential expression analysis of the GE from mild IgA-N specimens
Functional differences in the GE between mild IgA-N specimens and normal specimens were explored.
IgA-N is characterized by proliferative changes in the mesangial cells and matrix and deposition mainly of IgA in the mesangial area, which were observed in the present specimens (Supplementary Fig. 1). First, we assessed whether the pathway analysis using ingenuity pathway analysis (IPA) could capture the functional differences, which observed in histopathological analysis of IgA-N specimens, between IgA-N specimens and normal specimens in the mesangial cell population. The results showed that fibrosis-associated pathways were enriched in mesangial cells from IgA-N specimens (Fig. 5a, Supplementary Data 4, 5). Histopathological images also demonstrated mesangial matrix expansion, which indicates increased mesangial fiber components in the mesangial area19 (Supplementary Fig. 1), a finding consistent with the results of the pathway analysis. Thus, it was confirmed that the pathway analysis in the current study was capable of capturing functional differences between IgA-N specimens and normal specimens.

A pathway list was prepared for (a) mesangial cells and (b) GE using DEGs with a log-fold change of ≥0.25 in IgA-N specimens compared with normal specimens. The 30 pathways with -log10 (p-value) are listed in descending order. The pathways with no z-score determined were excluded. DEG differentially expressed gene, IgA immunoglobulin A.
A similar pathway analysis was performed using GE from IgA-N specimens and normal specimens. Inflammation-associated pathways such as interleukin-6 (IL-6) signaling were found to be enriched in GE from IgA-N specimens (Fig. 5b, Supplementary Data 4, 5). The differentially expressed genes with a z-score of >2 contained in the inflammation-associated pathways included representative inflammation-associated molecules such as C-C motif chemokine ligand 2 (CCL2) and C-X-C motif chemokine ligand 2 (CXCL2) (Supplementary Data 4). Additionally, immunohistochemistry (IHC) of CCL2 and CXCL2 was performed to confirm whether GE from the IgA-N specimens were inflamed. CCL2 (p-value < 0.001) and CXCL2 (p-value < 0.001) expression was found to be elevated in the GE from IgA-N specimens (Fig. 6a–d). scRNA-seq and IHC yielded consistent results. Among the differentially expressed genes contained in the inflammation-associated pathways that have a z-score of >2, increased expression of endothelin receptor type B (EDNRB), Interleukin 1 receptor-like 1 (IL1RL1), phospholipase C gamma 2 (PLCG2), and CCAAT enhancer binding protein beta (CEBPB), previously not known to be associated with human IgA-N, was noted (Supplementary Data 6). Visium data showed no increase in the expression of any of these genes (Supplementary Data 7).

a Double-stained images of EHD3 (green) and CCL2 (red); (b) Fluorescence intensity of CCL2; (c) Double-stained images of EHD3 (green) and CXCL2 (red); (d) Fluorescence intensity of CXCL2; (e) Double-stained images of EHD3 (green) and EDNRB (red); (f) Fluorescence intensity of EDNRB; (g) Double-stained images of EHD3 (green) and EDN1 (red); and (h) Fluorescence intensity of EDNRB. The box plot extends from the 25th to the 75th percentile, with a horizontal line marking the median, while the whiskers represent the range. Scale = 40 µm; **p < 0.001.
EDNRB, the most significantly differentially expressed gene, was examined for protein expression level and localization by immunostaining. Consistent with the scRNA-seq results, increased expression of EDNRB was confirmed in the GE from IgA-N specimens (p-value < 0.001) (Fig. 6e, f). Similarly, expression of endothelin 1 (EDN1), the ligand for EDNRB, was also significantly increased in the GE from IgA-N specimens (p-value < 0.001) (Fig. 6g, h).
Discussion
In this study, we identified the gene expression profile of GE using scRNA-seq and Visium in renal tissues from human specimens with mild IgA-N compared with normal specimens. Analysis of the cellular status in mild IgA-N may be informative for early therapeutic interventions, in that the pathogenesis and degree of progression becomes known at an early stage of the disease. We performed an integrated analysis of scRNA-seq and Visium to clearly define GE based on scRNA-seq data. Comparison of the gene expression profiles in GE between mild IgA-N specimens and normal specimens demonstrated that inflammation-associated pathways were activated in GE in mild IgA-N. Additionally, several inflammation-associated factors previously not known to be involved in IgA-N were identified. Thus, this study provides new insights into the mechanisms of GE in mild IgA-N in humans.
