
Abstract
Gestational diabetes during pregnancy is associated with an increased risk of developmental abnormalities in offspring, but the underlying mechanisms remain unclear. It is not known how maternal metabolism and gut microbes influence kidney development in the fetus. Here we show that gestational diabetes alters maternal gut microbiota and reduces the production of key fatty acids that normally support kidney development in offspring. We find that these changes impair a molecular pathway involving the receptor GPR43, which promotes the growth and migration of kidney cells. In a mouse model, restoring short-chain fatty acids or transferring gut bacteria from healthy donors improves kidney development in offspring, while blocking GPR43 reverses this effect. This study reveals a previously unknown link between maternal gut metabolism and fetal kidney formation and may guide future strategies to prevent congenital kidney disorders in children born to mothers with gestational diabetes.
Similar content being viewed by others
Introduction
Diabetes is a common metabolic disease worldwide, with an increasing incidence. In 2021, the global prevalence of diabetes was estimated at ~10.5%, and it is projected to rise to 12.2% by 20451,2,3. Gestational diabetes mellitus (GDM), as a specific type of diabetes mellitus occurring during pregnancy, has significant health implications for both the pregnant woman and the fetus. Compared to healthy pregnant women, those with GDM are at a higher risk of developing maternal and neonatal complications, as well as long-term metabolic disorders such as type 2 diabetes mellitus, cardiovascular disease, and dyslipidemia. Meanwhile, the offspring of GDM mothers are more likely to develop conditions such as fetal macrosomia, intrauterine growth restriction, and neonatal hypoglycemia4,5,6. Research indicates a complex relationship between GDM and alterations in the intestinal microbiota7. Throughout pregnancy, there are marked changes in the composition of the gut microbiota, characterized by a severe ecological imbalance in GDM patients, with beneficial taxa such as Bifidobacteria and Lactobacilli decreasing while opportunistic taxa like Enterococci increasing8. These changes can be detected early in pregnancy, hinting at potential early indicators of GDM risk9,10. However, the characteristics of GDM intestinal microbiota and metabolic deviations associated with the incidence of offspring congenital anomalies of the kidney and urinary tract (CAKUT) remain unclear.
The diversity of intestinal microbiota is a crucial indicator of the complexity of the gut microbial community11,12. Previous studies have demonstrated that gut microbiota dysbiosis is closely associated with metabolic diseases, including obesity, type 2 diabetes mellitus, cardiovascular diseases, and autoimmune disorders13. Pregnancy induces significant metabolic changes, which play a key role in the susceptibility to GDM14. Therefore, understanding the compositional and functional changes in the gut microbiota of GDM patients may provide novel perspectives for the prevention and treatment of this condition.
Studies have shown significant differences in gut microbiota diversity, abundance, and species composition between GDM patients and healthy pregnant women15,16. Metagenomic analyses indicate that gut microbiota diversity is lower in GDM patients. Specific bacterial taxa, including Ruminococcaceae, Parabacteroides distasonis, Prevotella, Desulfovibrio, Megamonas, and Phascolarctobacterium, are enriched in the gut microbiota of GDM patients17. Ye et al. further reported that GDM patients exhibit reduced gut microbiota abundance, characterized by an enrichment of Verrucomicrobia and a significant reduction in short-chain fatty acid (SCFA)-producing bacteria involved in dietary fiber fermentation18. Using Alpha diversity and Beta diversity analysis and employing 16S rRNA sequencing data from the EMBL-EBI database, researchers can further explore gut microbiota alterations in GDM19,20,21.
On the other hand, metabolomics allows for the precise detection of small-molecule metabolites, reflecting subtle metabolic changes in the body. Several studies have used metabolomic analysis to identify the roles of lipids, carbohydrates, and amino acids in the development and progression of GDM22,23,24. In this study, for the first time, we will utilize metabolomic analysis to explore changes in differential metabolites (DMs) related to CAKUT pathways in GDM patients. By leveraging databases such as KEGG and SMPDB, researchers can investigate the metabolic pathways associated with these differential metabolites25,26. Thus, this study will be the first to analyze the metabolic pathways involved in DMs of GDM patients using relevant databases, further uncovering the underlying mechanisms of GDM pathogenesis.
Gpr43, also known as FFAR2 (free fatty acid receptor 2), is a G protein-coupled receptor primarily activated by SCFAs. Some gut microbiota, such as acid-producing bacteria, generate SCFAs like butyric, propionic, and acetic acids through the fermentation process. These SCFAs act as natural ligands for Gpr43, binding to the receptor and activating it. Through Gpr43 activation, SCFAs reduce the migration and activation of inflammatory cells, helping to regulate inflammation levels in both the intestines and the body as a whole27,28. Furthermore, by signaling through Gpr43, SCFAs contribute to enhancing intestinal barrier function, reducing the invasion of external pathogens and harmful substances, thus safeguarding intestinal health29,30,31. Therefore, this study will identify key Gpr43-related genes through transcriptome sequencing to reveal the regulatory effects of GDM-associated DMs on Gpr43. Additionally, cell and animal experiments will be conducted to verify the impact of the Gpr43 pathway on renal cell proliferation and migration.
The ultimate objective of this study is to elucidate the distinctive characteristics of intestinal microbiota and metabolic variances in GDMGDM patients through bioinformatics analysis and in vitro and in vivo experiments, aiming to investigate their mechanisms affecting offspring’s CAKUT. The research findings hold significant scientific and clinical implications for a better understanding of the pathogenesis of GDM, intervening in pregnancy complications preventatively, and developing novel therapeutic strategies in the future. By comprehensively exploring the interplay between GDM and CAKUT, we can offer more precise diagnosis and treatment for GDM patients, as well as new research perspectives and treatment strategies for preventing CAKUT in offspring, ultimately enhancing the health status of pregnant women and their fetuses. This study proposes the following hypothesis: Alterations in the gut microbiota of GDM patients affect SCFA metabolism, leading to changes in Gpr43 activity in offspring. This impacts renal cell growth and migration, resulting in kidney damage, developmental abnormalities, and ultimately leading to congenital malformations of the kidney and urinary tract in offspring.
Results
Difference in intestinal microbiota composition and functional characteristics in GDM patients
Gut microbiota plays a crucial role in host health and disease, and its alterations in GDM patients were explored in this study. This study investigates the variations in intestinal microbiota among patients with GDM. In the absence of metagenomic sequencing data suitable for our research, we obtained GDM-related 16S rRNA sequencing dataset PRJNA963229 from the SRA database. Five GDM samples and five samples from healthy pregnant individuals were randomly selected for analysis. The decision to use five samples per group was based on resource constraints and experimental design considerations. Although a formal power analysis was not conducted, previous pilot experiments and published literature support that five samples can effectively capture key biological differences32,33. Future studies will incorporate larger sample sizes to enhance the generalizability and reliability of the findings.
The overall experimental workflow, including group allocation, treatment regimens, and sample collection timelines, is illustrated in Fig. S1.
Initially, by examining the α-rarefaction curve, it is evident that the richness of both the GDM and healthy groups gradually reaches saturation as the percentage of samples increases (Fig. S2A). This finding suggests that, at the current sequencing depth, the majority of microbial species have been detected. Furthermore, an assessment of species diversity between the two sample groups was carried out through alpha diversity analysis. Alpha diversity quantifies the species composition within a sample, incorporating both abundance and number. Commonly employed algorithms include Chao1, Invsimpson, Richness, Shannon, Simpson, and ACE indices. The results indicate a significant decrease in Chao1, ACE, and Richness indices in the healthy group compared to the GDM group, while the Shannon, Simpson, and Invsimpson indices display a clear decreasing trend (Fig. S2B–G). Subsequently, beta diversity among the samples was explored using principal coordinates analysis (PCoA) analysis. Beta diversity serves as an indicator of the differences in species composition between communities. The analysis reveals a certain level of separation between the two sample groups spatially, particularly evident in principal component 1 and principal component 2 (Fig. S2H), underscoring the notable distinctions in intestinal microbial structure between GDM patients and healthy individuals. Moreover, a Venn diagram analysis of operational taxonomic units (OTUs) obtained from the GDM and healthy groups showed that there were 87 distinct OTUs in the GDM group and 58 in the healthy group (Fig. 1A). This finding suggests both commonalities and significant differences in the intestinal microbiota between GDM and healthy individuals. In summary, the results demonstrate clear discrepancies in the diversity and abundance of intestinal microbiota between GDM patients and healthy pregnant individuals.

Note: A Venn diagram showing the overlap of intestinal microbiota OTUs between the GDM group and healthy group; B Clustered bar chart presenting the taxonomic distribution of intestinal microbiota species abundance in the GDM group and healthy group, where concentric circles from inner to outer represent taxonomic levels from phylum to genus, circle diameter indicates relative abundance, yellow nodes denote species with no significant difference, red nodes indicate microbial taxa with higher abundance in the GDM group, and green nodes represent taxa with higher abundance in the healthy group; C Bar plot displaying the LDA values distribution of species abundance in the gut microbiota of the GDM group and healthy group, green bars signify microbial taxa with higher abundance in the healthy group, red bars indicate taxa with higher abundance in the GDM group. GDM group, n = 5; Healthy group, n = 5.
To further investigate the disparities in intestinal microbiota between individuals with GDM and those with a healthy pregnancy, we conducted an analysis of the species composition at the levels of phylum, class, order, family, and genus in the GDM and healthy groups. Stacked bar graphs and chord diagrams were created to visualize the findings. The results indicate that at the Phylum level, the majority of microorganisms in both groups were found to be predominantly Firmicutes, Bacteroidetes, and Proteobacteria, with a notable increase in Firmicutes and a decrease in Proteobacteria in the GDM group compared to the Healthy group (Fig. S3A). At the Class level, most microorganisms belonged to Clostridia, Bacteroidia, and Alphaproteobacteria, demonstrating a significant increase in Clostridia and Alphaproteobacteria and a decrease in Bacteroidia in the GDM group compared to the Healthy group (Fig. S3B). At the Order level, the majority of microorganisms were classified into Clostridiales, Bacteroidales, and Lactobacillales, showing a marked increase in Clostridiales and a decrease in Lactobacillales in the GDM group compared to the Healthy group (Fig. S3C). At the family level, Prevotellaceae, Lachnospiraceae, and Peptoniphilaceae were the predominant families identified, with Lachnospiraceae significantly higher and Prevotellaceae and Peptoniphilaceae notably lower in the GDM group compared to the Healthy group (Fig. S3D). Finally, at the genus level, Prevotella, Blautia, Fournierella, and Other were the most abundant genera observed, with Blautia and Fournierella significantly higher and Fournierella noticeably lower in the GDM group compared to the Healthy group (Fig. S3E).
