
Abstract
Botulinum neurotoxins (BoNT) are the most potent protein toxins for humans, yet how BoNT-Light Chain/A1 (LC/A1) journeys to cleave intracellular SNAP-25 is understudied. Here we use a cell-based assay to measure cytosolic EGFP-LC/A1 intracellular trafficking and SNAP-25 cleavage in Neuro-2A cells. Intracellular LC/A1 associated on microtubules and co-localized with Rab GTPases involved in fast synaptic vesicles and endosome recycling. Multiple Dominant Negative (DN) Rabs GTPases involved in fast synaptic vesicles or endosome recycling inhibited LC/A1 trafficking to the intracellular plasma membrane and SNAP-25 cleavage. A cytosolic LC/A1 variant that bound the plasma membrane from the cytosol was insensitive to DNRab GTPases involved in fast synaptic vesicle recycling. LC/A1 traffics on fast synaptic vesicles to the intracellular plasma membrane to cleave SNAP-25. Our data suggest, like Heavy Chain host cell entry and LC catalysis, LC intracellular trafficking to target host substrates can contribute to bacterial toxin potency.
Similar content being viewed by others
Introduction
Botulinum neurotoxins (BoNT) are Category A Tier 1 Select Agents and potential bioweapons due to the severity and long duration of botulism in humans and vertebrate animals1. Currently, there are no licensed human vaccines against botulism2 and there is a limited therapy window to treat with heptavalent equine antiserum (BAT)3. BAT is effective when administered early in the disease before BoNT enters motor neurons. Once inside motor neurons, BoNT persists and cleaves membrane bound v or t- Soluble N-ethylmaleimide-sensitive-factor Attachment Protein REceptor (SNARE), resulting in long-lasting (weeks to several months) flaccid paralysis, characteristic of botulism1. Each botulism outbreak is a reportable public health emergency4. BoNTs comprise seven immunologically distinct serotypes (A-G) and over 40 subtypes5. Eight BoNT/A subtypes have been described, including A1-A85, where BoNT/A1 is an effective and widely used human therapy to treat movement disorders, reaching 6 million annual injections administered6,7. Several chimeric BoNTs and related homologs have also been identified in clostridia as well as more distantly related firmicutes8.
BoNT are a family of structurally conserved proteins which comprise a catalytic Light Chain (LC), a zinc metalloprotease, linked by a disulfide bond to a Heavy Chain (HC)9. HC has two functional domains, C-terminal HCC mediates BoNT binding to host cell receptors and N-terminal HCN mediates LC translocation into the cytosol of a motor neuron10. Early steps for BoNT/A1 entry into neurons include HCC initially binding extracellular ganglioside- synaptotagmin 1 complex11,12, subsequently binding host intracellular vesicle protein(s)13, and HCN mediating pH-triggered LC translocation from the lumen of a synaptic vesicle (SV) into the host cell cytosol14. However, mechanisms used by cytosolic LC/A1 to traffic to the intracellular plasma membrane to cleave Synaptosome Associated Protein of 25-kDa (SNAP-25) is understudied in BoNT pathogenesis15. Earlier, Aoki and coworkers correlated BoNT LCs with duration of action and plasma membrane association16,17, while Dolly and co-workers18 used BoNT serotype chimeras to implicate LC translocation as contributing to BoNT rate of action. Extending these studies, LC/A1 N terminus (N term) was found to contribute to LC/A1 binding to SNAP-2519.
Recently, we reported that BoNT/A3, a BoNT/A subtype, had a shorter duration of action in neurons than other BoNT/A subtypes and analyzing BoNT/A1-A3 chimeras, determined that BoNT/A duration of action was a LC property20. We also determined that Enhanced Green Fluorescent Protein (EGFP)-LC/A1 stably localized on intracellular vesicles and the intracellular plasma membrane, while EGFP-LC/A3 was localized on intracellular vesicles and in the cytosol21. While the LC/A1 and LC/A3 structures were superimposable22, alignment of LC/A1 and LC/A3 primary amino acids identified a common internal α-helix-turn-α-helix-turn region (LC/A1 residues 275-334), termed the Membrane Localization Domain (MLD) that possessed low primary amino acid homology between LC/A1 and LC/A3. Subsequent studies showed that the A1-MLD mediated stable LC/A1 binding to the intracellular plasma membrane, while the analogous A3-MLD mediated LC/A3 reversible binding to the intracellular plasma membrane21. During these studies, a LC/A3 variant, LC/A3V, was identified that possessed two amino acid substitutions within the N term of LC/A3 (Q7P and V14G) that shifted the intracellular location of LC/A3V to solely in the cytosol21, implicating a role for LC/A N term in intracellular membrane association. Table 1 and Supplementary Fig. 1 provide structure-function and sequence alignments information for LC/A1, LC/A3, LC/A3V, and a recombinant LC/A3V (A1 MLD) that are used in the current study. LC/A3V (A1 MLD) will be used as a control to measure LC/A binding to the intracellular plasma membrane and SNAP-25 cleavage from the cytosol21.
N2A cells are mouse-derived neuroblastoma cells previously used to study BoNT functions19,21,23,24, as well as general cytoskeleton properties, microtubules25 and actin26. N2A cells are morphologically useful for single cell analysis, and have been used to study cell signaling in response to Dominant Negative (DN) Rab GTPases27, and to study Rab-mediated viral intracellular trafficking28. The current study uses a eukaryotic CMV expression system21,24 to measure EGFP-LC/A1 localization and SNAP-25 cleavage in N2A cells. Given that the intracellular functions and localizations of individual Rab GTPases can overlap29,30; characterization of multiple Dominant Negative (DN) Rab GTPases showed that intracellular LC/A1 associated with several classes of intracellular vesicles and used fast synaptic vesicle recycling31 to traffic to the intracellular plasma membrane to cleave SNAP-25.
Results
LC/A1 utilized microtubules for intracellular localization in N2A cells
Initial experiments determined the role of actin and microtubules32,33,34 in LC/A intracellular localization. Two cytoskeleton inhibitors, cytochalasin D, which binds polymerizing ends of actin filaments to disrupt actin extension35, and nocodazole, which binds β-tubulin to disrupt microtubule depolymerization36 tested a role for actin or microtubules, respectively, in EGFP-LC/A1 and EGFP-LC/A3 intracellular localization. Note, LC/A1 is the catalytic LC of BoNT/A1 subtype with a high potency and a long duration of action, while LC/A3 is the catalytic component of BoNT/A3 subtype with a reduced potency and a short duration of action Table 1 and Supplementary Fig. 1. N2A cells were transfected overnight with pEGFP-LC/A1, where LC/A1 localized on intracellular vesicles and the intracellular plasma membrane, or pEGFP-LC/A3, where LC/A3 localized on intracellular vesicles and in the cytosol Fig. 1, Untreated. Cytochalasin D treatment caused actin disruption and yielded neurite sprouts containing microtubules (β-tubulin positive fibers), where LC/A1 was retained on the intracellular plasma membrane and on neurite sprouts Fig. 1-Cyto D A1-insert. Imaging 488nm (EGFP fluorescence) and 568nm (ß-tubulin fluorescence) showed EGFP-LC/A1 and ß-tubulin were colocalized on neurite sprouts. With Cytochalasin D treatment, EGFP-LC/A3 was retained on intracellular vesicles and was present in the cytosol Fig. 1-Cyto D A3-insert. Nocodazole treatment shifted EGFP-LC/A1 localization primarily to the plasma membrane in punctate structures Fig. 1nocodazole A1-insert, while nocodazole treatment shifted LC/A3 localization primarily to the cytosol Fig. 1nocodazole A3-insert. Nocodazole mediated LC/A3 shift to the cytosol, correlated with reversible binding of LC/A3 with the plasma membrane and subsequent release into the cytosol as observed earlier21. Overall, these data implicated a role for microtubules in LC/A1 trafficking to the intracellular plasma membrane. These findings supported earlier studies which showed transcytosis of iodinated BoNT serotypes increased when intracellular vesicle sorting was disrupted by brefeldin A and decreased when microtubules were disrupted with nocodazole37. Next, experiments analyzed N2A cells for expression of endogenous Rab GTPases and LC/A1 and LC/A3 steady state localization with these endogenous Rab GTPases.