IgA-N is a prevalent disease in the Asian region2 and is estimated to develop in 0.04% of the Japanese population20. The current analysis using specimens from Japanese patients with renal cell carcinoma showed that as high as ~17% of the patients had IgA-N (Fig. 1b). Previous studies have reported an incidence of 2.9%–18.3% based on glomerular analysis of isolated kidney samples21,22 and have also suggested the possibility of some forms of IgA-N being associated with renal cell carcinoma21. Although the incidence of IgA-N in association with renal cell carcinoma may have been high in the current study as well, the pathogenesis of IgA-N is considered to have followed the “four-hit theory”, in which a similar process develops as a result of various causes2. Furthermore, considering that the mean age at diagnosis of renal cancer in the general population is 65 years23, similar to that observed in the current analysis population, the time from onset of IgA-N in our study was much shorter than that observed in cases of mild IgA-N that occur at a young age and slowly progress23. Thus, the possibility that endothelial cell inflammation, described later, occurs early in IgA-N cannot be ruled out.
We attempted to understand the role of GE in the pathogenesis of IgA-N based on scRNA-seq data. The integrated analysis of scRNA-seq and ST-seq indicated that the GE clusters identified by the expression of the marker genes in scRNA-seq were predicted to localize at glomeruli. The IHC staining results of molecular markers using the same samples also showed expression of these markers in GE, establishing the cell group as the GE. We have newly identified that EHD3 and SOST as markers not only under normal conditions but also in the GE of mild IgA-N.
Furthermore, IL13RA2 was newly identified as a gene specifically expressed in GE. IL13RA2 has previously not been described in association with glomerular disease. Of note, IL13RA2 was expressed in a subset of GE, regardless of the presence or absence of IgA-N (Fig. 3c, d). Several publicly available normal human renal scRNA-seq datasets containing GE have also shown expression of IL13RA2 in GE, regardless of age (Supplementary Fig. 6a–e.), suggesting that IL13RA2 may serve as a GE marker in normal specimens. On the other hand, the heterogeneity in the IgA nephropathy group suggests that IL13RA2 expression may increase predominantly with the accumulation of more cases and further analysis of severe IgA nephropathy in the future.
Additionally, we showed that inflammatory responses were activated in the GE in mild IgA-N. Although previous studies have reported activated inflammatory responses in the GE in mild IgA-N, the findings pertained to only specific molecules such as IL-6, VCAM1, ICAM1 etc.5,24,25, and to the best of our knowledge, this is the first study to demonstrate activation of GE inflammatory responses based on expression analysis at single-cell resolution with spatial context.
IgA-N often presents with hematuria after upper respiratory tract infection, and its pathology is believed to involve inflammation associated with bacterial infection2. The current study also showed activation of the signal pathways associated with proinflammatory cytokines such as IL-6 (Fig. 5). Gd-IgA1 immune complexes promote migration of inflammatory cells by increasing adhesive factors such as intracellular adhesion molecule 1 (ICAM-1) and induce the production of proinflammatory cytokines by GE, thereby exacerbating intrarenal inflammation5. Moreover, both GE and mesangial cells associated with Gd-IgA1 immune complexes secrete IL-6 that induces loss of endothelial cells, increases endothelial permeability, and recruits inflammatory cells, resulting in enhancement of Gd-IgA1 deposition and inflammatory responses in the mesangial cell19,25. Several genes other than ICAM-1 that are known to be associated with mild IgA-N, including selectin E (SELE)26 and C-X3-C motif chemokine ligand 1 (CX3CL1)6, have shown increased expression in GE in a mouse model of mild IgA-N (gddY mice). These findings suggest that endothelial cell inflammation may also be involved in the mechanism of pathogenesis and progression of human IgA-N, consistent with the results in the mouse model of IgA-N6. Furthermore, given that the gene expression profiles of GE from gddY mice and those from humans with mild IgA-N appear to be partly similar, gddY mice may be useful for functional analysis of the newly identified genes and proteins.
We also identified EDNRB, CEBPB, IL1RL1, and PLCG2 as inflammation-associated genes previously not known to be associated with human IgA-N.
