
Abstract
Understanding the neurophysiological changes underlying conscious-unconscious transitions is a key goal in neuroscience. Using magnetic resonance neuroimaging, we investigate the network connectivity and neurovascular changes occurring as the human brain transitions from wakefulness to dexmedetomidine-induced hypnosis, and recovery. Hypnosis led to widespread decreases in functional connectivity strength and increased structure-function coupling, indicating functional patterns more constrained by the underlying anatomical connectivity. As individuals began to regain consciousness, both connectivity markers returned towards awake levels, with particularly prominent coupling changes across the cerebellum. Neurovascular dynamics were disrupted during hypnosis as well: cerebral blood flow decreased globally—most notably in the brainstem, thalamus, and cerebellum—and continued decreasing even as recovery commenced, except within the cerebellum. Notably, regions with higher functional connectivity strength during wakefulness exhibited greater blood flow reductions during hypnosis. Hypnosis also heightened the amplitude of low-frequency fluctuations in the hemodynamic signal, especially in visual and somatomotor regions. Critically, individuals who regained consciousness faster displayed higher baseline levels of both neurovascular, but not connectivity, markers. Together, these results reveal that the induction of, and emergence from, dexmedetomidine-induced unconsciousness involve widespread, coordinated changes in brain connectivity and neurovascular function; across our findings, we also highlight the recurrent role of cerebellum in conscious-unconscious transitions.
Similar content being viewed by others

Introduction
Unconsciousness is a multifaceted state; it can occur in a naturalistic setting, such as during sleep, in response to sedative/hypnotic drugs, or after a neurological event such as stroke, traumatic brain injury, or epileptic seizure. In addition to its clinical implications, understanding the neural, hemodynamic, and metabolic events taking place during loss of consciousness may provide further insights into the neural basis of consciousness. Anesthesia has emerged as an effective tool in this endeavor as it allows for the reversible manipulation of wakefulness in a highly controlled setting, under a variety of pharmacological agents.
One such agent, dexmedetomidine, has been particularly versatile in the clinical and research setting, due to its ability to induce hypnosis while still allowing for the easy awakening of the participant using gentle tactile or verbal stimulation1,2. It is typically used as a sedative/hypnotic and analgesic agent in the intensive care unit as well as during various medical and surgical procedures3,4. In the research setting, it has been used to pharmacologically induce a state that neurophysiologically resembles non-rapid eye movement (NREM) sleep5,6,7,8. An α2-adrenergic receptor agonist, dexmedetomidine, selectively binds to pre-synaptic α2-adrenergic receptors on neurons that project to major arousal centers across the brain, leading to a decrease in their firing rate and hence norepinephrine release6,9,10,11. Dexmedetomidine is also thought to induce hypnotic effects by engaging endogenous NREM sleep-promoting neural networks7,10,12,13.
Dexmedetomidine-induced loss of consciousness is accompanied by a cascade of neurophysiological changes spanning cortical and subcortical brain regions. Using tools from graph theory, prior work has shown that the dexmedetomidine-induced unconscious state is characterized by a decreased capacity for efficient information transmission, both locally and globally14. These changes result from brain regions reducing their overall synchronization strength with other regions, a feature that is particularly evident within strongly connected areas14. Interestingly, cortico-cortical functional connectivity within two higher-order association networks—the default mode network and the fronto-parietal network—defined using resting-state functional magnetic resonance imaging (fMRI) was found to be preserved during the unconscious state; functional connectivity between the thalamus and higher-order association networks, such as the default mode, executive control, and salience networks, however, was significantly disrupted12,15.
Previous work has also shown that dexmedetomidine affects regional cerebral blood flow (CBF) and metabolism, preferentially decreasing CBF and glucose metabolism in the thalamus, default mode, and bilateral fronto-parietal networks12. Recovery from dexmedetomidine-induced unconsciousness has been associated with (i) a sustained reduction in CBF12, (ii) restored thalamic functional connectivity to the default mode network12, (iii) increased functional connectivity between subcortical brain regions (i.e., locus coeruleus, thalamus) and the posterior cingulate cortex16, and (iv) increased functional activation of the hypothalamus, thalamus, brainstem, cerebellum, anterior cingulate cortex, and portions of the lateral orbital frontal and parietal lobes2. These findings underscore how interactions between posterior circulation regions and anterior regions give rise to volitional consciousness1,2.
Changes in blood flow have been known to accompany changes in neural activity—a process referred to as neurovascular coupling. By quantifying this relationship, recent work suggests a dynamical coupling between CBF and the amplitude of low-frequency (0.01–0.08 Hz) fluctuations (ALFF) in the blood oxygen level-dependent (BOLD) signal17,18,19,20. The power of low-frequency fluctuations has also been reported to change during anesthetic induction. Indeed, anesthetic agents including dexmedetomidine21,22, propofol22,23,24,25, sevoflurane23,26, and high-dose ketamine23,27,28, among others26,29,30, have all been reported to induce an increase in the power of slow/delta frequencies (0.1–4 Hz) in humans, based on electroencephalogram (EEG) recordings. Increases in slow wave activity (<1 Hz) are also evident during NREM sleep25,31 and coma (vegetative state)32,33. These findings collectively highlight the role of low-frequency oscillations in electrophysiologically characterizing unconsciousness (although see refs. 34,35,36 for interesting case studies wherein behavioral levels of consciousness were dissociated from EEG features such as slow wave oscillations).
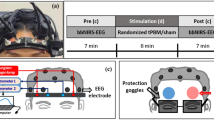
Building upon this work, we utilized multimodal non-invasive magnetic resonance neuroimaging to further characterize the regional connectivity and neurovascular alterations that occur in the human brain as it transitions from the awake state to a state of dexmedetomidine-induced unconsciousness and finally to the early-stage recovery of consciousness. To that end, we assessed two connectivity markers: functional connectivity strength and structure-function coupling (SFC)—a recently reviewed37 metric that quantifies the correlation between a region’s patterns of functional and structural connectivity to the rest of the brain—and two neurovascular markers: CBF and ALFF. We examined functional connectivity strength, SFC, and CBF using arterial spin labeling (ASL) imaging acquired across three awake time-points, two hypnotic time-points, and one recovery time-point, and ALFF using resting-state BOLD fMRI acquired across one awake and one hypnotic time-point (Fig. 1).

Schematic illustration of the experimental setup used in this study. Healthy individuals (n = 14) underwent a magnetic resonance imaging (MRI) battery consisting of structural (T1-weighted), resting-state (BOLD) functional MR, diffusion-weighted, and arterial spin labeling imaging, while awake. Each awake time-point is denoted, followed by the total number of individuals who completed it and the approximate duration. The MRI data collection during wakefulness lasted ~46 min. Each participant then received an intravenous dexmedetomidine bolus over 10 min (1 μg/kg), followed by a continuous dexmedetomidine intravenous infusion (1 μg/kg/h). Once individuals were behaviorally unconscious, the same MRI battery was repeated during hypnosis. For hypnotic time-points 1 and 2, data from all individuals (n = 14) were used. During the last experimental time-point (hypnotic time-point 3/recovery time-point), a subset of individuals began recovering from hypnosis (recovery sample: n = 7); the remaining individuals (unconscious sample: n = 7) remained unconscious. MRI data collection during hypnosis lasted ~40 min. BOLD blood oxygen level-dependent signal, DWI diffusion-weighted imaging, ASL arterial spin labeling.
Given the aforementioned decreased capacity for efficient information transmission during dexmedetomidine-induced loss of consciousness, we first asked whether functional connectivity strength and SFC respectively decrease and increase during the transition from wakefulness to hypnosis, and reverse towards recovery. Moreover, given the reported decreases in CBF and glucose metabolism across select brain regions during loss of consciousness, we next asked whether CBF decreases across the brain during pharmacological hypnosis. Given the dynamic relationship between blood flow and low-frequency fluctuations, as well as the previously reported increases in the power of low-frequency fluctuations in EEG during various states of unconsciousness, we then asked whether ALFF also increases during dexmedetomidine-induced hypnosis. To assess whether dexmedetomidine-induced changes in connectivity and neurovascular dynamics are site-specific, we tested these hypotheses across cerebellar, subcortical, and cortical networks and regions spanning the sensory-association axis of cortical organization, ranging from primary sensory and motor cortices to higher-order association cortices such as the fronto-parietal and default mode networks38,39,40. Lastly, we investigated whether there is a relationship between changes in connectivity and neurovascular dynamics as the brain loses and recovers consciousness.
Results
Connectivity changes from wakefulness to hypnosis
We first examined how functional connectivity strength—estimated via spectral coherence from ASL imaging (see “Methods”)—and structure-function coupling (SFC) change across the brain as it transitions from wakefulness to dexmedetomidine-induced unconsciousness. Functional connectivity strength and SFC were defined using ASL imaging41,42, and assessed across 3 awake and 2 hypnotic time-points (Fig. 1).
Functional connectivity strength remained similar across the 3 awake time-points, and separately across the 2 hypnotic time-points. During the transition from the 3rd awake time-point to the 1st hypnotic time-point, we observed a significant decrease in overall functional connectivity strength (Repeated measures [rm]ANOVA; Tukey’s post-hoc pairwise comparisons: 6% decrease; adjusted pHSD = 0; Fig. 2A). An overall decrease in functional connectivity strength from the average awake to the average hypnotic state was also observed across brain regions (5.1% decrease; paired samples Wilcoxon test: p = 2.1 × 10−23; Fig. 2B) and across individuals (5% decrease; two-sided paired t-test: p = 0.008; Fig. 3A).

A Changes in whole-brain functional connectivity strength across the 3 awake and 2 hypnotic time-points (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). B Changes in whole-brain functional connectivity strength across the average awake state and the average hypnotic state (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). C Changes in whole-brain structure-function coupling across the 3 awake and 2 hypnotic time-points (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). D Changes in whole-brain structure-function coupling across the average awake state and the average hypnotic state (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). p-value annotation legend: non-significant (ns): 0.05 < p ≤ 1, *: 0.01 < p ≤ 0.05, **: 0.001 < p ≤ 0.01, ***: 10−4 < p ≤ 10−3, ****: p ≤ 10−4; p-values in (A, C) correspond to repeated measures ANOVA tests followed by post-hoc correction for multiple comparisons (Tukey’s honestly significant difference [HSD] test), while p-values in (B, D) correspond to paired samples Wilcoxon tests.