N2A cells were incubated overnight alone or after transfection with pEGFP, pEGFP-LC/A1, or pEGFP-LC/A3. Cells were incubated for 1 h alone or with the actin inhibitor cytochalasin D (Cyto D), or the microtubule inhibitor Nocodazole. Cells were fix in 4% paraformaldehyde and stained for actin (phalloidin, cyan fluorescence), microtubules (β-tubulin, red fluorescence), and the nucleus (Hoechst stained, retained as a dark hole). (LEFT images) Representative immunofluorescent images (average panel of 5 randomly chosen panels) show the steady-state intracellular localization of EGFP (GFP), EGFP-LC/A1 (LC/A1), and EGFP-LC/A3 (LC/A3), alone (Untreated) or treated with Cyto D or Nocodazole. To enhance detection of EGFP, the perimeter (magenta box) of an Untreated, Cyto D treated, or Nocodazole treated pEGFP-LC/A1 or pEGFP-LC/A3 transfected N2A cells were enlarged (50%), brightened (25%) and placed below the respective transfected cell LC/A1 (A1-Insert) or LC/A3 (A3-insert). Within the magenta boxes, intracellular EGFP was marked with magenta arrows. (RIGHT graphs) As described in the methods21,24, pEGFP, pEGFP-LC/A1, or pEGFP-LC/A3 transfected N2A cells untreated (green), or treated with cytochalasin D (cyan), or Nocodazole (red) were scored for % of cells with intracellular EGFP localized (Intracellular Fluorescence, vertical axis) on intracellular plasma membranes (Membrane), intracellular vesicles (Vesicles), or cytosol (Cytosolic) (horizontal axis). For each transfection, ten random fields of ~100 N2A cells were scored for EGFP-LC/A localization. Note, total LC/A1 and LC/A3 intracellular localization were >100% due the multiple sites where intracellular LC/A1 and LC/A3 localized. Results were averaged from three independent experiments in triplicate and data were graphed with GraphPad Prism 10.1.2. Statistical analysis showed that LC/A1 colocalization in untreated N2A cells shifted from the plasma membrane and intracellular vesicles to primarily on the plasma membrane with Nocodazole treatment (***p < 0.005) (A1-insert), and LC/A3 colocalization in untreated N2A cells shifted from intracellular vesicles and the cytosol to primarily on the cytosol upon Nocodazole treatment (****p < 0.001) (A3-insert).
N2A cells express endogenous Rab GTPases involved in endosome- and synaptic vesicle- trafficking
A subset of Rab GTPases, which functioned in endosome and synaptic vesicle trafficking29,30 were analyzed for expression in N2A cells. N2A cells transfected overnight with pEGFP-LC/A1 or pEGFP-LC/A3 and probed with antibody to several endogenous Rab GTPases detected expression of Rab3a (late synaptic vesicles)38, Rab4 (fast anterograde endosomes)39, Rab5 (endosome recycling)29,40, Rab7 (retrograde endosomes)41, Rab11 (slow anterograde endosomes)42, and Rab27a (late synaptic vesicles)43 Fig. 2. Note, the Rab27a isoform was used for this analysis based upon availability of a α-Rab27a commercial antibody.

(UPPER SCHEMATIC ILLUSTRATIONS) (A) EGFP-LC/A1 and (B) EGFP-LC/A3 used in this experiment. After overnight transfections with (A) pEGFP-LC/A1 or (B) pEGFP-LC/A3, N2A cells were fixed with 4% paraformaldehyde and probed with rabbit antibody to individual Rab GTPases: (Rab3a (3a), Rab4 (4), Rab5 (5), Rab7 (7), Rab11 (11), or Rab27a (27a)). Bound rabbit antibodies were identified with goat α-rabbit with Alexa-fluor-conjugated secondary 568 nm. (MIDDLE IMAGES) Among five randomly screened transfected cells possessing alexa-fluor-conjugated secondary 568 nm signal, a representative cell is shown for individual Rab GTPase expression (Left column, Rab), EGFP-LC/A1 or EGFP-LC/A3 expression (Middle column GFP-LCA1 or GFP-LCA3), which were then merged and scored for colocalization (Right column Merged/Colocalized). White dots in the Merged/Colocalized lane indicate colocalization of the indicated endogenous Rab GTPases (red) and LC/A (green), using ImageJ at yellow color threshold 39−46. (LOWER GRAPHS) Pearson’s Colocalization Coefficient (PCC) of (A) LC/A1 or (B) LC/A3 with individual endogenous Rab3a (3a), Rab4 (4), Rab5 (5), Rab7 (7), Rab11 (11), or Rab27a (27a) was obtained by ImageJ where PCC values > 0.6 defined colocalization, PCC values of 0.3 - 0.6 defined partial colocalization, and PCC values of <0.3 defined segregation45. Standards: colocalization (PCC: 0.844, Rat-α-GFP IgG/EGFP-LC/A1) and segregation (PCC: -0.091, DNA (Hoechst stain)/EGFP-LC/A1) determined independent of Fig. 2 shown in Fig. S2. Data were graphed with GraphPad Prism 10.1.2. Results were averaged from three independent experiments in triplicate.
Subsequent experiments tested for the co-localization of LC/A1 and LC/A3 with these endogenous Rab GTPases. First, Pearson’s Colocalization Coefficients (PCC) established steady state co-localization (EGFP-LC/A1, PCC = 0.844, measuring Rat α-GFP IgG 568 / EGFP-LC/A1) and segregation (DNA-EGFP-LC/A1, PCC = -0.091, DNA (Hoechst) /EGFP-LC/A1) Supplementary Fig. 244. Earlier studies quantified PCC values > 0.6 as colocalization, PCC values of 0.3-0.6 as partial colocalization, and PCC values of <0.3 as segregation45. At steady state, LC/A1 and LC/A3 showed partial colocalization (PCC 0.3-0.6) with each endogenous Rab GTPase in the descending order: Rab4 > Rab27a > Rab5 > Rab11 = Rab7 > Rab3a Fig. 2. A qualitative measurement for LC/A1 and LC/A3 co-localization with endogenous Rabs, LC/A1 and LC/A3 were scored with the program Colocalized (Image J Function), where LC/A1 colocalized with Rab3a, Rab27a, and Rab4 along the plasma membrane, Rab5 and Rab11 in the cell interior, and Rab7 in the perinuclear region Fig. 2. Since rabbit polyclonal antibodies were used in this analysis, determination of the amount of each Rab GTPase expressed in N2A cells was not possible. Since physical interactions do not link to function, the next experiments tested if DNRab GTPases affected LC/A1 localization to the intracellular plasma membrane and SNAP-25 cleavage.
LC/A1 utilized fast synaptic vesicles and endosome recycling to localize to the intracellular plasma membrane
DNRab GTPases sequester Rab guanine nucleotide exchange factors, interfering with Rab GTPase-nucleotide exchange, which halts Rab GTPase-mediated intracellular vesicle movement46,47. A N2A single cell co-transfection of pDsRed-LC/A1 or pDsRed-LC/A3V (A1 MLD) with individual pEGFP-DNRab GTPases tested the trafficking pathway of LC/A1 to the intracellular plasma membrane. LC/A3V (A1 MLD) is a recombinant LC/A1/A3 chimera that that can traffic from the cytosol to the intracellular plasma membrane independent of intracellular vesicles21 Table 1 and Supplementary Fig. 1. DNRab5 assessed a role for endosome recycling, DNRab7 assessed a role for retrograde trafficking, DNRab4 and DNRab11 assessed a role for fast and slow endosome cycling, respectively, and DNRab3a and DNRab27b isoforms assessed late stage exocytic vesicle secretion to the intracellular plasma membrane47,48,49,50.
LC/A1 co-expression with DNRab3a, DNRab4, DNRab5, or DNRab27b inhibited LC/A1 localization to the intracellular plasma membrane on average Fig. 3A. DNRab5 and DNRab27b exhibited the greatest inhibition, on average but the inhibition levels were not statistically different from that observed by DNRab3a or DNRab4. The inability to discern the varying degrees of inhibition elicited by these four DNRab GTPases may be attributed to a single trafficking pathway for LC/A1 to the intracellular plasma membrane where each Rab GTPase functioned in an early or late stage of vesicle trafficking, with unique potency. Overall, this analysis indicated that fast synaptic vesicles and endosome recycling31 were involved in LC/A1 trafficking to the intracellular plasma membrane. Neither DNRab7 nor DNRab11 inhibited LC/A1 trafficking to the plasma membrane, indicating that neither retrograde trafficking nor slow endosome recycling were involved in the physical trafficking of LC/A1 to the plasma membrane.

