
Abstract
Therapeutical application of hydrogen sulfide (H2S) is limited due to the lack of delivery routes for specific organs and the rapid and wide dispersal of H2S in vivo. While H2S shows adipogenic effects in vitro, its in vivo impacts on obesity remain unclear. This study applies a H2S-slow-releasing hydrogel (H2S gel) to deliver H2S locally in subcutaneous adipose tissue and examines local lipid accumulation in mice. H2S is released from H2S gels within 6 h and lasts for 72 h, elevating H2S levels in local adipose tissue but not in the plasma. Localized H2S gel delivery causes significant lipid accumulation and larger lipid droplet diameter in mouse adipose tissues. The expressions of sterol regulatory element-binding protein, peroxisome proliferator-activated receptor-γ, adiponectin, and perilipin are all upregulated by H2S gel injections. Local delivery and retention of H2S in adipose tissues increase lipid accumulation more in wild-type than in cystathionine-γ-lyase knockout mice. This study confirms the feasibility of selectively delivering H2S via injectable hydrogels and their effectiveness in regulating targeted tissue functions. Furthermore, this work deepens our understanding of the role of H2S in obesity development under physiological conditions and offers a practical implementation strategy for H2S-based therapeutic interventions.
Similar content being viewed by others
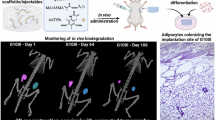


Introduction
Obesity is one of the most common morbidities, characterized by excessive body fat accumulation that adversely affects health. It is manifested with an overabundance of malfunctioning adipose tissues due to genetic and epigenetic factors as well as unhealthy lifestyles1,2. It can also lead to other disorders, including dyslipidemia, diabetes, cardiovascular diseases, osteoarthritis, neurodegenerative diseases, and certain types of cancer3,4,5. Adipocytes or fat cells play a crucial role in regulating energy metabolism and maintaining healthy body weight. The number of these cells are tightly controlled, and any disruption to this regulation can lead to obesity and other metabolic disorders6. Adipocyte hypertrophy (an increase in size) or hyperplasia (an increase in quantity) also contribute to the development of obesity. During periods of excess calorie intake, adipocytes tend to grow in size and produce adipokines, which attract additional pre-adipocytes and stimulate their maturation into adipocytes. When the capacity to recruit and expand adipocytes is exceeded, lipid accumulates in areas such as visceral depots, the liver, and skeletal muscle. These changes can lead to inflammation, insulin resistance, and other symptoms of metabolic syndrome6,7,8.
H2S contributes to the homeostatic regulation of the cardiovascular, pulmonary, gastroenterological, neurological, immunological, and endocrine systems9,10,11. Previously, we showed that exogenous H2S increased adipogenesis and lipid accumulation in mouse adipocytes12,13,14. Cystathionine γ-lyase (CSE) is the major enzyme involved in the production of H2S in adipose tissue. Upon knocking out the CSE gene (KO) in mice, we observed a significant decrease in H2S production in primary mouse adipocytes14. When CSE-KO preadipocytes were stimulated to differentiation, they showed a significantly lower adipogenesis and lipid accumulation than WT preadipocytes. Furthermore, the addition of exogenous H2S to the culture media increased adipogenesis and lipid accumulation in adipocytes in vitro12. However, in vitro cellular and molecular studies can never fully replicate in vivo physiological conditions. The physiological effects of H2S in vivo would be significantly impacted by the metabolism of this gasotransmitter, including its tissue/organ-specific production, absorption, distribution, and excretion. Interactions of different types of cells and tissues in the presence of circulation and numerous endogenous factors would determine the net effect of H2S on its targets in vivo. It is thus merited to investigate whether an elevated H2S level under in vivo conditions can induce adipogenesis and lipogenesis, one critical evidence for establishing a pro-obesity role of H2S.
The physiological effects of endogenous H2S can be mimicked by exogenously applied H2S salts and donors like NaHS and GYY413715,16. While selective over-expression of H2S-generating enzymes in regional adipose tissue in vivo is a daunting challenge, systemic administration of H2S-delivering compounds would generate system-wide alteration of H2S levels, which cannot fulfill the mission to produce regionalized delivery and retention of exogenous H2S for the targeted tissues or organs. H2S gel is a thiol-triggered H2S-releasing aromatic peptide amphiphile hydrogel17 (Fig. 1). In the present study, we utilized H2S gel as the means for directly and restrictively delivering H2S to subcutaneous adipose tissues in mice. We investigated whether this administration approach yielded the desired target-specific H2S release and retention. The development of adipogenesis and lipogenesis in the injected region in vivo was subsequently examined.

H2S gel in the presence of CaCl2, forms a biodegradable hydrogel (1). The SATO group, upon contact with thiols such as cysteine, undergoes chemical transformations, forming arylidenethiooxime and N-benzoylated cysteine (2). Arylidenethiooxime decomposes, releasing H2S (3). Created with BioRender. Verma, R. (2025) https://BioRender.com/dbuw3sa.
Results
In vitro assessment of H2S release kinetics and gel degradation of H2S gel
Twelve hours after H2S gel injection into the collected WT mouse adipose tissue lysate in vitro, both HD-H2S and LD-H2S gel released H2S to the maximal level, and it lasted more than 48 h (Fig. 2a). CD injections did not result in H2S release. The maximum amounts of H2S released from HD-H2S and LD-H2S were 21 ± 2 µM/mg and 12 ± 1 µM/mg, respectively (Fig. 2b). On Day 4, the HD-H2S injected pale-coloured area disappeared, indicating complete degradation of the injected HD-H2S (Fig. 2c).

a In vitro H2S release study of three peptide amphiphiles—HD-H2S, LD-H2S, CD compared to control samples (C-L) without injections, assessed via HPLC. n = 5 independent replicates for each time point. *p < 0.05 vs C-L. b Net H2S released from peptide gels by subtracting H2S levels in C-L tissues from those in HD-H2S, LD-H2S, CD-injected adipose tissues. *p < 0.05 vs. CD, and #p < 0.05 vs. LD-H2S. c Morphological changes of WT adipose tissues injected with or without HD-H2S. Dashed circles mark the original area with HD-H2S gel injection, while solid-line contours with the pale coloration indicate the actual visualization of the injected HD-H2S gel. d H2S levels in adipose tissues and plasma of WT mice receiving HD-H2S, LD-H2S, and CD injections for 30 days or no injection (Ctrl). n = 6 independent mouse tissue replicates. Statistical significance is shown as *p < 0.05 vs. Ctrl and CD. Abbreviations: HD-H2S-R: High dose (78 µmol/kg) H2S-releasing gel injected in right flank adipose tissue; LD-H2S-R: Low dose (39 µmol/kg) H2S-releasing gel; CD-R: Control gel (non-H2S-releasing) injected in right flank; C-L: Uninjected left flank adipose tissue control used as an internal control for each mouse; Ctrl: mouse given no injection. The error bars in this figure represent standard error.
H2S concentration in H2S gel-injected WT adipose tissue and surrounding tissues
After 30 days of H2S gel injections in vivo, WT mouse adipose tissue injected with LD-H2S and HD-H2S showed a local H2S concentration of 8 ± 3 µM/mg, and 6 ± 2 µM/mg significantly higher than those of non-injected (Ctrl) and CD-injected adipose tissues. H2S concentrations in the liver, spleen, and plasma of WT mice did not show any significant difference among the tested groups (Fig. 2d).
H2S levels and CSE expression in H2S gel-injected WT adipose tissue in vivo
For comparing the changes in total H2S levels in the tissue after 30 days of study, we utilized the lead acetate method. WT adipose tissues exhibited significantly higher H2S levels than that of CSE-KO tissues, as shown by the intensity of darkening on the lead acetate papers. Untreated WT adipose tissues exhibited a 56% higher H2S level than that of untreated CSE-KO adipose tissues. H2S gel injections increased H2S levels in WT adipose tissues, but not in CSE-KO, in vivo (Fig. 3a). CSE, CBS, and MST proteins were all expressed in WT adipose tissues. HD-H2S and LD-H2S treatments upregulated CSE expression in WT adipose tissues compared with the tissues that received non-injection (Ctrl) or CD. MST protein was abundant, but CSE protein was completely undetectable in CSE-KO mice adipose tissues (Fig. 3b, c). There were no significant changes in MST or CBS expression in any group following H2S gel injections, and the expression levels of CSE, MST, and CBS proteins in the adipose tissue of H2S gel-injected CSE-KO mice remained unaltered (Fig. 3b, c).