A Changes in whole-brain functional connectivity strength for each individual across the average awake state and the average hypnotic state (n = 14 datapoints for each state, denoting each individual). Out of the 14 individuals, 10 displayed decreased magnitude. B Changes in whole-brain structure-function coupling for each individual across the average awake state and the average hypnotic state (n = 14 datapoints for each state, denoting each individual). Out of the 14 individuals, 12 displayed increased magnitude. C Changes in whole-brain functional connectivity strength across the last hypnotic time-point and the recovery time-point (n = 132 brain regions/datapoints for each boxplot, averaged across the 7 individuals that recovered consciousness before the end of the experiment). D Changes in whole-brain functional connectivity strength for each individual across the last hypnotic time-point and the recovery time-point (n = 7 datapoints for each state, denoting each individual that recovered consciousness before the end of the experiment). Out of the 7 individuals, 6 displayed increased magnitude. E Changes in whole-brain structure-function coupling across the last hypnotic time-point and the recovery time-point (n = 132 brain regions/datapoints for each boxplot, averaged across the 7 individuals that recovered consciousness before the end of the experiment). F Changes in whole-brain structure-function coupling for each individual across the last hypnotic time-point and the recovery time-point (n = 7 datapoints for each state, denoting each individual that recovered consciousness before the end of the experiment). Out of the 7 individuals, 5 displayed decreased magnitude. p-value annotation legend: non-significant (ns): 0.05 < p ≤ 1, *: 0.01 < p ≤ 0.05, **: 0.001 < p ≤ 0.01, ***: 10−4 < p ≤ 10−3, ****: p ≤ 10−4; p-values in (A, B, D, F) correspond to paired t-tests, while p-values in (C, E) correspond to paired samples Wilcoxon tests.
Narrowing our focus from the whole-brain level to the network level, we focused on previously established38 resting-state functional networks spanning the sensory-association axis of the human brain: the visual, somatomotor, dorsal attention, ventral attention, limbic, fronto-parietal, default mode, subcortical, and cerebellar networks. Changes in functional connectivity strength were starkly evident across all networks, with each network decreasing its strength as the brain entered hypnosis (Supplementary Table 1; Supplementary Fig. 1). Out of all networks considered, the visual and somatomotor networks displayed the largest percent decreases in magnitude (visual: 9.1% decrease; paired samples Wilcoxon test with Benjamini-Hochberg correction for multiple comparisons: pBH = 3.4 × 10−5, somatomotor: 6% decrease; pBH = 2.3 × 10−5).
We next examined how individual brain regions within these networks altered their functional connectivity strength during the shift from wakefulness to hypnosis. Functional connectivity strength decreases were most prominent within regions involved in visual and auditory processing, including the cuneal cortex, intracalcarine cortex, supracalcarine cortex, and lingual gyrus (Supplementary Table 2).
We then assessed how the relationship between a region’s functional connectivity and underlying structural connectivity changes as the human brain transitions from wakefulness to dexmedetomidine-induced unconsciousness. For that purpose, we turned to SFC, a metric assessing the statistical similarity between a brain region’s structural and functional connectivity patterns towards the rest of the brain. Although similar in magnitude across the 3 awake time-points and across the 2 hypnotic time-points, SFC significantly increased during the transition from the last awake time-point to the first hypnotic time-point (rmANOVA; Tukey’s post-hoc pairwise comparisons: 22.8% increase; adjusted pHSD = 0.003; Fig. 2C). Furthermore, the average hypnotic state was characterized by higher SFC than the average awake state across brain regions (18.7% increase; paired samples Wilcoxon test: p = 1.5 × 10−13; Fig. 2D) and across individuals (19.5% increase; two-sided paired t-test: p = 0.002; Fig. 3B).
On the network level, SFC increased in magnitude in unimodal visual and motor cortices (visual: 18% increase; paired samples Wilcoxon test with Benjamini-Hochberg correction for multiple comparisons: pBH = 2.1 × 10−4, somatomotor: 10.1% increase; pBH = 0.013), higher-order transmodal association cortices (fronto-parietal: 15.1% increase; pBH = 0.028, default mode: 13.7% increase; pBH = 0.002), and cerebellar networks (51.6% increase; pBH = 2.7 × 10−7) (Supplementary Fig. 2). Across these networks, regions within the cerebellum displayed the most pronounced increases in SFC (average 134.1% increase in SFC across individuals; pBH = 0.023), with certain cerebellar regions increasing their SFC more than three-fold (Supplementary Table 3). SFC increases across these regions indicate that, during hypnosis, they displayed functional connectivity patterns that were less dynamic and more similar to their underlying structural connectivity, compared to wakefulness.
Connectivity changes from hypnosis to early-stage recovery
Next, we examined how functional connectivity strength and SFC changed during the transition from hypnosis to early-stage recovery (i.e., between the 2nd hypnotic time-point and the recovery time-point; Fig. 1) on a subset of the population that started regaining consciousness towards the end of the hypnosis protocol (recovery sample: n = 7), while still receiving a steady infusion of dexmedetomidine.
Across the whole brain, there was a significant increase in functional connectivity strength across brain regions towards awake levels (4% increase; paired samples Wilcoxon test: p = 5.1 × 10−21; Fig. 3C). This increase persisted—although as a trend—across individuals (4.4% increase; two-sided paired t-test: p = 0.074; Fig. 3D). Increases in functional connectivity strength were evident across all networks examined, especially across the visual network (6.3% increase; paired samples Wilcoxon test with Benjamini-Hochberg correction for multiple comparisons: pBH = 4.6 × 10−5) (Supplementary Table 4; Supplementary Fig. 3). As a reference, we also examined the change in functional connectivity strength between the 2nd hypnotic time-point and the 3rd hypnotic time-point on the subset of individuals that remained unconscious until the end of the experimental protocol. In contrast to the recovery sample, functional connectivity strength across brain regions within this sample continued decreasing with sustained hypnosis (1.3% decrease; paired samples Wilcoxon test: p = 4.5 × 10−9; Supplementary Figs. 4A and D).
Moreover, although there was no change in whole-brain SFC across individuals towards the early-stage recovery time-point (10.5% decrease; two-sided paired t-test: p = 0.11; Fig. 3F), decreases in whole-brain SFC were pronounced across brain regions (12.1% decrease; paired samples Wilcoxon test: p = 2.4 × 10−8; Fig. 3E), with SFC magnitude reverting back towards awake levels. These decreases were most prominent within the cerebellar network (26.4% decrease; paired samples Wilcoxon test with Benjamini-Hochberg correction for multiple comparisons: pBH = 3.7 × 10−5; Supplementary Fig. 5); notable decreases in SFC towards awake levels were also observed across the default mode network (11.1% decrease; pBH = 0.037). Similar to the recovery sample, the whole-brain SFC of the subset of the population that remained unconscious during the 3rd hypnotic time-point also began reverting towards awake levels during the transition from the 2nd to the 3rd hypnotic time-point, although to a lesser extent (9.4% decrease; paired samples Wilcoxon test: p = 2.6 × 10−6; Supplementary Figs. 4B and E).
Neurovascular changes from wakefulness to hypnosis
To investigate the neurovascular changes that take place as the brain transitions from wakefulness to dexmedetomidine-induced unconsciousness, we next focus on two markers: the amplitude of low-frequency fluctuations (ALFF; 0.01–0.08 Hz) within the resting-state BOLD signal, and the CBF obtained from ASL imaging. To that end, ALFF was assessed during awake time-point 1 and during hypnotic time-point 3, as the resting-state BOLD fMRI had been acquired at only those time-points; the ASL-derived CBF was instead assessed across all awake and hypnotic time-points (Fig. 1).
On average, brain regions’ ALFF increased during hypnosis (28.9% increase; paired samples Wilcoxon test: p = 5 × 10−19; Fig. 4A). This result was recapitulated across individuals (33% increase; two-sided paired t-test: p = 0.011; Fig. 4C). Increases in ALFF were evident across most networks (Supplementary Fig. 6), with the unimodal primary visual and motor, as well as dorsal attention and default mode networks displaying the most pronounced changes (Supplementary Table 5). After dividing each brain region’s ALFF signal by its overall BOLD signal power—a metric commonly referred to as fractional ALFF43—the directionality of results inverted, with fractional ALFF decreasing from wakefulness to hypnosis across brain regions (10.5% decrease; paired samples Wilcoxon test: p = 4.2 × 10−13; Supplementary Fig. 7A) and as a trend across individuals (9.5% decrease; two-sided paired t-test: p = 0.07; Supplementary Fig. 7B). Notably, these decreases were most prevalent across the same networks as ALFF (Supplementary Fig. 8; Supplementary Table 6). Within these identified networks, both ALFF increases and fractional ALFF decreases were particularly robust across occipital and temporal regions involved in visual and auditory processing, including the left planum temporale, lingual gyrus, right cuneal cortex, and posterior superior temporal gyrus (Supplementary Tables 7 and 8).

A Changes in whole-brain amplitude of low-frequency fluctuations (ALFF) across the awake state and the hypnotic state (n = 132 brain regions/datapoints for each boxplot, averaged across the 7 individuals that remained unconscious until the end of the experiment). B Changes in whole-brain cerebral blood flow (CBF) across the average awake state and the average hypnotic state (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). C Changes in whole-brain ALFF for each individual across the awake state and the hypnotic state (n = 7 datapoints for each state, denoting each individual that remained unconscious until the end of the experiment). Out of the 7 individuals, 6 displayed increased magnitude. D Changes in whole-brain CBF for each individual across the average awake state and the average hypnotic state (n = 14 datapoints for each state, denoting each individual). Out of the 14 individuals, 12 displayed decreased magnitude. p-value annotation legend: non-significant (ns): 0.05 < p ≤ 1, *: 0.01 < p ≤ 0.05, **: 0.001 < p ≤ 0.01, ***: 10−4 < p ≤ 10−3, ****: p ≤ 10−4; p-values in (A, B) correspond to paired samples Wilcoxon tests, while p-values in (C, D) correspond to paired t-tests.
Following the opposite directionality than ALFF, CBF decreased, overall, during the transition from wakefulness to hypnosis across brain regions (10.1% decrease; paired samples Wilcoxon test: p = 2.1 × 10−23; Fig. 4B) and across individuals (9.7% decrease; two-sided paired t-test; p = 0.001; Fig. 4D). When examined across the different awake and hypnotic time-points, CBF remained similar across the 3 awake time-points, and separately across the 2 hypnotic time-points. During the transition from the 3rd awake time-point to the 1st hypnotic time-point, however, there was a significant drop in CBF across brain regions (rmANOVA; Tukey’s post-hoc pairwise comparisons: 7.8% decrease; adjusted pHSD = 0.002; Fig. 5A).

A Changes in whole-brain cerebral blood flow across the 3 awake and 2 hypnotic time-points (n = 132 brain regions/datapoints for each boxplot, averaged across all individuals). B Changes in whole-brain cerebral blood flow across the last hypnotic time-point and the recovery time-point (n = 132 brain regions/datapoints for each boxplot, averaged across the 7 individuals that recovered consciousness before the end of the experiment). C Changes in whole-brain CBF for each individual across the last hypnotic time-point and the recovery time-point (n = 7 datapoints for each state, denoting each individual that recovered consciousness before the end of the experiment). p-value annotation legend: non-significant (ns): 0.05 < p ≤ 1, *: 0.01 < p ≤ 0.05, **: 0.001 < p ≤ 0.01, ***: 10−4 < p ≤ 10−3, ****: p ≤ 10−4; the p-value in Fig. 5A corresponds to a repeated measures ANOVA test followed by post-hoc correction for multiple comparisons (Tukey’s honestly significant difference [HSD] test), the p-value in (B) corresponds to a paired samples Wilcoxon test, and the p-value in (C) corresponds to a paired t-test.