(SCHEMATIC ILLUSTRATIONS) (A) pDsRed-LC/A1 and (B) pDsRed-LC/A3V (A1 MLD)21. After overnight co-transfections of (A) pDsRed-LC/A1 or (B) pDsRed-LC/A3V (A1 MLD) with individual pEGFP- DNRab3a (3a), DNRab4 (4), DNRab5 (5), DNRab7 (7), DNRab11 (11), or DNRab27b (27b), N2A cells were fixed and imaged for DsRed-LC (Red) and EGFP-DNRab GTPases (Green). (IMAGES) representative fluorescent images24 that represents the primary phenotype among the analyzed cells are show: (Left column, GFP-DNRab GTPase fluorescence) (Center column, EGFP-DNRab GTPase fluorescence, (Right column, Merge of Left and Center images) Imaging of DsRed-LC/A1 (Panel A) or DsRed-LC/A3V (A1 MLD) (Panel B) + individual DN GTPase are shown. Note, plasma membrane localization of DsRed-LC/A3V (A1 MLD) in the presence of each individual DNRab GTPase is marked with a Red Arrow. (GRAPHS) Percentage of cells with membrane localized DsRed-LC/A1 (Left Graph) or DsRed-LC/A3V (A1 MLD) (Right Graph) (Alone) or in the presence of individual DNRab GTPases: DNRab3a (3a), DNRab4 (4), DNRab5 (5), DNRab7 (7), DNRab11 (11), or DNRab27b (27b) were analyzed from ten random fields in three independent experiments. Mean and SEM were evaluated with GraphPad Prism 10.1.2 with an ordinary one-way ANOVA with Dunnett’s multiple comparisons test using % membrane localized DsRed-LC/A1 (alone) or DsRed-LC/A3V(A3-MLD) (alone) as the control column. ns not significant, ***p < 0.002.
LC/A3V (A1 MLD) co-expression with neither DNRab3a, DNRab4, DNRab5, nor DNRab27b inhibited LC/A3V (A1 MLD) localization to the intracellular plasma membrane Fig. 3B. This showed that LC/A3V (A1 MLD) localization on the plasma membrane was independent of DNRabs involved with fast synaptic vesicles or endosome recycling. This outcome is consistent with LC/A3V (A1 MLD) trafficking from the cytosol to the plasma membrane independent of vesicle trafficking21.
Neither LC/A3 nor LC/A3V were tested in this analysis since at steady state neither LC/A3 nor LC/A3V localized on the intracellular plasma membranes of N2A cells21 Table 1 Overall, these findings indicated that fast synaptic vesicle recycling31 was involved in LC/A1 trafficking to the intracellular plasma membrane. Next, the effect of DNRab GTPases on LC/A1 cleavage of SNAP-25 was tested.
DNRab GTPases associated with fast synaptic vesicle recycling inhibit LC/A1 cleavage of SNAP-25 in N2A cells
Next N2A single cell analysis tested if individual DNRab GTPases modulated LC/A1-mediated SNAP-25 cleavage. This experiment measured SNAP-25 cleavage with an antibody that bound only LC/A-cleaved SNAP-25. Control experiments established a dose-response for LC/A1 cleavage of SNAP-25, where transfection of 30 ng of pEGFP-LC/A1 yielded ~30% cleavage of total SNAP-25 Supplementary Fig. 3. Other controls showed the expression of carrier EGFP, EGFP-LC/A1, and each individual EGFP-DNRab GTPase, where expression of the individual DNRab GTPases did not affect LC/A1 expression Supplementary Fig. 4. In addition, transfection of 125 ng of individual pDNRab GTPases yielded saturating DNRab protein expression Supplementary Fig. 4. Addition of carrier EGFP in the co-transfection experiments allowed the initial identification of transfected cells, prior to assessing the amount of SNAP-25 cleavage.
LC/A1 co-transfection of pEGFP-A1 with carrier pEGFP yielded a range of total EGFP fluorescence of cells where in individual transfected cells cleaved SNAP-25 was proportional to EGFP with a slope of 0.9 (units SNAP-25 cleaved / unit expressed EGFP) Fig. 4Top panel, left. Co-transfection of individual pEGFP-DNRab GTPases had a range of effects on LC/A1-mediated cleavage of SNAP-25, where expression of DNRab27b, DNRab5, DNRab3a, and DNRab4 reduced the slope to -0.01, 0.07, 0.08, and 0.19, respectively, while DNRab11 and DNRab7 had smaller effects on LC/A1 mediated SNAP-25 cleavage. Statistics for the significance of the DNRab GTPase inhibition of LC/A1 elicited by each DNRab are shown in Supplementary Fig. 5. Examination of representative individual co-transfected cells, LC/A1 + an individual DNRab GTPase, from five randomly chosen cells with the same amount of total EGFP expression showed that co-expression of DNRab27b, DNRab5, DNRab3a, and DNRab4 reduced the amount of LC/A1-mediated SNAP-25 cleaved to below detection, while co-expression of DNRab11 and DNRab7 resulted in detectable amounts of LC/A1-mediated SNAP-25 cleavage Fig. 4, Top panel, right. Controls showed that cleaved SNAP-25 was detected in the positive control (co-expression EGFP and EGFP-LC/A1), while the negative control (expression of EGFP alone) did not yield detectable cleaved SNAP-25. Thus, LC/A1-mediated SNAP-25 cleavage was inhibited by DNRab GTPases involved in fast synaptic vesicle recycling.

UPPER SCHEMATIC ILLUSTRATION above each panel defines the LC/A used in each individual experiment, Top panel, LC/A/A1; Middle panel, LC/A3V (A1 MLD); and Bottom panel, LC/A3. After overnight co-transfections with plasmids encoding carrier EGFP (EGFP), carrier EGFP + LC/A, carrier EGFP + LC/A + individual DNRab3a (3a), DNRab4 (4), DNRab5 (5), DNRab7 (7), DNRab11 (11), or DNRab27b (27b), LEFT GRAPHS show a composite of individual cells scored for sum-total EGFP fluorescence (X axis, GFP Fluorescence (107 AU)) and then scored for cleaved SNAP-25, using an antibody that only bound LC/A-cleaved SNAP-25 (Y axis, (Cleaved SNAP-25 Fluorescence (107 AU)). Linear regression of the slope of cleaved SNAP-25 Fluorescence / EGFP fluorescence of sum total EGFP for carrier EGFP + LC/A alone was compared statistically to the slope of carrier EGFP (Alone) or the slope of carrier EGFP + LC/A1 + individual DNRab GTPase: where ns not significant, *p < 0.05, **p < 0.005, ***p < 0.001, or ****p < 0.0001. Statistical analysis of the slopes is shown in Fig S5. RIGHT IMAGES Among five randomly chosen individual N2A cells with ~3 ×107 AU sum-total EGFP fluorescence, representative cells: Top panel LC/A1 (A1); Middle panel LC/A3 (A1 MLD) (A3-1M), or Bottom panel LC/A3 (A3) were scored for SNAP-25 cleavage alone (-) or with the indicated DNRab GTPase. Left column shows sum-total EGFP fluorescence, Center column shows cleaved SNAP-25 (cleaved SNAP-25); and (Right column shows a Merge (Merge) of sum-total EGFP expression (Green) and cleaved SNAP-25 (Red). Above each panel is a negative control (EGFP), representing a N2A cell transfected with carrier EGFP alone (Left column), assayed for SNAP-25 cleavage (Center column), and a merge of EGFP expression (Green) and cleaved SNAP-25 (Red) (Right column).
LC/A3V (A1 MLD) co-transfection of 30 ng of pEGFP-A3V (A1 MLD) with carrier pEGFP yielded a range of total EGFP fluorescence in cells that was proportional to the amount of cleaved SNAP-25 and had a slope of 0.4 (units SNAP-25 cleaved / unit EGFP expressed) Fig. 4. Addition of individual pEGFP-DNRab GTPases had a range of effects on LC/A3V (A1 MLD) cleavage of SNAP-25 that were not statistically significant, where co-expression of only DNRab7 achieved a statistically significant lower slope than LC/A3V (A1 MLD) alone Supplementary Fig. 5. Examination of representative individual co-transfected cells, LC/A3V (A1 MLD) + an individual DNRab GTPase, from five randomly chosen cells with the same amount of total EGFP expression showed similar amounts of SNAP-25 cleavage as LC/A3V (A1 MLD) alone Fig. 4, Middle panel, right. Overall, LC/A3V (A1 MLD)-mediated SNAP-25 cleavage was not inhibited by DNRab GTPases involved in fast synaptic vesicle recycling. Note, the lower slope of LC/A3V (A1 MLD)-mediated cleaved SNAP-25 relative to LC/A1.