EDN1 is a bioactive peptide with a blood flow-regulating effect and has two types of receptors, type A and type B27. Endothelin receptor type A (EDNRA), located on vascular smooth muscle cells, is one of the most potent vasopressor substances in nature27, and Sparsentan, a dual antagonist of EDNRA and angiotensin receptors, has a therapeutic effect on IgA-N28. On the other hand, EDNRB, expressed in vascular endothelial cells, exerts vasodilatory effects29. The current scRNA-seq analysis showed no difference in the expression level of either EDN1 or EDNRA between IgA-N and normal tissues, but there was an increased expression of EDNRB in specimens from specimens with IgA-N (Supplementary Data 4). Tissue staining revealed elevated expression of EDN1 in the glomeruli from patients with IgA-N, as previously reported30. A possible reason is not the result of increased mRNA expression in GE, but rather reflects the result of serum EDN1 binding to EDNRB. In addition, the mild IgA-N cases in our study might have had increased expression of EDNRB, which has vascular protective effects, as a compensatory mechanism for glomerular damage.
Our IgA-N tissue specimens also showed increased expression of PLCG2 (Supplementary Data 4). Although the role of PLCG2 in IgA-N remains unknown, it has been reported that activation of PLCG2 activates the 1,2-diacylglycerol-protein kinase C (PKC) pathway30,31, which regulates vascular permeability32. Thus, it is also possible that enhancement in vascular permeability by PLCG2 may induce Gd-IgA1 deposition in the mesangial area.
Further, we observed elevated expression of the chemokine-related genes CCL2 and CXCL2 in the IgA-N specimens (Supplementary Data 4). Although CCL2 and CXCL2 are known to be associated with IgA-N33,34, elevated expression of these molecules in the GE in IgA-N has not been described previously. Chemokines in endothelial cells are involved in various kidney diseases as inflammatory mediators35. CCL2, also known as monocyte chemoattractant protein-1 (MCP-1), is a member of the chemokine family, and its expression is elevated in the glomeruli in IgA-N36,37. Hypertension and diabetes also increase CCL2 expression in renal endothelial cells37,38. However, considering that the study excluded patients with diabetes and that similar numbers of subjects had received antihypertensive drugs in the normal group and IgA-N group, the increase in CCL2 expression could be attributable to IgA-N. Although CXCL2 is elevated in renal tissue from patients with IgA-N39, its expression status in GE is unknown. One study reported that CXCL2 expression is also increased in human aortic endothelial cells (HAECs) stimulated by interleukin 17 A (IL-17), which is implicated in various inflammatory diseases40. Similar to this report, the current analysis revealed activation of the IL-17-related signaling pathway (Fig. 5b), suggesting that CXCL2 may also be involved in the progression of IgA-N. Thus, we have successfully validated the already known causal relationship between CCL2, CXCL2 and IgA-N in the pathogenesis of IgA-N using a novel method. This demonstrates the certainty of the method used in this study, and we also found that a pathway involving factors characteristic of mild IgA nephropathy (IL-17-related signaling pathway), which was obtained in the present comprehensive analysis, plays an important role in mild IgA nephropathy.
In diabetic nephropathy, a disease in which the role of GE dysfunction has attracted attention, platelet derived growth factor receptor beta (PDGFR) expression has been shown to be elevated in GE41. However, the current differentially expressed gene analysis detected no activation of either the vascular endothelial growth factor (VEGF) or platelet derived growth factor (PDGF) signaling pathway. Furthermore, although proinflammatory cytokines (interleukin 6 (IL-6) and interleukin 8 (IL-8)) are released in anti-MPO-ANCA antibody- or anti-PR3-ANCA antibody-associated crescentic glomerulonephritis, a representative glomerular disease caused by endothelial cell damage42, the current study found an increase in only IL-8. These data suggest that the signaling pathways in endothelial cell damage caused by IgA-N are distinct from those in specimens with diabetes or crescentic glomerulonephritis.
