
Abstract
Encapsulating peritoneal sclerosis (EPS) is a life-threatening fibrotic condition characterized by severe abdominal adhesions, chronic inflammation, and significant morbidity. The lack of effective treatments for EPS stems from a limited understanding of its underlying mechanisms. In this study, we developed a modified mouse model of PD-induced EPS and investigated the role of the STING signaling pathway in disease progression. Our findings reveal that STING activation in peritoneal mesothelial cells significantly increases the secretion of the macrophage chemokine CCL2, leading to enhanced macrophage infiltration and the formation of pathological adhesions. Notably, pharmacological inhibition of STING using the inhibitor H151 effectively reduced macrophage infiltration and fibrosis, demonstrating its therapeutic potential in alleviating EPS. These results identify the STING pathway as a critical mediator of EPS pathogenesis and suggest that STING inhibitors could offer a promising therapeutic strategy to prevent or reverse EPS, particularly in clinical settings such as peritoneal dialysis.
Similar content being viewed by others

Introduction
Globally, abdominal adhesions represent a significant cause of morbidity and mortality1,2. The pathogenesis of intra-abdominal adhesions is multifactorial, with causes ranging from surgical procedures to conditions such as peritoneal dialysis (PD), tuberculous peritonitis, radiation therapy, intra-abdominal infections, and chronic inflammatory diseases like Crohn’s disease3. While local application of hydrogels is commonly used to prevent post-surgical adhesions4,5,6, this strategy becomes impractical in cases involving diffuse adhesions. In such instances, pharmacological interventions that can be administered throughout the abdominal cavity or systemically are essential7.
Among the various causes of intra-abdominal adhesions, encapsulating peritoneal sclerosis (EPS) stands out as a particularly severe condition. EPS is characterized by extensive adhesions leading to intestinal obstruction and is often accompanied by peritoneal thickening, sclerosis, calcification, and the encasement of the small intestine8,9. One of the most common causes of EPS is PD, especially when complicated by peritonitis3. Thus, in this study, we selected PD-induced EPS as our pathological model to better understand the mechanisms of adhesion formation and to identify potential therapeutic strategies. While EPS modeling is challenging, especially given the resistance of mice to EPS, we refined our animal models to more accurately reflect human EPS pathology.
The cGAS-STING signaling axis, composed of cyclic GMP-AMP synthase (cGAS) and its downstream adaptor stimulator of interferon genes (STING), is critical in regulating the transcription of host defense genes, including type I interferons and pro-inflammatory cytokines. Recent research has implicated STING activation in the progression of fibrosis and inflammatory diseases through pathways involving NF-κB activation, cell death, and endoplasmic reticulum stress10,11. However, its role in abdominal adhesion formation, particularly in the context of PD-induced EPS, remains unexplored.
In this study, we developed a novel mouse model of PD-induced EPS and performed a series of in vivo and in vitro experiments to address key mechanistic questions. We first sought to identify the critical drivers of EPS pathogenesis, with a particular focus on macrophage chemotaxis and inflammation. We then evaluated whether pharmacological inhibition of this pathway could mitigate inflammatory infiltration and collagen deposition. Finally, we explored the cellular mechanisms underlying macrophage recruitment, highlighting the role of mesothelial cells in activating the STING pathway. Together, these findings offer new insights into cellular crosstalk at the mesothelial interface and uncover unexpected consequences of STING activation in the peritoneal environment.
Materials and methods
Study population
We conducted a retrospective analysis of clinical data from patients diagnosed with Encapsulating Peritoneal Sclerosis (EPS) who were admitted to The Third Affiliated Hospital of Sun Yat-sen University. Data collected included relevant medical history, clinical laboratory results, and abdominal ultrasound findings. The study was approved by the Medical Ethics Committee of The Third Affiliated Hospital of Sun Yat-sen University ([2022]02-268-01). Informed consent was obtained from all the patients and all ethical regulations relevant to human research participants were followed (Supplementary Table 1). The diagnosis of EPS was made based on the criteria established by the Special Committee of the International Society for Peritoneal Dialysis12.
