
Abstract
Bacteria usually possess more than one quorum sensing (QS) regulatory modules that sometimes form complex regulatory networks. These configurations have evolved through the integration of novel transcription factors into the native regulatory systems. However, the selective advantages provided by these alternative configurations on QS-related phenotypes is poorly predictable only based on their underlying network structure. Here, we show that the acquisition of extra regulatory modules of QS has important consequences on the overall regulation of microbial growth by significantly reducing the variability in the final cell density in Burkholderia. By mapping the distribution of horizontally transferred QS modules in extant bacterial genomes, we found that these tend to add up to already-present modules in the majority of cases. We then selected a strain harboring two intertwined QS modules and, using mathematical modeling, we predicted an intrinsic ability of the newly acquired module to buffer the variability in the final cell density. We validated this prediction choosing one strain possessing both systems, deleting one of the two and measuring parameters such as cell density and QS synthase promoter activity. Finally, using transcriptomics, we show that the de-regulation of metabolism likely plays a key role in differentiating the two configurations.
Similar content being viewed by others


Introduction
Quorum sensing (QS) is a communication system among bacterial cells that allows a coordinate collective behavior depending on cell density, growth rate, and on the composition of the surrounding microbial community1,2,3. It is based (i) on communication molecules of different chemical nature, called autoinducers (AI), that are produced and secreted by bacterial cells and (ii) on receptor regulators that, after the recognition of a given amount of a specific AI, can control the expression of several different genes4. In Gram-negative bacteria QS is typically regulated by N-acyl-homoserine lactone (AHL) molecules-mediated systems. This is the case of the model organism Vibrio fischeri where LuxI synthesizes an AHL signal and LuxR is an AHL receptor protein that activates or represses gene expression by binding to a consensus sequence in the promoter regions of target genes5. Many different cellular processes are under the control of QS, including virulence, pathogenesis, biofilm formation and fluorescence6. They are generally highly expensive from an energetic point of view, and bacterial cells activate them only when a large population can benefit from them. As a consequence, the regulation of this phenotype is tightly controlled by microorganisms that, in time, have evolved many different regulatory strategies that ultimately give origin to complex behaviors that include environment-dependent life-style switches7. Interestingly, in many cases, bacteria possess more than one QS system4,6,8.While in some cases the multiple QS systems are based on distinct types of signal molecules, in others they respond to the same class of compounds, thus giving rise to complex regulatory interactions whose outcomes still not understood. Arguably, these complex regulatory systems derive from simpler systems (e.g., a native LuxIR module) that were later modified by diverse evolutionary processes. Early studies on the origin and evolution of AHL-based systems, for example, have indicated HGT (Horizontal Gene Transfer) as a possible factor in the spreading of QS modules among Bacteria9,10. Nowadays, it is clear that QS genes are indeed often associated with mobile genetic elements (MGEs), that QS circuits can move through HGT11,12,13 and that upon transmission they will begin interacting with the residing ones. Much less clear and predictable are the effects of these non-native QS modules on the overall phenotype of the receiving cells, despite they might be of importance in the characterization of specific biological processes (e.g., infections, biodegradation, etc.) as shown by Bellieny-Rabelo et al. (2020). These authors showed that the HGT-mediated reprogramming of QS regulatory circuit, conferred specific selective advantages for the expression of crucial host adaptation- and fitness-oriented systems in the Pectobacteriumand Dickeya14. Similarly, representatives of the Pseudoalteromonas genus were shown to possess highly heterogeneous QS regulation schemes with QS modules that resemble those of other microorganisms (like Escherichia coli, Vibrio and Pseudomonasspecies). The addition of these modules has been hypothetically linked to the capability of colonizing complex ecological niches thanks to a more finely tuned, QS-mediated, collective behavior15. From a broader perspective, the inclusion of a regulatory module inside a mobile element and its further acquisition by a microorganism, represents an extraordinary example of the plasticity of cellular transcriptional networks and an exceptional occasion to study the effect of alternative regulatory architectures on the expression of some phenotypic traits. Indeed, as HGT and/or recombination events lead to the appearance of novel regulatory modules, these, to be maintained over evolutionary time scales, have (i) to integrate with the native one(s) and, eventually, (ii) increase the fitness of the recipient organism. As for the first point, as stated, recent analyses confirmed that some extant QS regulatory networks engaged in HGT during their evolution and have been maintained by present-day microorganisms. Regarding the second aspect, instead, our knowledge is lagging far behind. In other words, it remains unknown why the regulation of QS in some microorganisms is hierarchical (i.e., composed by multiple, interacting modules as shown, for example, in the genus Burkholderia16,17 or in Vibrio fischeri18) and which are the functional advantages of multi-input (QS) systems. Here, we first quantified the distribution of non-native LuxI and LuxR copies across Prokaryotes, validating the role of HGT in shaping the structure and dissemination of QS modules within microbial genomes. To gain a deeper understanding of the impact of acquiring an additional QS module on the phenotype of a receiving cell, we then selected one specific case among those identified in our genome-level analysis (the CepIR and CciIR systems in Burkholderia) and modeled the possible outcomes of introducing an extra QS module in a genome. To confirm the model predictions, we generated mutants harboring a reduced (core) regulatory circuit (mirroring the pre-HGT situation) and compared their QS response with the wild-type circuit (complete circuit, mirroring the post-HGT architecture). Finally, whole transcriptome sequencing and differential gene expression analysis identified key metabolic genes repression as a possible effector in diverting the phenotype in cells harboring the additional QS module. Overall, we observed the capability of the newly acquired module to reduce the variability in final cell density values over a 48 h growth period and, from a more general standpoint, we addressed the possible consequences of evolution-driven changes in the structure of a regulatory circuit.
Results
The occurrence of non-native QS-modules in prokaryotic genomes
We started by examining how often a QS module composed of a luxIR-like gene pair is found on genomic islands (GI) i.e., large chromosomal regions with evidence of HGT. This put our work into perspective since a large body of literature supports the idea that the members of the canonical QS module (i.e., a luxIR gene pair) share a common origin followed by co-evolution as regulatory modules19. Because many bacterial strains contain multiple QS modules in their genome, HGT from independent sources has been indicated as a possible way to expand/modify the underlying regulatory circuits9,12,19,20. To verify this hypothesis and quantify the contribution of HGT in this process we searched present day GIs for the presence of QS modules. Overall, we identified 259 microbial GIs containing at least one LuxI-like and one LuxR-like coding genes within close boundaries (Fig. 1, Supplementary Data 1 and https://doi.org/10.5281/zenodo.12796961). Conversely, we found 3087 LuxIR-like pairs that were (i) close to one another in their harboring genome and (ii) were not included in any of the GIs analyzed. This latter group of sequences likely represents a set of native LuxIR pairs, i.e., QS modules that did not originate from a transfer event, at least in evolutionary recent times. In terms of relative frequency that’s 8.0% of 3346 analyzed QS modules associated to a GI. Perhaps more importantly, in 63.32% of the cases a newly acquired QS module entered a chromosome that already owned a QS module, thus probably giving rise to a complex interplay between the native and the non-native regulatory systems. These results underscore the importance of HGT in determining the distribution of added QS modules in extant prokaryotic genomes. However, what remains to be addressed is the possible array of selective advantages provided by the acquisition of additional QS modules. In other words, why would multiple, horizontally transferred QS modules be maintained over the course of evolution? In the following paragraphs we will describe in detail one specific case of non-native QS modules integration among those shown in Fig. 1 and examine its possible contribution to specific cellular phenotypes.