LC/A3 co-transfection of 30 ng of pEGFP-A3 with carrier pEGFP yielded a range of total EGFP fluorescence of cells that was proportional to the amount of cleaved SNAP-25 and had a slope of 1.4 (units SNAP-25 cleaved / unit EGFP expressed) Fig. 4, Bottom panel, left. Co-transfection of each of the individual pEGFP-DNRab GTPases statistically reduced the amount of SNAP-25 (p < 0.0001) Supplementary Fig. 5. However, the amount of LC/A3 inhibition elicited from co-expression of the DNRabs GTPases associated with fast synaptic vesicles and for endosome recycling were less than observed for inhibition of LC/A1 by the same DNRabs. Examination of representative individual co-transfected cells, LC/A3 + an individual DNRab GTPase, from five randomly chosen cells with the same amount of total EGFP expression showed similar amounts of cleaved SNAP-25, except in the case of co-expression with DNRab11 which showed a reduced amount of cleaved SNAP-25, relative to LC/A3 alone Fig. 4, Bottom panel, right. Thus, LC/A3-mediated SNAP-25 cleavage was inhibited by DNRab GTPases involved in fast synaptic vesicle recycling, but the inhibition was less efficient than for LC/A1. This may be due to the ability of cytosolic LC/A3 to cleave SNAP-25 independent of vesicle trafficking. Unexpectedly, the slope of LC/A3-mediated SNAP-25 cleavage was similar to LC/A1.
Controls tested the activity of the DNRab GTPases used in this experiment on cholera toxin B subunit intracellular trafficking Supplementary Fig. 6. Only DNRab7 and DNRab11 interfered with cholera toxin B subunit trafficking, which showed that Rab7 and Rab11 were functional in N2A cells51,52. The inability of DNRabs GTPases involved in fast synaptic vesicles recycling to inhibit cholera toxin B subunit trafficking showed that these DNRab GTPases did not show off-target inhibition of CTB trafficking. LC/A3V was not tested in this analysis since LC/A3V showed limited ability to cleave SNAP-25 under these experimental conditions (Fig. 5). Overall, the outcomes of these experiments were consistent with the intracellular trafficking of LC/A1 via fast synaptic vesicle recycling to cleave SNAP-25 on the intracellular plasma membrane of N2A cells. Next, experiments directly tested the relative activity of the LC/A derivatives to cleave SNAP-25 in N2A cells.

N2A cells were transfected overnight with two-fold serial dilutions of DNA encoding EGFP-LC/A1 (4−125 ng), EGFP-LC/A3 (4−125 ng), EGFP-LC/A3V (A1-MLD) (4−125 ng), or pEGFP-LC/A3V (60−2000 ng). Note, LC/A3V need to be transfected with greater amounts of DNA to achieve 30% SNAP-25 cleavage at the highest amount of DNA transfected. Cell lysates were prepared and probed with α-SNAP-25 IgG to detect cleaved and uncleaved SNAP-25, and with α-EGFP to measure LC/A expression. A Densitometry from at least three independent experiments measured cleaved and uncleaved SNAP-25 to normalized units of LC/A protein expression, and data graphed using GraphPad Prism 10.1.2. B Shown is the average relative activity of LC/A1, LC/A3, LC/A3V (A1-MLD), or LC/A3V to cleave 30% of total SNAP-25, averaged from at least three independent experiments. Relative cleavage of SNAP-25 by LC/A1 and LC/A3V (*p < 0.05), and LC/A1 and LC/A3 (***p < 0.001) were statistically different, while relative cleavage of SNAP-25 by LC/A1 and LC/A3V (A1 MLD) were not statistically different.
LC/A3, LC/A1, and LC/A3V (A1 MLD) mediated greater SNAP-25 cleavage than LC/A3V in N2A cells
To assess intracellular LC/A potency, N2A cells were transfected overnight with serial dilutions of pEGFP-LC/A1, pEGFP-LC/A3, pEGFP-LC/A3V, or pEGFP-LC/A3V (A1 MLD) and monitored for intracellular SNAP-25 cleavage and LC/A expression by Western blots. This enabled dose-dependent measurements for intracellular LC/A potency. At 30% SNAP-25 cleavage, LC/A3 was ~2 fold more active than LC/A1 or LC/A3V (A1 MLD), and ~30-fold more active than LC/A3V Fig. 5A. LC/A1 cleavage of SNAP-25 was slower than LC/A3, faster than LC/A3V, and similar to LCA3V (A1 MLD) Fig. 5B. The apparent greater activity of LC/A3 relative to LC/A1 in cells was not anticipated, since BoNT/A1 has greater potency than BoNT/A321 and longer duration of action than BoNT/A3 in mice20 (see discussion).
Discussion
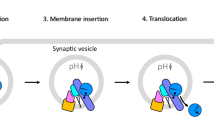
While early steps of BoNT/A1 entry into neurons are known11,13, how cytosolic LC/A1 traffics to the intracellular plasma membrane to cleave SNAP-25 is understudied15. The current study measured physical movement and catalytic activity of cytosolically expressed LC/A1 to extend our understanding of canonical BoNT/A intoxication. We propose that after pH-mediated LC translocation into the cytosol14, LC/A1 associates with intracellular vesicles to traffic via fast synaptic vesicle recycling to cleave SNAP-25 on the intracellular plasma membrane Fig. 6.

Using steady state- and live- imaging along with single cell co-transfection cell-based assays, a model is shown for how cytosolic LC/A1 (Green) traffics to the intracellular plasma membrane to cleave SNAP-25 via fast SV recycling, involving recycling endosomes (DNRab4) that can mature into enriched synaptic vesicles (DNRab3a and DNRab27b). LC/A1 intracellular trafficking to the intracellular plasma membrane sensitivity to DNRab5 may be due to the need for the system to recycle synaptic vesicles from the plasma membrane to facilitate LC/A1 intracellular trafficking. This model is incorporated to the canonical BoNT/A1 entry into motor neurons (reviewed in ref. 78) where EGFP-LC/A1 (Green) provides details for BoNT/A-LC/A1 (Red) trafficking upon translocation into the cytosol from an acidified vesicle. Created with BioRender.com.
Strengths of the current study include establishing a LC/A1 dose response Supplementary Fig. 5 as an intracellular quantitative measure of LC/A1 SNAP-25 cleavage in the cell-based assay, and the use of multiple DNRab GTPases to identify a role of fast synaptic vesicle and endosome recycling pathways to traffic LC/A1 from the cytosol to the intracellular plasma membrane and to cleave SNAP-25. A limitation of the study was that while assays were performed within a LC/A1-dose range for SNAP-25 cleavage, overall intracellular LC/A1 expression are expected to be greater than LC/A1 entering a cell during a natural BoNT/A intoxication.
Understanding how LC/A traffics within a neuron may identify new families of host proteins for therapeutic intervention of LC within the host cell cytosol. Therapeutic interventions may target LC/A1 N term, which is involved in LC association with intracellular vesicles and the LC/A1 MLD, which is involved in stable LC association with the intracellular plasma membrane, presumably via a component of SNAP-2519,24. Understanding the basis for association with intracellular vesicles and the plasma membrane may also identify inhibitors that block LC/A1 trafficking to SNAP-25 without interfering with host metabolism as a post BoNT/A1 intoxication therapy. Potential targets include: specific LC/A amino acid sequences such as a THR-triad (T306,T307, T312) within the R2 region Supplementary Fig. 1 of the MLD that may directly target SNAP-2524; Rab-GEFs and Rab-GAPs involved in the trafficking steps of LC/A1 along the fast synaptic vesicles pathway42; and Rab-GTP effectors, such as Rabphilin-3A53 or the Rim proteins54 that are uniquely involved in synaptic vesicles and recycling55.