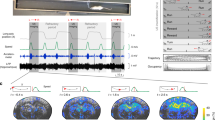
Changes in CBF were ubiquitous across the brain, with all networks diminishing their CBF magnitude during the transition from wakefulness to dexmedetomidine-induced unconsciousness (Supplementary Table 9; Supplementary Fig. 9). Within these networks, the thalamus (an average 18% decrease across individuals; pBH = 6.7 × 10−4), brainstem (an average 19.5% decrease across individuals; pBH = 5.8 × 10−4), and cerebellum (an average 17.4% decrease across individuals; pBH = 0.016) displayed the largest decreases in CBF magnitude during the transition from wakefulness to hypnosis (Supplementary Table 10; Fig. 6).

Top row: Cerebral blood flow (CBF) changes during the transition from wakefulness to hypnosis along axial brain slices (left column), sagittal slices (middle column), and on a 3D rendered standardized (MNI152) brain (right column), across all individuals (n = 14). Warm colors indicate brain regions where CBF is higher during wakefulness, compared to hypnosis. Bottom row: CBF changes during the transition from hypnosis to early-stage recovery of consciousness along axial brain slices (left column), sagittal slices (middle column), and on a 3D rendered standardized (MNI152) brain (right column), across individuals within the recovery sample (n = 7). Warm colors indicate brain regions where CBF is higher during hypnosis, compared to recovery. The orientation of the axial slices on the leftmost column follows neurological convention. All spatial maps were generated using FSL’s randomize tool, a non-parametric permutation inference tool (number of permutations for each analysis = 5000). test statistic: threshold-free cluster enhancement-based statistic110 derived from FSL’s randomize tool; pFWE: p-value corrected for family-wise error.
Neurovascular changes from hypnosis to early-stage recovery
Interestingly, instead of reverting back to awake levels, whole-brain CBF of the recovery sample (n = 7) continued decreasing during the transition from hypnosis to early-stage recovery, across brain regions (3.9% decrease; paired samples Wilcoxon test: p = 5.9 × 10−17; Fig. 5B) and across individuals (3.6% decrease; two-sided paired t-test; p = 0.053; Fig. 5C). This overall sustained decrease in CBF persisted across all networks examined, with the notable exception of the cerebellar network which began reverting its blood flow—albeit non-significantly—towards awake levels (Supplementary Table 11; Supplementary Fig. 10). Similar to the recovery sample, whole-brain CBF in the subset of individuals that remained unconscious during the 3rd hypnotic time-point also continued decreasing with sustained hypnosis (3.5% decrease; paired samples Wilcoxon test: p = 2.2 × 10−17; Supplementary Figs. 4C and F).
BOLD signal-derived ALFF could not be compared between the states of hypnosis and early-stage recovery across the recovery sample, as it was only acquired once at the beginning and once at the end of the experiment (Fig. 1; Supplementary Material: Methodological Limitations).
Individuals who remained unconscious versus individuals who regained consciousness
We next examined whether the aforementioned connectivity and neurovascular markers could distinguish between individuals in our sample who unexpectedly regained consciousness before the end of the experimental session (recovery sample: n = 7) and those who remained unconscious (unconscious sample: n = 7). To that end, we compared the two samples’ average connectivity (functional connectivity strength and SFC) and neurovascular metrics (ALFF and CBF) during the 3 awake time-points, before the onset of hypnosis. Notably, almost all (6/7) individuals within the recovery sample were female (see “Methods”).
In brain connectivity, there were no significant differences in functional connectivity strength and SFC between the two samples, either at the whole-brain or network level (Fig. 7A, B). In neurovascular dynamics, however, individuals who regained consciousness early had significantly higher levels of awake ALFF (percentage difference: 17.2% two-sided Mann-Whitney U test with Benjamini-Hochberg correction for multiple comparisons: pBH = 2.3 × 10−5; Fig. 7C) and CBF (9% increase; pBH = 1.6 × 10−4; Fig. 7D)—but not fractional ALFF—across the whole brain, compared to the individuals who remained unconscious until the end of the experimental session. Differences in ALFF between the two samples were localized within the visual (30.7%; pBH = 5.2 × 10−3), dorsal attention (33.2%; pBH = 0.018), and default mode (15.7%; pBH = 0.046) networks (Supplementary Fig. 11); instead, differences in CBF were pronounced within the somatomotor (9.9%; pBH = 0.014) and cerebellar (8.9%; pBH = 0.014) networks (Supplementary Fig. 12). Collectively, these results indicate that higher awake levels of the neurovascular—but not connectivity—markers examined were indicative of faster recovery of consciousness after dexmedetomidine administration.

A Differences in whole-brain functional connectivity strength between the two samples (n = 132 brain regions/datapoints for each boxplot). B Differences in whole-brain structure-function coupling between the two samples (n = 132 brain regions/datapoints for each boxplot). C Differences in whole-brain amplitude of low-frequency fluctuations between the two samples (n = 132 brain regions/datapoints for each boxplot). D Differences in whole-brain cerebral blood flow between the two samples (n = 132 brain regions/datapoints for each boxplot). p-value annotation legend: non-significant (ns): 0.05 < p ≤ 1, *: 0.01 < p ≤ 0.05, **: 0.001 < p ≤ 0.01, ***: 10−4 < p ≤ 10−3, ****: p ≤ 10−4; p-values here correspond to two-sided Mann-Whitney U tests.
Relationship between connectivity and neurovascular dynamics
In previous sections, we have established changes in connectivity and neurovascular dynamics taking place as the brain transitions between wakefulness, dexmedetomidine-induced unconsciousness, and the early stages of consciousness recovery. Is there, however, a correspondence between these two biological aspects? Do changes in connectivity correlate with changes in neurovascular dynamics? To address this question, we focus on the relationship between functional connectivity and SFC with the two neurovascular markers of interest: ALFF and CBF.
First, we observed a significant negative correlation between a brain region’s functional connectivity strength and ALFF (permutation test across subjects: mean Spearman’s r across subjects = −0.19; pSPICE = 0.007) during the awake time-points. This result indicates that brain regions with higher overall power of low-frequency oscillations tended to display lower overall functional connectivity with the rest of the brain, during wakefulness. This association did not persist during hypnosis.
In contrast to functional connectivity strength, a brain region’s SFC was negatively correlated with its ALFF (permutation test across subjects: mean Spearman’s r across subjects = −0.12; pSPICE = 0.027), only during hypnosis. Similarly, a brain region’s SFC was positively correlated with its fractional ALFF (permutation test across subjects: mean Spearman’s r across subjects = 0.31; pSPICE = 0.024) during hypnosis. This result indicates that during hypnosis, brain regions with higher relative power of low-frequency hemodynamic oscillations (adjusted for the overall BOLD signal power) tended to display functional connectivity patterns that deviate less from the underlying structural connectivity patterns.
Lastly, we observed that a brain region’s functional connectivity strength during wakefulness predicted the subsequent percentage change in CBF within that region, during the transition from wakefulness to hypnosis (permutation test across subjects: mean Spearman’s r across subjects = 0.08; pSPICE = 0.043).
Discussion
Characterizing the neurophysiological changes underlying loss and recovery of consciousness is a fundamental goal in neuroscience. In this work, we examine how the human brain alters its connectivity and neurovascular patterns across these transition periods by analyzing diffusion-weighted, resting-state functional MRI, and ASL imaging data acquired at multiple time-points during dexmedetomidine-induced unconsciousness. Our results indicate that the brain prominently decreased its overall functional connectivity strength during the transition from wakefulness to dexmedetomidine-induced hypnosis, pointing to brain regions becoming less functionally synchronized to each other. In parallel, brain regions—and especially the cerebellum—displayed functional connectivity patterns that were less variable and more reflective of the underlying structural connectivity, as evinced by an increase in SFC during hypnosis. As individuals began regaining consciousness, functional communication across brain regions and SFC returned towards awake levels. Besides changes in brain connectivity, the transition from wakefulness to hypnosis was also characterized by widespread changes in neurovascular dynamics. Cerebral blood flow robustly decreased across the whole brain—especially within the brainstem, thalamus, and cerebellum—and notably continued decreasing in magnitude even after the brain had begun regaining consciousness. Decreases in CBF during hypnosis were accompanied by increases in low-frequency fluctuations in the hemodynamic signal, especially within visual and somatomotor brain regions, collectively pointing towards the prevalence of less metabolically demanding dynamics during hypnosis. In assessing a potential relationship between changes in connectivity and neurovascular dynamics during loss of consciousness, we identified that brain regions with higher levels of functional connectivity strength during wakefulness tended to reduce their CBF more as they entered hypnosis, suggesting a potential mechanistic relationship between the two. Lastly, our results indicated that higher awake levels of the neurovascular—but not connectivity—markers examined were indicative of faster recovery of consciousness after dexmedetomidine administration (Fig. 8).

First box: Changes in connectivity (functional connectivity strength, structure-function coupling) and neurovascular dynamics (cerebral blood flow, amplitude of low-frequency fluctuations) in the human brain during the transition from wakefulness to dexmedetomidine-induced hypnosis. ↑: Marker increased during hypnosis, ↓: Marker decreased during hypnosis. Second box: Changes in connectivity (functional connectivity strength, structure-function coupling) and neurovascular dynamics (cerebral blood flow, amplitude of low-frequency fluctuations) in the human brain during the transition from dexmedetomidine-induced hypnosis to early-stage recovery of consciousness. ↑: Marker increased during recovery, ↓: Marker decreased during recovery. Third box: Faster recovery of consciousness was associated with increased levels of neurovascular dynamics, but not brain connectivity. CBF Cerebral Blood Flow, ALFF Amplitude of Low-Frequency Fluctuations. Fourth box: Relationship between the examined markers of connectivity and neurovascular dynamics. The function corr refers to the correlation between the examined markers, as reported in the manuscript. FC Functional Connectivity Strength, SFC Structure-Function Coupling, CBF Cerebral Blood Flow, ALFF Amplitude of Low-Frequency Fluctuations.
The transition from wakefulness to dexmedetomidine-induced unconsciousness and then recovery of consciousness was characterized by widespread changes in functional connectivity strength across the whole brain. During loss of behavioral responsiveness, functional connectivity strength—assessed via ASL imaging—dropped across all resting-state networks examined (visual, somatomotor, dorsal attention, ventral attention, fronto-parietal, default mode, limbic, subcortical, and cerebellar), indicating a breakdown in communication as networks entering hypnosis became less coherent with the rest of the brain across the frequency band assessed by resting-state fMRI. As individuals began regaining consciousness, whole-brain functional connectivity strength was restored to its awake levels. These findings are consistent with previous work utilizing a complementary definition of functional connectivity strength derived from resting-state BOLD fMRI alongside a set of functional networks (subcortical, sensory, default mode, attention/executive, language/memory), reporting a significant reduction followed by a non-significant increase in functional connectivity strength, during the wakefulness-to-unconsciousness and unconsciousness-to-recovery transitions, respectively14. Building on this work, we additionally report significant increases in functional connectivity strength—and thus synchronization—during the transition from hypnosis to recovery, especially across the visual, dorsal attention, and default mode networks. This increase in synchronization during hypnosis could indicate a restoration in functional communication among brain regions within unimodal sensory and higher-order association networks.