Animals and experimental design
Eight-week-old male C57BL/6 J mice were purchased from the Laboratory Animal Center of South China Agricultural University. All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council) and were approved by the Ethics Committee of South China Agricultural University (Approval No. [2022] D136). To establish a model of peritoneal dialysis-induced encapsulating peritoneal sclerosis (EPS), mice were randomly assigned to one of five groups. The control group received intraperitoneal injections of saline in volumes equivalent to the treatment groups. PD group received daily intraperitoneal injections of 100 mL/kg of 4.25% peritoneal dialysis solution (PDS). LPS group received intraperitoneal injection of 4 mg/kg lipopolysaccharide (LPS; Sigma, derived from E. coli O111) once-weekly. The SHS group was administered 0.3 mL of a surgical hygiene solution (SHS) daily, following a previously published protocol13. The EPS model group (PD + LPS + SHS) received daily intraperitoneal injections of 100 mL/kg of 4.25% peritoneal dialysis solution (PDS) combined with 0.3 mL SHS, and a once-weekly intraperitoneal injection of 4 mg/kg lipopolysaccharide (LPS; Sigma, derived from E. coli O111). Daily monitoring of mice included observation for intra-abdominal adhesion and other clinical symptoms. Following a 3-week exposure period, the animals were euthanized and samples collected for subsequent analysis. For the intervention study, mice received daily intraperitoneal injections of the STING inhibitor H151 (750 nmol per mouse, dissolved in 200 µL of 10% Tween-80 in PBS; MCE, HY-112693), administered 30 minutes prior to EPS model induction. Control animals in the treatment arm received the same volume of vehicle solution without H151.
Gross Adhesion scoring
Mice were anesthetized, and their abdominal cavities were carefully opened for objective evaluation of adhesion formation. The criteria for diagnosing EPS were adapted from previous studies13, with modifications for scoring. Specifically, Score 0: No adhesions. Score 1: Presence of 1-3 distinct adhesions. Score 2: More than three distinct adhesions. Score 3: Extensive, sheet-like adhesions. We examined key anatomical structures, including the peritoneum, gastrointestinal loops, mesentery, and liver, for signs of thickening, contraction, or adhesion formation. The total adhesion score for each mouse was calculated, with a maximum score of 42 representing cocoon-like adhesions. Adhesion data were recorded for statistical analysis, with details provided in Supplementary Table 2.
Histologic scoring of adhesion tissue
Histological assessment methods for adhesion were adopted from published research and appropriately modified14,15. Following the euthanasia of the mice, transverse sections of the entire peritoneal cavity were collected from consistent anatomical locations for embedding. Tissue sections were subsequently subjected to Masson’s trichrome staining. Six random microscopic fields of adhesive knots per slide were captured, and Masson-positive areas were quantified using ImageJ software.
Abdominal ultrasonography
At week 3, abdominal wall ultrasonography was performed using a high-resolution small animal ultrasound system (Vevo3100, Fujifilm Visual Sonics). Mice were anesthetized with 1% inhaled isoflurane at a flow rate of 0.6-1 L/min, maintaining a heart rate between 400–600 beats per minute. To facilitate ultrasonographic imaging, approximately 3 mL of physiological saline solution was injected into the abdominal cavity via an indwelling needle to create abdominal distension. The ultrasound scans were conducted by three experienced professionals specializing in small animal ultrasonography, ensuring consistent imaging results.
Elisa
For abdominal lavage fluid analysis, mice were anesthetized with isoflurane, and 2 mL of physiological saline solution was injected into the abdominal cavity for peritoneal lavage. The lavage fluid was collected, centrifuged at 3000 rpm and 4 °C for 10 minutes, and the supernatant was obtained for ELISA measurements according to the instructions provided in the respective ELISA kits. For cell culture supernatants and peritoneal dialysis effluent, collect the supernatant and perform the detection according to the instructions after centrifuging. All the ELISA kit were purchased from MeiMian, China, and the catalog numbers were listed as follow: Mouse CCL2: MM-0723M1; Mouse IL-6: MM-0163M1; Human CCL2: Human CCL2: MM-2261H1; Human cGAMP:MM-60090H1).