The finding that Rab5 encoded a necessary step in LC/A1 trafficking may be due to Rab5 endosome functions, such as the formation of downstream vesicular bodies, including Rab4-positive recycling endosomes56,57. Rab4 and Rab11 facilitate endosomal recycling prior to the formation of synaptic vesicles42. As such, kinesin-2-mediated endosome recycling is categorized into two populations where Rab4-mediates fast recycling and Rab11-mediates slow recycling39,42. DNRab4 was more efficient at inhibiting LC/A1 cleavage of SNAP-25 than DNRab11 Fig. 4. These data indicated that LC/A1 preferentially utilizes Rab4-mediated fast synaptic vesicles to reach the plasma membrane. This is in contrast to studies on ricin toxin, where toxin action is sensitive to DNRab1156 and Rab11 is required for lysosome exocytosis58.
Rab3 and Rab27 control SV docking, fusion, and exocytosis42. Rab3(a-d) are essential for fast kinesin1-mediated anterograde trafficking and vesicle assembly on microtubules59. While Rab3b, 3c, and 3 d are redundant, KO models showed that the removal of Rab3a was lethal post-birth60. Rab3 and Rab2754 are structurally related, partially colocalized, and share effector proteins such as Rabphilin3a, which binds to Rab3 and Rab27 and regulates synaptic vesicle cycling prior to trafficking and localization at the plasma membrane with SNAP-2538,61. Inhibition of SNAP-25 cleavage by LC/A1 in DNRab3a or DNRab27b co-transfected cells Fig. 4 is consistent with previous work showing Rab3 is necessary for SNARE complex assembly and cellular sensitivity to BoNT62. While present in overlapping pools, temporally, Rab3 and Rab27 transition in distinct SVs38. The observed ability of DNRab3a and DNRab27b to elicit near complete inhibition of LC/A1-mediated SNAP-25 cleavage supports function of Rab3a and Rab27b at sequential, late steps in LC/A1 trafficking to cleave SNAP-25.
An unexpected outcome was that LC/A3 possessed greater SNAP-25 cleavage activity than LC/A1 in the cell based assays Fig. 5, while prior in vitro studies showed LC/A3 and LC/A1 possessed similar catalytic activity22,63. In addition, full-length BoNT/A1 had greater potency21 and longer duration of action20 than BoNT/A3 in mice. Subtype LC composition defines the long duration of BoNT/A1 and short duration of BoNT/A320. We propose that cell-based assays and in vitro-based assays of catalysis and systems-based assays for BoNT potency and duration in mice have unique rate limiting steps. The unique amino acid compositions of the A1-MLD and A3-MLD may be responsible for the common catalytic actions of LC/A1 and LC/A3 in short-cell based assays, but unique BoNT/A1 and BoNT/A3 potencies and durations of action in mice. The LC/A1-MLD and LC/A3-MLD share only ~ 60% homology and align into two physically separated regions, R1 region (275-299) and R2 region (300-334) (PDB 1XTG) Supplementary Fig. 1. The R1 region is distanced from SNAP-25 and may recognize SNAP-25 accessory proteins, such as Rabphilin-3a64, while R2 has direct interaction with SNAP-25 where R2 encodes several direct contacts between SNAP-25 that are shared between LC/A1 and LC/A321. Thus, the unique components of the R1 region may define unique BoNT/A1 and BoNT/A3 potencies in mice20, while the R2 region may define similar LC catalytic potential for SNAP-25 observed in cell-based and in vitro assays.
Earlier studies have implicated Rab proteins in the intracellular trafficking of protein toxins65. Rab4 and Rab5 compartments have been implicated in Ricin56 trafficking and in targeting Yersinia ADP-ribosyltransferase, a glycosylating toxin from C. difficile66. In Drosophila, DNRab5 mediates homotypic fusion of SVs, increasing SV size and inhibiting neurotransmitter release67. Pseudomonas aeruginosa Type-III effectors and toxins such as ExoS and Exotoxin A, respectively, utilize specific retrograde trafficking pathways, involving several Rab GTPases, in intracellular movement68,69. Recently, Vibrio MARTX toxin has been shown to process and degrade Rab GTPases70. This study characterized multiple Rab GTPases to assign a candidate pathway for LC/A1 trafficking from the cytosol to the intracellular plasma membrane to cleave SNAP-25.
The current study used N2A cell-based assays to implicate fast synaptic vesicle cycling to traffic cytosolic LC/A1 to the intracellular plasma membrane to cleave SNAP-25. Defining late steps in intracellular LC/A1 trafficking in neurons and may identify new classes of host proteins to target to neutralize post-BoNT intoxication of neurons.
Materials and methods
Materials
Reagents were purchased from Life Technologies (Grand Island, NY) unless otherwise specified.
Engineering of EGFP-LC/A and DNRab constructs
pEGFP-C3 encoded DNA from LC/ A1, A3, A3V, and A3V (A1-MLD) were engineered21. EGFP or red fluorescent protein (DsRed) fusions with LC/A1 (GB ACS6681), LC/A3 (GB ACA57525), LC/A3V (GB ABY56337), were subcloned into the SacI-BamHI restriction sites of pEGFP-C371 or pDsRed72. EGFP- Rab3a, Rab4, Rab5, Rab7, Rab11, and Rab27b73 were subcloned into pEGFP-C371 or pDsRed72. Primers were designed and obtained from New England Biolabs® NEBaseChanger® (Ipswich, MA) to engineer DNRab proteins with the indicated mutation: Rab3a(T36N)74, Rab4(S22N)75, Rab5(S34N)75, Rab7(T22N)51, Rab11(S25N)76, and Rab27b(T23N)43.
Cell Culture and imaging
Neuro-2A (N2A) cells ATCC # CCL-131 (Manassas, VA). N2A cells were cultured in Complete Essential Media supplemented with 10% fetal bovine serum, 1x penicillin-streptomycin, 0.1% sodium bicarbonate, 1 mM sodium pyruvate, and 1% nonessential amino acids in humidified 5% CO2 (v/v) at 37°C and transfected21,24. N2A cells were seeded in a 24-well plate at 50,000 cells/well on acid-etched glass coverslips coated for 1 h with Poly-D lysine (1:500), cultured overnight, and then transfected with the indicated plasmids in Lipofectamine LTX; Invitrogen™ (Waltham, MA). After the procedures described below, cells were imaged for fluorescence on a Nikon Eclipse Ti-inverted microscope using a 60x oil-immersion objective (1.4 NA objective) with Eclipse software for data analysis. Four experiments were performed.
Testing the role of microtubules and actin in LC/A1 intracellular trafficking in N2A cells, Fig. 1. N2A cells were transfected overnight with 500 ng of either pEGFP-LC/A1 or pEGFP-LC/A3. Cells were treated for 60 min with 4 µM of cytochalasin D (C8273, Sigma-Aldrich, St. Louis) or 9 µM of nocodazole (M1404, Sigma-Aldrich, St. Louis). N2A cells were then fixed with 4% (w/v) paraformaldehyde for 15 min at RT, washed 2x with RT PBS and blocked for 60 min with 10% FBS (v/v) in 2.5% cold-water fish skin gelatin (w/v), 0.1% Triton-X (v/v), and 0.05% Tween-20 (v/v) in DBPS (14040182), Gibco™, IL) (incubation buffer). The following day, N2A cells were washed 3x (five min) with DPBS + 0.1% Tween-20 (v/v) and incubated with phalloidin-Alexa-fluor 568 nm (1:250) (A12380, Life Technologies, WI) or 1:5000 rabbit α-ß-3 tubulin (ab52623, Abcam, Cambridge, UK), in incubation solution for 60 min at RT, washed 2x in 0.5% Tween-20 (v/v) DPBS, and stained with Hoechst (1:10,000 in DPBS, H21492, Invitrogen™, MA) for 5 min. Coverslips were flipped onto microscope slides and cured with 8 µL of Prolong™ Gold Antifade Mountant (Invitrogen, MA). For each transfection, ten random fields of N2A cells were scored for EGFP-LC/A localization in a total of ~100 cells. An earlier study showed EGFP-A1 plasma membrane colocalized with wheat germ agglutinin on the plasma membrane24. Cells positive for membrane determined by localization on the plasma membrane were scored as a % of total cells utilizing the following equation: (% of cells expressing membrane localization/total number of cells with EGFP fluorescence) × 100. Immunofluorescence results were graphed utilizing GraphPad Prism 10.1.2 (San Diego, CA, USA) and subjected to a statistical test using ordinary one-way ANOVA with Dunnett’s multiple comparisons with LC/A3V as the control column. Vesicle association was scored when EGFP was associated with intracellular vesicles alone or in the presence of cytosolic EGFP-LC. Total values may be > 100% since each LC/A derivative may localize in multiple sites within one cell.