Thus, integration of scRNA-seq and Visium analyses revealed a tendency of more active inflammatory responses in GE than in mesangial cells in glomerular cells from specimens with mild IgA-N (Fig. 5, Supplementary Data 8). In gddY mice, GE have been reported to play a critical role in the early phase of intraglomerular inflammation in IgA-N6. In fact, treatment for IgA-N generally involves steroids with anti-inflammatory effects2. Thus, the activation of inflammatory signaling pathways in GE characteristic of IgA-N identified in the current study are not only valuable for understanding the progression of IgA-N, but also to choosing more effective therapeutic agents28.
Some limitations of the study need to be acknowledged. First, the absolute number of specimens available for analysis was small, particularly in the IgA-N group. As mild IgA-N lacks prominent symptoms such as proteinuria, subject recruitment depends on chance. To overcome the heterogeneity and individual differences in the pathology of IgA-N, large-scale recruitment is needed in future studies to ensure a larger sample size of specimens with mild IgA-N. Additionally, the study used organs resected from specimens with cancer after removal of cancer sites, which may still have had influences of cancer tissue. While the study successfully captured genes present in IgA-N by analyzing the differences between IgA-N specimens and normal specimens, determination of whether or not this gene group contributes to IgA-N needs further investigation, including in vivo and in vitro experiments and studies using tissues from specimens other than those with cancer. Second, some differentially expressed genes captured by scRNA-seq were not captured by Visium-based analysis. A possible reason is that the granularity of transcriptional profile data differs between Visium and scRNA-seq. As Visium spots contain multiple non-GE, differential expression in GE may have been obscured. Other recently available high-resolution spatial transcriptome technologies such as Xenium, VisiumHD, and CosMx may be useful for analysis of cells constituting complicated structures such as the glomeruli.
Nevertheless, the study demonstrated that inflammation and subsequent GE damage are critical even in patients with mild IgA-N, providing invaluable data for future studies on IgA-N. Moreover, we established a role of the inflammation‑associated molecules play a significant role in GE in mild IgA-N, thus highlighting the importance of understanding inflammation mechanisms. This new information is essential not only for elucidating the mechanisms of IgA-N pathogenesis but also for developing therapeutic strategies for suppression of inflammation.
Methods
Sample acquisition and diagnostics
All renal tissues were obtained from specimens who had undergone partial nephrectomy or radical nephrectomy for renal cancer at Kitasato University Hospital (Department of Urology) and had provided written informed consent. From these non-tumor areas, we obtained fresh samples, FFPE, and frozen sections. First, we diagnosed IgA nephropathy or normal samples using pathological diagnosis with FFPE and immunofluorescence with frozen sections (Fig. 1b). Using the samples diagnoses in this manner, we performed this study by integrating scRNAseq data obtained from fresh samples (Normal: 3, IgA-N: 3) with Visium data obtained from FFPE (Normal: 9, IgA-N: 6). (Fig. 1b).
Antibodies
Anti-IgG, IgA, IgM, C3, C4, C1q, fibrinogen, EHD3, EDN1, SOST, IL13RA2, CCL2, CXCL2, EDNRB, and fluorescence-labeled secondary antibody corresponding to the primary antibody were used to diagnose IgA-N and normal specimens and determine the expression levels of each antigen. Supplementary Table 1 provides detailed information on the antibodies used.
Tissue staining
Paraffin sections from FFPE (Normal: 9, IgA-N: 6) were subjected to periodic acid-Schiff’s staining, hematoxylin & eosin staining, periodic acid-methenamine silver staining (Jones’ stain), and Masson’s trichrome staining43,44. Immunofluorescence staining for each antigen was performed on frozen sections. Renal tissues were embedded in Tissue Tek OCT compound (Sakura Finetek Japan Co., Ltd., Tokyo, Japan) and sliced into 4-μm-thick sections. The sections were washed, incubated with the primary antibody, and stained with the secondary antibody. Pathologic images were visualized using an optical microscope (BX51; Olympus Corporation, Tokyo, Japan) and analyzed using ImageJ software (https://imagej.nih.gov/ij/). Fluorescence images were visualized using a confocal laser microscope (LSM710; Carl Zeiss AG, Oberkochen, Germany) and analyzed using ZEN imaging software (Carl Zeiss AG, Oberkochen, Germany).
Assessment of glomerular lesions
Renal tissues were evaluated by two nephrologists. Histologic lesions of IgA-N were classified according to the 2016 Oxford and JHC classifications45,46. Tissues showing no immunoglobulin or complement deposition in the glomeruli or morphological changes were considered normal and used for the analysis.