a Changes in H2S levels in adipose tissues of wild-type (WT) and CSE-knockout (KO) mice after 30 days of injection with HD-H2S, LD-H2S, and CD preparations relative to baseline H2S levels in untreated WT mouse adipose tissues (Ctrl). n = 6 mouse replicates. *p < 0.05 vs. Ctrl and CD, and #p < 0.05 vs. CSE-KO mouse tissues. b Expression levels of CSE, CBS, and MST proteins in WT and CSE-KO mouse adipose tissues after 30 days of treatments. c Relative expressions of CSE, CBS, and MST to β actin and α tubulin. n = 6; *p < 0.05 vs. Ctrl and CD, #p < 0.05 vs. KO mouse tissues. Cys stands for Cysteine; P5P stands for Pyridoxial 5′-phosphate. The error bars in this figure represent standard error.
The effects of H2S gel on lipid accumulation in WT adipose tissue in vivo
After 30 days of local injections of H2S gel, significant increases in lipid content were observed in CSE-KO adipose tissue injected with HD-H2S, but not with LD-H2S (Fig. 4a, b). In both WT adipose tissues, HD-H2S and LD-H2S induced significantly higher lipid accumulation (Fig. 4a, b). Mice injected with CD showed no change in adipose tissue weight or lipid content in the injected adipose tissue of CSE-KO and WT mice (Fig. 4a, b). No change in mouse overall body weights was observed in any animal groups (Fig. 4c).

a Lipid accumulation in CSE-KO and WT subcutaneous adipose tissues outlined by dotted contours. b Changes in lipid content/adipose tissue weight in injected vs. non-injected controls for CSE KO and WT mice. c Changes in mouse body weights of CSE KO and WT mice. n = 6 mouse replicates. *p < 0.05 vs. % change in CD. I stands for Injected side; NI stands for Non-injected side of the same mice; AT stands for Adipose tissue. The error bars in this figure represent standard error.
The effects of H2S gel on the expressions of adipogenic and lipogenic marker proteins in vivo
HD-H2S injections upregulated the expression of PPARγ in CSE-KO mouse adipose tissues (Fig. 5a, b). HD-H2S injection upregulated the expression of mature SREBP (mSREBP) in CSE-KO adipose tissues (Fig. 5a, c). HD-H2S and LD-H2S upregulated PPARγ and mSREBP expression in WT adipose tissues (Fig. 5a, c). An increased expression of adiponectin by LD-H2S, but not by HD-H2S, was noted in WT adipose tissues (Fig. 5a, d). In contrast, HD-H2S significantly decreased adiponectin expression in WT adipose tissues (Fig. 5a, d). PPARγ expressions was higher in WT Ctrl adipose tissue than in CSE KO Ctrl adipose tissue. CD injection did not cause any change in the expression levels of adipogenic or lipogenic marker proteins in both WT and KO adipose tissues (Fig. 5a–d).

a Western blotting of PPARγ, m-SREBP, and adiponectin proteins in adipose tissues from WT and CSE-KO mice. b Relative expression levels of PPARγ protein. c Relative expression levels of m-SREBP protein. d Relative expression levels of adiponectin protein. Open columns and filled columns in (b–d) represent the tissues from WT and CSE-KO mice, respectively, after 30 days of CD injections. *p < 0.05 vs. WT CD-R and C-L; #p < 0.05 vs. CSE-KO CD-R and C-L. n = 6 for each group. m-SREBP stands for mature SREBP. e Oil Red O staining of mouse primary preadipocytes (n = 5). The red staining highlights intracellular lipid accumulation, indicative of adipogenic differentiation. ADP: adipogenic media; Betulin: SREBP inhibitor. Scale bar - 50 µm scale. The error bars in this figure represent standard error.
H2S promotes adipogenesis through an SREBP-dependent pathway in vitro
To determine whether H2S-induced lipid accumulation in adipocytes depends on SREBP activation, primary mouse preadipocytes were treated with adipogenic media (ADP), 20 µM H2S, 10 µM Betulin (a pharmacological SREBP inhibitor), or a combination of H2S and Betulin, and assessed by Oil Red O staining (Fig. 5e). Cells treated with ADP alone exhibited moderate lipid accumulation, as indicated by intracellular red staining. H2S treatment significantly enhanced lipid accumulation compared to the ADP group, confirming its pro-adipogenic effect. In contrast, Betulin-treated cells showed markedly reduced lipid staining, consistent with SREBP inhibition suppressing adipogenic differentiation. Notably, co-treatment with H2S and Betulin partially restored lipid accumulation compared to Betulin alone, suggesting that exogenous H2S can counteract the inhibitory effects of SREBP suppression. These findings support the hypothesis that H2S promotes adipogenesis at least partially through SREBP-dependent mechanisms.
The effects of H2S gel on lipid accumulation in vivo
No significant change in adipocyte diameter was observed in adipocytes from CSE-KO mice that received LD-H2S injections (Fig. 6a, b). In HD-H2S injected WT adipose tissue, and CSE-KO tissues, hypertrophic adipocytes were observed with the cell diameter of 89.61 ± 3 µm and 81.56 ± 5 µm, significantly larger than those from non-injected (Ctrl) and the CD-injected WT adipose tissues (Fig. 6a, b).

a Cryosection images of adipose tissue from CSE-KO and WT mice, showing adipocyte morphology. Scale bars, 200 µm. b The cell diameters of adipocytes from CSE-KO and WT mice treated with CD-R, LD-H2S-R or HD-H2S-R, or no treatment (C-L). *p < 0.05 vs. WT CD-R and C-L. #p < 0.05 vs. CSE-KO CD-R and C-L. c Representative images of adipose tissue stained with Oil Red O to visualize lipid accumulation (red, lipid droplet marker), along with immunofluorescence staining for Perilipin (green, lipid droplet coating protein), and DAPI (blue, nuclear stain). Merged images were used to assess lipid droplet morphology and size. Scale bars, 50 µm. Frequency distribution of lipid droplet size from CSE-KO (d) and WT (e) mice using ImageJ software. n = 6 independent replicates, each replicate is the average of 10 images. *p < 0.05 vs. respective C-L. The error bars in this figure represent standard error.
The frequency distribution of lipid droplet diameters in KO and WT adipose tissues reveals distinct trends following H2S gel injection. HD-H2S significantly increased lipid droplet sizes, particularly in WT adipocytes. About 41% of lipid droplets in HD-H2S injected WT mouse adipocytes fell in the diameter range of 100–150 µm, whereas non-treated WT cells only had about 1.7% of lipid droplets in the same diameter range (Fig. 6c, e). The most lipid droplets in non-treated CSE KO adipocytes had the diameter in the range of 60–80 µm (Fig. 6c, d) whereas the HD-H2S-treated KO cells exhibited about 22.1% of lipid droplets in the diameter range of 100–150 µm. However, the increase in lipid droplet size was less pronounced in CSE KO mice treated with LD-H2S, which aligns with lower lipid accumulation observed in these mice (Fig. 4b). These observations reveal that H2S increase adipocyte size as well as lipid droplet size with a more pronounced impact on WT mice.
The effects of H2S gel on perilipin expression and HSL activation in vivo
HD-H2S and LD-H2S injection significantly increased perilipin expression in CSE-KO adipose tissues (Fig. 7a, b). While Perilipin expression remained the same in WT adipose tissues following injections of LD-H2S and HD-H2S (Fig. 7a, b). In contrast, LD-H2S and HD-H2S had no effect on the phosphorylation of HSL (pHSL) in CSE-KO adipose tissues (Fig. 7a, c). HD-H2S injection significantly increased pHSL level in WT adipose tissues (Fig. 7a, c). There were no discernible changes in perilipin or pHSL expressions in CSE-KO and WT adipose tissues that received CD injection or in the tissues from non-injected sides of CSE-KO and WT mice.

a The expression of perilipin, HSL, and pHSL proteins in adipose tissue from WT and CSE-KO mice after CD-R, LD-H2S or HD-H2S injections and their controls (C-L). b The expression levels of perilipin. c The expression levels of pHSL/HSL in subcutaneous adipose tissue from CSE-KO and WT mice after 30 days of LD-H2S or CD injections. *p < 0.05 vs. respective C-L and CD-R; #p < 0.05 vs. CSE-KO CD-R and C-L, n = 6 for each group. HSL stands for hormone-sensitive lipase; pHSL stands for phosphorylated HSL. The error bars in this figure represent standard error.