The direction of each arrow indicates whether that feature is annotated on the forward strand (pointing right) or reverse strand (pointing left). Marked by an asterisk are those that have at least another LuxIR couple elsewhere on the native genome. The fourth and fifth GIs from the top are the same species yet come from two distinct assemblies (GCF_003443555.1 and GCF_000154745.2, respectively). LuxIs are shown in purple and LuxRs in red, whereas protein-coding genes involved in genetic mobilization are displayed in shades of orange and green. The native luxIR counterpart is not represented in this figure. The representation of the entire set of 259 GIs identified in this study are available as Dataset S1.
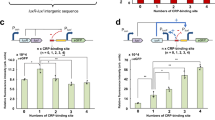
Two concurrent QS regulations in the Burkholderia genus
Among the QS modules with a history of HGT identified in the first part of the analysis, we picked as study case the well characterized CepIR and CciIR systems in Burkholderia21. Besides the standard LuxIR-like system that goes under the name of CepIR, some of the strains in this genus possess an integrated regulatory module named CciIR that influences (and is influenced by) CepIR expression, as schematically shown in Fig. 2A. Specifically, the integration of CciIR and CepIR systems occurs through the implementation of several positive and negative control loops (Fig. 2B). The overall layout of the CepIR and CciIR QS modules resembles that of the classical LuxIR circuit, except for the negative autoregulation of CepR and CciR (LuxR-like proteins are commonly found as positively autoregulated). Furthermore, being associated to a genomic island (cci island), this additional module is prone to being horizontally transferred, giving rise to a potentially patchy genomic distribution and, in turn, to a complete (CepIR and CciIR systems together) vs. core (CepIR only) organization of QS regulation in this group of microorganisms. We assessed the distribution of the complete and core architecture across all reference species of Burkholderia by mapping in silico the presence/absence patterns of the cci genomic island genes. Despite part of the cci genomic island being used as an epidemic strain marker21, its actual distribution inside Burkholderia has not been investigated at the genus level. For this reason, we probed the presence of the cci island encoded genes (55) in all the available complete Burkholderia genomes and combined this information with the genomic relatedness of the representatives of this genus. We measured the relatedness in terms of Average Nucleotide Identity (ANI, see Methods) for each pair of Burkholderia genomes included in the dataset. As reported in Supplementary Fig. 1, the distribution of the genes by the cciisland follows a pattern that matches the one obtained through ANI computation. More in detail, in the cluster comprising B. mallei, B. pseudomallei and B. thalilandiensis species, the complete set of cci genes is never found. Instead, the distribution of cci genes in the other cluster (hereinafter cci-group, that includes, among the others, the Burkholderia cepacia complex (Bcc) and the members of the B. glumae/gladioli/plantarii group) is patchy and includes (22) microbes that harbor more than 50% of the reference cci genes and others (93) possessing less than 50% of the reference cci genes (Fig. 2C). This pattern suggests a complex evolutionary history for this genomic island, mainly guided by gain/loss events that resulted in some strains harboring a completearchitecture (e.g., Burkholderia cenocepaciaJ2315, K56, ST32) and others a core one (e.g., B. cenocepaciaAU1054, H111, etc.), as schematically shown in Fig. 2C. Having defined the taxonomic boundaries of the QS modules distribution in Burkholderia, we next asked which may have been the consequences and advantages (if any) provided by the acquisition of a novel, additional QS control system and its integration into the native regulatory circuit.

A Schematic representation of the relationship betweeen CepIR and CciIR QS modules. B Detailed representation of the interconnections between CepIR and CciIR QS modules. C Phylogenetic distribution of the cci genes in the Burkholderia genus with a focus on the CepIR and CciIR distribution in B. cenocepacia species. Yellow and purple sqares indicate the presence and the absence of the corresponding genes, respectively.
Modeling the effects of core and complete architectures
To address this point, we first formalized the two QS regulatory networks (core vs. complete) into a comprehensive mathematical model. The QS model implemented in this work is schematically represented in Fig. 2B and detailed in Supplementary Information. The model takes into account the main players shown in Fig. 2B, i.e., 4 genes (cepI, cepR, cciI and cciR), 4 proteins (CepI, CepR, CciI and CciR), 2 activated transcription factors (CepR* and CciR*) and 3 metabolites (C8HSL, C6HSL, O-ACP, octanoyl acyl carrier protein). The latter represents the most likely substrate for the production of AI molecules22. The entire model (hereinafter termed complete) recapitulates the architecture of the Burkholderia representatives possessing both the CepIR and CciIR modules (Fig. 2C), whereas the architecture of those species harboring only the CepIR regulation is accounted for by the core model (the blue circuit of Fig. 2B). The completemodel contains 18 parameters (Supplementary Tables 1 and 2) and 11 of them are shared with the model accounting for the core architecture, thus making the corea subset of the completemodel. Growth and cepI/cciI expression were linked through a Hill-like kinetic in which the growth of the bacterial cells is gradually stopped by the increase of the autoinducer concentration produced by the population (see Supplementary Information). Then, we performed an exploratory experiment to parametrize this model. In particular, we selected one of the strains that naturally bears a complete configuration (B. cenocepacia K56-2) and removed its cciIR module. Hereinafter, we will refer to these two strains as the complete(wt) strain (i.e., the strain harboring both QS regulatory modules, cepIRand cciIR) and the core(mutant) strain (i.e., the strain in which the cciIR module has been deleted). For these two strains, we measured both their cepIpromoter activity and their cell density over a growth period of 48 h. These preliminary experiments (including details on whole-genome sequencing to confirm gene deletion) are described in Supplementary Information, Supplementary Figs. 2 and 3. We then used these experimental data (namely cepI promoter activity, a proxy for cepI expression) and OD600 (a proxy for cellular growth) from the mutant strain to derive the most likely values for the core model parameters using a stochastic curve-fitting method, as described in the Methods section (Fig. 3A, C). The set of the fitted parameters of the core model is reported in Supplementary Table 1. We then proceeded to fit the completemodel. Initially, we constrained the parameters of the completemodel shared with the core model to their fitted values. However, this produced an unsatisfactory fit (dashed line in Fig. 3C) and, for this reason, we decided to remove the constraints of the values obtained when fitting the core model. By doing so, the quality of the fit improved and the models accurately recapitulated the overall dynamics of the system (Fig. 3C, D). More specifically, we computed the goodness-of-fit in both cases by resampling 1000 times the array of the predicted values (to have arrays of simulated and experimental data of the same length) and calculating the coefficient of correlation (r) between the experimental data and the prediction for each of the resamplings. When considering all the resamplings, we found a better (and statistically significant) fit for the model with unconstrained parameters (r = 0.90 vs 0.88, t-test, p < 0.0001). This is compatible with a deep rewiring of the QS regulatory circuit following the deletion of one of the two modules in B. cenocepaciaK56-2, thus confirming the strong integration and feedback between the two systems. The values of the parameters together with their confidence intervals for both the completeand core models are reported in Supplementary Tables 1 and 2. While the deterministic approach is key to understand whether the model is capable of reproducing the main features of these regulatory circuits, it cannot take into account one of the key characteristics of QS, i.e., the capability of cells to sense the correct signal concentration and behave in a coordinate fashion, in the presence of fluctuations. Thus, we simulated the probabilistic model described by equations (1)-(26) in Supplementary Information using the algorithm described in ref. 23, specifically focusing our attention on cell density at stationary level. We computed the fluctuations of the predicted cell density in respect to the end-point predicted by the deterministic coreand complete models (Fig. 3E–G). The results of the simulations revealed that fluctuations are much more pronounced in the core model, while the trajectories of the complete one span over a much narrower range (Fig. 3E–G). Overall, the model seems to suggest that the steady-state concentration of the species in the complete model is less affected by fluctuations. On the contrary, the core model appears to be more sensitive to fluctuations, reflected by a larger variability around their stationary values.