Preparation of a library for Visium analysis
For Visium analysis, a library was constructed using resected renal tissues from 32 adult specimens with cancer after removal of the cancerous part.
The sequencing library was prepared using Visium Spatial for FFPE Gene Expression Kit, Human Transcriptome (10x Genomics, Inc., California, U.S.) according to the manufacturer’s instructions (CG000407_Rev_C). A histological image of each sample was acquired using an inverted microscope (Nikon Eclipse Ti; Nikon Solutions Co., Ltd. Tokyo, Japan). The library was sequenced using Illumina NextSeq2000 (Illumina, Inc. California, U.S.).
Tissue dissociation for scRNA-seq
For scRNA-seq, renal tissues were collected from 14 adult specimens with cancer after removal of the cancerous part. Fresh renal tissue was sectioned using a vibrating-blade microtome (Leica Microsystems, Wetzlar, Germany), dissociated in Dulbecco’s modified Eagle medium (Nacalai Tesque, Inc., Kyoto, Japan) containing 100 U/mL Collagenase D (F. Hoffmann-La Roche Ltd; [1108886600]) and 5 U of DNaseI (Takara Bio Inc., Shiga, Japan) by shaking at 37 °C for 60 min, and then filtered through a 40 μm cell strainer (Corning Inc., New York, U.S.). The resultant cell suspension was subjected to density-gradient centrifugation using Optiprep (Abbott Diagnostics Technologies AS, Oslo, Norway) and 1% (w/v) bovine serum albumin containing phosphate-buffered saline (Nacalai Tesque, Inc., Kyoto, Japan) to isolate cells and then filtered through a 35 μm cell strainer (Corning Inc., New York, U.S.).
Preparation of a library for scRNA-seq
For each sample, the quantity and viability of cells were determined, and microscopy was used to confirm that there were no aggregated cells or debris. An scRNA-seq library was prepared using Chromium Next GEM Automated Single Cell 3′ Reagent Kits v3.1 (10x Genomics, Inc., California, U.S) according to the manufacturer’s instructions (CG000286 Rev_E). The library was sequenced using Illumina NextSeq2000(Illumina, Inc., California, U.S.).
Data processing of the Visium dataset
Sequence results (BCL files) were converted to FASTQ files and mapped to the human reference genome (GRCh38) using Space Ranger (v.1.3.1; 10 × Genomics; https://www.10xgenomics.com/support/software/space-ranger/downloads/previous-versions). Downstream analysis was performed using R package, Seurat (v.3.2.2; https://satijalab.org/seurat/articles/install.html). Outputs from Space Ranger were converted to Seurat objects and preprocessed using the SCTransform47 function. The objects of all samples were integrated using the Seurat anchor-based integration workflow48, followed by principal component analysis, dimensional reduction, and unsupervised clustering.
Data processing of the scRNA-seq dataset
Sequence results (BCL files) were converted to FASTQ files and mapped to the human reference genome (GRCh38) using Cell Ranger (v.4.0.0; 10 × Genomics; https://www.10xgenomics.com/support/software/cell-ranger/downloads/previous-versions). Downstream analysis was performed using R package, Seurat (v.3.2.2). The output matrices from Cell Ranger were transformed into Seurat objects, and the barcodes with <200 genes were excluded as part of primary quality control to exclude low-quality cells or cell-free droplets. After primary quality control, each object was normalized and scaled according to the Seurat standard workflow, followed by principal component analysis, uniform manifold approximation and projection (UMAP), dimensional reduction, and unsupervised clustering, based on the top 2000 highly differentially expressed genes. Among the resultant clusters, those with a high proportion (%) of mitochondrial genes (median of mt >85%) and a low gene number (median of nFeature <1500) were excluded, and dead cells were removed as part of secondary quality control. After secondary quality control, all objects were reprocessed according to the Seurat standard workflow and subsequently integrated by the Seurat anchor-based integration workflow, followed by principal component analysis, dimensional reduction, and unsupervised clustering (ndim34; granularity, 0.8). Cell type was identified on the basis of unsupervised clustering and known marker gene expression. Differentially expressed genes in the IgA-N group as compared with the normal group were identified in FindMarkers (logfc.threshold = 0.25) with the default parameter.