The effects of H2S gel on macrophage infiltration
CD68 immunofluorescence staining was used to assess macrophage infiltration in adipose tissue from wild-type (WT) and CSE-KO mice (Fig. 8). In WT mice, LD-H2S and HD-H2S treatment increased CD68 fluorescence intensity compared to the control and C-E4-treated groups (Fig. 8a, b). Quantification showed a significant increase in corrected total cell fluorescence in LD-H2S and HD-H2S -treated groups (p < 0.05). In CSE-KO mice, LD-H2S did not significantly alter CD68 fluorescence, whereas HD-H2S treatment led to a significant increase (Fig. 8c, d). Control and C-E4 groups showed no significant differences in fluorescence intensity.

a Representative immunofluorescence images of adipose tissue sections from WT and CSE KO mice stained for Caveolin-1 (green, marking lipid raft domains), CD68 (red, a macrophage marker), and DAPI (blue, staining nuclei). Scale bars: 50 μm. b Quantification of red fluorescence intensity for CD68 in WT and CSE KO adipose tissue (n = 5). *p < 0.05 vs. respective C-L and CD-R. c Immunohistochemistry images of adipocytes marked by CD68 macrophages (dark brown). Scale bars: 50 μm. The error bars in this figure represent standard error.
The effects of H2S gel on collagen accumulation and fibrosis
Picrosirius Red staining was used to evaluate collagen deposition in adipose tissue. Treatment with exogenous H2S induced fibrosis in a dose-dependent manner. In WT mice, both HD-H2S gel and LD-H2S gel increased collagen accumulation, with a more pronounced effect observed at higher H2S concentrations (Fig. 9a, b). In CSE-KO mice, LD-H2S did not induce significant fibrosis, whereas HD-H2S resulted in a mild increase in collagen deposition (Fig. 9a, c). Collagen deposition was higher on the Right side compared to the Left, suggesting localized effects of treatment on ECM remodeling. WT and CSE-KO control groups showed comparable ECM composition, with no significant differences in collagen deposition (Fig. 9a, b).

a Representative images of adipose tissue stained with Picrosirius Red. The left images show regular light microscopy, while the right images show polarized light images, where collagen fibers appear as red or yellow. Red collagen represents thicker, older collagen fibers, while yellow collagen indicates thinner, newly formed collagen fibers. Scale bars = 50 μm. b The ratio of red/yellow collagen fibers in WT and KO mouse adipose tissue sections, analysed based on polarized image. c The total collagen area (%) in the adipose tissue of WT and KO mice. *p < 0.05 vs. WT C-L and CD-R; #p < 0.05 vs. CSE-KO C-L and CD-R, n = 5 for each group. The error bars in this figure represent standard error.
Discussion
A plethora of physiological and pathophysiological effects of H2S has been reported. Increased H2S concentration is beneficial for the health of numerous systems, such as the cardiovascular, respiratory, neuronal, or reproductive systems7,8,9,13. On the other hand, over-production of H2S may constitute a pathogenic factor for diabetes18,19,20,21, obesity10,12, and certain types of inflammation or cancers22. These differential effects of H2S on specific targets render the interpretation of the outcome of systematic H2S administration perplex and inconclusive. Administration of H2S salts, such as NaHS and Na2S, would deliver H2S that would distribute to and disappear from the whole body. The frequently employed H2S-releasing compound, GYY4137, is a water-soluble salt that liberates H2S in a slow process23,24. It is administered through daily injection over many days or weeks, and the released H2S would disperse widely and enter the circulation. Other available means for H2S delivery, such as H2S-prodrugs or H2S hybrids with other known compounds, face the same challenge of reaching the specific targets without the system-wide spill-over effect24. Our results from the current study offer one solution for this challenge for localized delivery and retention of H2S while achieving regional effect.
The SATO-Ile-Ala-Val-Glu-Glu (SATO-IAVE2) gelator is a peptide sequence linked to an S-aroylthiooxime (SATO), forming an aromatic peptide amphiphile (APA) hydrogel17,25,26,27. The gelation of H2S gel was initiated by calcium ions that crosslink the peptide units through salt bridges, thus aiding the formation of hydrogel17,25,26,27. H2S gel is a biodegradable gel, and as cysteine reacts with the SATO unit, H2S is released, and the gel degrades (Fig. 1)17,25,26,27. The 3D network forms a depot in the form of a hydrogel near the site of action, ensuring that the drug is delivered precisely where it is needed. This technique limits the delivery of H2S to a specific localized area, which is the adipose tissue in our case. Moreover, the localized H2S gel can provide continuous and efficient drug delivery. A control peptide hydrogel of a similar chemical structure but with an oxygen atom in place of the sulfur was also studied (CD) as a material control that was incapable of H2S release.
H2S gel was confirmed to be safe in our current study as it showed no adverse effects on mice behavior or mortality rate, neither causing any change in the body weights of WT and CSE-KO mice. H2S gel was absent in or near adipose tissue after 30 days of injections, indicating that the gel had degraded before the subsequent injection. This aligns with our in vitro H2S gel release studies, which predicted that the gel would degrade before the next injection given to the mice in vivo (6 days). In vitro studies revealed that H2S gel provided a controlled slow release of H2S for up to 72 h upon contact with cysteine, whether endogenously or exogenously supplied. The H2S concentration was found to be highest in adipose tissue after 12 h of injection during in vitro testing. Similarly, in a 30-day in vivo study on mice, adipose tissue exhibited a high H2S concentration, while no significant increase was observed in H2S concentration in the plasma. These results indicate that H2S release was restricted to adipose tissue, effectively targeting the delivery and retention of H2S to this specific site. Elevated exogenous H2S levels in regional adipose tissues in mice led to the accumulation of lipids in adipocytes, validating the effectiveness of H2S gel in achieving localized H2S delivery and localized functional changes.
Adipose tissue plays a critical role in regulating lipid metabolism and energy storage, but excess body fat, often associated with obesity, can disrupt this balance, leading to metabolic disorders, inflammation, insulin resistance, and conditions like type 2 diabetes and cardiovascular disease1,2,3,4,5. The three key stages of lipid metabolism in adipose tissue are adipogenesis, lipogenesis, and lipolysis. Adipogenesis involves the differentiation of preadipocytes into mature adipocytes, primarily driven by insulin and transcription factors. These mature adipocytes esterify fatty acids and glycerol to form triacylglycerol (TAG). Lipogenesis encompasses the synthesis of fatty acids and their incorporation into various lipids. De novo lipogenesis, a subset of this process, occurs primarily in the liver during energy surplus, synthesizing fatty acids from non-lipid precursors like glucose6,7,8. Lastly, lipolysis involves the enzymatic breakdown of stored TAGs in adipocytes, releasing free fatty acids and glycerol into the bloodstream, with HSL playing a crucial role in initiating triglyceride hydrolysis8,28.
Our previous in vitro experiments proved that H2S increased the rate of adipogenesis and elevated lipid levels in cultured adipocytes12,14. To investigate the in vivo adipogenic and lipogenic effects of H2S, we injected H2S gel hydrogel into mouse subcutaneous adipose tissue (SAT). While other fat depots, such as visceral adipose tissues, have distinct endocrine functions and hormone secretion, SAT stands out quantitatively as the largest adipose depot with a significant contribution to lipid storage and release. This makes SAT an ideal target for studying the impact of H2S on adipose tissue dynamics29.
In WT mice, we observed an increase in adipose tissue lipid content after having injected low- and high- doses of H2S gel. In CSE-KO mice, only the high dose of H2S gel increased lipid content of adipose tissues. The major difference between WT and CSE-KO mice is the minimal level of endogenous H2S in the latter group, which may thus elevate the threshold level of exogenous H2S to increase local lipid levels. Previous in vitro cellular studies have indicated a specific concentration range where exogenous H2S exhibited the adipogenic effect10,30. Moreover, exogenous H2S may stimulate additional endogenous H2S release by increasing CSE expression as reported in the plasma, ischemic heart tissue31, and mammalian myocardial cells32. This exogenous H2S-induced reinforcement of endogenous H2S production was shown in the current study where H2S gel increased CSE expression and H2S production in WT adipose tissues, but not in CSE-KO adipose tissues due to their lack of CSE expression (Fig. 4).