A cepI and cciI expression in the coreand the completearchitectures (RLU stands for Relative Light Units). B OD600 values in the coreand complete architectures. C Model fit of the complete and coremodels for cepI (LU stands for Light Units). D Model fit of the complete and coremodels for OD. In (C, D), experimental data are represented by empty circles and error bars while simulations are represented by solid lines. E The comparison between the fluctuations about the steady state of OD600 in the complete and the core model. F A single stochastic realization implemented through the Gillespie algorithm23 for OD600 values in the complete model. G A single stochastic realization implemented through the Gillespie algorithm for OD600 values in the core model.
An additional feedback loop over QS regulation influences population-level growth variability
To validate the model predictions and to detect the possible variability, if any, in the final stages of Burkholderia mutant growth, we performed a further set of experiments. First, we assessed whether the cell denisty measured during the growth experiments was a reliable reporter of cell counts, since features such as biofilm formation and/or differences in cell volume may interfere with such measure. Evaluating the number of CFUs for different ODs, we confirmed the correlation (Linear regression model, adjusted R2 = 0.87, p = 0.0041) between the two (also when considering the log10-transformed values of cell counts, Adjusted R-squared = 0.911, p = 0.0019), thus confirming that, in these conditions, the optical density can be a reliable proxy of the number of cells within the population (Supplementary Fig. 4). Afterwards, we repeated the preliminary experiment described above for a larger number of replicates (36, Supplementary Fig. 5) and evaluated the final cell density (after 48 h) of the coreand completestrains. We found a larger and statistically significant variability (Levene’s test of variances, p < 0.0025, Fig. 4A) in the final cell density of the corestrain in respect to the completeone. Quite interestingly, the difference between the coreand completestrains variances resulted to be significant also at earlier time points (i.e., after 35 h. In that case the cell density values followed normal distributions (Shapiro-Wilk normality test, p = 0.8732 and 0.1409 for complete and core, respectively) and their variances were significantly different (i.e., the one of the core strain was higher, F-test, p = 1.438e-05). Prompted by this finding, we also tested whether, besides cell density, also cepI promoter activity displayed different values among the replicates. However, no statistically significant differences were found for the variance of cepI promoter activity between the coreand the completestrains (Fig. 4B). To further validate the observation that the variance of final cell density was greater in the core strain and to check whether it was differentially higher between technical and/or biological replicates, we performed two additional experiments (see Supplementary Figs. 6 and 7). In the first one, each growth curve represented a single biological replicate since each of them was obtained from a different pre-inoculum. In other words, we used ten different pre-inocula to inoculate ten different wells of the same plate. In a second experiment, instead, ten aliquots from the same pre-inoculum were used to inoculate ten different wells (i.e., the growth curves represented ten different technical replicates). In both cases, the growth of the cells was monitored for 48 hours and the variability of the final cell density was assessed for the coreand the completemutant strains. Data obtained (Supplementary Fig. 8) confirmed a larger and statistically significant variability in the final cell density values of the core strain, confirming the data shown in Fig. 4A. To check whether key growth parameters (i.e., temperature and growth medium) influenced such discrepancy between the core and the complete strains, we repeated the same overall experiment schematically represented in Supplementary Fig. 5 at 30 °C (instead of 37 °C as in the original set of experiments) and growing cells on a minimal medium (M9 + glucose as the sole C source) rather than a complex one (LB). Results obtained (Supplementary Fig. 9) overall confirmed the trends observed when cells were grown in the original conditions.

A Growth dynamics of strains harboring the completevs. the core QS regulatory configurations and boxplot showing the variation across the final (end-point) OD600 values in the strains harboring the completevs. the core configuration (the asterisk indicates a statistically significant comparison). B cepI expression in the complete and core configurations (data is shown starting from hour 5 due to extremily noisy reads of luciferase activity in the initial stages of growth). C Growth curves for different starting OD600 values for strains harboring the complete and D the core configurations. E Boxplot showing the variation across the final (end-point) OD600 values of plots in (C, D) panels (the asterisk indicates a statistically significant comparison according to the Levene’s test of homogeneity). All the data shown in this figure refer to n. 4 biological and n. 9 technical replicates.
From this whole set of results, we conclude that one of the differences between the strains harboring the core and the complete architectures resides in the capability of the latter to efficiently lock the population density around a well-defined value at the end of a 48 h growth period. Core circuit-harboring cells lack this fine tuning, thus showing a much more variable signal (Fig. 4A).
As QS is generally considered to be a cell density dependent process, we tested whether the capability of the complete circuit to stabilize the final cell density on a certain value was dependent on the initial number of cells of the population (similar to an inoculum effect). We evaluated, over three independent experiments, the growth of the two strains from a range of initial values of cell density. To homogenize the results, here we report normalized OD600 values in which each OD600 value has been divided by the maximum OD600 observed in the corresponding experiment (raw data from this experiment are reported in Supplementary Fig. 10). As shown in Fig. 4C, the final cell density of cell populations harboring the complete circuit was not influenced by the initial OD600 of the cultures. On the contrary, the core strains reached a much broader range of final OD600 values Fig. 4D. The robustness of this observation was successfully evaluated by testing the variance of the final endpoints of the two strains (core vs. complete, Levene’s test, p = 0.0001414, Fig. 4E). Such narrower range of final cell densities for the complete vs. the core strains was also observed when endpoints from the same starting OD600 were compared between the two configurations (Supplementary Fig. 11). This finding reinforces the idea that the acquisition of an extra QS module mitigates the variability of the final cell cell density, independently from the initial number of cells within the tested range, at least in the strains and in the conditions used herein. Finally, we checked whether the phenotype of strains naturally possessing one of the two regulatory schemes discussed before resembled those of the B. cenocepaciaK56-2 wt and mutant strains. To this purpose, we randomly selected two more strains from the B. cenocepacia group shown in the inlet of Fig. 2C and accounting for the core (B. cenocepacia AU1054) and complete (B. cenocepacia J2315) structures. We then tested the variability of their end-point cell density, in relation to the initial number of cells (i.e., repeating the experiment whose results for the B. cenocepacia K56-2 wt and mutant strains are shown in Fig. 4C–E). As shown in Fig. 5, B. cenocepaciaAU1054 displayed a significantly higher variability in the end-point OD600 in respect to B. cenocepaciaJ2315 (Levene’s test, p < 0.00001), a result that is in line with that obtained with B. cenocepaciaK56-2 wt and mutant strains and the predictions of the mathematical model. A narrower range of final cell densities for the AU1054 vs. J2315 architectures was also overall observed when endpoints from the same starting OD600 were compared between the two configurations (Supplementary Fig. 12).