Integration of Visium and scRNA-seq datasets
Integration of Visium and scRNA-seq datasets was performed according to Seurat’s anchor-based integration workflow48. Among the scRNA-seq datasets, only those of GE and endothelial cell clusters were used as reference for integration. The integrated analysis was performed using Visium and scRNA-seq datasets of the same specimens. Predictive scores for each cell type were calculated for all spots in the Visium dataset.
Canonical pathway analysis
Canonical pathway analysis was performed using QIAGEN IPA (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA)49.
Expression analysis of IL13RA2 using publicly available data
The figure/analysis was created with BBrowserX/Talk2Data software (Bio Turing Inc., San Diego, CA, USA). The following data, registered in BBrowserX/Talk2Data, were used for analysis: ID:LAKEETAL50; GSE14098917; ID:PMID31604275_Mature51; GSE16004852; and ID:GSE159115_6_BENIGN18.
Statistics and reproducibility
Baseline values were expressed as medians (25th percentile to 75th percentile). Sex ratio was compared using the chi-square test, and for other baseline values and expression levels determined by immunostaining, group medians were compared using the two-tailed Mann-Whitney U test. The sample size was determined based on the number of past nephrectomy cases at our hospital and the limitations of the research system. Statistical analysis was performed used StatFlex (v.7; Artech Co., Ltd., Tokyo, Japan). P < 0.05 was considered as statistical significance.
Approval for research
Renal tissues were obtained from specimens who had undergone nephrectomy following written informed consent under the approval of the Kitasato University Ethics Committee (KMEO) (#KMEO B20-263) and Kyowa Kirin Co., Ltd. Research Ethics Committee (# 2020_033). Subjects were identified by number, not by name, and all data including results of histological diagnosis (IgA nephropathy, Normal) were anonymized. The study was conducted in compliance with the principles of the Declaration of Helsinki. All ethical regulations relevant to human research participants were followed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data supporting the results and histology results of Visium in the study are deposited in the Japanese Genotype-phenotype Archive (JGA) with accession number JGAS000736. The use of these data is subject to restrictions approved by the Ethics Committee and is registered in JGA, a restricted public database. Data are, however, available from JGA upon reasonable request and with the permission of Human Data Review Committee of the NBDC Human Database, which was established by Database Center for Life Service (DBCLS). Source data for the figures presented in the manuscripts can be found in Supplementary Data 9.
References
Wyatt, R. J. & Julian, B. A. IgA nephropathy. N. Engl. J. Med. 368, 2402–2414 (2013).
Stamellou, E. et al. IgA nephropathy. Nat. Rev. Dis. Prim. 9, 67 (2023).
Huang, M. et al. Endothelial injury by circulating IgA1 complex through the activation of mesangial cells in IgA nephropathy. Clin. Nephrol. 97, 173–182 (2022).
Kusano, T. et al. Endothelial cell injury in acute and chronic glomerular lesions in patients with IgA nephropathy. Hum. Pathol. 49, 135–144 (2016).
Makita, Y. et al. Glomerular deposition of galactose-deficient IgA1-containing immune complexes via glomerular endothelial cell injuries. Nephrol. Dial. Transpl. 37, 1629–1636 (2022).
Zambrano, S. et al. Molecular insights into the early stage of glomerular injury in IgA nephropathy using single-cell RNA sequencing. Kidney Int 101, 752–765 (2022).
Pitcher, D. et al. Long-Term Outcomes in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 18, 727–738 (2023).
Kashima, Y. et al. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 52, 1419–1427 (2020).
Hanai, A. et al. Single-cell RNA sequencing identifies Fgf23-expressing osteocytes in response to 1,25-dihydroxyvitamin D(3) treatment. Front Physiol. 14, 1102751 (2023).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Zheng, Y. et al. Single-cell transcriptomics reveal immune mechanisms of the onset and progression of IgA nephropathy. Cell Rep. 33, 108525 (2020).
Tang, R. et al. A partial picture of the single-cell transcriptomics of human IgA nephropathy. Front Immunol. 12, 645988 (2021).
Zeng, H. et al. Single-cell RNA-sequencing reveals distinct immune cell subsets and signaling pathways in IgA nephropathy. Cell Biosci. 11, 203 (2021).