To delve deeper into the differences in intestinal microbiota between GDM patients and healthy pregnant individuals and identify characteristic bacterial species, our study focused on the genus level using LEfSe analysis. The analysis revealed that in fecal samples from individuals in the Healthy group, the relative abundance of Proteobacteria, Comamonas, Comamonadaceae, Burkholderiales, and Betaproteobacteria was significantly higher than in the GDM group, with a linear discriminant analysis (LDA) score (log 10) > 2 as the threshold. In contrast, fecal samples from GDM patients exhibited significantly higher relative abundances of Agathobacter, Extibacter, Fournierella, Ruthenibacterium, Ruminococcaceae, Faecalibacillus, Erysipelotrichaceae, Erysipelotrichales, and Erysipelotrichia compared to the Healthy group (Fig. 1B, C). Previous studies have reported that Agathobacter uses acetate as a substrate to produce butyrate, a type of SCFA that serves as an energy source for colonic cells and has anti-inflammatory properties34. Additionally, it is associated with the metabolism of unsaturated fatty acids35. Ruminococcaceae are renowned for their ability to break down plant fiber, contributing to the production of SCFAs, which are believed to account for ~70% of acetate in the intestines36.
These findings highlight significant differences in the species composition of intestinal microbiota between GDM patients and healthy pregnant individuals.
Inhibition of SCFAs metabolism in GDM mice
Subsequently, we conducted a high-throughput analysis of metabolites in urine and serum samples of the mice using LC-MS/MS technology. The obtained data were processed using MassLynx software to generate metabolite spectra for each sample. Following normalization and standardization of all sample data, preparation was made for subsequent multivariate statistical analysis. The OPLS-DA analysis confirmed significant differences between the two groups. Based on VIP scores and S-plot screening, we identified 144 distinct metabolites, with 115 downregulated in the GDM group and 29 upregulated (Fig. 2A). Through enrichment analysis using the KEGG and SMPDB databases, we investigated these DMs to reveal the enriched biological processes. The results indicated that the DMs were primarily enriched in metabolic pathways including fatty acid biosynthesis, fatty acid degradation, butanoate metabolism, pyruvate metabolism, propanoate metabolism, ketone body metabolism, and butyrate metabolism (Fig. 2B, C), with the most significant changes observed in pathways related to SCFAs such as butyric acid, isobutyric acid, and propionic acid synthesis. Subsequently, we delved into the profiles of the three major SCFAs, revealing a significant decrease in levels of butyric acid, propionic acid, and acetic acid compared to the Ctrl group (Fig. 2D–G). These findings indicate that SCFA metabolism is inhibited in GDM mice.

Note: A Volcano plots of DMs under positive ion mode (top) and negative ion mode (bottom); Functional enrichment analysis results of DMs in the MetaboAnalyst database, where (B) represents KEGG and C represents SMPDB; D GC-MS measurement of acetic acid metabolism levels in mice; E GC-MS measurement of propionic acid metabolism levels in mice; F GC-MS measurement of butyric acid metabolism levels in mice; G GC-MS measurement of total acid metabolism levels in mice. n = 6 for GDM group and Ctrl group mice, * denotes significance compared to Ctrl group at P < 0.05.
Impact of FMT on blood glucose levels and SCFA production in GDM mice
To explore the influence of intestinal microbiota on GDM and SCFA production, we analyzed the metabolic and glycemic changes in GDM mice after FMT. Blood glucose levels in the FMT group were significantly reduced compared to the GDM group (Fig. S4A). Furthermore, GC-MS analysis of three crucial SCFAs in mouse serum indicated a significant increase in levels of butyric acid, propionic acid, and acetic acid in the FMT group compared to the GDM group (Fig. S4B–D).
Increased risk of CAKUT in GDM
MR analysis identified 1080 SNPs significantly associated with GDM, among which 11 SNPs met the criteria for instrumental variables (IVs) (Table S1). The IVW method revealed a statistically significant causal association between GDM and CAKUT (p < 0.05). The beta coefficients from MR-Egger, weighted median, and weighted mode methods were all greater than 0 and directionally consistent with IVW. All four methods produced odds ratios >1, supporting that GDM increases the risk of CAKUT (Table S2, Fig. 3A).

Note: A Scatter plot of SNP correlation between GDM and congenital urinary system anomalies. The plot illustrates the magnitude of the association effects between SNP-GDM and SNP-congenital urinary system anomalies, with a 95% confidence interval. Different colored lines represent distinct MR methods. B Funnel plot for heterogeneity assessment, where light blue represents evaluation by the Inverse Variance Weighted method, and dark blue denotes evaluation by MR Egger method. C Forest plot of the causal effect of SNPs associated with GDM on congenital urinary system anomalies. The red line represents the results of the MR Egger test and the MR results of the inverse variance weighted method. Human GDM and congenital urinary system anomalies GWAS data originate from the GWAS public database. This study conducted a two-sample analysis with GDM as the exposure factor and congenital urinary system anomalies as the outcome factor.
To further validate the reliability of the results, we conducted tests for heterogeneity and pleiotropy. The results showed that both the MR Egger and inverse variance weighted (IVW) methods had heterogeneity testing p-values greater than 0.05, indicating no heterogeneity among the samples (Fig. 3B). The pleiotropy testing result with a p-value of 0.5102596 suggested that there was no pleiotropy among the IV SNPs. Subsequently, we performed a leave-one-out analysis by sequentially excluding each selected IV SNP and conducting an MR analysis. The red dots and their confidence intervals remained consistent, indicating that the analyzed results were not strongly influenced by any single SNP (Fig. 3C), thus confirming the reliability of the conclusions.
Key factors in identifying CAKUT in offspring of GDM mothers
Differential expression analysis revealed 217 significantly downregulated genes and 501 significantly upregulated genes in kidney tissues of GDM offspring compared to Ctrl offspring (Fig. 4A).

Note: A Volcano plot depicting differential analysis of high-throughput transcriptomes in offspring kidneys of Ctrl and GDM mice, where blue represents upregulated genes, red represents downregulated genes, and gray denotes genes with no significant differences. B Venn diagram showing the intersection of differentially expressed genes and those related to fatty acid metabolism. C Protein interaction network diagram of differentially expressed fatty acid metabolism genes in GDM offspring mouse kidney tissues, with connecting lines indicating interaction relationships among genes. D Statistical graph of the number of connected gene nodes, where a higher number indicates a higher degree of centrality. E T-test evaluating the expression levels of differentially expressed fatty acid metabolism genes in GDM offspring mouse kidney tissues. F RT-qPCR analysis of mRNA expression levels of differentially expressed fatty acid metabolism genes in GDM offspring mouse kidney tissues. G Western Blot examination of protein expression levels of differentially expressed fatty acid metabolism genes in GDM offspring mouse kidney tissues, ** indicating P < 0.01 compared to Ctrl group, and *** indicating P < 0.001 compared to Ctrl group. High-throughput sequencing of mouse samples, n = 3.
In the analysis of 16S rRNA sequencing data and metabolite analysis data, we observed significant changes in fatty acid metabolism pathways in GDM. Subsequently, we searched for fatty acid metabolism-related genes in the GeneCard online database, selecting genes with a relevance score greater than 50. By intersecting these genes with differentially expressed genes in the offspring mouse kidneys, we identified 5 fatty acid metabolism-related genes that displayed significant differential expression in GDM offspring mice (Fig. 4B). Using the String online database, a protein interaction network map was constructed for these five genes, with Il6 and Ptgs2 showing higher core degrees (Fig. 4C, D). T-tests evaluated the expression of these five genes in the offspring mouse kidneys of Ctrl and GDM groups based on the sequencing data, revealing that in GDM offspring mice kidney tissues, Il6 and Ptgs2 were significantly upregulated while Gpr43, Faah, and Slc27a2 were significantly downregulated compared to Ctrl group offspring mice (Fig. 4E–G). These results indicate that Gpr43 is a key factor in regulating the development of CAKUT in the offspring of GDM mothers through the involvement of the intestinal microbiota of parent GDM patients.
Association between Gpr43 expression and renal cell injury in offspring of GDM mothers
Acetate salt treatment significantly upregulated Gpr43 mRNA and protein expression in renal cells in a dose-dependent manner under high glucose conditions (Fig. 5A, B). Furthermore, GPR43 expression was successfully silenced in HK-2 cells under high glucose conditions, and the sequence with the highest knockdown efficiency was used in subsequent experiments (Fig. S5A, B). The expression of Gpr43 in the renal cells in each treatment group was assessed through RT-qPCR and Western Blot experiments. The results showed a significant increase in Gpr43 mRNA and protein expression levels in the SCFA group compared to the Control group, while a significant decrease was observed in the SCFA+sh-Gpr43 group compared to the SCFA+sh-NC group (Fig. 5C, D). Cell viability was measured using the CCK-8 assay and cell migration capacity was assessed through scratch experiments. The results revealed a significant increase in cell viability and migration capacity in the SCFA group compared to the Control group. Conversely, the SCFA+sh-Gpr43 group exhibited significantly reduced cell viability and migration capacity compared to the SCFA+sh-NC group (Fig. 5E–G).