Testing Rab GTPases expression on endosomes and synaptic vesicles and co-localization with LC/A1 in N2A cells, Fig. 2. N2A cells were transfected with 0.5 µg of pEGFP-LC/A1. After an overnight incubation, N2A cells were fixed with 4% (w/v) paraformaldehyde for 15 min at RT, washed 2x with RT PBS and blocked for 60 min in 10% FBS (v/v) in (incubation buffer). Rab detection: primary rabbit antibodies (1:1000) from Abcam, UK: α-Rab3a (ab3335), α-Rab4 (ab109009), α-Rab5 (ab218624), α-Rab7 (ab137029), α-Rab11 (ab180504), or α-Rab27a (ab223044) were added with overnight shaking at 4°C in incubation solution. The following day, N2A cells were washed 3x (five min each) with 0.05% Tween-20 (v/v) in DPBS and incubated with Alexa-fluor 488 nm-conjugated secondary or phalloidin-Alexa-fluor 568 nm (1:250) (A12380, Life Technologies, WI) with 10% FBS (v/v) in incubation solution for 60 min at RT, washed 2x in 0.05% Tween-20 (v/v) DPBS, and stained with Hoechst (1:10,000 in DPBS, H21492, Invitrogen™, MA) for 5 min. Coverslips were flipped onto microscope slides and cured with 8 µL of Prolong™ Gold Antifade Mountant (Invitrogen, MA). For each transfection, ten random fields of N2A cells were scored for EGFP-LC/A derivative localization in a total of ~100 cells. Pearson’s Colocalization Coefficient (PCC) of EGFP-LC/A1 or EGFP-LC/A3 with individual endogenous Rab3a, Rab4, Rab5, Rab7, Rab11, or Rab27a with bound rabbit antibodies identified with goat α-rabbit with Alexa-fluor-conjugated secondary 568 nm were analyzed with ImageJ where PCC values > 0.6 defined colocalization, PCC values of 0.3 – 0.6 defined partial colocalization, and PCC values of <0.3 defined segregation45. Data were graphed with GraphPad Prism 10.1.2. Results were averaged from three independent experiments in triplicate. White dots presented in merged/colocalized (Fig. 2) EGFP-LC/A with individual Rab GTPases 568 nm were obtained using the color threshold feature in Image J (Image → Adjust → Color Threshold) defining the lower threshold at 39 and the upper threshold at 46, the yellow colocalized region. Note, PPCs, measure the degree of linear relationship between the changes in pixel intensity of two images, not the absolute amount of intensity or overlap, while “Colocalization” visualizes the correlation of the pixel intensity77.
Testing the effect of DNRab GTPases on LC/A1 or LC/A3V (A1MLD) localization to the intracellular plasma membrane, Fig. 3. N2A cells were transfected with 500 ng of pDsRed-LC/A1 or pDsRed-LC/A3V (A1 MLD) alone or with individual 500 ng of individual pEGFP-DNRab GTPase (DNRab3a, DNRab4, DNRab5, DNRab7, DNRab11, or DNRab27b). After an overnight incubation, N2A cells were fixed with 4% (w/v) paraformaldehyde for 15 min at RT, washed 2x with RT PBS and blocked for 60 min with 10% FBS (v/v) in incubation solution. For each transfection, ten random fields of N2A cells were scored for pDS-Red-LC/A derivative localization in a total of ~100 cells. DsRed fluorescence was scored membrane-bound when fluorescence colocalized with wheat germ agglutinin. Cells positive for membrane determined by localization on the plasma membrane were scored as a % of total cells utilizing the following equation: (% of cells expressing membrane localization/total number of cells with EGFP fluorescence) × 100. Immunofluorescence results were graphed utilizing GraphPad Prism 10.1.2 (San Diego, CA, USA) and subjected to a statistical test using ordinary one-way ANOVA with Dunnett’s multiple comparisons with LC/A3V as the control column.
Testing the effects of DNRab GTPases to inhibit LC/A1, LC/A3V (A1 MLD), and LC/A3 mediated SNAP-25 cleavage, Fig. 4. N2A cells were transfected with pEGFP-LC/A1 (30 ng), carrier pEGFP (600 ng) alone or with an individual pEGFP-DNRab GTPase (125 ng of DNRab3a, DNRab4, DNRab5, DNRab7, DNRab11, or DNRab27b)21,24. After an overnight transfection, N2A cells were fixed with 4% (w/v) paraformaldehyde for 15 min at RT, washed 2x with RT PBS and blocked for 60 min with 10% FBS (v/v) in incubation buffer. SNAP-25 cleavage (Fig. 4Left Graph). SNAP-25 cleavage was detected with a 1:1500 dilution of a primary mouse α-LC/A1 antibody that recognized only cleaved-SNAP-25 (ABIN236419, Antibodies-Online (Limerick, PA)) by shaking overnight at 4°C in incubation solution. The following day, N2A cells were washed 3x (five min) with 0.05% Tween-20 (v/v) in DPBS and incubated with goat α-mouse IgG Secondary Antibody, Alexa Fluor™ 568 (1:1000) (A11004, Life Technologies, WI) in incubation solution for 60 min at RT, washed 2x in 0.05% Tween-20 (v/v) DPBS, and stained with Hoechst (1:10,000 in DPBS, H21492, Invitrogen™, MA) for 5 min. Coverslips were prepared and individual cells were initially analyzed for total expressed EGFP and then analyzed for SNAP-25 cleavage measuring fluorescence at 568 nm. For LC/A1, EGFP-LC/A1 alone + carrier EGFP or EGFP-LC/A1 + the indicated DNRab GTPase + carrier EGFP were analyzed where the Linear Translation of Pixel Values (LUTS) for the immunofluorescent panels for total EGFP were between (453 min and 3251 max) and for cleaved SNAP-25 were between (736 min and 1344 max). The experiment was repeated three times and ~ 30 individual cells that were randomly chosen for analysis based upon the cells possessing EGFP fluorescence within the established LUTS values. Imaging representative N2A cells for LC/A1-mediated SNAP-25 cleavage in the presence of individual Rab GTPases (Fig. 4. Right Images). Among five randomly chosen individual N2A cells with a sum-total EGFP fluorescence of ~3 x 107 AU, a representative cell imaged to show total EGFP fluorescence alone (negative control), EGFP co-transfected with EGFP-LC/A1 (positive control) or EGFP co-transfected with EGFP-LC/A1 and the indicated individual EGFP-DNRab GTPase (test). The representative cell was imaged for EGFP expression (left column), cleaved SNAP-25 (center column), and merged sum-total EGFP (green) and cleaved SNAP-25 (red) (right column). Top panel showed the negative control a N2A cell transfected with carrier EGFP alone and scored for cleaved SNAP-25 and merged sum-total EGFP (green) and cleaved SNAP-25 (red). Potency of LC/A3V (A1 MLD) and LC/A3 to cleave SNAP-25 in the presence of individual pEGFP-DNRab GTPases (DNRab3a, DNRab4, DNRab5, DNRab7, DNRab11, or DNRab27b) was measured as described above for LC/A1.
Cholera Toxin-B subunit detection
After an overnight transfection of DNRab GTPases (Rab3a, Rab4, Rab5, Rab7, Rab11, or Rab27b), N2A cells were washed 2x times with 4°C DPBS and treated with 200 μM Cholera Toxin Subunit B-Alexa Fluor 594 Conjugate (C34777, Thermo, IL) for 5 min at 37 °C and 5% CO2. N2A cells were then washed 2x with DPBS, fixed with 4% (w/v) paraformaldehyde for 15 min at RT, washed 2x with DPBS, and treated with Hoechst (1:10,000 in DPBS, H21492, Invitrogen™, MA) as a nuclear marker for 5 min at RT. Coverslips were prepared as described above Supplementary Fig. 6.