Onoda, N. et al. Spatial and single-cell transcriptome analysis reveals changes in gene expression in response to drug perturbation in rat kidney. DNA Res. 29, dsac007 (2022).
Kaihan, A. B. et al. The Japanese histologic classification and T-score in the oxford classification system could predict renal outcome in Japanese IgA nephropathy patients. Clin. Exp. Nephrol. 21, 986–994 (2017).
Sato, R. et al. Validation of the Japanese histologic classification 2013 of immunoglobulin A nephropathy for prediction of long-term prognosis in a Japanese single-center cohort. Clin. Exp. Nephrol. 19, 411–418 (2015).
Menon, R. et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 5, e133267 (2020).
Zhang, Y. et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 118, e2103240118 (2021).
Zhang, H., Deng, Z. & Wang, Y. Molecular insight in intrarenal inflammation affecting four main types of cells in nephrons in IgA nephropathy. Front Med.10, 1128393 (2023).
Kiryluk, K. et al. Global incidence of IgA nephropathy by race and ethnicity: a systematic review. Kidney360 4, 1112–1122 (2023).
Magyarlaki, T. et al. Renal cell carcinoma and paraneoplastic IgA nephropathy. Nephron 82, 127–130 (1999).
Kläger, J. P. et al. More than ancillary records: clinical implications of renal pathology examination in tumor nephrectomy specimens. J. Nephrol. 34, 1833–1844 (2021).
Padala, S. A. et al. Epidemiology of renal cell carcinoma. World J. Oncol. 11, 79–87 (2020).
Trimarchi, H. & Coppo, R. Glomerular endothelial activation, C4d deposits and microangiopathy in immunoglobulin A nephropathy. Nephrol. Dial. Transpl. 36, 581–586 (2021).
Yang, Y. C., Fu, H., Zhang, B. & Wu, Y. B. Interleukin-6 Downregulates the expression of vascular endothelial-cadherin and increases permeability in renal glomer. Inflammation 45, 2544–2558 (2022).
Hession, C. et al. Endothelial leukocyte adhesion molecule 1: direct expression cloning and functional interactions. Proc. Natl Acad. Sci. USA 87, 1673–1677 (1990).
Schiffrin, E. L. & Pollock, D. M. Endothelin system in hypertension and chronic kidney disease. Hypertension 81, 691–701 (2024).
El Karoui, K., Fervenza, F. C. & De Vriese, A. S. Treatment of IgA nephropathy: a rapidly evolving field. J. Am. Soc. Nephrol. 35, 103–116 (2024).
Drakopoulos, A., Goumenos, D. S., Vlachojannis, J. G. & Tsakas, S. Endothelin receptors in the kidney of patients with proteinuric and non-proteinuric nephropathies. Ren. Fail 28, 461–467 (2006).
Zanatta, C. M. et al. Endothelin-1 and endothelin a receptor immunoreactivity is increased in patients with diabetic nephropathy. Ren. Fail 34, 308–315 (2012).
Wilde, J. I. & Watson, S. P. Regulation of phospholipase C gamma isoforms in haematopoietic cells: why one, not the other?. Cell Signal 13, 691–701 (2001).
Das Evcimen, N. & King, G. L. The role of protein kinase C activation and the vascular complications of diabetes. Pharm. Res 55, 498–510 (2007).
Zhou, X., Wang, N., Zhang, Y. & Yu, P. Expression of CCL2, FOS, and JUN may help to distinguish patients with iga nephropathy from healthy controls. Front Physiol. 13, 840890 (2022).
Yuan, J., Wang, Z. & Wang, Y. P. Identification of common key genes associated with Crohn’s Disease and IgA nephropathy. Eur. Rev. Med. Pharm. Sci. 26, 3607–3620 (2022).
Oates, J. C., Russell, D. L. & Van Beusecum, J. P. Endothelial cells: potential novel regulators of renal inflammation. Am. J. Physiol. Ren. Physiol. 322, F309–f321 (2022).
Gao, J. et al. Genetic variants of MCP-1 and CCR2 genes and IgA nephropathy risk. Oncotarget 7, 77950–77957 (2016).