A Growth curves for different starting OD600 values for B. cenocepacia AU1054 (core-like architecture). B Growth curves for different starting OD600 values for B. cenocepacia J2315 (complete-like architecture). C Boxplot showing the variation across the final (end-point) OD600 values of plots in (A, B) (the asterisk indicates a statistically significant comparison).
Core architecture leads to down-regulation of metabolism in the mutant strain
Next, we investigated which cellular processes might influence the differing phenotypes observed between the core and complete strains (B. cenocepaciaK56-2), and performed RNA-seq over growing populations of the two strains. We sampled the transcriptomes of the strains harboring the two architectures at three different time points of their growth curves, at 20, 25 and 30 h. We focused on three time points closer to the end of the growth curves since that’s where most of the differences in terms of growth dynamics reside, i.e., the variability of the core strain starts to be larger than the one harboring the complete architecture (Fig. 4A). The results of this experiment are shown in Fig. 6. After applying variance stabilization on the gene expression matrix, we explored how well the different architecures over time explain patterns across all samples. Figure 6A shows a clear distinction of the samples in a time-specific manner. Moreover, the deletion strain consistently localizes on top of / separated from the wild type. Some minor, local attractions are also observable. This is in line with the phenotypic differences among the strains that are particularly evident in the final stages of their growth curves (Fig. 4C). In all three cases the density is strong around a fold change of 0 (Fig. 6B, meaning that most of the genes are not differentially expressed or that they exhibit a mild change in gene expression). Samples at T3 show a more relaxed distribution with a slight offset towards negative log fold changes compared to time points T1 and T2 but similar decay around an absolute LFC of 1. We then filtered the genes by significance threshold (Wald significance test, adjusted p < 0.05) and required that the absolute LFC be greater than 1. Table 1 summarizes the upregulated and downregulated genes in the mutant, at each time point, filtered by statistical significance (Wald significance test, adjusted p < 0.05) and by absolute LFC be greater than 1.

A Principal component analysis on the scaled gene expression matrix of all samples. PC1: principal component 1. Principal components were calculated using plotPCA function from DESeq2 package implemented in R. B Log2 fold change density chart of six samples per time point. Higher values indicate genes overexpressed in the deletion strain compared to the wild type. The figure cuts the LFC in the range [-2,2] since most of the data are centered in this interval. C Venn diagram of DEGs at three time points, significant at adjusted p < 0.05 (Wald significance test) and ∣lfc∣ > 1 (D) Functional categories of the upregulated genes in the mutant strains. E Functional categories of the downrelgulated genes in the mutant strain.
We then compared the set of DEGs of each time point and Fig. 6C summarizes this analysis. Among the 91 distinct genes that passed our statistical test, 9 are shared by all three sets of DEGs. At 20 h and 30 h the majority of differentially expressed genes are proprietary to that specific time point, and the set of genes at T2 seems to be a transition state from a gene expression point of view (Fig. 6C). Interestingly, 5 out of the 9 genes shared by all samples, are involved in metabolism and, more specifically, in energy and carbohydrate metabolism (Table 2). Focusing on T3 (i.e., when, according to RNAseq and phenotypic data, the separation between core and complete strains is maximal), the involvement of metabolism is even clearer (Fig. 6D, E, Supplementary Information, Supplementary Table 3). More than half of the genes from both up-regulated and down-regulated categories belong to metabolic processes, with a predominance of energy metabolism among up-regulated ones and a more even distribution among metabolic functions in down-regulated ones. A similar scenario emerged also when DEGs shared between the two strains after 20 h of growth were analysed (Supplementary Fig. 13). While the fraction of genes with unknown function is remarkable among down-regulated genes, these data indicate that one of the cellular features leading to a reduced variability in end-point cells counts in the strain harboring the complete architecture could be the fine-tuned regulation of metabolism and, in particular, in the down-regulation of those involved in energy production. Indeed, these genes were found to be overexpressed following the removal of the cciIRmodule. The regulation of metabolism by means of QS has been widely described in microbes and it has been shown to play important roles in sustaining stable communities and shaping cooperation24,25. Similarly to26, our results suggest that the downregulation of key metabolic processes in a QS-dependent manner may represent a mechanism through which population-level homeostasis is achieved under crowded conditions. A possible scenario connecting gene expression and growth patterns observed here could envision the capability of the complete architecture to exert a tighter control over key nodes in energy metabolism (e.g., acnB aconitate hydratase 2 or D-isomer specific 2-hydroxyacid dehydrogenase only to cite the two top-ranking among upregulated DEGs) leading to a more uniform collective growth phenotype, possibly optimizing energy and resource utilization by individuals living in crowded environments. On the contrary, in the core harboring cells, the absence of such metabolic breaks on the production of key intermediates might cause the loss of a coordinate response at increasingly higher cell densities. We emphasize that a mechanistic understanding of this specific process is lacking, and the data generated in this study will serve as a foundational contribution.
Discussion
QS is known to regulate hundreds of different traits in a given bacterial species27. QS regulatory circuits often display a complex organization that results in several feedback and feedforward loops16,28,29,30,31. Both the evolutionary history and the functional understanding of these complex structures are largely unknown10. Here we have shown that HGT is a major force driving the occurrence of multiple, integrated QS modules in the same genome14. In particular, the inclusion of additional regulatory systems (CciIR in the case of the Burkholderiagenus) inside a genomic island represents an extraordinary example of the plasticity of the cellular transcriptional network and an exceptional occasion to study the effect of alternative regulatory architectures on the expression of specific phenotypic traits. Indeed, HGT and/or recombination events can lead to the appearance of novel regulatory modules (CciIR in our case) that must exhibit some degree of compatibility with pre-existing ones in order to be maintained over evolutionary time scales14. To understand the functional implications of this increased structural complexity we modeled the additional control of B. cenocepacia K56-2 over the native cepIRsystem and simulated the functioning of this regulatory circuit. Guided by this theoretical framework, we experimentally showed that the simultaneous presence of the two QS modules can stabilize the end-point level of the population around a well-defined value. The lack of the non-native module, instead, introduces significant variability in cell density, starting from soon after the end of the exponential phase. Since it is known that QS functioning can be associated with the low cell density state of bacterial populations32, we confirmed the robustness of the complete circuit for initial cell density levels as low as OD600 = 0.008. While we reckon that using OD600 as a proxy for cell density could have some limitations and the risk of artefacts may, in some cases, become significant (due, for example, to variables such as cell shape, size, clumping, and/or the accumulation of by-products) we nevertheless demonstrated that there is a significant correspondence between the number of cells and OD600, at least in our experimental conditions. Interestingly, we didn’t notice any significant difference in the variability of cepI promoter activity, at least at the population level. Whole transcriptome sequencing in the two strains, led to the identification of cellular energy metabolism as a likely key factor in determining the two growth phenotypes observed here. Our analyses seem to suggest the capability of the cepIR regulatory module to fine tune key energetic pathways in the cell, whose deregulation may contribute to a non-cooperative growth behavior. Existing evidence shows that an imbalanced cellular metabolism could lead to variable cell density patterns (and vice versa) and this, in turn, may be caused by a plethora of different mechanisms. Metabolic upregulation, for example, may generate end products that could interfere with overall gene expression and cellular activity thus introducing feedback mechanisms that introduce fluctuations that contribute to cell density noise33,34. Moreover, feedback regulation of glycolitic enzymes can lead to oscillations in metabolic activity that influence growth rates and cell density stability35. Furthermore, metabolic dysregulation could impact the biosynthesis of QS autoinducers, thereby disrupting coordinated and homogeneous population-level growth phenotypes.