Histological examination and immunohistochemistry
Histological and immunohistochemical analyses were performed as previously described16,17. After intraperitoneal lavage, the visceral and parietal peritoneum, along with adherent tissues, were harvested following cardiac perfusion. The tissues were immediately fixed in 4% polyformaldehyde for 24 hours, followed by gradient dehydration in ethanol. The tissues were embedded in paraffin and sectioned into 4 µm slices using a microtome. For general histological evaluation, the sections were deparaffinized and stained with hematoxylin and eosin (H&E) using Harris’ stain. Fibrosis was assessed using Masson’s trichrome staining.
For immunohistochemistry, deparaffinized sections were blocked with 5% bovine serum albumin (BSA) and incubated overnight at 4°C with the following primary antibodies: anti-Fibronectin (Abcam, ab2413, 1:100), anti-COL1A1 (CST, 72026S, 1:100), anti-α-SMA (Sigma-Aldrich, A5228, 1:100), anti-IL-1β (CST, 12242, 1:100), anti-IL-6 (CST, 12912, 1:100), anti-TNF-α (CST, 11948, 1:100), anti-CD31(Servicebio, GB120005, 1:300), anti-CD45 (Servicebio, GB113886, 1:300), anti-F4/80 (Servicebio, GB113373, 1:500), anti-Ly6G (Servicebio, GB12229, 1:500), and anti-CD3 (Servicebio, GB111337, 1:500), anti-STING (CST, 13647S, 1:100), anti-CCL2 (Servicebio, GB11199, 1:500), anti-Cytokeratin 7 (Santa Cruz, sc-23876, 1:50). The sections were then incubated with species-specific secondary antibodies and visualized using the DAB chromogen system. The secondary antibodies: anti-mouse Alexa Fluor 488 (4408S, 1:1000), anti-rabbit Alexa Fluor 594 (8889S, 1:1000).
Primary culture of human peritoneal mesothelial cells and treament
Human primary mesothelial cells were obtained and cultured as previously described17. They were maintained in Earle’s M199 medium (Gibco, C11150500BT) supplemented with 20% FBS, 1% penicillin-streptomycin (Gibco, 15140122), and 1% insulin-transferrin-selenium (ThermoFisher, 41400-045).
Cell culture
Human primary mesothelial cells were obtained and cultured as previously described16,17. They were maintained in Earle’s M199 medium (Gibco, C11150500BT) supplemented with 10% FBS, 1% penicillin-streptomycin (Gibco, 15140122). Met-5A cell line were purchased from ATCC and were cultured in M199 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in a 5% CO2 atmosphere. In this study, Met-5A cells were used only in experiments involving cell transfection, while all other experiments were conducted using primary human peritoneal mesothelial cells. To simulate EPS conditions, 0.1% SHS (containing 0.1% chlorhexidine gluconate and 15% ethanol) and 1 µg/mL lipopolysaccharide (LPS) were added to the culture medium. THP-1 cells were differentiated into macrophages by treatment with 100 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma, P1585) for 48 hours. As for H151 treatment, the drug was added to the mesothelial cells 30 min before adding SHS + LPS, with a working concentration of 2 μM.
Cell Transfection
The Met-5A cells were seeded into 6-well plates and transfected with si-IRF3 or control siRNA (GenePharm, suzhou, China) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). The target sequences for the preparation of siRNAs of human IRF3 are listed in Supplementary table 3. The siRNA and Lipofectamine 2000 were gently mixed, incubated at room temperature for 20 min, and transferred into cells. After 6 h incubation, the cells were washed twice with PBS and cultured in 10% FBS-containing M199. The effect of siRNA-induced gene silencing was verified by Western Blot.