We then asked whether H2S gel can upregulate the expression of other H2S-generating enzymes in addition to CSE. H2S can be produced by at least three enzymes, i.e., CSE, cystathionine beta synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (MST) in vivo9,10,11. In mouse adipose tissue, CSE was the most abundantly expressed, followed by MST and CBS14. Under laboratory conditions, the cystathionine aminotransferase-MST pathways produce H2S with optimal alkaline conditions and high cysteine concentration10. However, under more physiological conditions, MST fails to produce meaningful H2S in rat liver and kidney10,33. Moreover, the unstable nature of 3-mercaptopyruvate poses a significant challenge as it is the only sulfur donor for MST, and its presence in the body is not yet directly detected10. Although MST and CBS expressions were observed in CSE-KO mouse adipose tissues (Fig. 4), H2S gel injections did not change their expression nor their H2S production rates (Figs. 2a and 4a).
Consistent with our previous in vitro studies12, the current study demonstrated that increased local H2S concentration stimulated adipogenesis and lipogenesis, leading to larger lipid droplets and increased lipid content in mouse adipose tissue. High doses of H2S gel led to higher local H2S concentration and increased hypertrophic adipocytes with larger lipid droplets in the injected mouse adipose tissues.
The current study revealed that low doses of H2S gel had no effect on lipid accumulation in CSE-KO adipose tissue in vivo. However, our previous in vitro experiments showed increased adipogenesis at NaHS concentrations ranging from 10–100 µM in CSE-KO adipocytes12. This discrepancy may stem from the slowly delivered H2S not reaching the necessary concentration threshold to exert its physiological effects in vivo. Furthermore, our current in vivo study shows that WT adipocytes injected with HD-H2S exhibited hypertrophy and reduced pHSL/HSL expression, but this effect was not observed in H2S-induced hypertrophic adipocytes in vitro12. This difference may be related to the diverse effects of H2S on matured adipocytes, preadipocytes, and other types of cells in vivo.
The mechanisms for H2S-induced local lipid accumulation revealed in the current in vivo study aligns well with our previous in vitro research12. The expression levels of PPARγ, a key adipogenic marker and master regulator of adipogenesis and lipid storage34,35, are upregulated in both WT and CSE-KO mice by H2S gel injections. SREBP belongs to the family of lipogenic transcription factors governing the expression of various lipogenic enzymes. Full SREBPs (fSREBPs) are synthesized and bound to the rough endoplasmic reticulum membrane as full-length precursor proteins. Upon activation, fSREBPs undergo sequential cleavage yielding matured SREBP (mSREBP), which enters the nucleus to activate transcription by binding to specific sterol response elements in the promoter regions of lipogenic genes35,36,37. The activation of SREBP to mSREBP serves as an indicator of the tissue’s lipogenic activity. Using mSREBP inhibitors in mouse primary preadipocytes, we found that H2S treatment significantly increased lipid accumulation (via Oil Red O staining), which was abolished by pre-treatment with the inhibitor. This suggests H2S promotes lipid biosynthesis through mSREBP reactivation. Although our focus remains on the pharmacological effects of localized H2S delivery via H2S gel, these findings propose a potential mSREBP-dependent mechanism underlying H2S-induced lipid accumulation. The similar upregulation of mSREBP by H2S has been shown in the previous studies on cultured 3T3L1 cells in vitro32.
Adiponectin, a hormone with significant roles in promoting insulin sensitivity, energy balance, and inflammation, is upregulated by PPARγ through direct binding to conserved cis-acting regulatory DNA elements in the promoter region of the adiponectin gene34,35,38,39. In conditions like obesity or when adipocytes undergo hypertrophy, the normal upregulation of adiponectin by PPARγ is hindered, leading to reduced adiponectin expression. This disruption primarily stems from chronic low-grade inflammation within adipose tissue in obesity, marked by the release of inflammatory cytokines such as TNF-α and IL-639. These cytokines can disrupt PPARγ‘s function and its capacity to promote adiponectin expression by competing with PPARγ for coactivators that are essential for its transcriptional activity. The injections of low-dose H2S gel upregulated adiponectin expression in WT adipose tissues. The stimulatory effect of H2S on adiponectin expression was previously shown with the cultured primary adipocytes12 as well as in another study on 3T3L1 cells40. In contrast, HD-H2S injected WT adipose tissue with both larger lipid droplets and larger average adipocyte diameter exhibited downregulated adiponectin expression, indicating putative toxic effects of high doses of exogenous H2S in the presence of endogenous H2S production39,41.
Perilipin coats lipid droplets, preventing their breakdown under normal conditions. When energy deficits occur, protein kinase A (PKA) phosphorylates perilipin, dissociating it from the lipid droplets. This action enables HSL and adipose triglyceride lipase (ATGL) to breakdown the droplets into fatty acids and glycerol28,42,43,44,45,46,47. H2S-induced lipid accumulation involves the formation of large lipid droplets and inhibit perilipin1-mediated lipolysis42,43,44,45,46,47. The presence or increase in perilipin indicates the maturation, enlargement, and fusion of adipocyte lipid droplets. In our previous cell study in vitro12, H2S treatment during adipocyte differentiation did not affect free fatty acid concentration or HSL phosphorylation, suggesting that H2S-stimulated adipogenesis and lipogenesis are independent of lipolysis. However, the current in vivo mouse study showed that a high dose of H2S gel significantly increased HSL phosphorylation in WT mouse adipose tissues, which could be due to the significant increase in lipid accumulation in WT adipose tissue. HSL is an indicator of lipolysis since it is the primary enzyme responsible for mobilizing free fatty acids from adipose tissue28,46. In CSE-KO mouse adipose tissue, perilipin levels increased with H2S gel injections but not in WT adipose tissue (Fig. 7). One reason for these divergent results between in vitro and in vivo studies may be the complex regulation of lipolysis, influenced by factors such as the nervous system, hormones, and paracrine/autocrine factors, which may not be completely recapitulated in vitro. Furthermore, the enlargement of adipocytes causes higher storage of triglycerides. Yet, the expression level of perilipin does not necessarily increase, because perilipin predominantly controls lipolysis, not triglyceride/lipid synthesis44.
Our results demonstrate that H2S influences macrophage infiltration and fibrosis in a concentration-dependent manner in mouse adipose tissues. WT tissues exhibited greater macrophage presence compared to CSE-KO tissues, suggesting that endogenous CSE-derived H2S promotes immune cell recruitment. Picrosirius red staining revealed that increased H2S levels were associated with enhanced collagen deposition and fibrosis, but only at higher concentrations. Specifically, in WT mice, both LD-H2S and HD-H2S increased collagen accumulation, with a more pronounced effect at higher H2S concentrations. This increase was accompanied by a higher red/yellow collagen ratio in WT adipose tissues, particularly in the HD-H2S group, indicating enhanced collagen crosslinking and maturation. Red collagen represents newly synthesized, loosely packed fibers, while yellow collagen corresponds to mature, highly crosslinked fibers that contribute to fibrosis. In contrast, CSE-KO mice treated with LD-H2S showed no significant changes in fibrosis, while HD-H2S induced a mild increase, suggesting that high-dose exogenous H2S may trigger ECM remodeling. However, the lower red/yellow collagen ratio in KO mice reinforces the idea that endogenous H2S is necessary for robust collagen crosslinking and ECM remodeling.
Immunohistochemical staining for CD68 showed that H2S gel treatment increased macrophage infiltration in WT mice, with a stronger effect at higher doses, suggesting a dose-dependent inflammatory response to exogenous H2S. In contrast, CSE-KO control mice exhibited lower macrophage presence, indicating that endogenous H2S plays a role in basal macrophage regulation. While LD-H2S did not significantly alter macrophage infiltration in CSE-KO mice, HD-H2S led to a moderate increase, suggesting that exogenous H2S at high doses can partially restore macrophage recruitment in the absence of CSE.
Previous studies suggest that H2S regulates ECM remodeling and immune cell recruitment in a dose-dependent manner48,49. The lack of significant fibrosis differences between WT and CSE-KO control groups suggests that CSE-derived H2S does not actively regulate ECM composition under normal physiological conditions. Instead, H2S primarily modulates immune cell recruitment, as reflected in the higher macrophage presence in WT control mice. The red/yellow collagen ratio findings further support this, as WT mice exhibited a clear dose-dependent increase, whereas KO mice did not show a significant response.
Our findings suggest that at physiological levels, H2S primarily influences immune homeostasis, while at higher concentrations, it drives immune activation and ECM remodeling. Potential mechanisms include oxidative stress modulation, macrophage polarization shifts toward a pro-inflammatory M1 phenotype, and interactions with cytokine-ECM signaling pathways50.