Through computational analysis, mathematical modeling, in vitro experiments and whole genome/transcriptome sequencing we have identified a connection between QS, metabolism and fine-tuning of bacterial cell density in Burkholderia, yet some aspects of the more intimate relationship of these three players remain obscure. For example, although we didn’t observe any population-level heterogeneity of cepI promoter activity, it remains to be addressed whether the expression of this AI-synthase occurs stochastically at the single cell resolution (as observed in other organisms in refs. 36,37) and/or whether ii) this is somehow affected by the two different QS regulations configurations. It would also be intriguing to quantify the concentration of key intracellular energetic metabolites and assess whether the observed differences in specific metabolic genes propagate throughout the entire metabolic network, and if so, to what extent. Finally, since it has been shown that cepRand cciRare capable of jointly tuning the expression of many virulence factors38, we acknowledge the relevance in pointing future work toward the characterization of key pathogenic phenotypes in relation to the two distinct architectures investigated in this study, and whether or not they represent actionable target for new therapeutic strategies. Further work is necessary to address these specific questions.
Materials and methods
Retrieval of genomic islands
We retrieved genomic islands information from the IslandViewer4 database39, maintained by the Brinkman lab at Simon Fraser University, Canada. The pre-computed genomic island coordinates for all sequenced genomes until March 2022 were downloaded from IslandViewer at pathogenomics. sfu. ca/islandviewer/ on July 29, 2023. After much research, we have chosen this framework because of its consistency in providing updated GI predictions over several years and because it offers information based off three diverse prediction methods. Briefly, SIGI-HMM40 identifies alien genes using a generative model on codon usage bias, bypassing the highly expressed genes. IslandPath-DIMOB41 couples dinucleotide bias and mobility gene presence in a window of at least eight genes as a sensitive way to detect GI regions, whereas IslandPick42 finds probable islands and non-island regions through a flexible comparative genomic approach. We downloaded the data set that is the union of SIGI-HMM, IslandPath-DIMOB and IslandPick to try capturing as much variability as possible. We refer to this as the IslandViewer4 data set, that is a table of genomic coordinates (see Supplementary Data 1).
Retrieval of genome sequences and annotations
The IslandViewer4 data set contains chromosome names, but it does not tie them to the assembly accession they belong to. We therefore first downloaded the assembly structure reports files sourced by RefSeq from https://www.ncbi.nlm.nih.gov/assemblyby searching ’Bacteria’ and applying the filters “Complete genome" and “Chromosome", to be consistent with39. On Sept 12th 2023 this search produced 40255 results, which we downloaded and copied to our local server. These files were parsed and a conversion table associating assembly accession and chromosome names was extracted. Crossing the conversion table with the actual chromosome names in the IslandViewer4 data set allowed us to retrieve a unique list of 17868 assembly accession numbers used in the prediction of the genomic islands by the above mentioned third party. We used the NCBI datasets standalone application to download the sequences and annotations of the bacterial genomes present in the unique list of accessions derived from the IslandViewer4 data set. The list of accessions is available in Supplementary Data 1.
Identification of LuxIR-like sequences and data validation
After the pre-identification with HMMER 3.3.2 we further validated our data having all analyses active on the InterProScan 5.60 software, and filtering for an E value of at most 0.001. We labeled as P (passed), the LuxI-like proteins predicted as IPR001690 (Autoinducer synthase), IPR016181 (Acyl-CoA acetyltransferase) or IPR018311 Autoinducer synthase CS). We also labeled as P the LuxR-like representatives predicted to have both the autoinducer-binding and the helix-turn-helix domain, formally IPR005143+IPR000792, or IPR036693+IPR016032 or IPR036693+IPR036388. We did not discard the entries that did not pass the InterProScan validation, but we labeled them as F (failed). The data table is available in Supplementary Data 1. Using these filters, we obtained an F/P ratio of 0.40. Throughout the manuscript we refer to LuxIR-like as only the proteins that passed the InterProScan validation.
ANI calculation
The relatedness of Burkholderia genomes was measured using Average Nucleotide Identity (ANI) as implemented in FastANI 1.3343.
Statistical tests and reproducibility
Unless otherwise specified, we used the Levene’s test of variance44 for assessing whether the corestrain had a higher variance in end-point cell density values in respect to complete. We used this test because our datasets of cell densities end-points did not follow a normal distribution (according to a Shapiro-Wilk normality test). In case where the data to be tested actually followed a normal distribution (again according to a Shapiro-Wilk normality) we used a parametric test, i.e., the F-test as implemented in R. The statistical tests to evaluate significant differentially expressed genes was the Wald test45, adjusting the p-value for multiple tests. To assess the correlation between OD600 and cell counts we used a linear regression as implemented in the lm function in R. As for replicates, we considered technical replicates the growth curves and cepI promoter activity profiles obtained from the same pre-inoculum but from different wells of the same multi-well plate. We instead considered biological replicates those data coming from different pre-inocula (the main experiment reported in Supplementary Fig. 5 was repeated 4 times, i.e., four biological replicates).
Bacterial strains, media and culturing conditions
The bacterial strains and plasmids used in this work are reported in Supplementary Table 4. Unless stated otherwise bacteria were grown under aerobic condition at 37 °C in Luria-Bertani (LB) agar or broth. Antibiotic concentration used were 40 μg/ml kanamycin, 50 μg/ml trimethoprim, 35 μg/ml chloramphenicol and 15 μg/ml tetracycline for E. coli, 100 μg/ml trimethoprim, 200 μg/ml chloramphenicol, 200 μg/ml ampicillin and 150 μg/ml tetracycline for B. cenocepacia K56-2. All antibiotics were purchased from Merk Life Science S.r.l.