Note: A RT-qPCR analysis of Gpr43 mRNA expression in HK-2 cells after 24 h of treatment with different concentrations of acetic acid; B Western Blot analysis of GPR43 protein expression in HK-2 cells after 24 h of treatment with different concentrations of acetic acid; C RT-qPCR analysis of Gpr43 mRNA expression in cells in different treatment groups; D Western Blot experiment to detect GPR43 protein expression in cells in different treatment groups; E Assessment of HK-2 cell viability using CCK-8 assay in different treatment groups; F Scratch assay to evaluate the migratory ability of HK-2 cells in different treatment groups; G Statistical chart of migration distances in the scratch assay; # indicates P < 0.05 compared to the SCFA+sh-Gpr43 group; all cell experiments were repeated three times.
To investigate renal damage in offspring mice and the expression of Gpr43, immunofluorescence staining was conducted to explore the expression of renal injury marker protein WT-1 and GPR43. The results showed a significant decrease in the levels of WT-1 and GPR43 expression in the GDM group mice compared to the Ctrl group (Fig. 6A, B). Further investigation through RT-qPCR and Western Blot experiments on their mRNA and protein expression levels corroborated with the immunofluorescence results (Fig. 6C, D). Thus, it is suggested that there is a correlation between the development of renal damage in offspring mice and the expression of Gpr43.

Notes: A Immunofluorescence staining showing the expression of WT-1 and GPR43 in renal tissues of offspring mice, scale bar = 50 μm; B Statistical chart of fluorescent intensity in immunofluorescence staining; C RT-qPCR analysis of mRNA expression of WT-1 and Gpr43 in renal tissues of offspring mice; D Western Blot experiment detecting protein expression of WT-1 and GPR43 in renal tissues of offspring mice; * indicates P < 0.05 compared to the Ctrl group or control group; n = 6 mice per group.
These findings indicate that the metabolism of SCFAs can promote the growth and migration of offspring renal cells by activating the Gpr43 pathway in renal tissue.
Protective roles of SCFA and Gpr43 in renal injury induced by GDM
To investigate whether changes in SCFA levels in GDM mediate renal damage in offspring mice, LC-MS technology was used to measure SCFA levels in the blood. The results showed: There were no significant differences in acetate, butyrate, and propionate metabolism levels between the NC group and the Ctrl group, indicating that sodium intake at this level did not significantly affect SCFA metabolism in mice. Compared to the Ctrl group, the GDM group showed significantly reduced levels of acetate, butyrate, and propionate. Compared to the GDM group, the GDM + SCFA group exhibited significantly increased levels of acetate, butyrate, and propionate. No significant differences in acetate, butyrate, or propionate metabolism levels were observed between the GDM + SCFA + Gpr43-inh group and the GDM + SCFA group (Fig. 7A–C). To assess Gpr43 expression levels, RT-qPCR and Western Blot experiments were conducted. The results showed: There was no significant difference in Gpr43 expression between the NC group and the Ctrl group, indicating that sodium intake did not affect Gpr43 expression. Compared to the Ctrl group, Gpr43 expression was significantly reduced in the GDM group. Compared to the GDM group, Gpr43 expression was significantly increased in the GDM + SCFA group. No significant differences in Gpr43 expression were observed between the GDM + SCFA + Gpr43-inh group and the GDM + SCFA group (Fig. 7D, E). To assess renal injury, we performed: PAS staining to evaluate the glomerular basement membrane thickness and overall kidney structure. Immunohistochemistry (IHC) to detect WT-1 and CD68 expression, assessing podocyte number and inflammatory cell infiltration. PSR staining to evaluate renal fibrosis. The results showed: no significant morphological changes were observed in the Ctrl group compared to the NC group, suggesting that sodium intake did not negatively impact renal structure. Compared to the Ctrl group, the GDM group exhibited thickened glomerular basement membranes, reduced podocyte numbers, increased renal fibrosis, and increased inflammatory cell infiltration. Compared to the GDM group, the GDM + SCFA group exhibited thinner glomerular basement membranes, increased podocyte numbers, reduced renal fibrosis, and decreased inflammatory cell infiltration. Compared to the GDM + SCFA group, the GDM + SCFA + Gpr43-inh group exhibited thickened glomerular basement membranes, reduced podocyte numbers, increased renal fibrosis, and increased inflammatory cell infiltration (Fig. 7F–M).

Note: A LC-MS detection of acetic acid metabolism levels in the blood of offspring mice in each group; B LC-MS detection of propionic acid metabolism levels in the blood of offspring mice in each group; C LC-MS detection of butyric acid metabolism levels in the blood of offspring mice in each group; D RT-qPCR analysis of Gpr43 mRNA expression in renal tissues of offspring mice in each group; E Western Blot analysis of GPR43 protein expression in renal tissues of offspring mice in each group; F PAS staining assessment of changes in the glomerular basement membrane and volume in offspring mouse renal tissues, scale bar = 50 μm; G Statistical graph of changes in glomerular volume in PAS staining; H Immunohistochemistry staining of WT-1 protein in offspring mouse renal tissues, scale bar = 50 μm; I Statistical graph of the average number of podocytes per glomerulus; J Immunohistochemistry staining of CD68 to evaluate inflammatory cell infiltration in offspring mouse renal tissues, scale bar = 50 μm; K Statistical graph of the number of CD68-positive cells; L PSR staining assessment of fibrosis degree in offspring mouse renal tissues, scale bar = 50 μm; M Relative area percentage of collagen protein in glomeruli detected by Picrosirius Red (PSR) staining, units in percentage (%). Each group consisted of n = 6 mice, * indicates P < 0.05 compared to the Ctrl group; # indicates P < 0.05 compared to the GDM + SCFA group; & indicates P < 0.05 compared to the GDM + SCFA group.
These findings indicate that Gpr43 plays a significant role in renal injury in the offspring of GDM mothers, while SCFAs can effectively alleviate renal damage through the Gpr43 pathway.
Discussion
GDM refers to the elevated blood glucose levels occurring during pregnancy37,38. CAKUT are structural anomalies in the kidneys and urinary tract that arise during fetal development39. Previous studies have indicated a certain association between GDM and CAKUT39,40. The prevalence of GDM is estimated to be ~2–10%, while CAKUT affects around 1–2% of pregnancies41. Past research has predominantly focused on exploring the relationship between GDM and CAKUT, leaving substantial uncertainties regarding the specific mechanisms42. GDM is one of the most common pregnancy complications, defined as glucose intolerance first detected during pregnancy. It is associated with multiple adverse maternal and fetal outcomes, including gestational hypertension, cesarean delivery, fetal macrosomia, shoulder dystocia, and neonatal hypoglycemia43. Although GDM is typically a transient condition, and glucose homeostasis often returns to normal after delivery, it can have long-term health consequences for both mothers and offspring. Studies have shown that 20%-50% of GDM patients develop type 2 diabetes mellitus within 10-20 years. Furthermore, compared to offspring of non-GDM mothers, children of GDM mothers have a 2–8 times higher risk of obesity, metabolic syndrome, type 2 diabetes mellitus, and impaired insulin secretion and sensitivity44,45. Identifying optimal prevention and treatment strategies for GDM remains a crucial area of research. However, the underlying pathological mechanisms of GDM remain largely unknown. CAKUT have a significant impact on health, accounting for 50% of pediatric chronic kidney disease cases46. Furthermore, undiagnosed CAKUT in childhood or adolescence increases the risk of end-stage renal disease in adulthood47. Effective preventative strategies for neonatal CAKUT could significantly reduce the disease burden. Studies suggest that maternal factors, genetic factors, and environmental factors contribute to CAKUT development48,49. Understanding modifiable maternal risk factors associated with CAKUT is critical. While GDM is considered a major contributor to congenital anomalies, limited evidence supports the direct role of diabetes as a risk factor for congenital urinary system anomalies50.
Earlier studies have highlighted the significant role of intestinal microbiota in GDM7,51,52. Intestinal microbiota plays a crucial role in carbohydrate metabolism and immune regulation, which may be closely linked to the occurrence and progression of GDM7,43. Nevertheless, current research on the association mechanisms and impacts of intestinal microbiota with GDM remains insufficient18,53,54.
This study conducted a detailed analysis of intestinal microbiota in patients with GDM. The results revealed significant differences in microbial diversity, abundance, and species composition between GDM patients and healthy pregnant women. Specifically, the relative abundance of Proteobacteria, and Comamonas in fecal samples from healthy individuals was markedly higher compared to the GDM group. On the other hand, GDM patients exhibited significantly higher levels of Agathobacter, Extibacter, Fournierella, and Ruthenibacterium in their fecal samples than the healthy group. Additionally, this study identified significant differences in pathways related to fatty acid metabolism and the metabolism of cofactors and vitamins between the healthy and GDM groups. Studies have shown that Agathobacter utilizes acetate as a substrate to produce butyrate, an SCFA that serves as an energy source for colonic cells and possesses anti-inflammatory properties34. Additionally, butyrate metabolism is linked to unsaturated fatty acid metabolism35. Ruminococcaceae is a key bacterial family known for its ability to degrade plant fibers, contributing to the production of SCFAs, which account for ~70% of acetate in the gut36. Our study demonstrated that the relative abundance of Comamonas and other bacteria was significantly higher in the Healthy group compared to the GDM group. However, the role of Comamonas in GDM progression remains unclear and requires further investigation. Consistent with our findings, multiple studies have reported that the relative abundance of Lachnospiraceae (including Agathobacter and Ruthenibacterium), Enterobacteriaceae (including Extibacter), and Clostridiaceae (including Ruthenibacterium) was significantly increased in GDM patients43,55,56,57. These results confirm that alterations in the relative abundance of Agathobacter, Extibacter, Fournierella, and Ruthenibacterium are significant features of gut microbiota dysbiosis in GDM patients.