Western Blot N2A cells were transfected overnight with two-fold serial dilutions of DNA encoding EGFP-LC/A1 (4-125 ng), EGFP-LC/A3 (4-125 ng), EGFP-LC/A3V (A1 MLD) (4-125 ng), or pEGFP-LC/A3V (60-2000 ng) and lysed in 150 µL of 2x PSB. Individual N2A cell lysates (10 μL) were resolved on a 13.5% SDS-PAGE and transferred to an Immobilon-P polyvinylidene difluoride membrane (PVDF) (Millipore, Billerica, MA). PVDF membranes were rehydrated in methanol, washed in reverse osmosis water, stained with Ponceau (P3504, Sigma-Aldrich, St. Louis), and incubated in blocking solution (2% powder milk (w/v) in TBS + 0.1% T). PVDF membranes were incubated with 1:4000 – 1:25000 rat α-EGFP-monoclonal IgG (3H9, Chromotek, Planegg, DE), 1:4000 mouse α-SNAP-25 monoclonal IgG (111011, SYSY, Gottingen, DE), and 1:2000 mouse α-actin (loading control, A2228 Sigma) shaking, overnight, at 4°C. Bound primary antibodies were recognized as 1:10,000 α-rat or 1:10,000 α-mouse secondary antibodies conjugated with horseradish peroxidase (Life Technologies, MA). Secondary antibodies were visualized using Super Signal™ West Pico PLUS Chemiluminescent Substrate (34578, Thermo, IL) on an Azure C600 Imaging System (Dublin, CA), using a 60-second exposure Fig. 5.
Statistics and reproducibility
All data was collected from at least three independent biological replicates with two technical replicates (separate wells within a culture experiment) and statically analyzed by one-way ANOVA, two-way ANOVA, or Dunnett’s multiple comparison test utilizing GraphPad Prism 9.3.1. Bar graph report as means ± standard error of the mean. Panels presenting statistical analysis presented p-values as *p < 0.05, **p < 0.005, ***p < 0.001, or ****p < 0.0001. Linear regression for slopes of the images + standard deviation among the three experiments is shown in Supplementary Fig. 5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Johnson, E. A. & Montecucco, C. Botulism. Handb. Clin. Neurol. 91, 333–368 (2008).
Centers for Disease, C. & Prevention Notice of CDC’s discontinuation of investigational pentavalent (ABCDE) botulinum toxoid vaccine for workers at risk for occupational exposure to botulinum toxins. MMWR Morb. Mortal. Wkly Rep. 60, 1454–1455 (2011).
Drug, U. S. F. a. BAT (Botulism Antitoxin Heptavalent (A, B, C, D, E, F, G) - (Equine). https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/bat-botulism-antitoxin-heptavalent-b-c-d-e-f-g-equine (2018).
Shapiro, R. L., Hatheway, C., Becher, J. & Swerdlow, D. L. Botulism surveillance and emergency response. A public health strategy for a global challenge. JAMA 278, 433–435 (1997).
Hill, K. K. & Smith, T. J. Genetic diversity within Clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Curr. Top. Microbiol. Immunol. 364, 1–20 (2013).
Dressler, D. & Johnson, E. A. Botulinum toxin therapy: past, present and future developments. J. Neural Transm. (Vienna) 129, 829–833 (2022).
Rasetti-Escargueil, C. & Popoff, M. R. Engineering botulinum neurotoxins for enhanced therapeutic applications and vaccine development. Toxins 13, 1 (2020).
Doxey, A. C., Mansfield, M. J. & Montecucco, C. Discovery of novel bacterial toxins by genomics and computational biology. Toxicon 147, 2–12 (2018).
Lacy, D. B. & Stevens, R. C. Sequence homology and structural analysis of the clostridial neurotoxins. J. Mol. Biol. 291, 1091–1104 (1999).
Montal, M. Botulinum neurotoxin: a marvel of protein design. Annu. Rev. Biochem. 79, 591–617 (2010).
Joensuu, M. et al. Presynaptic targeting of botulinum neurotoxin type A requires a tripartite PSG-Syt1-SV2 plasma membrane nanocluster for synaptic vesicle entry. EMBO J. 42, e112095 (2023).
Stenmark, P., Dupuy, J., Imamura, A., Kiso, M. & Stevens, R. C. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathog. 4, e1000129 (2008).
Dong, M. et al. SV2 is the protein receptor for botulinum neurotoxin A. Science 312, 592–596 (2006).
Fisher, A. & Montal, M. Characterization of Clostridial botulinum neurotoxin channels in neuroblastoma cells. Neurotox. Res. 9, 93–100 (2006).
Varela-Chavez, C., Blondel, A. & Popoff, M. R. Bacterial intracellularly active toxins: membrane localisation of the active domain. Cell Microbiol. 22, e13213 (2020).
Fernandez-Salas, E., Ho, H., Garay, P., Steward, L. E. & Aoki, K. R. Is the light chain subcellular localization an important factor in botulinum toxin duration of action?. Mov. Disord. 19, S23–S34 (2004).
Fernandez-Salas, E. et al. Plasma membrane localization signals in the light chain of botulinum neurotoxin. Proc. Natl. Acad. Sci. USA 101, 3208–3213 (2004).
Lawrence, G. W., Ovsepian, S. V., Wang, J., Aoki, K. R. & Dolly, J. O. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochem. J. 441, 443–452 (2012).
Chen, S. & Barbieri, J. T. Association of botulinum neurotoxin serotype A light chain with plasma membrane-bound SNAP-25. J. Biol. Chem. 286, 15067–15072 (2011).
Pellett, S. et al. The light chain defines the duration of action of botulinum toxin serotype A subtypes. mBio 9, e00089-18 (2018).
Gardner, A., Tepp, W. H., Bradshaw, M., Barbieri, J. T. & Pellett, S. Resolution of two steps in botulinum neurotoxin serotype a1 light chain localization to the intracellular plasma membrane. Int. J. Mol. Sci. 22, 11115 (2021).
Leka, O., Wu, Y., Li, X. & Kammerer, R. A. Crystal structure of the catalytic domain of botulinum neurotoxin subtype A3. J. Biol. Chem. 296, 100684 (2021).
Wang, J. et al. Novel chimeras of botulinum neurotoxins A and E unveil contributions from the binding, translocation, and protease domains to their functional characteristics. J. Biol. Chem. 283, 16993–17002 (2008).
Gardner, A. P., Barbieri, J. T. & Pellett, S. How botulinum neurotoxin light chain A1 maintains stable association with the intracellular neuronal plasma membrane. Toxins (Basel) 14, 814 (2022).
Dehmelt, L., Smart, F. M., Ozer, R. S. & Halpain, S. The role of microtubule-associated protein 2c in the reorganization of microtubules and lamellipodia during neurite initiation. J. Neurosci. 23, 9479–9490 (2003).
Ge, Y. et al. Cadmium induces actin cytoskeleton alterations and dysfunction in Neuro-2a cells. Environ. Toxicol. 34, 469–475 (2019).
Li, Z. et al. Porcine hemagglutinating encephalomyelitis virus enters neuro-2a cells via clathrin-mediated endocytosis in a Rab5-, cholesterol-, and PH-dependent manner. J. Virol. 91, e01083-17 (2017).
Desingu, P. A. et al. PARP1 inhibition protects mice against Japanese encephalitis virus infection. Cell Rep. 42, 113103 (2023).
Kiral, F. R., Kohrs, F. E., Jin, E. J. & Hiesinger, P. R. Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 28, R471–R486 (2018).
Zerial, M. & McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 (2001).
Ivanova, D. & Cousin, M. A. Synaptic vesicle recycling and the endolysosomal system: a reappraisal of form and function. Front Synaptic Neurosci. 14, 826098 (2022).
Straight, A. F. & Field, C. M. Microtubules, membranes and cytokinesis. Curr. Biol. 10, R760–R770 (2000).
Etienne-Manneville, S. Actin and microtubules in cell motility: which one is in control?. Traffic 5, 470–477 (2004).
Akhshi, T. K., Wernike, D. & Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton 71, 1–23 (2014).
Brenner, S. L. & Korn, E. D. Substoichiometric concentrations of cytochalasin D inhibit actin polymerization. Additional evidence for an F-actin treadmill. J. Biol. Chem. 254, 9982–9985 (1979).
Vasquez, R. J., Howell, B., Yvon, A. M., Wadsworth, P. & Cassimeris, L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell 8, 973–985 (1997).
Maksymowych, A. B. & Simpson, L. L. Binding and transcytosis of botulinum neurotoxin by polarized human colon carcinoma cells. J. Biol. Chem. 273, 21950–21957 (1998).
Pavlos, N. J. et al. Quantitative analysis of synaptic vesicle Rabs uncovers distinct yet overlapping roles for Rab3a and Rab27b in Ca2+-triggered exocytosis. J. Neurosci. 30, 13441–13453 (2010).