Conclusion
This study demonstrates that localized, slow-releasing delivery of H2S via the injectable H2S hydrogel promotes lipid accumulation, adipocyte hypertrophy, and tissue remodeling in subcutaneous adipose tissue in vivo. By comparing WT and CSE-KO, we highlight the physiological role of endogenous H2S in adipose regulation and confirm a dose-dependent threshold effect for exogenous H2S. While our study emphasizes the pharmacological impact of localized H2S delivery, mechanistic insights—particularly involving adipogenic regulators like SREBP—were primarily supported by in vitro analyses. Overall, our findings offer a platform for targeted H2S modulation in metabolic tissues and suggest that precise control of H2S levels may be critical for maintaining adipose tissue homeostasis and preventing obesity-related dysfunction.
Impaired H2S metabolism is related with metabolic disorders, including obesity, insulin resistance, and inflammation12,14,40. At physiological levels, H2S supports adipogenesis and energy homeostasis, but excessive accumulation of H2S disrupts lipid metabolism and dysregulate adipocyte phenotype. Previous studies have shown that high-fat diet (HFD) feeding of C57BL/6 J mice increases CSE expression and H2S production in adipose tissue, underscoring the metabolic importance of the CSE/H2S system14.
To our knowledge, our study is the first to demonstrate that localized, slow-releasing H2S delivery via the injectable H2S hydrogel induces obesity-like remodeling of adipose tissue in vivo, including lipid accumulation, adipocyte hypertrophy, macrophage infiltration, and fibrosis (Fig. 10). We show that total H2S levels—both endogenous and exogenous—correlate with increased lipid content in subcutaneous adipose tissue, and that hypertrophy occurs at the highest concentrations of H2S. While previous cellular studies reported similar adipogenic effects of H2S in vitro12,14,40, this work provides direct in vivo evidence of site-specific H2S-driven adipose expansion.

Subcutaneous injections of H2S-releasing hydrogel release H2S in subcutaneous adipose tissue, which increases adipogenesis and lipogenesis by upregulation of PPARγ and SREBP. Consequently, adipocyte hypertrophy and tissue remodeling are promoted, resulting in adipose tissue dysfunction characterized by increased CD68+ macrophage infiltration, fibrosis, elevated perilipin and pHSL levels, and altered adiponectin expression. Created with BioRender. Verma, R. (2025) https://BioRender.com/1hrgiek.
Importantly, our comparative analyses using CSE-KO and WT mice reveal that endogenous H2S is required to sensitize adipose tissue to exogenous H2S, supporting a threshold effect in adipogenesis and lipogenesis9,12,14. CSE-KO mice showed decreased lipid accumulation, and downregulated expression of adipogenic transcription factors (PPARγ and SREBP), highlighting the multifaceted role of endogenous H2S in maintaining adipose tissue homeostasis9,12,14.
Moreover, our findings reveal that high concentrations of H2S induce a pathological shift, including downregulation of adiponectin and promotion of macrophage infiltration and fibrosis (Fig. 10) mirroring conditions seen in dysfunctional adipose tissue in obesity51. This biphasic effect of H2S, where low doses promote homeostasis and high doses drive dysfunction, has also been observed in cancer biology52. Previous studies suggest that in obese adipose tissue, despite increased CSE expression, H2S bioavailability is reduced due to oxidative consumption, leading to enhanced Ca2+ influx and M1 macrophage polarization53. Our results suggest that exogenous H2S at high doses may similarly exacerbate inflammation and immune activation in adipose tissue.
Clinically, these findings carry substantial implications. The ability to precisely modulate H2S levels in adipose tissue using localized, biodegradable hydrogels like H2S gel opens therapeutic avenues for obesity and related metabolic diseases. Tissue-specific H2S inhibition or scavenging strategies could help restore adipose tissue function in the presence of excess of H2S, while controlled delivery could support regeneration where H2S is deficient. Moreover, the platform’s adaptability offers potential for organ-targeted interventions in other H2S-linked conditions beyond adipose biology.
The study further suggests that H2S-regulated adipogenic pathways are conserved across species and may be relevant to human adipose biology. However, additional studies in diverse animal models and human tissues are needed to confirm the broader applicability and therapeutic potential of targeting the CSE/H2S axis.
Ultimately, our study provides critical insight into the dual physiological and pathological roles of H2S in adipose tissue, while laying a strong foundation for precision-based therapeutic strategies that harness or restrain H2S in a tissue-specific and clinically translatable manner.
Material and Methods
Preparation procedure for H2S gel, control gel, and their H2S release in the presence of adipose tissues
An aromatic peptide amphiphile (APA) hydrogel was used, featuring an S-aroylthiooxime (SATO) functional group at the N-terminus of the IAVE2 peptide sequence Ile-Ala-Val-Glu-Glu (Fig. 1). The peptide was synthesized in a similar manner to a related peptide25. When combined with 10% CaCl2, the APA forms a biodegradable hydrogel inside the tissue. The SATO unit of the APA releases H2S upon contact with thiols like cysteine and glutathione26. A control APA, called Control-IAVE4 (CD), has similar rheological properties to SATO-IAVE2 (H2S gel) but has an oxygen atom in place of the sulfur and cannot release H2S25,26. CD and two doses of SATO-IAVE2 were used, SATO-IAVE2-Low (LD-H2S; 39 µmol/kg H2S) and SATO-IAVE2-High (HD-H2S; 78 µmol/kg H2S).
Animal and tissue preparation
Wild-type (WT) mice were maintained on C57BL/6J × 129SvEv background. In-house-bred homozygous CSE-KO mice were generated after backcrossing of at least 10 generations of heterozygous of CSE-KO mice with WT mice with the same genetic background54. All WT and CSE-KO mice were verified by genotyping and used in this study. Each group of WT and their matching CSE-KO mice were from the same generations. WT and CSE-KO mice were maintained on standard rodent chow and had free access to food and water. Mice were monitored daily for general health, including activity level, grooming behavior, body weight, and signs of distress. No adverse effects or signs of pain or illness were observed throughout the study duration. All animal experiments conducted were in compliance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and were approved by the Animal Care Committees of York University and Laurentian University in Canada.
Only male mice aged 5 weeks were included to reduce variability due to sex and developmental stage. No additional inclusion or exclusion criteria were applied. Male mice of 5 weeks old received injections at intervals of every 6 days over a 30-day period. To minimize pain, stress, and discomfort, mice were lightly anesthetized using isoflurane during each injection procedure, in accordance with approved animal care protocols. We have complied with all relevant ethical regulations for animal use.
H2S gel was prepared as previously described17 by mixing 63 µL of H2S gel solution (0.01 mg/mL in saline) and 7 µL of 10 wt.% CaCl2 solution in saline. H2S gel CaCl2 solution was quickly injected into the subcutaneous adipose tissue (SAT) located on the right rear flank side of the mouse body, between the ribcage and the hind legs. The SAT on the left rear flank side served as a non-injected control. At the end of the experiment, the mice were euthanized. Regional SAT from both the left and right rear flank at the injection sites were removed, weighed, and immediately frozen in liquid nitrogen and stored at −80 °C.
Measurement of H2S levels under different conditions
In vitro H2S release from aromatic peptide amphiphile
A Reaction Mixture was prepared by mixing 63 µL of H2S gel solution and 7 µL of CaCl2 solution, prepared as described above, with 2 mL of 50 mM sodium carbonate buffer (pH 8.8) containing 400 mg adipose tissues and protease inhibitors and 10% penicillin-streptomycin solution (Sigma-Aldrich). A control for this reaction mixture had identical mixture components except for the absence of H2S gel solution.
At predetermined intervals (0, 6, 12, 24, 48, and 72 h), 200 µL of adipose tissue lysate were withdrawn from the Reaction Mixture, named as Measure Unit here. To each Measure Unit, 250 µL of 1% zinc acetate and 50 µL of double-distilled water were added and mixed. Then, 50 µL of N, N-dimethyl-p-phenylenediamine sulphate (DPD) and 50 µL of FeCl3 were added to the fully mixed Measure Unit in the dark for 20 min. The released sulfide reacted with DPD in the presence of FeCl3 under acidic conditions, yielding methylene blue. Subsequently, 50 µL of 10% trichloroacetic acid (TCA) was added to denature and precipitate all the protein in the Measure Unit, which was then centrifuged at 14000 RPM for 5 min. The supernatant was used for measuring the formation of methylene blue at an absorbance of 670 nm.