This study analyzed the metabolic pathways related to GDM. The results revealed significant alterations in the activity of certain metabolic pathways in patients with GDM. These metabolic pathway abnormalities may be associated with the onset and progression of GDM. Our findings are consistent with those of Wang et al., who demonstrated that SCFA levels (butyrate, propionate, and acetate) were significantly reduced in GDM patients compared to healthy pregnant women, indicating that SCFA metabolic pathways are disrupted. Their study suggested that SCFAs have potential anti-diabetic and anti-inflammatory effects, influencing placental immune metabolism and fetal development via G-protein-coupled receptors (GPRs) or histone deacetylases (HDACs)58. Additionally, the study identified a critical role of the Gpr43 pathway in CAKUT in the offspring of GDM mothers. Previous research by Yang et al. demonstrated that β-glucan supplementation activates GPR41 and Gpr43, improving gut chemical barrier function, immune cell activity, cytokine responses, and nutrient metabolism in both mothers and fetuses59. Compared to previous research, this study conducted a more detailed investigation and analysis of the relationship between metabolic pathways and GDM, providing new insights into the pathological. Furthermore, we identified and elucidated the role of GPR43 in CAKUT for the first time, shedding light on its molecular mechanisms. Our transcriptomic data revealed that Gpr43, Faah, and Slc27a2—key regulators of fatty acid metabolism—were significantly downregulated in the kidneys of GDM offspring, while Il6 and Ptgs2, inflammatory markers known to influence lipid metabolism, were markedly upregulated. These molecular alterations suggest that both inflammation and impaired SCFA-GPR43 signaling may contribute to metabolic disruption in the developing kidney. Combined with the reduced SCFA levels observed in maternal metabolomics, these findings further support the hypothesis that maternal gut microbiota dysbiosis impairs offspring renal development via the GPR43 pathway.
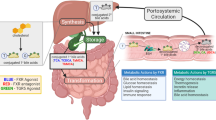
In summary, this study unveiled the relationship between GDM intestinal microbiota and offspring CAKUT, analyzing the differential characteristics and functions of intestinal microbiota between GDM patients. Moreover, it discovered the changes in metabolic pathways associated with GDM and the significant role of the Gpr43 pathway. The study revealed that GDM leads to a decrease in the metabolism levels of SCFAs in the intestinal microbiota, resulting in reduced SCFAs in the body, subsequently lowering offspring Gpr43 activation, inhibiting renal cell growth and migration, causing renal damage and developmental abnormalities in the offspring’s kidneys and urinary tracts (Fig. 8). These findings provide crucial clues for further exploring the pathogenesis of GDM-induced congenital kidney malformations in offspring and developing new treatment strategies.

Influence of GDM intestinal microbiota on the molecular mechanism of CAKUT in offspring (Created by BioRender).
A key innovation of this study lies in revealing the relationship between GDM intestinal microbiota and offspring CAKUT. By conducting a comprehensive analysis of the intestinal microbiota and metabolic pathways of GDM patients, the study uncovered new features and mechanisms. These novel findings are of significant importance for understanding the pathogenesis and intervention strategies of diseases related to GDM.
This study holds important scientific value for understanding the pathological mechanisms and intervention measures of diseases related to GDM. By elucidating the association between intestinal microbiota and GDM and its related diseases, this study offers fresh perspectives and approaches for further exploration of disease mechanisms and the pursuit of effective intervention methods. Nevertheless, the study’s data and sample size have certain limitations, and the differences between animal models and humans also need further consideration. Future research should delve deeper into the detailed mechanistic relationship between intestinal microbiota and CAKUT, and conduct clinical application studies.
Materials and methods
Acquisition of 16S rRNA sequencing data
Using the EMBL-EBI database (https://www.ebi.ac.uk/ena/browser/search), we retrieved relevant sequencing projects with the keyword “Gestational diabetes mellitus”. The selected project, identified as BioProject PRJNA963229, contains over 100 samples, including gut microbiota sequencing data from both GDM patients and healthy individuals. To ensure data representativeness and analytical feasibility, we randomly selected five fecal samples from healthy pregnant individuals (Healthy group) and five fecal samples from GDM patients (GDM group) for analysis. These datasets were downloaded from the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/) and prepared for downstream analysis60.
Microbial relative abundance analysis
To ensure high data quality, we conducted quality control and preprocessing using multiQC for sequence quality assessment and knead-data to remove host and contaminant sequences. The microbial species tree was annotated using GraPhlAn, and microbial taxonomy relative abundance was obtained. Diversity Analysis: Alpha diversity: The InvSimpson index was used to assess within-sample microbial species complexity.Beta diversity: Bray-Curtis dissimilarity was employed for PCoA to visualize community differences between groups. Differential analysis: to evaluate differences in microbial abundance and diversity: Wilcoxon rank-sum test and Welch’s t-test were used for statistical comparisons. The R package edgeR was used to compute differential abundance between groups. Volcano and Manhattan plots were generated for visualization. LDA effect size (LEfSe) was conducted, setting the LDA score threshold at 2.0, a widely accepted criterion in gut microbiota research55.
Mendelian randomization (MR) analysis
GDM and congenital genitourinary system anomalies-related genome-wide association study (GWAS) data were obtained from the public database FinnGen (https://www.finngen.fi/en). SNPs associated with GDM were selected as exposure factors using a significance threshold of p < 5e-08, while SNPs unrelated to the outcome were further screened as IVs. The TwoSampleMR package was utilized for MR analysis to assess the effectiveness of IVs and to estimate the magnitude and statistical significance of causal effects. To enhance the reliability of the analysis results, methods such as “mr_ivw,” “mr_egger_regression,” “mr_weighted_median,” and “mr_weighted_mode” were employed to compute causal effect-related indicators61.
To investigate the causal relationship between GDM and CAKUT, we performed a two-sample Mendelian randomization (MR) analysis using the TwoSampleMR package in R. GWAS summary statistics for GDM and CAKUT were obtained from the FinnGen database. SNPs associated with GDM (p < 5 × 10⁻8) were selected as exposure variables, and those related to known confounding traits were excluded using the PhenoScanner database. We then applied LD clumping to ensure independence of SNPs and identified 11 valid IVs for analysis. Before causal inference, we harmonized the effect alleles of exposure and outcome datasets. Causal estimates were computed using multiple MR methods, including IVW, MR-Egger regression, weighted median, and weighted mode, to improve the robustness of the findings.
Establishment of a GDM mouse model
Forty-two 6-week-old C57BL/6J mice and 14 six-week-old male C57BL/6J mice (catalog number: 219) were purchased from Weton Life Science Technology Co., Ltd. in Beijing, China. The mice were housed in standard cages under controlled conditions of constant room temperature (23 ± 1 °C), 12-h light/dark cycles, with ad libitum access to food and water. Prior to experimentation, a 1-week acclimatization period was provided. The experimental procedures and animal protocols were approved by the Institutional Animal Ethics Committee.
To establish the GDM mouse model, the mice were fed a diet comprising 60% high-fat feed (D12492, Research Diets). The control group received standard rodent chow (containing 18% protein, 5% fat), while the GDM group was fed a high-fat diet (containing 20% protein, 60% total fat, 32.1% saturated fat). Following allocation to their respective dietary groups, the mice underwent a 1-month dietary intervention. Prior to the experiment, fasting blood glucose (FBG) levels were measured via tail vein sampling, with normal glucose levels ranging from 3 to 5 mmol/L. After 1 month of dietary intervention, the mice were allowed to reproduce. The estrous cycle of female mice was determined using vaginal smears analysis, and females in early estrus were paired with male mice at a female-to-male ratio of 3:1. The following morning, vaginal plugs were checked to confirm pregnancy, with the presence of a vaginal plug marking gestational day 1. During pregnancy, the high-fat diet was continuously administered. Mice with FBG levels ≥11.1 mmol/L after one month of dietary intervention were considered to have successfully established the GDM mouse model62,63.
Blood glucose measurements were conducted using a blood glucose test kit (YX-XS-Q12253, Yuanxin Biotech) following the manufacturer’s instructions. Subsequently, GDM mice were randomly divided into four groups, each consisting of six mice for the experiments. The animal groups included donor mice (normal mice, n = 6), Ctrl group (healthy pregnant mice, n = 6), GDM group (GDM mice, n = 6), GDM + SCFA group (GDM mice + drinking water containing SCFA, n = 6), GDM + SCAF + Gpr43-inh group (GDM mice + drinking water containing SCFA + intravenous injection of Gpr43 active inhibitor, n = 6), and fecal microbiota transplantation (FMT) group (GDM mice + FMT − details in the experimental section “FMT”, n = 6).
For the administration of SCFA drinking water, sodium acetate (SA, Cat#: S431683) 100 mM, sodium butyrate (SB, Cat#: S102954) 50 mM, and sodium propionate (SP, Cat#: S100121) 100 mM were dissolved in the drinking water and provided ad libitum to the mice. These reagents were all purchased from Aladdin (Shanghai, China). The Ctrl group mice received water matched for pH and sodium content, while the NC group mice received sodium-free regular drinking water. The feed and water solutions were refreshed three times a week64.
The Gpr43 active inhibitor was administered by quickly injecting 10 μg Gpr43 inhibitor (CATPB, Cat#: HY-116263, Sigma-Aldrich) dissolved in 2.5 ml of saline into the tail vein of the mice. Injections were carried out every other day from the beginning of the experiment until the offspring were produced. Mice in the other groups received injections of an equivalent volume of saline62,65. The experimental grouping and flow chart are shown in Fig. S1.
RNA extraction and sequencing
In this study, we collected six kidney tissue samples from 1-week-old mice in the Ctrl group and GDM group, respectively. To minimize the impact of individual differences and optimize sequencing resources, we employed a random sampling method within each experimental group, pairing every two samples randomly to create one sequencing sample. Consequently, each experimental group ultimately formed three sequencing samples for subsequent RNA sequencing analysis. Total RNA extraction was performed using Trizol reagent (15596026, Invitrogen, Carlsbad, CA, USA), followed by quantification of RNA sample concentration and purity using a Nanodrop2000 spectrophotometer (1011U, Nanodrop, USA). Only total RNA samples meeting the following criteria were selected for further experiments: RNA integrity number (RIN) ≥ 7.0 and 28S:18S ratio ≥ 1.5.