We next turned to changes in the coupling between functional connectivity and the underlying structural connectivity, as individuals traversed from wakefulness to hypnosis and finally to early-stage recovery. SFC robustly increased during loss of consciousness, especially across the unimodal sensory and motor, higher-order transmodal (fronto-parietal and default mode), and cerebellar networks. As individuals began recovering consciousness, whole-brain SFC decreased towards awake levels; such decreases were predominantly robust across the cerebellum (for a more expansive discussion on the putative role of the cerebellum in conscious-unconscious transitions, see the corresponding section in our Supplementary Material).
Intuitively, a brain region’s SFC captures the extent to which its functional connectivity patterns mirror the underlying structural connectivity patterns44. Thus, the observed increase in whole-brain SFC from wakefulness to hypnosis may reflect a decrease in the number of functional configurations the brain can attain during loss of consciousness, given the underlying anatomical connectivity. Our results complement prior work identifying a smaller number of functional configurations and functional connectivity patterns that were progressively more similar to the underlying structural connectivity in patients with disorders of consciousness (vegetative state/unresponsive wakefulness syndrome or in minimally conscious state)45,46,47, in macaque monkeys anesthetized with propofol48,49, sevoflurane49, or ketamine49, in humans transitioning from the awake to propofol-induced unconscious state47,50, and in humans transitioning from wakefulness to deeper levels of sleep50,51.
We also assessed changes in neurovascular dynamics as individuals transitioned from wakefulness to dexmedetomidine-induced unconsciousness, and finally, early-stage recovery. To that end, we focused on two markers: CBF (derived from ASL imaging) and ALFF (derived from resting-state fMRI). ASL-derived CBF provides a direct measure of brain perfusion, which reflects a combination of cerebrovascular integrity, cerebral autoregulation, and neurovascular coupling52,53. In our cohort of young, healthy subjects in whom cerebrovascular disease is highly unlikely, changes in CBF should predominantly reflect neural activity. ALFF provides a measure of neurovascular dynamics as assessed by fluctuations in the BOLD signal, which is sensitive to changes in CBF, cerebral blood volume, and cerebral oxygen metabolism54. Although CBF and ALFF have been shown to be correlated, they may also become decoupled17.
We observed significant decreases in CBF during dexmedetomidine-induced unconsciousness, across all networks and almost all brain regions examined. Regions where CBF displayed the most pronounced decreases in magnitude included the brainstem, thalamus, and cerebellum. Interestingly, CBF continued decreasing across all brain networks during hypnosis and even during the early stages of recovery of consciousness—with the notable exception of the cerebellar network; this is in stark contrast to the connectivity markers (functional connectivity strength and SFC), which started reverting back to their awake levels, even before behavioral arousal. The cerebellum was the only brain network wherein blood flow began reverting—albeit non-significantly—towards awake levels, during the transition from hypnosis to early-stage recovery of consciousness. This finding could point towards the potential early involvement of the cerebellum in restoring consciousness, perhaps by coordinating motor programs.
Our results recapitulate and expand upon complementary studies identifying widespread decreases in cortical and subcortical CBF across a variety of different dosages of dexmedetomidine administration55,56. Furthermore, our findings strongly align with the results of a previous study, which not only reported significant decreases in CBF across the thalamus and higher-order transmodal networks (i.e., default mode and fronto-parietal) during dexmedetomidine-induced unconsciousness, but also revealed that these decreases in CBF did not immediately revert to their awake levels during recovery of consciousness12.
To better understand why dexmedetomidine reduces CBF, we turn to its role as an α2-adrenergic receptor agonist. Agonists, such as dexmedetomidine, have been reported to decrease CBF by (i) activating centrally and peripherally located α2A-adrenergic receptors mediating sympatholytic and vagomimetic effects, causing a decrease in heart rate and arterial pressure57,58, and/or (ii) activating α2B-adrenergic receptors in vascular smooth muscle, causing cerebral vasoconstriction55,57. In addition to decreasing during the transition from wakefulness to hypnosis, our analyses indicated that overall CBF continued decreasing in magnitude even during the initial stages of recovery of consciousness. This finding is physiologically supported by work demonstrating that dexmedetomidine weakens cerebral autoregulation—the ability of cerebral vasculature to maintain stable CBF despite changes in arterial pressure—causing a delay in CBF restoration back to awake levels during conditions of already reduced CBF (such as during dexmedetomidine-induced unconsciousness)59. Besides a potentially weakened cerebral autoregulation, sustained reductions in CBF might also reflect altered neurovascular coupling due to the reduced noradrenergic tone. By decreasing norepinephrine release in arousal-promoting regions, dexmedetomidine could be momentarily weakening the temporal coupling between oxygen demand and blood supply across these regions60, thus delaying the return of CBF to awake levels.
We next turn to the second neurovascular marker of interest, ALFF. Much like CBF61,62,63,64, ALFF is a metric with high test-retest reliability65,66. ALFF captures the local power of low-frequency fluctuations within the 0.01–0.08 Hz range of the resting-state BOLD signal; these fluctuations are thought to reflect each brain region’s intrinsic, spontaneous neural activity, relatively devoid of cognitive load43,67. In contrast to CBF, ALFF robustly increased across the whole brain as individuals transitioned from wakefulness to hypnosis. Fractional ALFF, on the other hand, which measures the relative power of low-frequency fluctuations compared to the overall BOLD signal, decreased across the whole brain. On the regional level, ALFF increases (and similarly fractional ALFF decreases) were dominant within unimodal regions involved in visual, somatomotor, and auditory processing—similar to functional connectivity strength.
Our ALFF results align with previous studies reporting increases in the power of low-frequency BOLD oscillations within a comparable frequency range (0.03–0.10 Hz) across unimodal sensory and motor regions, during the transition from wakefulness to sleep31,68. Similarly, two other studies reported a significant increase in spontaneous, low-frequency BOLD fluctuations (0.01–0.08 Hz) from resting wakefulness to light sleep, especially within primary sensory areas, including visual, motor, and auditory cortices, with the visual cortex displaying the most robust increases69,70. In fact, the magnitude of low-frequency BOLD signal fluctuations observed in the visual cortex during sleep was comparable to that observed during the performance of a visual task by the individuals while they were awake70. The ALFF increases observed during both dexmedetomidine-induced unconsciousness and sleep may reflect changes in neural oscillations—and specifically increases in slow wave amplitudes associated with clearance of metabolic waste from the brain—a process more commonly referred to as glymphatic clearance71—and could provide further evidence of dexmedetomidine acting as a potential glymphatic clearance enhancer72,73,74,75 in humans.
Interestingly, another study examining changes in ALFF using a different anesthetic—propofol—reported overall decreases in both ALFF and fractional ALFF during light and deep sedation, compared to wakefulness, and especially within transmodal frontal cortices76. Collectively, these findings could point towards dexmedetomidine and natural sleep imprinting similar effects on the cortical BOLD signal during the unconscious state—especially within unimodal sensory cortices—as well as highlight the contrasting effects between dexmedetomidine/sleep and propofol in inducing unconsciousness, with the latter preferentially altering the BOLD signal of transmodal cortices.
Collectively, the widespread increases in ALFF, decreases in fractional ALFF, and decreases in CBF observed during dexmedetomidine-induced hypnosis could support the notion that the brain enters a metabolically less-demanding state. Arriving at this conclusion requires revisiting the physiological basis of the BOLD signal. The BOLD signal obtained from resting-state functional MRI is sensitive to local changes in the concentration of de-oxygenated hemoglobin in the blood, which acts as a paramagnetic contrast agent, causing inhomogeneities in the magnetic field and decreasing MRI signal intensity77,78,79. Given that the BOLD signal fundamentally captures changes in brain blood de-oxyhemoglobin, it can indirectly infer changes in local neuronal activation, via the processes of neurovascular and neurometabolic coupling: under normal physiological conditions, increases in neural activity lead to proportionately larger increases in blood flow to the active region relative to the increase in oxygen consumption, resulting in a relative decrease in venous de-oxyhemoglobin concentration. Therefore, the balance between oxygen consumption and oxygen supply modulates the BOLD signal. Prior work in humans has demonstrated that this balance shifts during both NREM sleep80 and after administration of dexmedetomidine56, with oxygen consumption decreasing proportionately more than oxygen supply, compared to wakefulness. This leads to a decrease in paramagnetic de-oxyhemoglobin, and, in turn, an increase in the BOLD signal. The increased BOLD-derived ALFF and decreased CBF observed in our work could thus similarly suggest a decrease in oxygen supply accompanied by an even larger decrease in oxygen consumption taking place during dexmedetomidine-induced hypnosis, consistent with the brain attaining a metabolically less-demanding state. This notion is also corroborated by the decreased fractional ALFF we observed during hypnosis, considering recent work reporting strong correlations between fractional ALFF with oxygen and glucose metabolism81.
Given that a subset of the individuals in our sample unexpectedly regained consciousness before the end of the experimental session while still receiving a maintenance infusion of dexmedetomidine dosed by weight (see Methodological Limitations section in our Supplementary Material), we attempted to identify whether any of the aforementioned connectivity and neurovascular markers differed between them (recovery sample) and the individuals who remained unconscious (unconscious sample).
Although there were no differences in connectivity (functional connectivity strength and SFC) between the two samples, they differed in neurovascular dynamics. More specifically, individuals within the recovery sample displayed higher levels of awake ALFF and CBF across the whole brain, compared to individuals within the unconscious sample. Differences in ALFF were localized across the visual, dorsal attention, and default mode networks, while differences in CBF were localized across the somatomotor and cerebellar networks. These differences could intuitively shed light on why the recovery group might have been able to arouse earlier: a decrease in these individuals’ CBF during hypnosis would still confer them with higher neurovascular tone compared to the rest of the individuals, due to their higher baseline values.
Interestingly, almost all (6/7) individuals within the recovery sample were females, an observation in accordance with recent work in the field indicating that females regain consciousness and recover cognition faster than males after anesthesia—a process seemingly modulated by testosterone’s conversion to estradiol in males82,83,84. More generally, dexmedetomidine has been shown to differentially impact females versus males, producing, for instance, a weaker morphine-sparing effect in controlling post-operative acute pain in females85. Critically, investigating the sex differences between commonly used anesthetic agents—and how their differential impact depends on other factors known to alter induction and emergence timescales, such as age and health status—would be a particularly fruitful clinical direction.
Finally, we examined whether the overall changes in connectivity observed during the transition from wakefulness to dexmedetomidine-induced unconsciousness, and then early-stage recovery, correlated with the reported changes in neurovascular dynamics. Indeed, brain regions characterized by higher levels of spontaneous BOLD fluctuations during wakefulness—in the form of higher awake ALFF—tended to be less functionally synchronized with the rest of the brain. This could indicate that during wakefulness, regions with higher levels of spontaneous activity tend to be more functionally segregated, engaging in specialized processing rather than participating in distributed functionally connected networks. During hypnosis, brain regions with higher levels of ALFF displayed functional connectivity patterns that deviated more from the underlying anatomical connectivity, in the form of lower SFC. This finding could point to these regions’ higher overall intrinsic activity during unconsciousness, decoupled from anatomical constraints.