H2S levels in mouse adipose tissues
Adipose tissues, the liver, and spleen of 100 mg each were removed from WT mice 30 days after H2S gel injection and homogenized in 0.5 mL of 50 mM sodium carbonate buffer (pH 10). Blood samples were also obtained from the same mice. We mixed 200 µL of tissue lysates or 200 µL of plasma with 250 µL of 1% zinc acetate and 50 µL of double-distilled water. DPD of 50 µL and FeCl3 of 50 µL were then added in the dark for 20 min. Trichloroacetic acid (TCA) of 50 µL at 10% was added to denature and precipitate proteins, followed by centrifugation at 14000 RPM for 5 min. The supernatant containing methylene blue was filtered through a 20 µm filter for H2S measurement.
HPLC measurement of H2S level
A Dionex Ultimate 3000 U-HPLC with a diode array detector set to 660 nm (Thermo Scientific) allows a lower detection limit of 650 nM of methylene blue. Aliquots (200 µL) of the filtered sample solutions were injected into a C18 column (250 × 4.6, 5 μm, Thermo Scientific Hypersil GOLD). A 20 min gradient elution, using acetonitrile and 0.1% (v/v) trifluoroacetic acid in water as a mobile phase, was run at a flow rate of 1 mL/min. The gradient elution protocol used acetonitrile and water in various proportions, ranging from 10% to 50% acetonitrile and 50% to 90% water with 0.1% trifluoroacetic acid. Standard solutions of NaHS were prepared from stock solution with appropriate dilution in 1% zinc acetate solution. DPD and FeCl3 were then added and kept in the dark for 20 min to produce methylene blue. TCA was added to keep the conditions consistent with tissue samples. The methylene blue peak was detected on the HPLC chromatogram at a retention time of 12.33 min. The area under each peak was plotted against corresponding NaHS standards to construct the H2S calibration curve.
H2S gel in vitro degradation study
Adipose tissue samples were extracted from WT mice and incubated in Dulbecco’s Modified Eagle’s Medium (DMEM, 4.5 g/L glucose, SA) supplemented with 10% fetal bovine serum (FBS) and 1% pen/strep at 37 °C in vitro. We injected H2S gel, prepared as described above, into the tissue in vitro, and photographs were taken to document the changes in the physical appearance of the injected H2S gel.
Changes in total H2S levels of mouse adipose tissues
Adipose tissues were sonicated in an ice-cold PBS buffer. In a 96-well plate, 60 μL of cell lysates with an equal amount of protein (200 μg) were mixed with 10 μl of cysteine (10 mM), 10 μl of pyridoxial 5′-phosphate (2 mM), and 20 μl of double-distilled water. A lead acetate paper prepared by soaking filter paper in a 1% lead acetate solution followed by drying was placed over the well for 2 h at 37 °C in the dark. The intensity of the darkening on the lead acetate paper, reflecting H2S level55,56, was analyzed using Image J software. The percent increase/decrease in H2S generated from our H2S gel injected tissues was relative to the WT control (WT Ctrl) multiplied by 100.
Lipid extraction from adipose tissue
The SAT fat pads from both the right (injection site) and left (control site) rear flank regions were removed, and the Bligh and Dyer extraction method30,57 was used to extract the lipid, which was then normalized by adipose tissue weight. Tissue samples of 300 mg were vortexed with 300 µL chloroform and 600 µL methanol five times, periodically after every minute. Then, 300 µL chloroform was added, and the mixture was homogenized for five minutes (1:1, v/v chloroform/methanol ratio) on ice. Next, 300 µL double-distilled water was added and the mixture filtered using a 40 µm filter. The filtrate was incubated on ice for 10 min, and then centrifuged at 2000 rpm for 5 min at 4 °C. The lower organic phase containing chloroform was transferred to a new glass vial. The lipid from the aqueous layer and the leftover adipose tissue from the filter were re-extracted with 500 µL of 1:1 chloroform/methanol (v/v). The chloroform layers were pooled and dried with a nitrogen stream. The lipid content was weighted and normalized using total tissue weight = Lipid extracted (mg)/adipose tissue weight (mg). The lipid content of the injected (right side) vs. non-injected control (left side) subcutaneous adipose tissue (AT) was calculated using the following equation.
(Right AT, H2S gel/CD injected side; Left AT, non-injected control side.)
The aqueous upper layer of tissue homogenates was centrifuged again at 2000 rpm for 5 min at 4 °C, and supernatant was collected for western blot analysis.
Adipocyte size and lipid droplet size measurement
Adipose tissues were first collected and stored at −80 °C until processing. Prior to imaging and staining, tissues were cryosectioned into 5 µm thick sections and mounted on adhesive-coated Superfrost Plus slides. The sections were then thawed at room temperature for 10 min, after which images of unstained sections were captured using an IX71 Inverted Microscope (Olympus) for later analysis. These images were stored for subsequent processing in ImageJ. Immediately after imaging, the tissue sections were fixed in 4% paraformaldehyde (PFA) at 4 °C for one hour, followed by three PBS washes (5 min each) to remove residual fixative.
Cell size analysis
Stored images of unstained sections were processed in ImageJ by converting them to grayscale. To enhance contrast, the “Enhance Contrast” function was applied, normalizing brightness and setting saturated pixels to 0.3%. If the images exhibited uneven illumination, the rectangular selection tool was used to divide the image into quadrants for separate analysis. Thresholding was applied to distinguish cells from the background, with adjustments to the red-highlighted regions for optimized segmentation. The images were then converted to binary, and morphological processing, including hole filling, erosion, and dilation, was applied to refine cell structures. To separate overlapping cells, the Watershed function was used, ensuring clear boundary definition. Cell size was measured using the Analyze Particles function, with appropriate size and circularity filters. When necessary, pixel-based measurements were converted to micrometer units using a known scale for accurate quantification.
Perilipin and Oil red O immunofluorescence
Following imaging of unstained sections, immunofluorescence staining was performed to visualize macrophages and adipocyte membranes. After fixation, sections were blocked with 5% bovine serum albumin (BSA) in PBST (PBS with 0.1% Tween-20) for one hour to prevent non-specific binding. Primary antibody incubation was performed using Perilipin (Cell Signaling Technology, Danvers, MA, USA) at a 1:100 dilution in 5% BSA in PBST, followed by another PBS rinse. Secondary antibody incubation was conducted using Goat anti-Rabbit IgG (H + L) (Thermo Fisher, Waltham, MA, USA) at a 1:00 dilution in 5% BSA in PBST for 1 h.
For lipid accumulation assessment, Oil Red O staining was performed after immunofluorescence imaging. Sections were incubated in Oil Red O solution (saturated in 70% ethanol) for 15 min, followed by gentle rinsing. Finally, the stained sections were mounted in 10% glycerol in PBS with DAPI and covered with a coverslip. Sections were visualized under a fluorescence microscope, where Perilipin-labeled adipocyte lipid droplet (green), Oil red O (red) and nuclei (blue) were identified. To maintain fluorescence signal integrity, all procedures were performed in the dark, and slides were protected from light exposure. Lipid droplet morphology was analyzed using DP2-BSW Version 2.1q (Olympus), and droplet diameters were measured using ImageJ software (National Institutes of Health, USA.
Immunofluorescence detection of macrophages in adipocyte cryosections
Macrophage detection in adipocyte cryosections was performed using immunofluorescence staining with an anti-CD68 antibody, while Caveolin was used as a cell membrane marker to delineate adipocyte boundaries. Adipose tissue cryosections (5–10 µm thickness) were mounted on adhesive-coated slides (Superfrost Plus) and thawed at room temperature for 10–15 min. The sections were fixed in 4% PFA for 1 h at 4 °C, followed by three PBS washes (5 min each) to remove residual fixative. To minimize non-specific binding, sections were incubated with a blocking buffer containing 5% BSA in PBST for 1 h at room temperature in a humidified chamber.
Primary antibody incubation was performed using CD68 (1:100 dilution; Cell Signaling Technology, Danvers, MA, USA) and Caveolin (1:100 dilution), both diluted in 5% BSA in PBST. The slides were incubated overnight at 4 °C in a humidified chamber to enhance specificity. Following incubation, the slides were washed three times with PBS (5 min each). For Immunofluorescence, the secondary antibodies, Alexa Fluor 594 Goat anti-Mouse IgG (H + L) (1:200 dilution) for CD68; Alexa Fluor 488 Goat anti-Rabbit IgG (H + L) (1:200 dilution) for Caveolin, and, for CD68 immunohistochemistry, Peroxidase-linked Goat anti-Mouse antibody (Calbiochem) (1:200 dilution) were diluted in 5% BSA in PBST and applied to the sections. The slides were incubated for 1 h at room temperature in a humidified chamber, followed by three PBS washes (5 min each) to remove excess antibody.