The sequencing libraries were prepared and sequenced by CapitalBio Technology (Beijing, China). Each sample was processed with 5 μg of RNA. In brief, Ribo-Zero™ magnetic kit (MRZE706, Epicentre Technologies, Madison, WI, USA) was used to deplete ribosomal RNA (rRNA) from total RNA. NEB Next Ultra RNA Library Prep Kit (#E7775, NEB, USA) was utilized to construct a library suitable for Illumina sequencing. Subsequently, the RNA fragments were converted into ~300 base pair (bp) fragments using NEB Next First Strand Synthesis Reaction Buffer (5×), first-strand cDNA was synthesized with reverse transcriptase primers and random primers, and second-strand cDNA was synthesized in second-strand synthesis reaction buffer with dUTP Mix (10×). The cDNA fragments underwent end-repair, including polyadenylation and sequencing adapter ligation. After adapter ligation, the second strand of cDNA was digested using the USER enzyme (#M5508, NEB, USA) to construct strand-specific libraries. The library DNA was amplified, purified, and PCR-enriched. Finally, library identification was performed using Agilent 2100 and quantification with KAPA Library Quantification Kit (KK4844, KAPA Biosystems). The sequencing was carried out on the NextSeqCN500 (Illumina) platform using paired-end sequencing66.
To identify the core factors regulating the development of CAKUT in offspring of GDM mothers, kidney tissues were collected from offspring mice in both the Ctrl and GDM groups for RNA sequencing. The RNA integrity was confirmed with 260/280 ratios between 1.8 and 2.0 and RIN values above 8. Sequencing was performed using the Illumina Novaseq platform, yielding an average of ~30 million paired-end reads per sample. Differential gene expression analysis was conducted using the “limma” package in R.
FMT
Fecal samples from healthy C57BL/6 N mice were collected and diluted in cold phosphate-buffered saline (PBS) at a ratio of 100 mg feces per 1 mL buffer solution. The samples were homogenized for 10 min to a paste-like consistency, followed by vortexing for 1 min and centrifugation at 800 × g for 5 min. The supernatant was collected, aliquoted, and stored in 80% glycerol-PBS solution at −20 °C until transplantation.
To reduce bacterial load in recipient mice, an antibiotic mixture consisting of vancomycin (50 mg/kg; 1404-93-9, Sigma-Aldrich, USA), neomycin (100 mg/kg; 1405-10-3, Sigma-Aldrich, USA), metronidazole (100 mg/kg; 13182-89-3, Sigma-Aldrich, USA), and amphotericin B (1 mg/kg; 1397-89-3, Sigma-Aldrich, USA) was administered. The treatment was given orally through gavage for 7 consecutive days. Ampicillin (1 g/L; A5354, Sigma-Aldrich, USA) was added to the drinking water for supplementation67. Subsequently, after the antibiotic treatment, the recipient mice received 1 mL of fecal microbiota supernatant from the donor mice through gavage every other day for 2 weeks. The mice were euthanized 1 week after successful transplantation68,69.
After the completion of FMT treatment, blood samples were collected from recipient mice. The serum was isolated for downstream analyses. Serum levels of acetic acid, butyric acid, and propionic acid were determined using gas chromatography-mass spectrometry (GC-MS).
Application of liquid chromatography-mass spectrometry (LC-MS)
Urine samples were collected using metabolic cages, and serum samples were obtained by centrifuging whole blood collected from the retro-orbital sinus. Both were analyzed using LC-MS, with 300 μL of each sample transferred to a 1.5 mL polypropylene tube. The samples were mixed with 900 μL of 80% methanol (67-56-1, Sigma-Aldrich, USA) and 0.1% formic acid (64-18-6, Sigma-Aldrich, USA) by vortexing for 2 min, followed by centrifugation at 12,000 × g for 10 min. The supernatant was transferred to an autosampler vial.
Fecal metabolomics analysis was conducted using an LC20 ultrahigh-performance liquid chromatography system (SHIMADZU, Japan) coupled with a Triple TOF-6600 mass spectrometer (AB Sciex, USA). A Waters ACQUITY UPLC HSS T3 C18 column (100 × 2.1 mm, 1.8 μm) was employed for chromatographic separation. The column temperature was maintained at 40 °C with a flow rate of 0.4 mL/min. The mobile phase consisted of acetonitrile (75-05-8, Sigma-Aldrich, USA) water solution containing 0.1% formic acid. The gradient elution program for mobile phase B was as follows: 5% from 0.0 to 11.0 min; 90% from 11.0 to 12.0 min; 5% from 12.1 to 14 min. The eluate was directly introduced to the mass spectrometer without splitting70.
For positive/negative ion mode mass spectrometry, the conditions utilized were as follows: ionization voltage at 5500 V, capillary temperature at 550 °C, nebulizer gas flow rate at 50 psi, and auxiliary heating gas flow rate at 60 psi. To prevent overfitting, preprocessing data was analyzed using Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) and permutation tests (performed 100 times). Metabolites with VIP score above 1, P-value < 0.05 from the OPLS-DA model, as well as metabolites with fold change ≥2 or ≤0.5 from univariate analysis and P-value < 0.05 from Student’s t-test, were identified as DMs. MetaboAnalyst (Version 5.0) was used for pathway analysis of the identified metabolites71.
Quality control of sequencing data and alignment to reference genome
The quality of paired-end reads in the raw sequencing data was assessed using FastQC software version 0.11.872. Subsequently, preprocessing of the raw data was performed using Cutadapt software version 1.1873 to eliminate Illumina sequencing adapters and poly(A) tail sequences. A perl script was utilized to remove reads with an N content exceeding 5%. Reads with a base quality over 20 and comprising at least 70% of the bases were extracted using FASTX Toolkit software version 0.0.13 (https://biocontainers.pro/tools/fastx_toolkit). BBMap (https://sourceforge.net/projects/bbmap/) software was employed to repair the paired-end sequences. Lastly, alignment of the filtered high-quality reads to the mouse reference genome was conducted using hisat2 software version 0.7.1274.
Differential gene analysis
Differential gene expression analysis was conducted on sequencing data using the “limma” package in the R software75. The criteria for selecting differentially expressed genes were set at │logFC│ > 1 and P < 0.05. Visualization of the differential analysis results was performed using the “ggplot2” package in R, which generated a volcano plot76. The “VennDiagram” package in R was then utilized to identify the intersection between the differentially expressed genes and genes related to fatty acid metabolism, followed by the creation of a Venn diagram for the intersecting genes77. Genes associated with fatty acid metabolism were obtained by searching the “Fatty acid metabolism” term in the GeneCards database (https://www.genecards.org/).
Construction of protein-protein interaction networks (PPI) and target gene prediction
The STRING database contains experimental data, results obtained through text mining of PubMed abstracts, and predictions derived from combining data from other databases and bioinformatics methods. These data are utilized to study protein interactions. In this study, we conducted a PPI analysis of factors related to fatty acid metabolism in the renal tissues of GDM mouse offspring using the STRING database (http://www.string-db.org/) with the species limited to mice and a confidence score set at 0.15. The interconnection node counts between each protein were statistically analyzed and visually represented using R software78.
Gas chromatography-mass spectrometry analysis (GC-MS)
Mouse serum from either parent or offspring were collected. Each 1 mL of serum was mixed with 10 μL of ultrapure water, vigorously vortexed, and left to stand for 30 min. Subsequently, the mixture was centrifuged at 12,000 rpm for 20 min at 4 °C, the supernatant was aspirated, passed through a 0.22 μm membrane filter, and transferred to the gas chromatography vial for analysis. The chromatographic column used was a DB-WAX 30 M (I.D. 0.32 mm, 5μm, Agilent, USA), with the injection port and detector temperatures set at 250 °C. The temperature program was as follows: initially held at 50 °C for 3 min, then increased at a rate of 6 °C per min to 120 °C, held for 0.5 min, further increased at a rate of 6 °C per min to 220 °C, and maintained for 5 min. The nitrogen flow rate was 3 mL/min, the hydrogen flow rate was 47 mL/min, and the airflow rate was 400 mL/min, with a split ratio of 1:3. The injection volume was 1.0 μL. Quantitative analysis was performed using an external standard method. The content of various SCFAs in the samples was calculated based on the ratio of peak areas between standard samples and the samples analyzed79,80,81.
Immunofluorescence staining
The kidney tissue samples of offspring mice underwent the following procedures: fixation in 4% paraformaldehyde, dehydration, clarification, embedding in paraffin, and tissue sectioning. For immunofluorescence staining, after dewaxing and rehydration of the sections, they were blocked with 2% BSA. Subsequently, primary antibodies WT-1 (catalog number: MA5-32215, Thermofisher, 1:200, USA) or Gpr43 (catalog number: HK9643, Guidechem, 1:200, China) were added and incubated overnight at 4 °C. The next day, the sections were washed with PBS and incubated at room temperature for 1 h with the secondary antibody, Alexa Fluor™ 555-conjugated goat anti-mouse IgG (A-21137, 1:2000, Thermofisher, USA). Following the incubation, the sections were rinsed with PBS and stained with DAPI solution (C1002, Beyotime, China) for 5 min to label the nuclei. Subsequent washes with PBS were performed to remove excess DAPI, repeated three times for 5 min each. Finally, the cells were carefully removed from the cell culture plate and placed on glass slides coated with an anti-fading mounting medium (cell-side down). Observations and imaging were conducted using a fluorescence microscope (FV-1000/ES, Olympus, Japan), and the fluorescence coverage area was quantitatively measured under a 40× objective. Six fields were selected per group, and the average value was calculated82. WT-1 immunofluorescence staining was employed to assess the number of podocytes in the renal glomeruli.