Lastly, baseline (awake) levels of functional connectivity strength predicted the percentage change in CBF during the transition from wakefulness to hypnosis. In other words, brain regions that were more strongly functionally synchronized to the rest of the brain tended to decrease their CBF more as they entered hypnosis. This finding could indicate how functionally integrated and topologically central brain regions might be entering a “resting” phase during unconsciousness, as a means to recalibrate and restore synaptic homeostasis86.
In summary, we analyzed MRI data collected across multiple time-points to characterize how the human brain’s connectivity and neurovascular dynamics change as it transitions from wakefulness to dexmedetomidine-induced unconsciousness and early-stage recovery of consciousness. During hypnosis, brain regions became less functionally synchronized to each other; they also attained a smaller number of functional configurations compared to wakefulness, and displayed functional connectivity patterns that were more similar to the underlying structural connectivity. Furthermore, cerebral blood flow significantly decreased across the whole brain, and low-frequency fluctuations in the hemodynamic signal became more prominent. Collectively, loss of consciousness was accompanied by widespread connectivity and neurovascular changes in the brain, characteristic of less metabolically demanding dynamics.
Methods
Dataset
Healthy individuals (n = 14; mean age: 28.3 ± 5.5 years; age range: 22–39 years; 10 female) were recruited from the local community and scanned at the Hospital of the University of Pennsylvania using a Siemens Prisma 3 T scanner (64-channel head/neck coil). They specifically underwent a battery of MRI sequences, including T1-weighted (voxel size: 0.8 mm isotropic; repetition time [TR]: 2400 ms; echo time [TE]: 2.24 ms; scan duration: 6 min:36 s), diffusion-weighted (voxel size: 3 mm isotropic; TR: 2500 ms; TE: 65 ms; 61 non-colinear directions; 8 b = 0 acquisitions; multiband acceleration factor: 2; scan duration: 2 min:54 s), resting-state functional (700 volumes; voxel size: 2 mm isotropic; TR: 720 ms; TE: 37 ms; scan duration: 9 min:23 s), and arterial spin labeling (single-shot background-suppressed pseudo-continuous arterial spin labeling [ASL]: 60 volumes; 30 label-control pairs; voxel size: 3.75 mm isotropic; TR: 4000 ms; TE: 10.3 ms; scan duration: 3 min:57 s, along with a reference image to estimate the equilibrium magnetization of blood [M0]) imaging, as previously reported87. Informed consent was obtained from all individuals, and the procedures were approved by the University of Pennsylvania Institutional Review Board. All ethical regulations relevant to human research participants were followed.
The experimental protocol followed is schematically illustrated in Fig. 1 and described below. Baseline scans (one T1-weighted scan, one resting-state functional MRI scan, and three pairs of diffusion-weighted scans, each followed by an ASL sequence) were first acquired while each subject was awake. Each participant then received a bolus administration of the anesthetic agent dexmedetomidine (1 μg/kg) via an intravenous catheter over 10 min followed by a continuous dexmedetomidine infusion (1 μg/kg/h) that began immediately after the bolus had ended and lasted for the duration of the remaining experiment. Dexmedetomidine was used to pharmacologically induce a state resembling NREM sleep. To ascertain wakefulness levels, a camera within the magnet room was entrained on each volunteer’s eyes; intermittent pre-recorded verbal commands were also issued during the scan, asking each volunteer to squeeze a ball that had been taped to their palm. We considered each participant to have entered a hypnotic state if they displayed 1) spontaneous eye closure and 2) a single non-response to the verbal command. After each individual became behaviorally unresponsive, the same MRI sequences (with the exception of the T1-weighted sequence) were re-acquired.
Out of the total of 14 individuals who completed the experiment described above, 7 (mean age: 27.6 ± 6.9 years; age range: 22–39 years; 6 female) began prematurely waking up or displaying signs of behavioral arousal (i.e., squeezing the alarm ball, opening eyes, moving extremities) during the ongoing dexmedetomidine infusion towards the end of the experiment, starting at the beginning of their 3rd pair of “hypnotic” diffusion-weighted scans (Fig. 1). To further study this small sub-sample of individuals who recovered consciousness early, we used their data in this “3rd hypnotic” time-point to define an “early-stage recovery” time-point.
To summarize, data from all 14 individuals were used for the first 5 time-points (the 3 awake and first 2 hypnotic time-points). During the 6th time-point (hypnotic time-point 3/recovery time-point: Fig. 1), a subset of individuals began recovering from hypnosis (recovery sample: n = 7), while the remaining individuals (unconscious sample: n = 7) remained unconscious until the end of the experiment. As denoted in our Results section, data collected during this last time-point were used to: (i) assess how the connectivity and neurovascular variables of interest changed during the transition from hypnosis to early-stage recovery of consciousness (using a recovery sample of n = 7), and (ii) assess how the BOLD-derived ALFF changed during the transition from wakefulness to hypnosis (using an unconscious sample of n = 7).
Processing pipelines
For all analyses considered, cortical, subcortical, and cerebellar brain areas were defined using a combination of the Harvard-Oxford atlas and the Automated Anatomical Labeling (AAL) atlas. This Harvard-Oxford-AAL atlas incorporated 91 cortical regions of interest (ROIs) defined from FSL’s88 Harvard-Oxford maximum likelihood cortical atlas (HarvardOxford-cort-maxprob-thr25-1mm.nii), 15 subcortical ROIs from the Harvard-Oxford maximum likelihood subcortical atlas (HarvardOxford-sub-maxprob-thr25-1mm.nii), and 26 cerebellar ROIs from the AAL atlas, for a total of 132 brain regions89,90,91,92,93.
We applied two pipelines: a (1) connectivity pipeline, wherein changes in functional connectivity and the statistical correlation between structural and functional connectivity—more commonly referred to as SFC—were assessed; and a (2) neurovascular dynamics pipeline, wherein changes in two markers—ALFF and CBF—were estimated. We describe both pipelines in detail below:
Connectivity pipeline
Structural connectivity
Structural connectivity was assessed using the acquired diffusion-weighted imaging sequences. Diffusion data were processed using the MRtrix3 toolbox94. The data were first denoised, and the residual maps (difference maps between the denoised and original diffusion images) were manually examined to ensure that no particular brain region was disproportionately impacted by artifactual noise. Eddy current-induced distortion and motion correction were then applied on the denoised diffusion data using the dwifslpreproc command. Afterwards, the percentage of outlier slices for each diffusion scan was output and manually inspected; all diffusion scans used contained less than 2% of slices classified as outliers. Finally, B1 field inhomogeneity correction was applied on the diffusion data, and an accurate brain mask was generated to restrict all analyses to brain voxels.
The orientation of diffusion within each brain voxel was then determined. First, a basis response function was generated from each individual’s diffusion scans using the dwi2response command and “dhollander” algorithm. The purpose of this step was to decompose the diffusion signal within each tissue type (gray matter, white matter, and CSF) into a set of fiber orientations across different directions. These basis response functions were subsequently used to estimate fiber orientation densities using multi-shell, multi-tissue constrained spherical deconvolution using the dwi2fod command. The resulting fiber orientation densities were then normalized to ensure that any observed differences were not due to intensity contrasts in the diffusion image.
Lastly, the white matter tracts (i.e., streamlines) connecting the different gray matter brain regions were reconstructed using anatomically constrained probabilistic tractography. For this purpose, an initial whole-brain tractogram containing 10 million streamlines was first generated using the tckgen command; the “-act” flag was supplied to the command to only focus on streamlines constrained to the white matter. The streamlines were then refined by assigning them weights to reduce known biases (SIFT2 method95; command tcksift2). Finally, each subject’s refined streamline map was registered to a parcellated brain atlas (Harvard-Oxford-AAL atlas: n = 132 brain regions) that had been registered to the subject’s native anatomical space. Subject-specific, symmetric, weighted structural connectivity matrices of size 132 × 132 were generated for each diffusion scan; each row and column of the matrix corresponded to a brain region, and each edge corresponded to the SIFT2-weighted sum of the streamlines connecting any two given regions divided by the sum of the regions’ gray matter volumes.
Functional connectivity
Functional connectivity was assessed during the 6 different experimental time-points using the acquired ASL sequences, as previously validated41,42.
ASL data were processed using MATLAB (version R2021a: The MathWorks, Inc.) and SPM (version 12)96. The ASL images were first motion corrected using the spm_realign (additional parameter used: 5 mm full-width half-maximum Gaussian kernel smoothing applied) and spm_reslice commands. The motion-corrected ASL images, as well as the reference image M0 acquired at each time-point, were co-registered to the anatomical space (i.e., the individual’s T1-weighted image) using trilinear interpolation, and then to the standardized MNI (2 mm) space using non-linear registration (FSL’s fnirt command).
Using the processed ASL images, we constructed functional connectivity matrices for each of the 6 time-points considered. The mean ASL regional time-series (size: 1 × 60 volumes) was computed for each brain region of interest (for a total of 132 brain ROIs, as per the Harvard-Oxford-AAL atlas); the coherence between two brain regions’ ASL regional time-series was calculated97. From a technical standpoint, coherence measures how strongly synchronized two time-series are in the frequency domain. The coherence between brain region i and brain region j at frequency f is defined as:
where \({P}_{{ij}}(f)\) is the cross power spectral density between the time-series corresponding to brain regions i and j at frequency \(f\)97,98. This metric measures the amount of cross-correlation between these time-series at frequency \(f\), after adjusting for their individual powers at that frequency. The average coherence between two brain regions across all frequencies greater than 0.01 Hz was computed using MATLAB’s mscohere command, and was used to define the functional connectivity between these two brain regions. A lower frequency threshold of 0.01 Hz was applied to remove signals of non-biological origin99. After computing the coherence between all pairs of brain regions for each ASL scan acquired, we constructed the corresponding functional connectivity matrices. A Fisher r-to-z transform was applied on the functional connectivity matrices, using MATLAB’s atanh function, to normalize their values98.
After the functional connectivity matrices were constructed for each individual, each brain region’s functional connectivity strength was defined as the sum of all edges between that brain region and all other brain regions in the functional connectivity matrix.
Structure-function coupling
Pairs of interspersed DWI and ASL sequences were acquired at multiple time-points during both wakefulness and dexmedetomidine-induced hypnosis. This enabled us to respectively assess both structural and functional connectivities during each awake and hypnotic time-point. Given that there were three pairs of structural connectivity matrices generated at the awake state and three pairs at the hypnotic state, while there were only three functional connectivity matrices, we selected the three structural connectivity matrices at each state corresponding to the diffusion scans with the least amount of noise (defined by each scan’s percentage of outlier slices, as defined above). To examine the statistical relationship between structural and functional connectivity across each of these time-points, we computed their SFC. A brain region’s SFC was defined as the Pearson’s correlation coefficient between its structural connectivity vector (size: 1 × 132 ROIs) and its functional connectivity vector (size: 1 × 132 ROIs) towards all other brain regions, after excluding the self-connection and any other entries where either the regional structural or functional connectivity was equal to zero, as previously defined44.