Co-culture trans-well migration assay
Trans-well migration assays were performed using 24-well chambers with an 8 μM pore size (Corning, USA). In the upper chamber, 1 × 105 THP-1 cells were seeded and differentiated into macrophages with PMA. HPMCs were seeded into the lower chamber and treated with 0.1% SHS and 1 µg/mL LPS. After 24 hours, the medium in the lower chamber was replaced with 750 µL M199 medium containing 10% FBS, and the upper chamber was filled with 500 µL RPMI 1640 containing 1% FBS. After a further 24 hours of incubation, non-migrating cells on the upper surface of the membrane were removed with cotton swabs. The membrane was fixed in 4% paraformaldehyde and stained with 0.5% crystal violet. Migrating cells were counted under a microscope, with results averaged from three independent experiments. As for H151 treatment, the drug or equivalent volume of solvent was added to mesothelial cells 30 min before adding SHS + LPS, with a working concentration of 2 μM. And for anti-CCL2 treatment, 30 ug/mL of Carlumab (HY-P99188, MCE) or anti-IgG was added to mesothelial cells 30 min in advance.
Western blot
Cell extracts were prepared using RIPA buffer containing protease and phosphatase inhibitors (Beyotime, China). For tissue samples, peritoneal tissues were minced into small pieces and homogenized in cell lysis buffer. Protein concentrations were determined using a BCA protein assay kit. The proteins were boiled in loading buffer for 10 minutes and separated by 10% or 12.5% SDS-PAGE, followed by transfer to a PVDF membrane (Roche, Switzerland). The membranes were blocked in 5% non-fat milk and incubated overnight at 4°C with primary antibodies, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (Boster, BA1054, 1:5000). Protein bands were visualized using enhanced chemiluminescence (ECL) and analyzed using ImageJ software.
The primary antibodies used for western blot were anti-STING (CST, 13647S, 1:1000), anti-c-GAS (CST, 79978, 1:1000), anti-IRF3 (CST, 11904, 1:1000), anti-p-IRF3 (CST, 29047, 1:1000), anti-P65 (CST, 8242, 1:1000), anti- p-P65 (CST, 3033, 1:1000), anti-β-actin (CST, 8457S, 1:2000).
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from mesothelial cells and mouse peritoneal tissues using the RNA extraction kit (GOONIE, China). cDNA was synthesized using a reverse transcription kit (Takara) following the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green Premix (Takara), with β-actin as the housekeeping gene. Gene expression levels were analyzed using the delta-delta Ct method. Primer sequences are listed in Supplementary Table 4.
Flow cytometry
Mouse visceral peritoneum tissue from intestine were excised and dissected into small fragments, followed by enzymatic digestion using a cocktail of enzymes to generate a single-cell suspension. The suspension was then incubated with fluorescently labeled antibodies specific for the following markers: PerCP/Cyanine5.5 anti-mouse CD45 (Biolegend, 103131), FITC anti-mouse F4/80 (Biolegend, 123107), APC anti-mouse/human CD11b (Biolegend, 101211), PE anti-mouse Ly-6G (Biolegend, 127607), APC/Cyanine7 anti-mouse CD19 (Biolegend, 1115529), Alexa Fluor® 700 anti-mouse TCR β (Biolegend, 109223). Incubation was carried out for 1 hour at 4°C. After incubation, cells were washed to remove unbound antibodies and resuspended in staining buffer. Flow cytometry was performed using a FACSCalibur flow cytometer (BD Biosciences, USA), and data were analyzed using FlowJo software (TreeStar, Inc).
RNA sequencing
After euthanizing the mice, the mesentery tissue of the intestine was swiftly dissected to obtain visceral peritoneum tissue from intestine, and it was then placed in cryovials; this entire process was performed on ice. The tissue was then immediately transferred to liquid nitrogen for flash freezing and subsequently stored at -80 °C until sequencing. Total RNA was extracted from the tissue, and its concentration and purity were assessed using an Agilent 2100 Bioanalyzer for quality control. mRNA was then purified and fragmented, followed by cDNA synthesis using the RNA as a template. After the library construction was completed, the libraries underwent a second quality assessment using the Agilent 2100 Bioanalyzer, which included an evaluation of the total library concentration and effective library concentration. Finally, Next-Generation Sequencing was conducted using the Illumina sequencing platform, where paired-end (PE) sequencing of the libraries was performed.