Immunohistochemical staining
Mouse kidney tissues were embedded and sectioned after baking at 60 °C for 20 min. The sections were then sequentially soaked in xylene solution, with each xylene change followed by a 15-min soak. Subsequently, they were immersed in anhydrous alcohol for 5 min and then hydrated in 95% and 70% alcohol for 10 min each. Each section was treated with 3% H2O2 and incubated at room temperature for 10 min to inhibit endogenous peroxidase activity. Following this, the citric acid buffer was added, and the samples were microwaved for 3 min. Antigen retrieval solution was applied, left at room temperature for 10 min, and washed three times with PBS. Normal goat serum blocking solution (catalog number: E510009, Shanghai Bioengineering Co., Ltd) was then added and incubated at room temperature for 20 min. The sections were then separately treated with diluted WT-1 (catalog number: MA5-32215, Thermo Fisher, 1:200, USA) or CD68 (catalog number: 14-0681-82, Thermo Fisher, 1:200, USA) primary antibodies and left overnight at 4 °C. The next day, the sections were washed three times in PBS. Subsequently, goat anti-rabbit IgG (A-11011, 1:500, Thermo Fisher, USA) or rabbit anti-mouse IgG (42-4817-80, 1:20, Thermo Fisher, USA) secondary antibodies were applied and incubated for 30 min. After PBS washes, DAB chromogenic reagent (DAB150, Sigma, USA) was used for staining. Drops of chromogenic reagents A, B, and C were added to the specimen and left for 6 min. Then, counterstaining was performed using hematoxylin for 30 s. The sections were dehydrated in 70%, 80%, 90%, 95% ethanol, and absolute ethanol, each for 2 min. Finally, they were immersed twice in an xylene clearing agent for 5 min each, followed by sealing with neutral resin64. Observations of the sections were made under a brightfield microscope (BX63, Olympus, Japan). Aperio Scanscope System (Vista, CA) was used for quantifying the areas of protein expression, and ImageJ software was employed for grayscale analysis to estimate protein expression levels. Statistical analysis was conducted to compare differences between different groups, and the quantitative results of cell or tissue slice fluorescent staining were visually presented64.
For blind evaluation of macrophage (CD68) staining, 20 consecutive high-power fields (×400 magnification) in the renal cortex of each section were analyzed. CD68-positive cells were counted using a gridded eyepiece and expressed as the number of cells per field. WT-1 positive cells were counted in 20 glomerular cross-sections (high-power fields, ×400 magnification). The formula N+podglom = NVpodglom × Vglom was employed to represent the number of podocytes per glomerulus64,83.
Periodic acid-schiff (PAS) staining
The PAS staining kit (Catalog No: M035, Shanghai Gefan Biotechnology Co., Ltd, China) was utilized to assess renal tissue damage. The methodology involved the following steps: renal tissue sections (embedded in renal paraffin) were deparaffinized twice for 20 min in xylene, followed by hydration in a series of alcohol solutions for 10 min each at varying concentrations—100% ethanol, 95% ethanol, 90% ethanol, 80% ethanol, and 70% ethanol. Subsequently, the sections were rinsed in tap water, stained in hematoxylin-eosin for 10 min, and then washed in tap water for 5 min. Next, the sections reacted in Schiff’s reagent for 10 min, followed by a 5-min water rinse. The sections were then stained in Sudan III for 1 min and rinsed in tap water for 5 min. Finally, dehydration was conducted in 95% ethanol for 5 min, followed by two 5-min washes in 100% ethanol, and then two 5-min treatments in xylene for transparency. After these steps, the neutral resin was utilized for mounting. All the necessary reagents were provided in the kit.
The extent of damage was graded based on the percentage of damaged tubules, with scores ranging from 0 to 5: 0 denoting normal, 1 indicating less than 10% tubular injury, 2 indicating 10–20% damage, 3 indicating 20–30% damage, 4 indicating 30–40% damage, and 5 representing over 40% damage. Ten random fields were selected per slide and blindly assessed under a Nikon (Tokyo, Japan) light microscope. The fibrosis-to-tubule ratio was determined using the ImageJ analysis software64.
Using DP2-BSW software v2.2 (OLYMPUS), the area of 20 glomerular tufts per mouse was measured to calculate glomerular volume (VG) using the formula: VG = (β/κ) × (AG)3/2, where β = 1.38 (shape coefficient of a sphere), and κ = 1.1 (size distribution coefficient). The extracellular matrix of glomeruli was defined as PAS-positive areas quantified by image analysis software (ImagePro Premier 9). Collagen content in the tubulointerstitium was evaluated by point counting on a grid at ×400 magnification in 20 consecutive fields64.
PSR staining
For PSR staining, kidney sections were first deparaffinized using Citrisolv (Fisher Scientific) and rehydrated through a graded ethanol series (100%, 70%, and 30%). The sections were then immersed in a PSR staining solution containing 0.1% w/v Sirius Red F3BA (Sigma-Aldrich) dissolved in a saturated aqueous solution of picric acid (Sigma-Aldrich) and incubated at room temperature for 40 min. After staining, the slides underwent either four washes in 0.5% glacial acetic acid (7 min each) or a 90-s incubation in 0.05 M hydrochloric acid (Fisher Scientific). Excess acidified water was carefully removed, and the sections were rapidly dehydrated in 100% ethanol through three consecutive 30-s incubations. The slides were then cleared in Citrisolv for 5 min and mounted with Cytoseal XYL (Fisher Scientific). To ensure consistency in staining intensity, all slides within each experiment were processed simultaneously84.
Western blot
Protein samples were extracted using a protein extraction kit (Bestbio, BB3101, Shanghai, China), and the protein concentration was determined using a BCA assay kit (Beyotime, P0012S, Shanghai, China). A 10% SDS-PAGE gel (Beyotime, P0012A, Shanghai, China) was prepared, and 50 μg of protein sample was loaded per well. Electrophoresis was conducted at a constant voltage of 80 V for 2 h, then increased to 120 V, and followed by a constant current transfer of 250 mA for 90 min onto a PVDF membrane (Merck, IPVH00010, Germany). The PVDF membrane was blocked with 5% non-fat milk in TBST for 2 h at room temperature, followed by three washes with TBST for 10 min each. Primary antibodies were incubated overnight at 4 °C (antibody information provided in Table S3), followed by three washes with TBST for 10 min each. The membrane was then incubated with Goat anti-rabbit IgG (1:2000, Abcam, ab6721, UK) or Goat anti-mouse IgG (1:2000, Abcam, ab6789, UK) at room temperature for 1 h, with subsequent three washes with TBST for 10 min each. Protein bands were visualized using an ECL detection kit (Beyotime, P0018FS, Shanghai, China), exposed in a dark box, and imaged85. Each sample was tested in triplicate. All the uncropped and unedited blot/gel images can be found at the Supplementary Information.
Gene expression analysis using RT-qPCR
Total RNA extraction from samples was performed using Trizol (16096020, Thermo Fisher, USA). The cDNA was synthesized using a reverse transcription kit (RR047A, Takara, Japan). The cDNA was generated by reverse transcription. Subsequently, the reaction system was prepared using the One Step TB Green® PrimeScript™ RT-PCR Kit (RR066A, Takara, Japan), and RT-qPCR was carried out in an ABI 7500 real-time PCR system (Thermo Fisher, USA). The reference gene used was β-actin. The PCR program was set as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s, annealing at 60 °C for 30 s, and extension at 95 °C for 15 s, with a final extension at 60 °C for 60 s and a further extension at 90 °C for 15 s. Amplification curves were plotted, and each RT-qPCR experiment was performed in triplicate. The primer sequences can be found in Table S4. The ratio of gene expression between the experimental and control groups was calculated using the 2−ΔΔCt method, where ΔΔCt = ΔCt test − ΔCt control, and ΔCt = Ct target − Ct reference. Ct refers to the cycle threshold, indicating the number of amplification cycles required for the real-time fluorescence signal to exceed the set threshold86. The experiments were repeated three times for data reliability.
Cell culturing and treatment
The human renal tubular epithelial cell line HK-2 (Catalog Number: CRL-2190) was obtained from ATCC (USA). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Catalog Number: 11965092, Thermofisher) supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum (Gibco, Catalog Number: 16140071, USA) and maintained in a 5% CO2, 37 °C humidified incubator87.
For the concentration gradient experiment, HK-2 cells were cultured in DMEM containing high glucose (30 mmol/L D-glucose, HG) supplemented with various concentrations of acetate (3, 5, 10, 20, and 40 mmol/L) for 24 h before cell collection for further experimentation31.
Lentiviral transduction
Lentiviral transduction is based on the construction of corresponding cell lines, including the human HK-2 cell line with silenced Gpr43 (sh-Gpr43) and its control cell line (sh-NC). Plasmids carrying a single luciferase reporter gene were transfected into 293T cells (CL-0469, Procell) together with helper plasmids. After validation, amplification, and purification, packaged lentivirus was obtained. The method for lentivirus-mediated cell transduction is as follows: 5 × 105 cells were seeded in a 6-well plate, and when the cell confluence reached 70–90%, the culture medium containing an appropriate amount of packaged lentivirus (MOI = 10, working titer ~5 × 106 TU/mL) and 5 μg/mL polybrene (Merck, TR-1003, USA) was added for transduction. After 4 h, an equal amount of fresh medium was added to dilute polybrene. After 24 h, the fresh medium was replaced, and after 48 h, transduction efficiency was observed through the luciferase reporter gene. The selection of stable transduced cell lines was performed using 60 μg/mL puromycin (Sangon Biotech, E607054-0001, Shanghai, China).
For lentiviral infection, HK-2 cells in the exponential growth phase were taken to prepare a cell suspension of 5 × 104 cells/mL, which was seeded into a 6-well plate at 2 mL per well and cultured at 37 °C overnight. Subsequently, each well was inoculated with lentivirus at a final concentration of 1 × 108 TU/mL for silencing or overexpression. After 48 h of infection, the infection efficiency was assessed using qRT-PCR, and cell lines showing good effects were selected for further experiments88. The silencing sequences were as follows: sh-Gpr43-1: 5’-CGTGGTGTTCAGTTCACTCAA-3’; sh-Gpr43-2: 5’-GCTACGAGAACTTCACCGATA-3’; sh-NC: 5’-CCTAAGGTTAAGTCGCCCTCG-3’. Each experiment was repeated three times.
CCK-8 experiment
HK-2 cells were seeded in a 96-well plate at a density of 5 × 103 cells per well. Subsequently, 10 μL of CCK-8 reagent solution (C0038, Beyotime, Shanghai, China) was added to each well, and the plate was incubated at 37 °C in a humidified culture chamber. After 1 h, the absorbance values of each well were measured at 450 nm using a Microplate Reader (abx700005, Beijing Qiwiyicheng Technology Co., Ltd)89.