For nuclear visualization in immunofluorescence, sections were counterstained with 10% glycerol in PBS with DAPI (1 µg/mL in PBS) and covered with a coverslip. The stained sections were examined using a fluorescence microscope, where CD68-positive macrophages were identified based on their Alexa Fluor 594 (red) fluorescence. Caveolin-labeled cell membranes were visualized with Alexa Fluor 488 (green), and DAPI-stained nuclei were detected under the DAPI filter (blue). To prevent dehydration, slides were kept hydrated throughout the procedure, particularly during washing and incubation steps, using a humidified chamber. Tissue detachment was minimized by using adhesive-coated slides (Superfrost Plus) and performing gentle washes. Background staining was reduced by optimizing blocking conditions and antibody dilutions.
a. Macrophage detection in adipose tissue cryosections was performed using ImageJ by isolating CD68-positive cells through color thresholding and particle analysis. The fluorescence image was first loaded into ImageJ and split into separate channels, with the red channel selected for analysis. To enhance specificity, a color threshold was applied, adjusting the hue, saturation, and brightness to highlight CD68+ macrophages while minimizing background fluorescence.
Following thresholding, the binary mask was refined using morphological operations to remove noise and separate overlapping macrophages. Watershed segmentation was used for further object separation. To quantify macrophages, particle analysis was performed, setting an appropriate size threshold (10–Infinity µm2) and circularity filter (0.4–1.0) to exclude artifacts. The number, area, and distribution of CD68+ cells were recorded, and an overlay was generated to visualize detected macrophages on the original image.
b. To quantify cell size in adipose tissue sections stained with fluorescence markers, ImageJ was used for image processing, segmentation, and measurement. The fluorescence image was first loaded into ImageJ, and the green fluorescence signal, corresponding to cell boundaries, was isolated followed by selecting the Caveolin-1 green (S) channel. Using the freehand selection tool, individual adipocyte boundaries were manually traced and measured. For each cell, parameters such as area, perimeter, and Feret’s diameter were recorded. Additionally, lipid droplet size was quantified using Oil Red O and Perilipin co-stained sections, allowing for a detailed, cell-by-cell analysis.
Next, a color thresholding method was applied to extract the green-stained regions representing adipocyte membranes. The Hue (H) range was set between 35–90, and the Saturation (S) and Brightness (B) levels were adjusted to eliminate background noise and highlight cell structures. After applying the threshold, the resulting binary mask was refined using morphological operations. Specifically, dilation and erosion were performed to smooth the segmented regions, followed by hole filling to ensure complete cell detection. To further separate overlapping cells, the watershed transformation was applied.
For accurate size measurements, the scale bar present in the image was used to set the pixel-to-micrometer ratio. Cell size quantification was conducted through particle analysis, with size thresholds set to 10-Infinity µm2 to exclude artifacts and non-cellular components. The circularity range was adjusted to 0.3–1.0 to focus on adipocyte structures while excluding elongated artifacts. The analysis provided measurements including cell area (µm2), perimeter, and equivalent diameter.
Picrosirius Red staining for fibrosis in adipose tissue
Adipose tissue cryosections were prepared and stained with Picrosirius Red to assess collagen deposition and fibrosis. Fresh-frozen adipose tissue was sectioned at 5 µm thickness and mounted onto glass slides. The sections were fixed overnight in 4% PFA at 4 °C, followed by three PBS washes (5 min each) to remove residual fixative. After fixation, sections were briefly rinsed in distilled water before proceeding to staining.
To enhance collagen visualization, the sections were incubated in 0.2% phosphomolybdic acid for 5–10 min to remove non-collagenous components. The sections were then stained with 0.1% Picrosirius Red (Direct Red 80 in saturated picric acid) for 60 min at room temperature, ensuring full immersion in the staining solution. After staining, excess dye was removed with a brief rinse in 0.01 N hydrochloric acid (HCl).
For optimal visualization, slides were rinsed in acidified water (0.5% acetic acid in distilled water) and mounted using an aqueous-based or resinous mounting medium. Coverslips were applied carefully to prevent air bubbles. Stained sections were examined under polarized light microscopy, where collagen fibers appeared red or orange, indicating fibrosis, while non-collagenous components appeared yellow or green. The intensity and distribution of Picrosirius Red staining were analyzed to assess the degree of adipose tissue fibrosis.
Western blotting
The aqueous layer of tissue homogenates containing protein was treated with a lipid removal agent (Sigma-Aldrich, St. Louis, MO, USA) and placed on a rocker/orbital shaker at 4 °C for 30 min. Samples were then centrifuged at 15,000 rpm for 15 min, and the supernatant was collected for further analysis. Equal amounts of protein (20 μg in 20 μL/well) were denatured and resolved by SDS-PAGE, then transferred onto nitrocellulose membranes (Pall Corporation, Pensacola, FL). Membranes were blocked in 5% milk in TBST and incubated overnight at 4 °C on a shaker with primary antibodies (1:1000 dilution unless otherwise stated). Following washing, membranes were incubated with HRP-conjugated secondary antibodies (1:2000) and developed using enhanced chemiluminescence (GE Healthcare, Amersham, UK).
The following primary antibodies were used:
-
1.
Hormone-Sensitive Lipase (HSL)—Mouse monoclonal antibody, Santa Cruz Biotechnology, sc-74489 (1:1000)
-
2.
Phospho-HSL (Ser563)—Rabbit polyclonal antibody, Cell Signaling Technology, #4139 T (1:1000)
-
3.
Adiponectin (C45B10)—Rabbit monoclonal antibody, Cell Signaling Technology, #2789, Lot 2 (1:1000)
-
4.
PPARγ (C26H12)—Rabbit monoclonal antibody, Cell Signaling Technology, #2435 (1:1000)
-
5.
SREBP-1 (E9F4O)—Rabbit monoclonal antibody, Cell Signaling Technology, #95879 (1:1000)
-
6.
Perilipin 1 (K117)—Rabbit monoclonal antibody, Cell Signaling Technology, #3467S (1:1000 for WB, 1:100 for Immunofluorescence(IF))
-
7.
α-Tubulin—Rabbit monoclonal antibody, Cell Signaling Technology, #2144S (1:1000)
-
8.
β-Actin—Mouse monoclonal antibody, Sigma-Aldrich, #A2228 (1:1000)
-
9.
CD68—Mouse monoclonal antibody, Invitrogen, #2923549 (1:100 for IF)
-
10.
Caveolin-1—Rabbit monoclonal antibody, Cell Signaling Technology, #3238 (1:400 for IF)
Secondary antibodies compatible with rabbit and mouse IgG were used as appropriate for Western blotting and immunofluorescence.
Cell culture to observe the role of H2S in SREBP activation
After primary preadipocytes were cultured and differentiated into mature adipocytes over a period of seven days12, treatments were administered. It was essential to ensure that terminal differentiation was complete before beginning any further analyses. Adipocytes were divided into four treatment groups to assess the effects of SREBP inhibition and H2S exposure during differentiation:
-
1.
Control Group: Adipocytes were differentiated under standard conditions without any additional treatment, serving as a negative control12.
-
2.
H2S Differentiation Group: 60 µM NaHS was added throughout differentiation (days 0–7) to examine the effects of H2S exposure on adipogenesis.
-
3.
Differentiation Inhibition Group: 10 µM betulin was added throughout the differentiation period (days 0–7) to assess the effect of early SREBP inhibition on adipogenesis.
-
4.
Differentiation + H2S Group: 10 µM betulin was added throughout differentiation (days 0–7), and 60 µM NaHS was added during differentiation (days 0–7) to determine whether H2S can modulate the effects of early SREBP inhibition.
After the treatment period, adipocytes Oil Red O staining was conducted to quantify triglyceride accumulation, providing a functional measure of lipid storage12.
Statistical analysis and reproducibility
All data were analyzed using Microsoft Excel Version 2403 (Redmond, WA, USA) and GraphPad Prism 10.2.1 software (La Jolla, CA, USA), with results expressed as mean ± standard error of the mean (SEM). Criteria for exclusion were established beforehand, excluding extreme outliers beyond the 99% confidence interval of the mean and those exceeding three times the SEM; however, no outliers were identified. Significance was determined at p < 0.05. Sample sizes (n = 6 per group) were chosen based on prior studies showing consistent effects, ensuring statistical reliability while balancing ethical and cost considerations. Animal experiments utilized six independent replicates, while the HPLC method employed five replicates. Unpaired two-tailed Student’s t-tests were employed for comparisons between two groups, as appropriate. For analyses involving three or more groups, either one-way or two-way analysis of variance (ANOVA) was conducted, followed by Tukey’s multiple comparison post hoc test. In animal studies, sample sizes were determined to balance statistical power with cost and ethical considerations for animal welfare. Random allocation of samples/animals into experimental groups was performed at the study’s outset.