Construction of deletion strains
Deletion strains were constructed as described in refs. 46, 47. Briefly, two DNA regions flanking the CciIR system genes were amplified from the B. cenocepacia K56-2 genome using the primer pairs cciI_L_for-cciI_L_rev and cciR_R_for- cciR_R_rev (Table S5). PCR amplifications were performed using PhusionTM High-Fidelity DNA Polymerase (Thermo Fisher Scientific) with specific amplification conditions for each primer pair. The obtained PCR products were digested with XbaI-BamHI and BamHI-KpnI (Thermo Fisher Scientific) respectively and ligated (T4 DNA Ligase, Thermo Fisher Scientific) into the pGPI-SceI-XCm suicide vector (containing a unique restriction site for the endonuclease I-SceI and the xylE reporter gene, Supplementary Table 4)46 digested with XbaI-KpnI. The obtained plasmids were introduced by transformation in E. coli SY327 and then mobilized by conjugation to B. cenocepacia K56-2. Exconjugants were selected in the presence of trimethoprim, chloramphenicol and ampicillin at the concentrations reported above for each strain. Then, a second plasmid, pDAI-SceI-SacB (encoding the I-SceI endonuclease)46 was introduced by conjugation, producing site specific double-strand breaks at the I-SceI recognition site. Resulting exconjugants were tetracycline resistant (due to the presence of pDAI-SceI-SacB) and trimethoprim and chloramphenicol susceptible (indicating the loss of the integrated mutagenic plasmid) and were selected using tetracycline and ampicillin at the concentrations reported above for each strain. The loss of the integrated plasmid was confirmed by plating in presence of trimethoprim and chloramphenicol. Finally, the plasmid pDAI-SceI-SacB was cured, and cured mutants were selected on LB plates without salt and supplemented with 5% (wt/vol) sucrose (Oxoid S.p.A.) and then screening the resulting colonies for loss of tetracycline resistance. The desired gene deletions were first confirmed by PCR amplification using primer pair E_cciI_for- E_cciR_rev (Supplementary Table 5) and then by genome sequencing.
Construction of reporter strains
The promoter regions of cepI (K562_21904 in B. cenocepacia K56-2) were amplified from the genome of B. cenocepacia K56-2 using the primer pair pcepI_for- pcepI_rev (Supplementary Table 5). The obtained PCR products were digested with BamHI and KpnI (Thermo Fisher Scientific) and ligated (T4 DNA Ligase, Thermo Fisher Scientific) into the mini-ctx-lux plasmid48, Supplementary Table 4) digested with the same restriction enzymes. The obtained plasmids were introduced by transformation in E. coli SM10 and then mobilized by conjugation to B. cenocepacia K56-2 and K56-2 ∆cciIR. Exconjugants were selected in the presence of tetracycline and ampicillin at the concentrations reported above for each strain. Successful chromosomal insertion of the plasmids was first confirmed by PCR using the primer pairs tRNA_for-mini_ctx_1 and tRNA_rev-mini_ctx_2 (Supplementary Table 5) and by genome sequencing.
Genome sequencing
Genomic DNA of B. cenocepacia K56-2, K56-2 ΔcciIR, K56-2 pcepI-lux and K56-2 ΔcciIR pcepI-lux was extracted using the DNeasy UltraClean Microbial Kit (Qiagen S.r.l). Whole genome shotgun sequencing (2 × 150 bp) was performed by BMR genomics S.r.l. The sequencing reads were submitted to the NCBI on Oct-30-2023, and are available under the SRA experiment identifier PRJNA1033778.
Measurement of the activity of pcepI-lux reporters
To determine the levels of expression from the promoter regions of cepI and growth dynamics in the B. cenocepacia K56-2 pcepI-lux and K56-2 ΔcciIR pcepI-lux strains, three overnight cultures of each strain were diluted until an OD600 of 0.01 and 200 μl of these dilutions were aliquoted in triplicate in a white assay 96 wells microplate with clear bottom (VWR International S.r.l.) and incubated at 37 °C in an Infinite 200 PRO Tecan plate reader. Both OD600 and luminescence were measured every hour for 48 h, with an orbital shaking of 10 s before each measurement. Luminescence was expressed in relative light units per optical density of the culture (RLU/OD600). The experiment was repeated four times.
Growth curve experiments for strains K56-2, K56-2 ΔcciIR, AU1054 and J2315
For growth curves of B. cenocepacia K56-2, K56-2 ΔcciIR, AU1054 and J2315 strains at different initial ODs, overnight cultures of each strain were diluted until an OD600 between 0.008 and 0.02 and 200 μl of these dilutions were aliquoted in triplicate in a 96 wells microplate (Sarsted AG and Co.) and incubated at 37 °C in an Infinite 200 PRO Tecan plate reader. OD600 was measured every hour for 48 h, with an orbital shaking of 10 s before each measurement. The experiment was repeated three times.
For growth curves of B. cenocepacia K56-2 and K56-2 ΔcciIR strains in minimal medium and at 30 °C, three overnight cultures of each strain were diluted until an OD600 of 0.01 in M9 medium (Merck Life Science S.r.l.) added with Glucose (Oxoid S.p.A.) at final concentration of 0.5% or LB. 200 μl of these dilutions were aliquoted in triplicate in a 96 wells microplate (Sarsted AG and Co.) and incubated at 37 °C (minimal medium) or 30 °C (LB) in an Infinite 200 PRO Tecan plate reader. OD600 was measured as stated above. The experiment was repeated three times.
For growth curves of B. cenocepacia K56-2 and K56-2 ΔcciIR strains with different biological and technical replicates, in the first case both strain cells were grown overnight in ten different pre-inocula, adopting the same overall growth conditions of the original set of experiments. After dilutions, 200 μl from each of the pre-inocula were inoculated into ten different wells of a 96 wells microplate. This experiment was repeated three times. In the second experiment, instead, cells of both strains were grown in a single pre-inoculum that, after dilutions, was used to inoculate ten different wells, each with the same amount of cell culture volume. Also in this case, the experiment was repeated three times.
Tecan calibration curve
To evaluate the correspondence between the OD600 values measured by an Infinite 200 PRO Tecan plate reader and the number of cells we used the following procedure: (i) we prepared 3 overnight cultures of B. cenocepacia K56-2, (ii) these cultures were then diluted into 6 different cultures, with an OD600 ranging from ~0.2 to ~1.2, (iii) the OD600 of these dilutions were evaluated using the same instrument, volumes and microplates of the original experiments described in the text, (iv) serial dilutions of diluted cultures of point (iii) were prepared, (v) 100 μL of each of these serial dilutions were plated on LB plates, (vi) plates were incubated for 48 h at 37 °C, vii) CFU/mL were determined for each of these dilutions, viii) the results were plot as OD600 vs. log10(CFU/mL) (Fig. S3).
Mathematical modeling and parameters fitting
The mathematical model (both deterministic and stochastic) of the QS regulation in B. cenocepacia K56-2 is reported in detail in Supplementary Information. The deterministic model described therein was implemented using MATLAB® 2022b. The ode45 solver was used to solve the set of differential equations accounting for the dynamics of the system. To estimate the parameters of the model from experimental data we used a stochastic curve-fitting in-house MATLAB software. The algorithm is based on the paper by Cardoso et al.49 and consists in the combination of the non-linear simplex and the simulated annealing approach to minimize the squared deviation function. The set of fitted parameters are reported in Supplementary Tables 1 and 2, together with their confidence intervals (CI). 95% CI of the fitted parameters were computed by performing 100 bootstrap resamplings of the original data matrices and estimating, for each of them, the best parameters set. Values presented are averages of these 100 resemplings. Finally, the stochastic simulations were performed implementing the Gillespie algorithm23 in C. The codes used to perform the simulations reported in this work are available at DOI: 10.5281/zenodo. 12796961.