These processes allowed us to compute 6 types of metrics for each connectivity variable (functional connectivity strength and SFC) examined: (i) the metric corresponding to each of the 6 time-points, (ii) the metric during the average awake state, (iii) the metric during the average hypnotic state, (iv) the metric during the recovery state, (v) the percent change in the metric from the average awake to the average hypnotic state, and (vi) the percent change in the metric from the last hypnotic time-point to the recovery time-point. The latter two were calculated using the formulae:
and
where X denotes the connectivity variable of interest (functional connectivity strength or SFC).
Neurovascular dynamics pipeline
Amplitude of low-frequency fluctuations (ALFF)
Changes in ALFF for each individual were assessed using the resting-state functional MRIs acquired during their awake and hypnotic states at the beginning and end of the experimental setup, respectively (Fig. 1). The resting-state functional MRIs were processed with CONN (https://web.conn-toolbox.org/home; version 20.b)100,101, using its “default pre-processing pipeline for volume-based analyses (direct normalization to MNI-space).” A more detailed description of the pipeline has been previously reported44. A brief listing of the processing steps is also provided here: (i) Each subject’s functional scans were co-registered to a reference image and (ii) corrected for any temporal misalignment between slices that may have had occurred during acquisition, (iii) the structural (T1-weighted) scans were segmented into gray matter, white matter, and CSF tissue classes, while both structural and functional scans were normalized into standardized MNI space, and (iv) the functional images were smoothed using an 8 mm full-width half-maximum Gaussian kernel to increase the BOLD signal-to-noise ratio101. After these pre-processing steps had taken place, CONN’s default denoising pipeline was utilized to remove any potential confounders identified in the BOLD signal. Confounders included noise components from white matter and cerebrospinal areas, estimated subject motion parameters (i.e., 3 rotation and 3 translation parameters, and their 6 associated first-order derivatives), flagged outlier scans, and session-related effects such as constant and linear BOLD signal trends. Frequencies below 0.008 Hz were also removed to mitigate the presence of low-frequency drifts and signals of non-biological origin. B0 field inhomogeneities in each subject’s resting-state functional MRI were also corrected, using double-echo gradient echo sequences (2 magnitude volumes and 1 phase difference volume [voxel size: 3 mm isotropic; TR: 580 ms; TE difference: 2.46 ms]) acquired during the MRI battery.
After extracting each individual’s denoised resting-state BOLD signal time-series, the ALFF was estimated at each experimental time-point. The ALFF captures the power of low-frequency (0.01–0.08 Hz) fluctuations within the BOLD signal, and was computed by bandpass filtering the denoised BOLD signal time-series of each brain region over the frequency range 0.01–0.08 Hz101,102. This allowed us to estimate an ALFF value for each brain region (as defined by the Harvard-Oxford-AAL atlas: n = 132 brain regions), for each individual, and at each experimental time-point.
Similarly, a brain region’s fractional ALFF was defined as the ratio between its ALFF and the power of the overall BOLD signal (frequencies ≥ 0.008 Hz). In other words, fractional ALFF quantifies the power of the BOLD signal within the low-frequency range, relative to the power of the overall BOLD signal.
There were 6 types of metrics extracted for each brain region and for each individual: (i) the (fractional) ALFF during the awake state, (ii) the (fractional) ALFF during the hypnotic state, and (iii) the percent change in (fractional) ALFF from the awake to the hypnotic state. The latter was calculated using the formula:
Awake levels of (fractional) ALFF were computed across all individuals (n = 14), and hypnotic levels of (fractional) ALFF were computed only across the individuals who remained unconscious until the end of the experimental session (n = 7; see “Dataset” section). Given that there were only two resting-state functional MRI sequences that were acquired for each individual during the experiment—once during wakefulness and once at the end of the experimental setup (Fig. 1)—we were not able to extract the percent change in (fractional) ALFF from hypnosis to recovery (see Supplementary Material: Methodological Limitations).
Cerebral blood flow
CBF within each brain region was estimated using the processed ASL sequences (see “Functional Connectivity” section above). A CBF map corresponding to each of the 6 experimental time-points was generated using the equation:
where \(\lambda\) is the brain/blood partition coefficient measured in mL/g (set to 0.9), \({{SI}}_{{control}}\) and \({{SI}}_{{label}}\) are the time-averaged signal intensities in the control and label images extracted from the processed ASL image (note: the ASL volumes consist of control and label images acquired in pairs and identically scaled), PLD refers to the post-labeling delay in the ASL scan measured in seconds (set to 1.5), \({T}_{1,{blood}}\) is the longitudinal relaxation time of blood measured in seconds (set to 1.65), \(\alpha\) is the labeling efficiency (scalar; set to 0.72), \({{SI}}_{{PD}}\) is the time-averaged signal intensity of the proton density-weighted image (M0 reference image), and τ is the label duration measured in seconds (set to 1.5)103. The 6000 factor in the numerator is included to convert the units of the above equation from mL/g/seconds to the more conventionally used mL/100 g/min103.
After the above calculation was performed for each time-point, 6 types of metrics were extracted for each brain region and for each individual: (i) the CBF corresponding to each of the 6 time-points, (ii) the CBF during the average awake state, (iii) the CBF during the average hypnotic state, (iv) the CBF during the recovery state, (v) the percent change in CBF from the average awake to the average hypnotic state, and (vi) the percent change in CBF from the last hypnotic time-point to the recovery time-point. The latter two were calculated using the formulae:
and
Statistical analyses
Statistical analyses were performed using MATLAB (version R2021a: The MathWorks, Inc.) and Python (version 3.7). A p-value < 0.05 was used as the threshold for significance.
Whole-brain level analyses
Repeated measures one-way analysis of variance (rmANOVA) tests were used to statistically compare the mean values of functional connectivity strength, SFC, and CBF across the 5 experimental time-points (3 awake time-points and the first 2 hypnotic time-points) described in the “Whole-brain level” sections of our Results. The rmANOVA tests were followed by post-hoc correction for multiple comparisons (Tukey’s honestly significant difference [HSD] test) to identify which pairs of time-points significantly differed in their mean values (as shown in Figs. 2A, C and 5A). To compare the mean values of the metrics of interest between the overall awake state (i.e., the average metric value across the 3 “awake” time-points) and the overall hypnotic state (i.e., the average metric value across the first 2 “hypnotic” time-points) (as shown in Figs. 2B, D and 4A, B), we used a Wilcoxon test for paired measurements, a non-parametric paired test bearing no assumption on the normality of the data distribution. We used the same test to compare brain regions’ functional connectivity strength, SFC, and CBF between the 2nd hypnotic time-point and recovery time-point (as shown in Supplementary Fig. 4 and Figs. 3C, E and 5B). Data from the 3rd hypnotic time-point were not included in the rmANOVA analyses, as a subset of the individuals (n = 7) did not remain unconscious during that time-point.
Lastly, paired t-tests were used to compare the global functional connectivity strength, SFC, ALFF, and CBF values between each individual’s average awake vs. average hypnotic, and last hypnotic time-point vs. recovery time-point (as shown in Figs. 3A, B, D, F, 4C, D and 5C). For each comparison, we report the percent change in the variables’ magnitude and the corresponding p-value.
Network-level analyses
To compare the mean functional connectivity strength, SFC, ALFF, and CBF metrics between the awake, hypnotic, and recovery states (as shown in Supplementary Figs. 1, 2, 3, 5, 6, and 8–10) across the 9 networks examined (visual, somatomotor, dorsal attention, ventral attention, limbic, fronto-parietal, default mode, subcortical, and cerebellar networks)38, we once again used paired samples Wilcoxon tests, with an additional Benjamini-Hochberg correction for multiple comparisons to control for the false discovery rate across these 9 comparisons. For each comparison, we report the percent change in the variables’ magnitude between the three states across each network and the corresponding p-value.
Regional analyses
Paired t-tests were once again used to compare regional differences in functional connectivity strength, SFC, ALFF, and CBF between the awake, hypnotic, and recovery states. For each comparison, we report the percent change in the variables’ magnitude between the three states and the corresponding p-value.
Individuals who remained unconscious versus individuals who regained consciousness
To compare the mean values of functional connectivity strength, SFC, and CBF between the subset of individuals who started regaining consciousness before the end of the experiment (recovery sample: n = 7) and the rest of the individuals who remained unconscious (unconscious sample: n = 7), we used a two-sided Mann-Whitney U test, a non-parametric test bearing no assumption on the normality of the data distribution (as shown in Fig. 7). The same test with a Benjamini-Hochberg correction for multiple comparisons to control for the false discovery rate was used to compare the two samples across the 9 networks examined (as shown in Supplementary Figs. 11 and 12). In contrast to the previous sections in our statistical analyses where we report percent changes in the variables’ magnitude, here we report the percent difference between the variables’ magnitude, since the two samples compared do not consist of the same individuals. The formula used to compute the percent difference for variable X (where X can be functional connectivity strength, SFC, ALFF, or CBF) was:
Relationship between connectivity and neurovascular dynamics
The comparisons reported in the “Relationship between connectivity and neurovascular dynamics” section of our Results were carried out using a previously established “simple permutation-based test of intermodal correspondence” (SPICE test) that assesses how two brain maps correlate at the individual level, on average104. We briefly describe the SPICE test below.
Let us assume that we want to correlate variables (i.e., brain maps) X and Y across multiple individuals. First, we format X and Y to each be of size <number of individuals × number of brain regions considered>, which in our case was 14 × 132. Each entry in these matrices corresponds to the value of the respective variable for a given individual and a given brain region. The permutation test then performs the following steps: (1) First, it computes the Spearman’s correlation coefficient between entries of each row of X and those in each row of Y (i.e., brain regions for each individual); (2) the average correlation coefficient across all individuals is then estimated, and designated as the observed correlation coefficient Cobs; (3) the rows of Y are randomly sampled without replacement, generating a shuffled version of variable Y denoted Ypermuted; what this step does is shuffle the order of individuals within variable Y, while maintaining the order of individuals in variable X (original order of rows of X). This step also maintains inherent spatial dependencies within both X and Y (i.e., original order of columns of both X and Y); (4) the new average correlation coefficient across individuals between variables X and Ypermuted is re-estimated as per steps 1 and 2, and designated as a null correlation coefficient Cnull; (5) steps 3 and 4 are repeated K times (here, K = 10,000) to generate a distribution of null coefficients; (6) a p-value (pSPICE) is finally estimated by counting the number of permutations where the (absolute) magnitude of Cnull exceeds that of Cobs, divided by the total number of permutations (here 10,000) plus 1, which accounts for the identity permutation104.
In the statistical comparisons mentioned in the section “Relationship between connectivity and neurovascular dynamics” of our Results, we report (i) the average Spearman’s correlation coefficient r across subjects for each comparison (variable Cobs described above) and (ii) the corresponding pSPICE value.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The human dataset analyzed in this study was recruited from the local community and scanned at the Hospital of the University of Pennsylvania. Data are available from the corresponding authors upon request. Source data (Supplementary Data 1) are provided with this paper.