Scratch migration assay
The kidney cells of each group were separately cultured in a 24-well plate. Using sterile 200 μL pipette tips, scratches were made in the middle of each well. Subsequently, PBS was used to wash away the damaged cellular debris. Cell images migrating into the scratched areas were captured at various time intervals (6 h, 12 h, and 24 h). Finally, the percentage of area occupied by the migrating cells was measured using Image J software90.
Statistics and reproducibility
This study utilized R software Version 4.2.1, compiled using the integrated development environment RStudio (RStudio Version 4.2.1). File processing was conducted using Perl language (Perl Version 5.30.0). Additionally, Cytoscape Version 3.7.2 and SPSS statistical software Version 21.0 (IBM SPSS Statistics, Chicago, IL, USA) were employed. Quantitative data were presented as mean ± standard deviation. Independent-sample t-tests were used for intergroup comparisons. For comparisons among different time points within each group, repeated measure analysis of variance was applied, followed by the Bonferroni post-hoc test91,92. A significance level of P < 0.05 was used to determine statistical differences.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The plasmids used in this study have been deposited in the Addgene repository and are publicly available as follows: pLKO.1 puro-shGPR43-1 (Addgene plasmid #239860) – https://www.addgene.org/239860/ pLKO.1 puro-shGPR43-2 (Addgene plasmid #239956) – https://www.addgene.org/239956/ pLKO.1 puro-shNC (Addgene plasmid #239957) – https://www.addgene.org/239957/ The metabolomics data have been deposited in the iProX database (iProX ID: IPX0012142000) and are publicly accessible at https://www.iprox.cn/page/project.html?id=IPX0012142000. The same dataset is also available via ProteomeXchange with the identifier PXD064555 at. The sequencing data have been submitted to the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1271241 (submission ID: SUB15362169). SRA accession numbers are as follows: Control group: SRR33798569, SRR33798568, SRR33798567 GDM group: SRR33798566, SRR33798565, SRR33798564. The underlying source data for all graphs presented in the manuscript have been provided as Supplementary Data.
Code availability
No custom software or code was developed for this study. Standard publicly available tools and packages were used for data processing and statistical analysis, as detailed in the Methods section.
References
Sun, H. et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119 (2022).
Wang, L. et al. Prevalence and treatment of diabetes in China, 2013-2018. JAMA 326, 2498 (2021).
Tanase, D. M. et al. The intricate relationship between Type 2 diabetes mellitus (T2DM), insulin resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J. Diabetes Res. 2020, 1–16 (2020).
Laredo-Aguilera, J. A., Gallardo-Bravo, M., Rabanales-Sotos, J. A., Cobo-Cuenca, A. I. & Carmona-Torres, J. M. Physical activity programs during pregnancy are effective for the control of gestational diabetes mellitus. IJERPH 17, 6151 (2020).
OuYang, H., Chen, B., Abdulrahman, A. M., Li, L. & Wu, N. Associations between gestational diabetes and anxiety or depression: a systematic review. J. Diabetes Res. 2021, 1–10 (2021).
Lu, L., Wan, B. & Sun, M. Mendelian randomization identifies age at menarche as an independent causal effect factor for gestational diabetes mellitus. Diabetes Obes. Metab. 25, 248–260 (2022).
Hasain, Z. et al. Gut microbiota and gestational diabetes mellitus: a review of host-gut microbiota interactions and their therapeutic potential. Front. Cell Infect. Microbiol. 10. https://doi.org/10.3389/fcimb.2020.00188 (2020).
Mao, L., Gao, B., Chang, H. & Shen, H. Interaction and metabolic pathways: elucidating the role of gut microbiota in gestational diabetes mellitus pathogenesis. Metabolites 14, 43 (2024).
Pinto, Y. et al. Gestational diabetes is driven by microbiota-induced inflammation months before diagnosis. Gut 72, 918–928 (2023).
Kuang, Y. S. et al. Connections between the human gut microbiome and gestational diabetes mellitus. GigaScience 6. https://doi.org/10.1093/gigascience/gix058 (2017).
Hardin, B. I. & Keyes D. Enterohormonal and Microbiota Pathophysiology of Obesity. In: StatPearls (StatPearls Publishing, 2024).
Liu, H., Cheng, Y., Qu, Y. & Wu, G. Unraveling the gut microbiota and short-chain fatty acids characteristics and associations in a cancer cachexia mouse model. Microb. Pathog. 183, 106332 (2023).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Song, Z., Li, S. & Li, R. An investigation into the correlation of intestinal flora with obesity and gestational diabetes mellitus. Comput. Math. Methods Med. 2022, 1–15 (2022).
Qin, S., Liu, Y., Wang, S., Ma, J. & Yang, H. Distribution characteristics of intestinal microbiota during pregnancy and postpartum in healthy women. J. Matern. Fetal Neonatal Med. 35, 2915–2922 (2021).
Gao, Y. et al. Alterations of gut microbiota-derived metabolites in gestational diabetes mellitus and clinical significance. Clin. Lab. Anal. 36, https://doi.org/10.1002/jcla.24333 (2022).
Ye, D. et al. Integrative metagenomic and metabolomic analyses reveal gut microbiota-derived multiple hits connected to development of gestational diabetes mellitus in humans. Gut Microbes 15, https://doi.org/10.1080/19490976.2022.2154552 (2022).
Lou, M. et al. Deviated and early unsustainable stunted development of gut microbiota in children with autism spectrum disorder. Gut Published online December 20, 2021:gutjnl-2021-325115. https://doi.org/10.1136/gutjnl-2021-325115.
Huang, J. H. et al. The intratumoral bacterial metataxonomic signature of hepatocellular carcinoma. Microbiol. Spectr. 10, https://doi.org/10.1128/spectrum.00983-22 (2022).
Chai, X. et al. Intratumor microbiome features reveal antitumor potentials of intrahepatic cholangiocarcinoma. Gut Microbes 15, https://doi.org/10.1080/19490976.2022.2156255 (2022).
Tan, B., Ma, Y., Zhang, L., Li, N. & Zhang, J. The application of metabolomics analysis in the research of gestational diabetes mellitus and preeclampsia. J. Obstet. Gynaecol. 46, 1310–1318 (2020).
Zhan, Y. et al. Plasma metabolites, especially lipid metabolites, are altered in pregnant women with gestational diabetes mellitus. Clin. Chim. Acta 517, 139–148 (2021).
Zhang, H. et al. Potential biomarkers identified in plasma of patients with gestational diabetes mellitus. Metabolomics 17, https://doi.org/10.1007/s11306-021-01851-x (2021).
Fleming, C. A., Mohan, H. M., O’Leary, D. P., Corrigan, M. & Redmond, H. P. Metabolomic pathway activity with genomic single-nucleotide polymorphisms associated with colorectal cancer recurrence and 5-year overall survival. J. Gastrointest. Cancer 54, 247–258 (2022).
Dong, Y. et al. Alterations of fecal metabolome associated with BBIBP-CorV vaccines against the SARS-CoV-2 virus. Front. Biosci. 28, https://doi.org/10.31083/j.fbl2804065 (2023).
Lee, D. H., Kim, M. T. & Han, J. H. GPR41 and GPR43: from development to metabolic regulation. Biomed. Pharmacother. 175, 116735 (2024).
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
de Vos, W. M., Tilg, H., Van Hul, M. & Cani, P. D. Gut microbiome and health: mechanistic insights. Gut 71, 1020–1032 (2022).
Song, Q. et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 79, 1352–1365 (2023).
Lu, J. et al. GPR43 deficiency protects against podocyte insulin resistance in diabetic nephropathy through the restoration of AMPKα activity. Theranostics 11, 4728–4742 (2021).
Bowman, J. S. et al. Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene. ISME J. 6, 11–20 (2011).
Lou Z. et al. Multi-omics identified THDCA as a key contributor to hyperlipidemia and as a potential therapeutic agent. Rev. Cardiovasc. Med. 24, https://doi.org/10.31083/j.rcm2409248 (2023).
Tang, S. et al. Time-course alterations of gut microbiota and short-chain fatty acids after short-term lincomycin exposure in young swine. Appl. Microbiol. Biotechnol. 105, 8441–8456 (2021).
Wen, M. et al. Integrated analyses of gut microbiome and host metabolome in children with Henoch-Schönlein purpura. Front. Cell Infect. Microbiol. 11, https://doi.org/10.3389/fcimb.2021.796410 (2022).
Liu, Y. S. et al. Microbiota populations and short-chain fatty acids production in cecum of immunosuppressed broilers consuming diets containing γ-irradiated Astragalus polysaccharides. Poult. Sci. 100, 273–282 (2021).
Hou, W. H. et al. Associations of hyperglycemic emergency and severe hypoglycemia incidences with seasonality and ambient temperature among pregnant women with diabetes: a nested case-control study in Taiwan. Environ. Health Prev. Med 27, 11–11 (2022).
Catinelli, B. B. et al. Reversal of diabetic-induced myopathy by swimming exercise in pregnant rats: a translational intervention study. Sci. Rep. 12, https://doi.org/10.1038/s41598-022-10801-z (2022).
Plaud Gonzalez, A. M. et al. Fetal nephrology: a quaternary care center experience. Kidney360 4, 333–340 (2023).
Dyck, R. F. et al. Congenital anomalies of the kidney and urinary tract (CAKUT): an emerging relationship with pregestational diabetes mellitus among first nations and non-first nations people in saskatchewan—results from the DIP: ORRIIGENSS project. Can. J. Diabetes 45, 346–354.e1 (2021).
Nethery, E., Hutcheon, J. A., Law, M. R. & Janssen, P. A. Validation of insurance billing codes for monitoring antenatal screening. Epidemiology 34, 265–270 (2022).
Nasri, H. Z., Houde Ng, K., Westgate, M., Hunt, A. & Holmes, L. B. Malformations among infants of mothers with insulin-dependent diabetes: is there a recognizable pattern of abnormalities? Birth Defects Res. 110, 108–113 (2018).