Sampling for RNA sequencing experiment
For RNA-seq, overnight cultures of B. cenocepacia K56-2 and K56-2 ΔcciIR were diluted until an OD600 of 0.01 and 200 μl of this dilution were aliquoted in quadruple in a 96 wells microplate (Sarsted AG & amp; Co.) and incubated at 37 °C in an Infinite 200 PRO Tecan plate reader (Tecan Group Ltd.). OD600 was measured every hour. After 20 h, 25 h and 30 h, cells from each of 4 replicates of the same strain, were recovered and pooled, and 500 μl of this pool were treated with the RNA protect bacteria reagent (Qiagen S.r.l.) and conserved at −80 °C. Each experiment was repeated three times.
RNA extraction and sequencing
Total RNA was extracted using a RNeasy Mini Kit (QIAGEN S.r.l.) following manufacturer’s instructions, extending the incubation with the DNase enzyme (RNase-Free DNase Set, QIAGEN S.r.l.) to 1 h. RNA concentration and quality were assessed using a QUBIT RNA assay kit and a Qubit 4 Fluorometer (both from Invitrogen - Thermo Fisher Scientific Inc.) and an Agilent RNA 6000 Nano kit and an Agilent 2100 Bioanalyzer (both from Agilent Technologies Italia S.p.A.). RNA sequencing (2 × 150 bp) was performed by BMR genomics S.r.l. on Illumina platform (Illumina, Inc.). The sequencing reads were submitted to the NCBI and are available under the SRA experiment identifier PRJNA1033778.
Sequencing reads filtering and validation
Quality assessment and filtering using fastp 0.23.2 (parameters: –cut_tail –average_qual 20 –trim_poly_g –detect_adapter_for_pe –length_required 55 –overrepresentation_analysis –overrepresentation_sampling 1000) yielded most of the initial reads, with very little trimming and filtering needed. The overrepresentation flag enabled the sampling of one every 1000 sequencing reads, which were later confirmed to be mostly ribosomal RNAs.
Sequencing reads diagnostics
RefSeq (GCF_014357995.1) Burkholderia cenocepacia K56-2 reference genome, GFF3 annotation and transcripts files were accessed on 18th March 2024 via NCBI. Polished reads were mapped to the reference genome using bowtie2 2.4.4 in sensitive mode and enabling the end to end setting (entire read must align, parameters: –end-to-end –sensitive). Inspection of the SAM files showed >98% of reads mapping to the reference genome sequence with an equal balance between read pairs flagged as 99/147 (properly paired, R1 aligns to the forward strand) and 83/163 (properly paired, R2 aligns to the forward strand), consistent with the equal strandedness of the reference genome and our inward stranded reverse library preparation protocol. Half or more of the read pairs mapped to non-coding RNAs (mostly rRNA), and this was also confirmed by a manual check of the overrepresented reads after filtering with fastp. A median of 99.03% reads aligned well to the reference genome, confirming no major flaws in sequencing or in the experimental procedure.
Transcripts abundance estimation
An index file for quantification was built from the RefSeq transcript file using version 1.10.0 using k-mer=21 and with decoy awareness enabled (parameters -k and -d). Transcripts abundance estimation was performed with salmon 1.10.050 in selective alignment mode using the inward stranded reverse parameter “ISR”, consistent with our library preparation layout (parameters: -l ISR –seqBias –gcBias –posBias –incompatPrior 1 –minScoreFraction 0.8 –numGibbsSamples 10). We enabled these command line options to fine-tune the quantification process and account for various sequence-specific biases and improve accuracy in estimating transcript abundances. A total of 292,463,702 raw read counts were estimated across 18 samples and 7008 transcripts. The final number of genes effectively utilized (row sum over all samples below 20) was 6974, with locus tag prefixes “K562_RS”. An average of 44.16% (s.d 10.48) of the total reads were attributed to mRNA, which is expected since the samples were dominated by non-coding RNAs, which the NCBI transcripts file omits. The codes used for the analysis of the RNAseq data are available at DOI: 10.5281/zenodo. 12796961.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The experimental data and the simulation results that support the findings of this study are available in Zenodo under the following https://doi.org/10.5281/zenodo.12796961. Sequencing and RNAseq reads are available under the SRA experiment identifier PRJNA1033778.
Code availability
All the codes used to generate the figures of this manuscript are available at https://doi.org/10.5281/zenodo.12796961.
References
Abisado, R. G., Benomar, S., Klaus, J. R., Dandekar, A. A. & Chandler, J. R. Bacterial quorum sensing and microbial community interactions. mBio 9, 10–1128 (2018).
O’Grady, E. P., Viteri, D. F. & Sokol, P. A. A unique regulator contributes to quorum sensing and virulence in Burkholderia cenocepacia. PloS ONE 7, e37611 (2012).
Nickzad, A. & Déziel, E. Adaptive significance of quorum sensing-dependent regulation of Rhamnolipids by integration of growth rate in Burkholderia glumae: a trade-off between survival and efficiency. Front. Microbiol. 7, 1215 (2016).
Whiteley, M., Diggle, S. P. & Greenberg, E. P. Progress in and promise of bacterial quorum sensing research. Nature 551, 313–320 (2017).
Waters, C. M. & Bassler, B. L. QUORUM SENSING: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21, 319–346 (2005).
Papenfort, K. & Bassler, B. L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016).
Hense, B. A. & Schuster, M. Core principles of bacterial autoinducer systems. Microbiol. Mol. Biol. Rev. 79, 153–169 (2015).
Le Guillouzer, S., Groleau, M.-C. & Déziel, E. The complex quorum sensing circuitry of Burkholderia thailandensis Is both hierarchically and homeostatically organized. mBio 8, e01861–17 (2017).
Gray, K. M. & Garey, J. R. The evolution of bacterial luxi and luxr quorum sensing regulators. Microbiology 147, 2379–2387 (2001).
Rajput, A. & Kumar, M. In silico analyses of conservational, functional and phylogenetic distribution of the luxi and luxr homologs in gram-positive bacteria. Sci. Rep. 7, 6969 (2017).
Prescott, R. D. & Decho, A. W. Flexibility and adaptability of quorum sensing in nature. Trends Microbiol. 28, 436–444 (2020).
Hudaiberdiev, S. et al. Census of solo luxr genes in prokaryotic genomes. Front. Cell. Infect. Microbiol. 5, 20 (2015).
Yu, Z. et al. Dissemination of genetic acquisition/loss provides a variety of quorum sensing regulatory properties in pseudoalteromonas. Int. J. Mol. Sci. 19. https://www.mdpi.com/1422-0067/19/11/3636 (2018).
Bellieny-Rabelo, D., Nkomo, N. P., Shyntum, D. Y. & Moleleki, L. N. Horizontally acquired quorum-sensing regulators recruited by the PhoP regulatory network expand the host adaptation repertoire in the phytopathogen Pectobacterium brasiliense. mSystems 5, e00650–19 (2020).
Yu, Z. et al. Dissemination of genetic acquisition/loss provides a variety of quorum sensing regulatory properties in pseudoalteromonas. Int. J. Mol. Sci. 19, 3636 (2018).
Le Guillouzer, S., Groleau, M.-C. & Déziel, E. The complex quorum sensing circuitry of burkholderia thailandensis is both hierarchically and homeostatically organized. MBio 8, 10–1128 (2017).