RNA-seq analysis
Differentially expressed genes (DEGs) between experimental groups were identified using the edgeR R package (v3.28.1). Heatmaps and volcano plots were generated using the heatmap and ggplot2 R packages, respectively. Genes with an adjusted p-value < 0.05 were considered significantly differentially expressed. Gene Ontology (GO) and signaling pathway analyses were performed using the clusterProfiler R package (v3.14.3) to identify enriched biological processes and pathways associated with DEGs.
Deconvolution of RNA-seq data
To estimate cell-type composition, RNA-seq data were analyzed using the Multi-subject Single Cell Deconvolution (MuSiC) R package (v1.0.0). Reference single-cell RNA sequencing data from mouse peritoneum were obtained from the GEO database (GSE136636, GSE176254). Normalized single-cell RNA expression data from these datasets were used to train the deconvolution model to estimate the cell-type proportions in bulk RNA-seq samples.
Transcription factor activity inference in bulk RNA-seq
We used the DecoupleR R package (v2.9.7)18 to infer transcription factor (TF) activity from bulk RNA-seq data. The analysis was based on the OmniPath dataset19, a comprehensive collection of signaling pathways and regulatory interactions. Prior to running the analysis, RNA-seq data were normalized and transformed to log2 counts per million (CPM). We then applied the Univariate Linear Model (ULM) method in DecoupleR to calculate TF enrichment scores for each sample, with default parameters unless otherwise specified. The resulting activity scores were visualized using the ggplot2 R package (v3.5.1), allowing for a clear comparison of TF activity between different conditions.
Chromatin immunoprecipitation (ChIP)
The ChIP enzymatic chromatin IP protocol (Cell Signaling Technology, USA) was employed to conduct the ChIP assay. Three groups were established: Control group, SHS + LPS+si-NC group, and SHS + LPS+si-IRF3 group. Twenty-four hours post-transfection, cells were treated with SHS + LPS for 24 hours. Subsequently, 1×107 Met-5A cells from each group were collected for the ChIP assay. Following the manufacturer’s instructions, the chromatin was immunoprecipitated using an anti-IRF3 antibody (Proteintech, 11312-1-AP) or a negative control IgG antibody on rotators at 4 °C overnight. The primer set F: AAAGTGTCTCGTCCTGACC and R: AGGGCGGAGTCAAGCAGGAG was utilized for qPCR of CCL2 promoter regions. The PCR products were then subjected to 1% agarose gel DNA electrophoresis and documented using a Tanon gel documentation system.
Statistical Analysis and Reproducibility
All datasets were first assessed for normality using the Shapiro–Wilk test. For comparisons between two groups, variance homogeneity was tested using the F-test. If data were normally distributed with equal variances, a two-tailed unpaired Student’s t-test was applied. Otherwise, the Mann–Whitney U test was used for non-normally distributed data or when variances were unequal. For comparisons among more than two groups, one-way ANOVA followed by Tukey’s post hoc test was used for normally distributed data with equal variances, while the Kruskal–Wallis test with Dunn’s correction was used for non-parametric data. All values are presented as mean ± SEM, and statistical significance was defined as p < 0.05. Analyses were performed using GraphPad Prism 8 (GraphPad Software, Inc.). Experimental reproducibility is defined as the ability to consistently replicate results under the same conditions. Sample sizes are determined based on experimental requirements and resources. In vitro experiments were typically repeated three or more times, and in vivo for at least 6 biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Establishing a mouse model of EPS with rapid intra-abdominal adhesions
We aimed to develop a novel and more efficient mouse model of encapsulating peritoneal sclerosis (EPS), closely mimicking the condition observed in human patients. To achieve this, we combined several high-risk factors commonly associated with peritoneal dialysis-related EPS, including a 4.25% peritoneal dialysis solution (PD), a surgical hygiene solution (SHS) consisting of chlorhexidine gluconate and ethanol, and lipopolysaccharide (LPS) to replicate episodic peritonitis (Fig. 1A). While traditional models using SHS alone require up to 8 weeks for EPS formation [13], the addition of PD and LPS successfully accelerated the process, enabling significant intra-abdominal adhesion formation by the 3rd week.