To ensure reproducibility, all experimental protocols—including sample preparation, data acquisition, and statistical analysis follow standardized operating procedures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Supplementary information file contains uncropped and unedited blot/gel images. The source data for the graphs presented in this study are provided as an Excel file in Supplementary Data 1. Any additional data or supporting information will be made available from the corresponding author upon reasonable request.
References
Abdelaal, M. et al. Morbidity and mortality associated with obesity. Ann. Transl. Med. 5, 161 (2017).
Bray, G. A. Medical consequences of obesity. J. Clin. Endocrinol. Metab. 89, 2583–2589 (2004).
Jung, R. T. Obesity as a disease. Br. Med. Bull. 53, 307–321 (1997).
Flynn, M. A. et al. Reducing obesity and related chronic disease risk in children and youth: a synthesis of evidence with ‘best practice’ recommendations. Obes. Rev. 7, 7–66 (2006).
Swinburn, B. A. et al. Diet, nutrition and the prevention of excess weight gain and obesity. Public Health Nutr. 7, 123–146 (2004).
Spalding, K. L. et al. Dynamics of fat cell turnover in humans. Nature 453, 783–787 (2008).
Jo, J. et al. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput. Biol. 5, e1000324 (2009).
Haczeyni, F. et al. Causes and mechanisms of adipocyte enlargement and adipose expansion. Obes. Rev. 19, 406–420 (2018).
Wang, R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896 (2012).
Wang, R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter?. FASEB J. 16, 1792–1798 (2002).
Wang, R. The gasotransmitter role of hydrogen sulfide. Antioxid. Redox Signal. 5, 493–501 (2003).
Verma, R. et al. Hydrogen sulfide promotes adipocyte differentiation, hyperplasia, and hypertrophy. Eng 20, 36–48 (2023).
Untereiner, A. & Wu, L. Hydrogen sulfide and glucose homeostasis: a tale of sweet and the stink. Antioxid. Redox Signal. 28, 1463–1482 (2018).
Yang, G. et al. Cystathionine gamma-lyase/hydrogen sulfide system is essential for adipogenesis and fat mass accumulation in mice. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1863, 165–176 (2018).
Zhao, W. et al. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016 (2001).
Verma, R. et al. A review of patents on therapeutic potential and delivery of hydrogen sulfide. Recent Pat. Drug Deliv. Formul. 11, 114–123 (2017).
Carter, J. M. et al. Peptide-based hydrogen sulphide-releasing gels. Chem. Commun. 51, 13131–13134 (2015).
Ali, A. et al. Cystathionine gamma-lyase/H2S system suppresses hepatic acetyl-CoA accumulation and nonalcoholic fatty liver disease in mice. Life Sci. 252, 117661 (2020).
Verma, R. A novel particle-based in situ gelling system for delivering hydrogen sulfide at a sustained rate for potential application in glaucoma and retinal neurodegeneration. Dissertation.https://www.proquest.com/docview/1957431030?pqorigsite=gscholar&fromopenview=true (Creighton University, 2017).
Whitfield, N. L. et al. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1930–R1937 (2008).
Yang, G. et al. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J. Biol. Chem. 282, 16567–16576 (2007).
Wu, L. et al. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab. Investig. 89, 59–67 (2009).
Szabo, C. et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 110, 12474–12479 (2013).
Li, L. et al. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117, 2351–2360 (2008).
Longchamp, A. et al. Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments. Acta Biomater. 97, 374–384 (2019).
Foster, J. C. et al. S-aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics. Org. Lett. 16, 1558–1561 (2014).
Kaur, K. et al. The benefits of macromolecular/supramolecular approaches in hydrogen sulfide delivery: a review of polymeric and self-assembled hydrogen sulfide donors. Antioxid. Redox Signal. 32, 79–95 (2020).
Shen, W. J. et al. Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J. Lipid Res. 50, 2306–2313 (2009).
Patel P., Abate N. Role of subcutaneous adipose tissue in the pathogenesis of insulin resistance. J. Obes. 2013, 489187 (2013).
Iverson, S. J. et al. Comparison of the Bligh and Dyer and Folch methods for total lipid determination in a broad range of marine tissue. Lipids 36, 283–287 (2001).
Elrod, J. W. et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 104, 15560–15565 (2007).
Qipshidze, N. et al. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int. J. Biol. Sci. 4, 430 (2012).
Stipanuk, M. H. & Beck, P. W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 206, 267–277 (1982).
Van Beekum, O. et al. Posttranslational modifications of PPAR-[gamma]: fine-tuning the metabolic master regulator. Obes 17, 213 (2009).
Astapova, O. & Leff, T. Adiponectin and PPARγ: cooperative and interdependent actions of two key regulators of metabolism. Vitam. Horm. 90, 143–162 (2012).
Shimano, H. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J. 276, 616–621 (2009).
Raghow, R. et al. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 19, 65–73 (2008).
Eberlé, D. et al. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848 (2004).
Kadowaki, T. & Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 26, 439–451 (2005).
Tsai, C. Y. et al. Hydrogen sulfide promotes adipogenesis in 3T3L1 cells. PLoS One 10, 0119511 (2015).
Kondo, H. et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes 51, 2325–2328 (2002).
Sun, Z. et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. commun. 4, 1594 (2013).
Sztalryd, C. & Brasaemle, D. L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1862, 1221–1232 (2017).
Sztalryd, C. & Kimmel, A. R. Perilipins: lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 96, 96–101 (2014).
Brasaemle, D. L. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559 (2007).
Tansey, J. T. et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. USA 98, 6494–6499 (2001).
Martinez-Botas, J. et al. Absence of perilipin results in leanness and reverses obesity in Leprdb/db mice. Nat. Genet. 26, 474–479 (2000).
Han, Y. et al. Hydrogen sulfide inhibits abnormal proliferation of lymphocytes via AKT/GSK3β signal pathway in systemic lupus erythematosus patients. Cell. Physiol. Biochem. 31, 795–804 (2013).
Pozzi, G. et al. Buffering adaptive immunity by hydrogen sulfide. Cells 11, 325 (2022).
Whiteman, M. et al. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal. 12, 1147–1154 (2010).
Reggio, S., Pellegrinelli, V., Clément, K. & Tordjman, J. Fibrosis as a cause or a consequence of white adipose tissue inflammation in obesity. Curr. Obes. Rep. 2, 1–9 (2013).
Gao, W. et al. The potential role of hydrogen sulfide in cancer cell apoptosis. Cell Death Discov. 10, 114 (2024).
Velmurugan, G. V. et al. Depletion of H2S during obesity enhances store-operated Ca2+ entry in adipose tissue macrophages to increase cytokine production. Sci. Signal. 8, ra128 (2015).
Yang, G. et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 32, 587–590 (2008).
Hine, C. et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144 (2015).
Li, H. et al. The interaction of estrogen and CSE/H2S pathway in the development of atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 312, H406–H414 (2017).
Manirakiza, P. et al. Comparative study on total lipid determination using Soxhlet, Roese-Gottlieb, Bligh & Dyer, and modified Bligh & Dyer extraction methods. J. Food Compos. Anal. 14, 93–100 (2001).
Acknowledgements
This study has been supported by Discovery Grants from Natural Sciences and Engineering Research Council of Canada to LW (RGPIN-2021-02754) and RW (RGPIN-2023-05099). This work was also supported by the US National Institutes of Health (R01GM123508) to JBM. Figure 1 was created using ChemDraw (chemical structures by J.B. Matson and K. Kaur) and BioRender.com (visual assembly by R. Verma). Figure 10 was created by R. Verma using elements sourced from BioRender.com. Publication licenses have been obtained for both figures to confirm appropriate usage rights.
Author information
Authors and Affiliations
Contributions
R.V. designed and performed the experiments. M.F. assisted with data analysis and performed the mouse injections. H.A. developed the HPLC method. G.Y. contributed to data analysis and interpretation. I.S., K.K. and J.B.M. prepared the H2S-releasing hydrogel. L.W. and R.W. conceptualized and supervised the project and secured funding. R.V., L.W. and R.W. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verma, R., Fu, M., Arif, H.M. et al. Localized delivery and retention of hydrogen sulfide causing regional lipid accumulation in mouse adipose tissues in vivo. Commun Biol 8, 963 (2025). https://doi.org/10.1038/s42003-025-08353-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08353-9