Dey, S., Banker, G. & Ray, K. Anterograde transport of Rab4-associated vesicles regulates synapse organization in drosophila. Cell Rep. 18, 2452–2463 (2017).
Wucherpfennig, T., Wilsch-Brauninger, M. & Gonzalez-Gaitan, M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 161, 609–624 (2003).
Verma, K., Saito-Nakano, Y., Nozaki, T. & Datta, S. Insights into endosomal maturation of human holo-transferrin in the enteric parasite Entamoeba histolytica: essential roles of Rab7A and Rab5 in biogenesis of giant early endocytic vacuoles. Cell Microbiol. 17, 1779–1796 (2015).
Mignogna, M. L. & D’Adamo, P. Critical importance of RAB proteins for synaptic function. Small GTPases 9, 145–157 (2018).
Saxena, S. K. & Kaur, S. Rab27a negatively regulates CFTR chloride channel function in colonic epithelia: involvement of the effector proteins in the regulatory mechanism. Biochem Biophys. Res. Commun. 346, 259–267 (2006).
Granger, E., McNee, G., Allan, V. & Woodman, P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin Cell Dev. Biol. 31, 20–29 (2014).
Kroken, A. R., Blum, F. C., Zuverink, M. & Barbieri, J. T. Entry of botulinum neurotoxin subtypes A1 and A2 into neurons. Infect. Immun. 85, e00795-16 (2017).
Grosshans, B. L., Ortiz, D. & Novick, P. Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 103, 11821–11827 (2006).
Caviglia, S., Flores-Benitez, D., Lattner, J., Luschnig, S. & Brankatschk, M. Rabs on the fly: functions of Rab GTPases during development. Small GTPases 10, 89–98 (2019).
Koike, S. & Jahn, R. SNARE proteins: zip codes in vesicle targeting?. Biochem. J. 479, 273–288 (2022).
Yang, Y. et al. Rab3a attenuates spinal cord injury by mediating vesicle release. Brain Res. Bull. 208, 110884 (2024).
Gallo, L. I. et al. RAB27B requirement for stretch-induced exocytosis in bladder umbrella cells. Am. J. Physiol. Cell Physiol. 314, C349–C365 (2018).
Progida, C. et al. Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 123, 1480–1491 (2010).
Guichard, A. et al. Cholera toxin disrupts barrier function by inhibiting exocyst-mediated trafficking of host proteins to intestinal cell junctions. Cell Host Microbe. 14, 294–305 (2013).
Li, T., Cheng, Q., Wang, S. & Ma, C. Rabphilin 3A binds the N-peptide of SNAP-25 to promote SNARE complex assembly in exocytosis. Elife11, e79926 (2022).
Fukuda, M. Distinct Rab binding specificity of Rim1, Rim2, rabphilin, and Noc2. Identification of a critical determinant of Rab3A/Rab27A recognition by Rim2. J. Biol. Chem. 278, 15373–15380 (2003).
Watanabe, S. Slow or fast? a tale of synaptic vesicle recycling. Science 350, 46–47 (2015).
Moisenovich, M. et al. Endosomal ricin transport: involvement of Rab4- and Rab5-positive compartments. Histochem. Cell Biol. 121, 429–439 (2004).
Cormont, M., Mari, M., Galmiche, A., Hofman, P. & Le Marchand-Brustel, Y. A FYVE-finger-containing protein, Rabip4, is a Rab4 effector involved in early endosomal traffic. Proc. Natl. Acad. Sci. USA 98, 1637–1642 (2001).
Escrevente, C., Bento-Lopes, L., Ramalho, J. S. & Barral, D. C. Rab11 is required for lysosome exocytosis through the interaction with Rab3a, Sec15 and GRAB. J. Cell Sci. 134, jcs246694 (2021).
Szodorai, A. et al. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J. Neurosci. 29, 14534–14544 (2009).
Schluter, O. M., Schmitz, F., Jahn, R., Rosenmund, C. & Sudhof, T. C. A complete genetic analysis of neuronal Rab3 function. J. Neurosci. 24, 6629–6637 (2004).
Pavlos, N. J. & Jahn, R. Distinct yet overlapping roles of Rab GTPases on synaptic vesicles. Small GTPases 2, 77–81 (2011).
Johannes, L. et al. Evidence for a functional link between Rab3 and the SNARE complex. J. Cell Sci. 109, 2875–2884 (1996).
Henkel, J. S. et al. Catalytic properties of botulinum neurotoxin subtypes A3 and A4. Biochemistry 48, 2522–2528 (2009).
Ostermeier, C. & Brunger, A. T. Structural basis of Rab effector specificity: crystal structure of the small G protein Rab3A complexed with the effector domain of rabphilin-3A. Cell 96, 363–374 (1999).
Guadagno, N. A. & Progida, C. Rab GTPases: switching to human diseases. Cells 8, 909 (2019).
Ost, G. S. et al. Inverse control of Rab proteins by Yersinia ADP-ribosyltransferase and glycosyltransferase related to clostridial glucosylating toxins. Sci. Adv. 6, eaaz2094 (2020).
Shimizu, H., Kawamura, S. & Ozaki, K. An essential role of Rab5 in uniformity of synaptic vesicle size. J. Cell Sci. 116, 3583–3590 (2003).
Zhang, Y., Deng, Q. & Barbieri, J. T. Intracellular localization of type III-delivered Pseudomonas ExoS with endosome vesicles. J. Biol. Chem. 282, 13022–13032 (2007).
Michalska, M. & Wolf, P. Pseudomonas exotoxin A: optimized by evolution for effective killing. Front. Microbiol. 6, 963 (2015).
Herrera, A. et al. Vibrio MARTX toxin processing and degradation of cellular Rab GTPases by the cytotoxic effector Makes Caterpillars Floppy. Proc. Natl. Acad. Sci. USA 121, e2316143121 (2024).
Arnold, A. S. et al. Construction of a plasmid containing human SMN, the SMA determining gene, coupled to EGFP. Plasmid 47, 79–87 (2002).
Tezcan, G. et al. Rab GTPase mediating regulation of NALP3 in colorectal cancer. Molecules 25, 4834 (2020).
Stenmark, H. & Olkkonen, V. M. The Rab GTPase family. Genome Biol. 2, REVIEWS3007 (2001).
Burstein, E. S., Brondyk, W. H. & Macara, I. G. Amino acid residues in the Ras-like GTPase Rab3A that specify sensitivity to factors that regulate the GTP/GDP cycling of Rab3A. J. Biol. Chem. 267, 22715–22718 (1992).
Potokar, M., Lacovich, V., Chowdhury, H. H., Kreft, M. & Zorec, R. Rab4 and Rab5 GTPase are required for directional mobility of endocytic vesicles in astrocytes. Glia 60, 594–604 (2012).
Ren, M. et al. Hydrolysis of GTP on Rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. USA 95, 6187–6192 (1998).
Dunn, K. W., Kamocka, M. M. & McDonald, J. H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–C742 (2011).
Winner, B. M., Bodt, S. M. L. & McNutt, P. M. Special delivery: potential mechanisms of botulinum neurotoxin uptake and trafficking within motor nerve terminals. Int. J. Mol. Sci. 21, 8715 (2020).
Acknowledgements
The authors acknowledge helpful communications and suggestions from members of the Barbieri and Pellett laboratories, including Amanda Przedpelski, who contributed with technical and topic comments during the execution and writing of the manuscript. JTB is a member of the MCW Neuroscience Research Center. This study was funded by NIH AI139306 (SP, JTB). AG was partially supported by NIH TR001437.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.B. and S.P. Methodology, A.G., M.S., W.T., S.P. and J.B. Validation, A.G., M.S., W.T., S.P. and J.B. Formal Analysis A.G., M.S., W.T., S.P. and J.B. Investigation, A.G., M.S., W.T., S.P. and J.B. Resources, S.P. and J.B. Data Curation, A.G., M.S., W.T., S.P. and J.B. Writing—Original Draft Preparation, A.G. Writing—Review and Editing, A.G., M.S., W.H., S.P. and J.B. Visualization, A.G., M.S., W.T., S.P. and J.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Wayne Lencer and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gardner, A., Sneller, M., Tepp, W.H. et al. Botulinum neurotoxin Light Chain/A1 uses fast synaptic vesicle cycling to cleave plasma membrane bound SNAP-25. Commun Biol 8, 1383 (2025). https://doi.org/10.1038/s42003-025-08633-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08633-4