Code availability
The MRtrix3 toolbox94 used to process the diffusion data is freely available and can be found at https://www.mrtrix.org, along with extensive documentation. The CONN toolbox100,101 used to process the resting-state fMRI sequences is also freely available and can be found at https://web.conn-toolbox.org/home, along with extensive documentation. The code used to perform the SPICE test104 described in our statistical analyses section that assesses the correlation between two brain maps at the individual level can be found in https://github.com/smweinst/spice_test.
References
Hudetz, A. G. General anesthesia and human brain connectivity. Brain Connect. 2, 291–302 (2012).
Långsjö, J. W. et al. Returning from oblivion: imaging the neural core of consciousness. J. Neurosci. 32, 4935–4943 (2012).
Shehabi, Y. et al. Early sedation with dexmedetomidine in critically ill patients. N. Engl. J. Med. 380, 2506–2517 (2019).
Kaur, M. & Singh, P. M. Current role of dexmedetomidine in clinical anesthesia and intensive care. Anesth. Essays Res. 5, 128–133 (2011).
Huupponen, E. et al. Electroencephalogram spindle activity during dexmedetomidine sedation and physiological sleep. Acta Anaesthesiol. Scand. 52, 289–294 (2008).
Akeju, O. et al. Dexmedetomidine promotes biomimetic non-rapid eye movement stage 3 sleep in humans: a pilot study. Clin. Neurophysiol.129, 69–78 (2018).
Chamadia, S. et al. Oral dexmedetomidine promotes non-rapid eye movement stage 2 sleep in humans. Anesthesiology 133, 1234–1243 (2020).
Scott-Warren, V. L. & Sebastian, J. Dexmedetomidine: its use in intensive care medicine and anaesthesia. BJA Educ. 16, 242–246 (2016).
Brown, E. N., Purdon, P. L. & Van Dort, C. J. General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu. Rev. Neurosci. 34, 601–628 (2011).
Zhang, Z. et al. Neuronal ensembles sufficient for recovery sleep and the sedative actions of α2 adrenergic agonists. Nat. Neurosci. 18, 553–561 (2015).
Gilsbach, R. et al. Genetic dissection of α2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Mol. Pharmacol. 75, 1160–1170 (2009).
Akeju, O. et al. Disruption of thalamic functional connectivity is a neural correlate of dexmedetomidine-induced unconsciousness. eLife 3, e04499 (2014).
Nelson, L. E. et al. The α2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology 98, 428–436 (2003).
Hashmi, J. A. et al. Dexmedetomidine disrupts the local and global efficiency of large-scale brain networks. Anesthesiology 126, 419–430 (2017).
Guldenmund, P. et al. Brain functional connectivity differentiates dexmedetomidine from propofol and natural sleep. Br. J. Anaesth. 119, 674–684 (2017).
Song, A. H. et al. Pharmacological modulation of noradrenergic arousal circuitry disrupts functional connectivity of the locus ceruleus in humans. J. Neurosci. 37, 6938–6945 (2017).
Baller, E. B. et al. Developmental coupling of cerebral blood flow and fMRI fluctuations in youth. Cell Rep. 38, 110576 (2022).
Jin, M. et al. Disturbed neurovascular coupling in hemodialysis patients. PeerJ 8, e8989 (2020).
Chiacchiaretta, P., Cerritelli, F., Bubbico, G., Perrucci, M. G. & Ferretti, A. Reduced dynamic coupling between spontaneous BOLD-CBF fluctuations in older adults: a dual-echo pCASL study. Front. Aging Neurosci. 10, 115 (2018).
Archila-Meléndez, M. E., Sorg, C. & Preibisch, C. Modeling the impact of neurovascular coupling impairments on BOLD-based functional connectivity at rest. NeuroImage 218, 116871 (2020).
Akeju, O. et al. Spatiotemporal dynamics of dexmedetomidine-induced electroencephalogram oscillations. PLoS ONE 11, e0163431 (2016).
Akeju, O. et al. A comparison of propofol- and dexmedetomidine-induced electroencephalogram dynamics using spectral and coherence analysis. Anesthesiology 121, 978–989 (2014).
Lee, U. et al. Disruption of frontal-parietal communication by ketamine, propofol, and sevoflurane. Anesthesiology 118, 1264–1275 (2013).
Lewis, L. D. et al. Rapid fragmentation of neuronal networks at the onset of propofol-induced unconsciousness. Proc. Natl. Acad. Sci. USA 109, E3377–E3386 (2012).
Murphy, M. et al. Propofol anesthesia and sleep: a high-density EEG study. Sleep 34, 283–291A (2011).
Purdon, P. L., Sampson, A., Pavone, K. J. & Brown, E. N. Clinical electroencephalography for anesthesiologists: Part I: background and basic signatures. Anesthesiology 123, 937–960 (2015).
Akeju, O. et al. Electroencephalogram signatures of ketamine-induced unconsciousness. Clin. Neurophysiol.127, 2414–2422 (2016).
Li, D. & Mashour, G. A. Cortical dynamics during psychedelic and anesthetized states induced by ketamine. NeuroImage 196, 32–40 (2019).
Brown, E. N., Pavone, K. J. & Naranjo, M. Multimodal general anesthesia: theory and practice. Anesth. Analg. 127, 1246–1258 (2018).
Warnaby, C. E., Sleigh, J. W., Hight, D., Jbabdi, S. & Tracey, I. Investigation of slow-wave activity saturation during surgical anesthesia reveals a signature of neural inertia in humans. Anesthesiology 127, 645–657 (2017).
Fultz, N. E. et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 366, 628–631 (2019).
Schiff, N. D., Nauvel, T. & Victor, J. Large-scale brain dynamics in disorders of consciousness. Curr. Opin. Neurobiol. 0, 7–14 (2014).
Young, G. B. The EEG in coma. J. Clin. Neurophysiol. 17, 473 (2000).
Pal, D. et al. Level of consciousness is dissociable from electroencephalographic measures of cortical connectivity, slow oscillations, and complexity. J. Neurosci. 40, 605–618 (2020).
Pal, D. et al. Differential Role Of Prefrontal And Parietal Cortices In Controlling Level Of Consciousness. Curr. Biol. 28, 2145–2152.e5 (2018).
Davis, C. J., Clinton, J. M., Jewett, K. A., Zielinski, M. R. & Krueger, J. M. Delta wave power: an independent sleep phenotype or epiphenomenon?. J. Clin. Sleep. Med. 7, S16–S18 (2011).
Fotiadis, P. et al. Structure–function coupling in macroscale human brain networks. Nat. Rev. Neurosci. 1–17. https://doi.org/10.1038/s41583-024-00846-6 (2024).
Yeo, B. T. T. et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 106, 1125–1165 (2011).
Margulies, D. S. et al. Situating the default-mode network along a principal gradient of macroscale cortical organization. Proc. Natl. Acad. Sci. USA 113, 12574–12579 (2016).
Sydnor, V. J. et al. Neurodevelopment of the association cortices: patterns, mechanisms, and implications for psychopathology. Neuron 109, 2820–2846 (2021).
Li, Z. et al. Effects of resting state condition on reliability, trait specificity, and network connectivity of brain function measured with arterial spin labeled perfusion MRI. NeuroImage 173, 165–175 (2018).
Dai, W., Varma, G., Scheidegger, R. & Alsop, D. C. Quantifying fluctuations of resting state networks using arterial spin labeling perfusion MRI. J. Cereb. Blood Flow Metab. 36, 463–473 (2016).
Zou, Q.-H. et al. An improved approach to detection of amplitude of low-frequency fluctuation (ALFF) for resting-state fMRI: Fractional ALFF. J. Neurosci. Methods 172, 137–141 (2008).
Fotiadis, P. et al. Myelination and excitation-inhibition balance synergistically shape structure-function coupling across the human cortex. Nat. Commun. 14, 6115 (2023).
Demertzi, A. et al. Human consciousness is supported by dynamic complex patterns of brain signal coordination. Sci. Adv. 5, eaat7603 (2019).
Perl, Y. S. et al. Low-dimensional organization of global brain states of reduced consciousness. Cell Rep. 42, 112491 (2023).
Luppi, A. I. et al. Distributed harmonic patterns of structure-function dependence orchestrate human consciousness. Commun. Biol. 6, 1–19 (2023).
Barttfeld, P. et al. Signature of consciousness in the dynamics of resting-state brain activity. Proc. Natl. Acad. Sci. USA 112, 887–892 (2015).
Luppi, A. I. et al. Local orchestration of distributed functional patterns supporting loss and restoration of consciousness in the primate brain. Nat. Commun. 15, 2171 (2024).
Castro, P. et al. Dynamical structure-function correlations provide robust and generalizable signatures of consciousness in humans. Commun. Biol. 7, 1–12 (2024).
Tagliazucchi, E., Crossley, N., Bullmore, E. T. & Laufs, H. Deep sleep divides the cortex into opposite modes of anatomical–functional coupling. Brain Struct. Funct. 221, 4221–4234 (2016).
Dolui, S. et al. Reliability of arterial spin labeling derived cerebral blood flow in periventricular white matter. Neuroimage Rep. 1, 100063 (2021).
Detre, J. A. et al. Noninvasive MRI evaluation of cerebral blood flow in cerebrovascular disease. Neurology 50, 633–641 (1998).
Simon, A. B. & Buxton, R. B. Understanding the dynamic relationship between cerebral blood flow and the BOLD signal: Implications for quantitative functional MRI. NeuroImage 116, 158–167 (2015).
Prielipp, R. C. et al. Dexmedetomidine-induced sedation in volunteers decreases regional and global cerebral blood flow. Anesth. Analg. 95, 1052 (2002).
Drummond, J. C. et al. Effect of dexmedetomidine on cerebral blood flow velocity, cerebral metabolic rate, and carbon dioxide response in normal humans. Anesthesiology 108, 225–232 (2008).
Talke, P. & Anderson, B. J. Pharmacokinetics and pharmacodynamics of dexmedetomidine-induced vasoconstriction in healthy volunteers. Br. J. Clin. Pharmacol. 84, 1364–1372 (2018).
Mikkelsen, M. L. G. et al. The effect of dexmedetomidine on cerebral perfusion and oxygenation in healthy piglets with normal and lowered blood pressure anaesthetized with propofol-remifentanil total intravenous anaesthesia. Acta Vet. Scand. 59, 27 (2017).
Ogawa, Y. et al. Dexmedetomidine weakens dynamic cerebral autoregulation as assessed by transfer function analysis and the thigh cuff method. Anesthesiology 109, 642–650 (2008).
Bekar, L. K., Wei, H. S. & Nedergaard, M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J. Cereb. Blood Flow Metab. 32, 2135–2145 (2012).
Yang, F. N. et al. Test-retest reliability of cerebral blood flow for assessing brain function at rest and during a vigilance task. NeuroImage 193, 157–166 (2019).
Almeida, J. R. C. et al. Test-retest reliability of cerebral blood flow in healthy individuals using arterial spin labeling: Findings from the EMBARC study. Magn. Reson. Imaging 45, 26–33 (2018).
Binnie, L. R. et al. Test–retest reliability of arterial spin labelling for cerebral blood flow in older adults with small vessel disease. Transl. Stroke Res. 13, 583–594 (2022).