Ashizawa, M. et al. Detection of nuclear factor-kappaB in IgA nephropathy using Southwestern histochemistry. Am. J. Kidney Dis. 42, 76–86 (2003).
Kim, M. J. & Tam, F. W. Urinary monocyte chemoattractant protein-1 in renal disease. Clin. Chim. Acta 412, 2022–2030 (2011).
Yu, K. et al. APOC1 exacerbates renal fibrosis through the activation of the NF-κB signaling pathway in IgAN. Front Pharm. 14, 1181435 (2023).
Mai, J. et al. Interleukin-17A promotes aortic endothelial cell activation via transcriptionally and post-translationally activating p38 mitogen-activated protein kinase (MAPK) pathway. J. Biol. Chem. 291, 4939–4954 (2016).
Li, T. et al. Glomerular endothelial cells are the coordinator in the development of diabetic nephropathy. Front Med (Lausanne) 8, 655639 (2021).
Mazzariol, M., Camussi, G. & Brizzi, M. F. Extracellular vesicles tune the immune system in renal disease: a focus on systemic lupus erythematosus, antiphospholipid syndrome, thrombotic microangiopathy and ANCA-vasculitis. Int. J. Mol. Sci. 22, 4194 (2021).
Naito, S., Nakayama, K. & Kawashima, N. Enhanced levels of glycosphingolipid GM3 delay the progression of diabetic nephropathy. Int. J. Mol. Sci. 24, 11355 (2023).
Hassan, H. et al. Essential role of X-box binding protein-1 during endoplasmic reticulum stress in podocytes. J. Am. Soc. Nephrol. 27, 1055–1065 (2016).
Trimarchi, H. et al. Oxford classification of IgA nephropathy 2016: an update from the IgA nephropathy classification Working Group. Kidney Int. 91, 1014–1021 (2017).
Kawamura, T. et al. A histologic classification of IgA nephropathy for predicting long-term prognosis: emphasis on end-stage renal disease. J. Nephrol. 26, 350–357 (2013).
Repana, D. et al. The network of cancer genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 20, 1 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Krämer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2014).
Lake, B. B. et al. An atlas of healthy and injured cell states and niches in the human kidney. Nature 619, 585–594 (2023).
Stewart, B. J. et al. Spatiotemporal immune zonation of the human kidney. Science 365, 1461–1466 (2019).
He, B. et al. Single-cell RNA sequencing reveals the mesangial identity and species diversity of glomerular cell transcriptomes. Nat. Commun. 12, 2141 (2021).
Acknowledgements
The authors thank all the subjects who participated in this study. The authors also thank Dr. Ishii D., Dr. Ikeda M., and Dr. Kitajima K. for help with the nephrectomy procedure; Ms. Ishigaki N. for help with tissue staining; Dr. Murakami T. for support in establishing the experimental system of sample dissociation for scRNA-seq; Mr. Miyazawa T. for managing the server for the analysis; and Dr. Yoneya T. for data quality check. Editorial assistance was provided by Cactus Life Sciences (a part of Cactus Communications) and funded by Kyowa Kirin Co., Ltd.
Author information
Authors and Affiliations
Contributions
K.H., N.K., A.K., M.S., I.U., M.M., and S.N. contributed to the conception and design of the study. N.K. and S.N. performed sample preparation for scRNA-seq and Visium analysis. N.K. captured the histopathology images, and T.S. and S.N. performed pathological assessment of the tissues. S.N. provided specimen samples and clinical data. K.H., A.K., and M.S. performed scRNA-seq and Visium experiments. K.H., A.K., and M.S. performed the data processing, database construction, and data analysis. K.H., N.K., A.K., N.O., M.M., and S.N. drafted the manuscript. K.H. and S.N. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: K.H., A.K., M.S., N.O., I.U. and M.M. are employees of Kyowa Kirin Co., Ltd. N.K. T.S. and S.N. declare no competing interests. This paper was funded by Kyowa Kirin Co., Ltd.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Ashwani Gupta & Rosie Bunton-Stasyshyn. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hasegawa, K., Kawashima, N., Kawabata, A. et al. Inflammation-associated molecules in the glomerular-endothelium in mild IgA-nephropathy patients identified by single-cell and spatial transcriptome. Commun Biol 8, 915 (2025). https://doi.org/10.1038/s42003-025-08325-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08325-z