Le Guillouzer, S., Groleau, M.-C. & Déziel, E. Two rsam homologues encode central regulatory elements modulating quorum sensing in burkholderia thailandensis. J. Bacteriol. 200, 10–1128 (2018).
Pérez, P. D., Weiss, J. T. & Hagen, S. J. Noise and crosstalk in two quorum-sensing inputs of vibrio fischeri. BMC Syst. Biol. 5, 1–14 (2011).
Bez, C., Geller, A. M., Levy, A. & Venturi, V. Cell-cell signaling proteobacterial luxr solos: a treasure trove of subgroups having different origins, ligands, and ecological roles. Msystems 8, e01039–22 (2023).
Lerat, E. & Moran, N. A. The evolutionary history of quorum-sensing systems in bacteria. Mol. Biol. Evol. 21, 903–913 (2004).
Baldwin, A., Sokol, P. A., Parkhill, J. & Mahenthiralingam, E. The Burkholderia cepacia epidemic strain marker is part of a novel genomic island encoding both virulence and metabolism-associated genes in Burkholderia cenocepacia. Infect. Immunity 72, 1537–1547 (2004).
Montebello, A. N. et al. Acyl-ACP substrate recognition in Burkholderia mallei BmaI1 Acyl-Homoserine Lactone Synthase. Biochemistry 53, 6231–6242 (2014).
Gillespie, D. T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361 (1977).
Liu, L. et al. Ahl-mediated quorum sensing to regulate bacterial substance and energy metabolism: a review. Microbiol. Res. 262, 127102 (2022).
Tang, X. et al. Metabolomics uncovers the regulatory pathway of acyl-homoserine lactones based quorum sensing in anammox consortia. Environ. Sci. Technol. 52, 2206–2216 (2018).
An, J. H., Goo, E., Kim, H., Seo, Y.-S. & Hwang, I. Bacterial quorum sensing and metabolic slowing in a cooperative population. Proc. Natl. Acad. Sci. 111, 14912–14917 (2014).
Miranda, S. W., Asfahl, K. L., Dandekar, A. A. & Greenberg, E. Pseudomonas aeruginosa quorum sensing. Adv. Exp. Med. Biol. 1386, 95–115 (2022).
Ulrich, R. L. et al. Role of quorum sensing in the pathogenicity of burkholderia pseudomallei. J. Med. Microbiol. 53, 1053–1064 (2004).
Ulrich, R. L., DeShazer, D., Hines, H. B. & Jeddeloh, J. A. Quorum sensing: a transcriptional regulatory system involved in the pathogenicity of burkholderia mallei. Infect. Immunity 72, 6589–6596 (2004).
Lee, J. & Zhang, L. The hierarchy quorum sensing network in pseudomonas aeruginosa. Protein Cell 6, 26–41 (2015).
Goryachev, A. B. Design principles of the bacterial quorum sensing gene networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 45–60 (2009).
Grote, J., Krysciak, D. & Streit, W. R. Phenotypic heterogeneity, a phenomenon that may explain why quorum sensing does not always result in truly homogenous cell behavior. Appl. Environ. Microbiol. 81, 5280–5289 (2015).
Barkai, N. & Leibler, S. Robustness in simple biochemical networks. Nature 387, 913–917 (1997).
Elowitz, M. B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Hynne, F., Danø, S. & Sørensen, P. G. Full-scale model of glycolysis in saccharomyces cerevisiae. Biophys. Chem. 94, 121–163 (2001).
Bettenworth, V. et al. Frequency modulation of a bacterial quorum sensing response. Nat. Commun. 13, 2772 (2022).
Mellini, M. et al. Rsal-driven negative regulation promotes heterogeneity in pseudomonas aeruginosa quorum sensing. mBio 14, e02039–23 (2023).
O’Grady, E. P., Viteri, D. F., Malott, R. J. & Sokol, P. A. Reciprocal regulation by the CepIR and CciIR quorum sensing systems in Burkholderia cenocepacia. BMC Genom. 10, 441 (2009).
Bertelli, C. et al. Islandviewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35 (2017).
Waack, S. et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden markov models. BMC Bioinform. 7, 1–12 (2006).
Bertelli, C. & Brinkman, F. S. Improved genomic island predictions with islandpath-dimob. Bioinformatics 34, 2161–2167 (2018).
Langille, M. G., Hsiao, W. W. & Brinkman, F. S. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 9, 1–10 (2008).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ani analysis of 90k prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Levene, H. Robust tests for equality of variances. J. Am. Stat. Assoc. 69, 364–367 (1974).
Wald, A. Tests of statistical hypotheses concerning several parameters when the number of observations is large. Transac. Am. Math. Soc. 54, 426–482 (1943).
Hamad, M. A., Skeldon, A. M. & Valvano, M. A. Construction of aminoglycoside-sensitive burkholderia cenocepacia strains for use in studies of intracellular bacteria with the gentamicin protection assay. Appl. Environ. Microbiol. 76, 3170–3176 (2010).
Flannagan, R. S., Linn, T. & Valvano, M. A. A system for the construction of targeted unmarked gene deletions in the genus burkholderia. Environ. Microbiol. 10, 1652–1660 (2008).
Becher, A. & Schweizer, H. P. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29, 948–950 (2000).
Cardoso, M. F., Salcedo, R. L. & De Azevedo, S. F. The simplex-simulated annealing approach to continuous non-linear optimization. Comput. Chem. Eng. 20, 1065–1080 (1996).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Acknowledgements
F.D.P. acknowledges the funding from Project ‘Mathematical modeling for a sustainable circular economy in ecosystems’ (grant n. P2022PSMT7) funded by EU in NextGenerationEU plan through the Italian ‘Bando Prin 2022 - D.D. 1409 del 14-09-2022’ by MUR. M.F. acknowledges the funding from (i) Project “EXPLORE - EXploiting pathogens PLOidy to fight drug REsistance: towards a precision medicine approach" funded by EU in NextGenerationEU plan through the Italian ‘Bando Prin PNRR 2022 - D.D. 1409 del 14-09-2022’ by MUR and (ii) Project 20208LLXEJ_002 “Escaping the ESKAPEs: integrated pipelines for new antibacterial drugs" Funded by MUR. C.R. expresses his gratitude to Justin Cook at Simon Fraser University in relation to key additional details about IslandViewer4. F.D.P. thanks Gruppo Nazionale di Fisica Matematica of Istituto Nazionale di Alta Matematica for partial financial support.
Author information
Authors and Affiliations
Contributions
M.F. started and supervised the project, contributed to the development of the mathematical model and performed part of the simulations, wrote the draft of the manuscript. E.P. performed the majority of the experiments described herein, supervised the remaining experimental part of the project, contributed to the writing of the manuscript. F.V. and F.C. performed some of the experiments under the supervision of E.P. C.R. performed the comparative genomic and RNAseq analyses of the work under the supervision of M.F. A.M. contributed to the discussion on the work and to the writing of the manuscript. F.D.P. contributed to the development of the mathematical model, performed the stochastic simulations and contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Chang Ge and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Silvio Waschina and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fondi, M., Riccardi, C., Di Patti, F. et al. The acquisition of additional control over quorum sensing regulation reduces the variability of final cell density in Burkholderia. Commun Biol 8, 1167 (2025). https://doi.org/10.1038/s42003-025-08566-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08566-y