Kyrou, A. et al. Test-retest reliability of resting-state cerebral blood flow quantification using pulsed Arterial Spin Labeling (PASL) over 3 weeks vs 8 weeks in healthy controls. Psychiatry Res. Neuroimaging 341, 111823 (2024).
Küblböck, M. et al. Stability of low-frequency fluctuation amplitudes in prolonged resting-state fMRI. NeuroImage 103, 249–257 (2014).
Vedaei, F., Alizadeh, M., Romo, V., Mohamed, F. B. & Wu, C. The effect of general anesthesia on the test–retest reliability of resting-state fMRI metrics and optimization of scan length. Front. Neurosci. 16, 937172 (2022).
Wang, P. et al. Amplitude of low-frequency fluctuation (ALFF) may be associated with cognitive impairment in schizophrenia: a correlation study. BMC Psychiatry 19, 30 (2019).
Song, C., Boly, M., Tagliazucchi, E., Laufs, H. & Tononi, G. fMRI spectral signatures of sleep. Proc. Natl. Acad. Sci. USA 119, e2016732119 (2022).
Fukunaga, M. et al. Large-amplitude, spatially correlated fluctuations in BOLD fMRI signals during extended rest and early sleep stages. Magn. Reson. Imaging 24, 979–992 (2006).
Horovitz, S. G. et al. Low frequency BOLD fluctuations during resting wakefulness and light sleep: a simultaneous EEG-fMRI study. Hum. Brain Mapp. 29, 671–682 (2008).
Bohr, T. et al. The glymphatic system: Current understanding and modeling. iScience 25, 104987 (2022).
Persson, N. D. Å, Uusalo, P., Nedergaard, M., Lohela, T. J. & Lilius, T. O. Could dexmedetomidine be repurposed as a glymphatic enhancer?. Trends Pharmacol. Sci. 43, 1030–1040 (2022).
Lilius, T. O. et al. Dexmedetomidine enhances glymphatic brain delivery of intrathecally administered drugs. J. Control. Release 304, 29–38 (2019).
Benveniste, H. et al. Anesthesia with dexmedetomidine and low-dose isoflurane increases solute transport via the glymphatic pathway in rat brain when compared with high-dose isoflurane. Anesthesiology 127, 976–988 (2017).
Wang, S. et al. Dexmedetomidine improves the circulatory dysfunction of the glymphatic system induced by sevoflurane through the PI3K/AKT/ΔFosB/AQP4 pathway in young mice. Cell Death Dis. 15, 448 (2024).
Liu, X. et al. Propofol attenuates low-frequency fluctuations of resting-state fMRI BOLD signal in the anterior frontal cortex upon loss of consciousness. NeuroImage 147, 295–301 (2017).
Hillman, E. M. C. Coupling mechanism and significance of the BOLD signal: a status report. Annu. Rev. Neurosci. 37, 161–181 (2014).
Kim, S.-G. & Ogawa, S. Biophysical and physiological origins of blood oxygenation level-dependent fMRI signals. J. Cereb. Blood Flow Metab. 32, 1188–1206 (2012).
Pauling, L. & Coryell, C. D. The magnetic properties and structure of hemoglobin, oxyhemoglobin and carbonmonoxyhemoglobin. Proc. Natl. Acad. Sci. USA 22, 210–216 (1936).
McAvoy, M. P., Tagliazucchi, E., Laufs, H. & Raichle, M. E. Human non-REM sleep and the mean global BOLD signal. J. Cereb. Blood Flow Metab. 39, 2210–2222 (2019).
Deng, S. et al. Hemodynamic and metabolic correspondence of resting-state voxel-based physiological metrics in healthy adults. NeuroImage 250, 118923 (2022).
Wasilczuk, A. Z. et al. Hormonal basis of sex differences in anesthetic sensitivity. Proc. Natl. Acad. Sci. USA 121, e2312913120 (2024).
Lennertz, R. et al. Connected consciousness after tracheal intubation in young adults: an international multicentre cohort study. Br. J. Anaesth. 130, e217–e224 (2023).
Braithwaite, H. E. et al. Impact of female sex on anaesthetic awareness, depth, and emergence: a systematic review and meta-analysis. Br. J. Anaesth. 131, 510–522 (2023).
Li, Y.-Y., Ge, D.-J., Li, J.-Y. & Qi, B. Sex differences in the morphine-sparing effects of intraoperative dexmedetomidine in patient-controlled analgesia following general anesthesia: a consort-prospective, randomized, controlled clinical trial. Medicine95, e3619 (2016).
Tononi, G. & Cirelli, C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014).
Vidorreta, M. et al. Whole-brain background-suppressed pCASL MRI with 1D-accelerated 3D RARE Stack-Of-Spirals readout. PLoS ONE 12, e0183762 (2017).
Jenkinson, M., Beckmann, C. F., Behrens, T. E. J., Woolrich, M. W. & Smith, S. M. FSL. NeuroImage 62, 782–790 (2012).
Makris, N. et al. Decreased volume of left and total anterior insular lobule in schizophrenia. Schizophr. Res. 83, 155–171 (2006).
Frazier, J. A. et al. Structural brain magnetic resonance imaging of limbic and thalamic volumes in pediatric bipolar disorder. Am. J. Psychiatry 162, 1256–1265 (2005).
Desikan, R. S. et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 31, 968–980 (2006).
Goldstein, J. M. et al. Hypothalamic abnormalities in schizophrenia: sex effects and genetic vulnerability. Biol. Psychiatry 61, 935–945 (2007).
Tzourio-Mazoyer, N. et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 15, 273–289 (2002).
Tournier, J.-D. et al. MRtrix3: A fast, flexible and open software framework for medical image processing and visualisation. NeuroImage 202, 116137 (2019).
Smith, R. E., Tournier, J.-D., Calamante, F. & Connelly, A. SIFT2: Enabling dense quantitative assessment of brain white matter connectivity using streamlines tractography. NeuroImage 119, 338–351 (2015).
Friston, K. J., Ashburner, J. T., Kiebel, S. J., Nichols, T. E. & Penny, W. D. Statistical Parametric Mapping: The Analysis of Functional Brain Images (Elsevier, 2011).
Chen, S. et al. Global functional connectivity at rest is associated with attention: an arterial spin labeling study. Brain Sci. 13, 228 (2023).
Mahadevan, A. S., Tooley, U. A., Bertolero, M. A., Mackey, A. P. & Bassett, D. S. Evaluating the sensitivity of functional connectivity measures to motion artifact in resting-state fMRI data. NeuroImage 241, 118408 (2021).
James, O., Park, H. & Kim, S.-G. Impact of sampling rate on statistical significance for single subject fMRI connectivity analysis. Hum. Brain Mapp. 40, 3321–3337 (2019).
Whitfield-Gabrieli, S. & Nieto-Castanon, A. Conn: a functional connectivity toolbox for correlated and anticorrelated brain networks. Brain Connect. 2, 125–141 (2012).
Nieto-Castanon, A. Handbook of fcMRI Methods in CONN (Hilbert Press, 2020).
Yang, H. et al. Amplitude of low frequency fluctuation within visual areas revealed by resting-state functional MRI. NeuroImage 36, 144–152 (2007).
Alsop, D. C. et al. Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: a consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn. Reson. Med. 73, 102–116 (2015).
Weinstein, S. M. et al. A simple permutation-based test of intermodal correspondence. Hum. Brain Mapp. 42, 5175 (2021).
Mitchell, S. M., Lange, S. & Brus, H. Gendered citation patterns in international relations journals. Int. Stud. Perspect. 14, 485–492 (2013).
Dion, M. L., Sumner, J. L. & Mitchell, S. M. Gendered citation patterns across political science and social science methodology fields. Polit. Anal. 26, 312–327 (2018).
Caplar, N., Tacchella, S. & Birrer, S. Quantitative evaluation of gender bias in astronomical publications from citation counts. Nat. Astron. 1, 1–5 (2017).
Maliniak, D., Powers, R. & Walter, B. F. The gender citation gap in international relations. Int. Organ. 67, 889–922 (2013).
Dworkin, J. D. et al. The extent and drivers of gender imbalance in neuroscience reference lists. Nat. Neurosci. 23, 918–926 (2020).
Smith, S. M. & Nichols, T. E. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. NeuroImage 44, 83–98 (2009).
Acknowledgements
This work was supported by NIH grants T32EB020087 (P.F.), R35GM142712 (A.R.M.W.), R37MH125829 (T.D.S.), R01MH113550 (T.D.S. and D.S.B.), RF1MH116920 (T.D.S. and D.S.B.), R21MH106799 (T.D.S. and D.S.B.), R01MH112847 (R.T.S. and T.D.S.), R01MH123550 (R.T.S.), R01NS112274 (R.T.S.), R01GM088156 (M.B.K.), R01GM144377 (M.B.K.), R01EB031080 (J.A.D.), the Swartz Foundation (D.S.B.), and the John D. and Catherine T. MacArthur Foundation (D.S.B.). This work was further supported by the National Center for Research Resources (grant: UL1RR024134), now at the National Center for Advancing Translational Sciences (grant: UL1TR000003), and partially supported by the University of Pennsylvania’s Institute for Translational Medicine and Therapeutics’ Transdisciplinary Program in Translational Medicine and Therapeutics (M.B.K. and J.A.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors declare no competing interests.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.F., M.B.K., J.A.D., and D.S.B.; methodology: P.F., A.R.M.W., S.M.W., T.D.S., R.T.S., M.B.K., J.A.D., and D.S.B.; data collection: A.R.M.W., P.A.C., M.E., M.C., J.T.D., A.P., M.B.K., J.A.D.; data analyses: P.F.; writing—original draft: P.F.; writing—review and editing: A.R.M.W., S.M.W., P.A.C., M.E., M.C., J.T.D., T.D.S., R.T.S., A.P., M.B.K., J.A.D., and D.S.B.; funding: M.B.K., J.A.D., and D.S.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Citation diversity statement
Recent work in several fields of science has identified a bias in citation practices such that papers from women and other minority scholars are under-cited relative to the number of such papers in the field105,106,107,108,109. We obtained the predicted gender of the first and last author of each reference by using databases that store the probability of a first name being carried by a woman109. By this measure (and excluding self-citations to the first and last authors of our current paper), our references contain 3% woman(first)/woman(last), 12.9% man/woman, 29.7% woman/man, and 54.4% man/man. This method is limited in that a) names, pronouns, and social media profiles used to construct the databases may not, in every case, be indicative of gender identity and b) it cannot account for intersex, non-binary, or transgender people. We look forward to future work that could help us better understand how to support equitable practices in science.
Peer review
Peer review information
Communications Biology thanks Javeria Hashmi and Guillaume Becq for their contribution to the peer review of this work. Primary Handling Editors: Enzo Tagliazucchi and Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fotiadis, P., McKinstry-Wu, A.R., Weinstein, S.M. et al. Changes in brain connectivity and neurovascular dynamics during dexmedetomidine-induced loss of consciousness. Commun Biol 8, 1254 (2025). https://doi.org/10.1038/s42003-025-08577-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08577-9