Wang, X. et al. Altered gut bacterial and metabolic signatures and their interaction in gestational diabetes mellitus. Gut Microbes 12, 1840765 (2020).
Lauenborg, J. et al. Increasing incidence of diabetes after gestational diabetes. Diabetes Care 27, 1194–1199 (2004).
Vohr, B. R. & Boney, C. M. Gestational diabetes: the forerunner for the development of maternal and childhood obesity and metabolic syndrome?. J. Matern. Fetal Neonatal Med. 21, 149–157 (2008).
Saran, R. et al. US renal data system 2014 annual data report: epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 66, A7 (2015).
Calderon-Margalit, R. et al. History of childhood kidney disease and risk of adult end-stage renal disease. N. Engl. J. Med 378, 428–438 (2018).
Bartram, M. P. et al. Conditional loss of kidney microRNAs results in congenital anomalies of the kidney and urinary tract (CAKUT). J. Mol. Med 91, 739–748 (2013).
Franke, D. et al. Prematurity, small for gestational age and perinatal parameters in children with congenital, hereditary and acquired chronic kidney disease. Nephrol. Dial. Transplant. 25, 3918–3924 (2010).
Zhao, E., Zhang, Y., Zeng, X. & Liu, B. Association between maternal diabetes mellitus and the risk of congenital malformations: a meta-analysis of cohort studies. DDT 9, 274–281 (2015).
Li, H. Y. et al. Effects and mechanisms of probiotics, prebiotics, synbiotics, and postbiotics on metabolic diseases targeting gut microbiota: a narrative review. Nutrients 13, 3211 (2021).
Chen, T. et al. Gestational diabetes mellitus is associated with the neonatal gut microbiota and metabolome. BMC Med. 19, https://doi.org/10.1186/s12916-021-01991-w (2021).
Graham, M. E. et al. Gut and vaginal microbiomes on steroids: implications for women’s health. Trends Endocrinol. Metab. 32, 554–565 (2021).
Mora-Janiszewska, O., Faryniak-Zuzak, A. & Darmochwał-Kolarz, D. Epigenetic links between microbiota and gestational diabetes. IJMS 23, 1831 (2022).
Wei, J. et al. 16S rRNA gene amplicon sequencing of gut microbiota in gestational diabetes mellitus and their correlation with disease risk factors. J. Endocrinol. Investig. 45, 279–289 (2021).
Huang, S. et al. Lachnospiraceae -derived butyrate mediates protection of high fermentable fiber against placental inflammation in gestational diabetes mellitus. Sci. Adv. 9, https://doi.org/10.1126/sciadv.adi7337 (2023).
Wang, S. et al. Maternal gestational diabetes mellitus associates with altered gut microbiome composition and head circumference abnormalities in male offspring. Cell Host Microbe 32, 1192–1206.e5 (2024).
Wang, S., Liu, Y., Qin, S. & Yang, H. Composition of maternal circulating short-chain fatty acids in gestational diabetes mellitus and their associations with placental metabolism. Nutrients 14, 3727 (2022).
Yang, Q. et al. Oat β-glucan supplementation pre- and during pregnancy alleviates fetal intestinal immunity development damaged by gestational diabetes in rats. Food Funct. 14, 8453–8466 (2023).
Crusell, M. K. W. et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome 6, https://doi.org/10.1186/s40168-018-0472-x (2018).
He, Y., Zheng, C., He, M. H. & Huang, J. R. The causal relationship between body mass index and the risk of osteoarthritis. IJGM 14, 2227–2237 (2021).
Chen, S., Liu, X. & Peng, Y. MicroRNA-351 eases insulin resistance and liver gluconeogenesis via the PI3K/AKT pathway by inhibiting FLOT2 in mice of gestational diabetes mellitus. J. Cell. Mol. Med. 23, 5895–5906 (2019).
Liang, C., DeCourcy, K. & Prater, M. R. High–saturated-fat diet induces gestational diabetes and placental vasculopathy in C57BL/6 mice. Metabolism 59, 943–950 (2010).
Li, Y. J. et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid–mediated activation of G protein–coupled receptors GPR43 and GPR109A. JASN 31, 1267–1281 (2020).
Björkman, L. et al. The Neutrophil response induced by an agonist for free fatty acid receptor 2 (GPR43) is primed by tumor necrosis factor alpha and by receptor uncoupling from the cytoskeleton but attenuated by tissue recruitment. Mol. Cell. Biol. 36, 2583–2595 (2016).
Reuter, J. A., Spacek, D. V. & Snyder, M. P. High-throughput sequencing technologies. Mol. Cell 58, 586–597 (2015).
Li, J. M. et al. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: a benefit of short-chain fatty acids. Microbiome 7, https://doi.org/10.1186/s40168-019-0713-7 (2019).
Pu, Y. et al. A role of the subdiaphragmatic vagus nerve in depression-like phenotypes in mice after fecal microbiota transplantation from Chrna7 knock-out mice with depression-like phenotypes. Brain Behav. Immun. 94, 318–326 (2021).
Qin, S. et al. Gut microbiota in women with gestational diabetes mellitus has potential impact on metabolism in pregnant mice and their offspring. Front Microbiol. 13, https://doi.org/10.3389/fmicb.2022.870422 (2022).
Chi, N. F. et al. Untargeted metabolomics predicts the functional outcome of ischemic stroke. J. Formos. Med. Assoc. 120, 234–241 (2021).
Hou, J. Y. et al. Integrated transcriptomics and metabolomics analysis reveals that C3 and C5 are vital targets of DuZhi Wan in protecting against cerebral ischemic injury. Biomed. Pharmacother. 155, 113703 (2022).
Wingett, S. W. & Andrews, S. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res 7, 1338 (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Tan, L., Xu, Q., Wang, Q., Shi, R. & Zhang, G. Identification of key genes and pathways affected in epicardial adipose tissue from patients with coronary artery disease by integrated bioinformatics analysis. PeerJ 8, e8763 (2020).
Luo, H. & Ma, C. Identification of prognostic genes in uveal melanoma microenvironment. PLoS ONE 15, e0242263 (2020).
Shi, T. & Gao, G. Identify potential prognostic indicators and tumor-infiltrating immune cells in pancreatic adenocarcinoma. Biosci. Rep. 42, https://doi.org/10.1042/bsr20212523 (2022).
Li, H. B. et al. Faecalibacterium prausnitzii Attenuates CKD via Butyrate-Renal GPR43 Axis. Circ. Res. 131, e120–e134 (2022).
Hoving, L. R., Heijink, M., van Harmelen, V., van Dijk, K. W. & Giera, M. GC-MS analysis of short-chain fatty acids in feces, cecum content, and blood samples. Methods Mol Biol. 247–256. https://doi.org/10.1007/978-1-4939-7592-1_17 (2018).
Zhang, S., Wang, H. & Zhu, M. J. A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta 196, 249–254 (2019).
Ma, X. H. et al. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 124, 1406–1417 (2014).
White, K. E. & Bilous, R. W. Estimation of podocyte number: a comparison of methods. Kidney Int. 66, 663–667 (2004).
Briley, S.M. et al. Reproductive age-associated fibrosis in the stroma of the mammalian ovary. Reproduction 245–260 https://doi.org/10.1530/rep-16-0129 (2016).
Chen, X., Wen, J., Liu, C., Guo, D. KLF4 downregulates FGF21 to activate inflammatory injury and oxidative stress of LPS‑induced ATDC5 cells via SIRT1/NF‑κB/p53 signaling. Mol. Med. Rep. 25. https://doi.org/10.3892/mmr.2022.12680 (2022).
Tian, Y. et al. Activation of RARα receptor attenuates neuroinflammation after SAH via promoting M1-to-M2 phenotypic polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-κB pathway. Front. Immunol. 13, https://doi.org/10.3389/fimmu.2022.839796 (2022).
Yi, J., Zheng, Y., Miao, C., Tang, J. & Zhu, B. Desflurane preconditioning induces oscillation of NF-κB in human umbilical vein endothelial cells. PLoS ONE 8, e66576 (2013).
Croissant, C. et al. DDR1 and DDR2 physical interaction leads to signaling interconnection but with possible distinct functions. Cell Adhes. Migr. 1–11 https://doi.org/10.1080/19336918.2018.1460012 (2018).
Chen, L. et al. A necroptosis related prognostic model of pancreatic cancer based on single cell sequencing analysis and transcriptome analysis. Front. Immunol. 13, https://doi.org/10.3389/fimmu.2022.1022420 (2022).
Feng, S. et al. MiR-93 regulates vascular smooth muscle cell proliferation, and neointimal formation through targeting Mfn2. Int J. Biol. Sci. 15, 2615–2626 (2019).
Cong, P. et al. Identification of the role and clinical prognostic value of target genes of m6A RNA methylation regulators in glioma. Front. Cell Dev. Biol. 9, https://doi.org/10.3389/fcell.2021.709022 (2021).
Morris, R., Loftus, A., Friedmann, Y., Parker, P. & Pallister, I. Intra-pelvic pressure changes after pelvic fracture: a cadaveric study quantifying the effect of a pelvic binder and limb bandaging over a bolster. Injury 48, 833–840 (2017).
Acknowledgements
This study was supported by the Liaoning Provincial Department of Education Research Project Program (No. L2010612).
Author information
Authors and Affiliations
Contributions
H.W. and T.K. conducted the experiments, performed data analysis, and contributed to manuscript drafting. W.L. conceptualized the study, supervised the research, and revised the manuscript critically for important intellectual content. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical statement
All animal experiments were approved by the Animal Ethics Committee of First Hospital of China Medical University. We have complied with all relevant ethical regulations for animal use.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Dr Madhumita Basu and Dr Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Kang, T. & Li, W. Reduced maternal SCFAs in GDM diminish GPR43 signaling and induce offspring CAKUT. Commun Biol 8, 1063 (2025). https://doi.org/10.1038/s42003-025-08469-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08469-y