A Schematic of the experimental regimen for establishing the mouse model of EPS. B Gross macroscopic examination of the abdominal cavity. Control mice displayed smooth, well-expanded mesenteries and sharp liver edges; PD group and LPS group showed no observable changes; SHS-treated mice showed blunted liver edges (yellow arrow), but little interorgan adhesions. In the PD + LPS + SHS group, there were extensive adhesions between abdominal organs (blue arrow) and thickened liver margins caused by surface fibrosis. C Quantification of adhesion scores across Control group (n = 7), PD group (n = 6), LPS group (n = 7), SHS group (n = 16), and PD + SHS + LPS group (n = 25). D Dynamic body weight changes in mice over the course of the study. E Kaplan–Meier survival curve showing survival rates in each group. EPS mice obtained a survival rate of 83.3% (25/30) and a success rate of 100% (25/25). F Ultrasonographic comparison highlights the anatomical similarities between the murine EPS model and the human condition. a–d Ultrasound imaging of control mice revealed a smooth parietal peritoneum and normal gastrointestinal motility (a). In contrast, the EPS mice showed peritoneal thickening and calcification (b, arrows), adhesions between the parietal peritoneum and intestinal loops (c, arrows), and a characteristic “cauliflower-like” central clumping of small bowel loops (d). Supplementary Video 1 provides dynamic ultrasonography of the abdominal cavity in both control and EPS mice. e–h In human patients, control subjects demonstrated a smooth peritoneal surface (e), while EPS patients exhibited peritoneal thickening (f, arrows), calcification (g, arrows), and significant adhesions between the parietal peritoneum and intestinal loops (h, arrows). ***p < 0.001 by Student’s t test or ANOVA test. Fig. 1A, and the ultrasound, human, mouse drawing elements in Fig. 1F were created in BioRender. Sun, J. (2025) https://BioRender.com/vbajaol.
Macroscopic evaluation revealed distinct differences between the experimental groups. Control mice showed smooth hepatic edges and well-expanded mesenteries with no visible adhesions (Fig. 1B). In contrast, the PD + LPS + SHS group exhibited prominent signs of adhesion, including thickened liver margins with fibrous deposits (Fig. 1B, yellow arrow) and extensive adhesions between the abdominal organs, particularly between the liver, intestines, and parietal peritoneum (Fig. 1B, blue arrow). Additionally, these mice displayed severe mesentery contraction and intestinal dilation, with some cases forming an abdominal “cocoon” structure, indicative of advanced EPS pathology (Supplemental Fig. 1). The adhesion scores of the PD + LPS + SHS group were significantly higher than those of the single control groups, demonstrating the robustness of the model in replicating EPS (Fig. 1C).
To assess the physiological impact of EPS, we monitored body weight and survival rates. Mice in the PD + LPS + SHS group showed significant weight loss compared to other groups, suggesting a progression toward malnutrition or cachexia, a common complication in EPS (Fig. 1D). Despite the severity of the disease, the survival rate in this group remained relatively high at 83.3%, allowing us to reliably conduct further analyses (Fig. 1E).
Since invasive exploration of the abdominal cavity is impractical in clinical settings, we employed ultrasonography to non-invasively assess the structural changes in the peritoneum, as commonly done in patients20. Comparing to the control group (Fig. 1F-a, Supplementary Video 1), ultrasonography of EPS mice revealed key pathological features, including peritoneal thickening, calcification, and characteristic intestinal dilation (Figs. 1F-b and 1F-c), resembling the “concertina-like” appearance often observed in EPS patients. The mouse model also demonstrated strong adhesions between the parietal peritoneum and intestinal loops, which were confirmed by static and dynamic imaging (Fig. 1F-d, Supplementary Video 2). To validate the clinical relevance of our model, we compared these findings with ultrasonographic features from human EPS patients. The human subjects exhibited similar pathological traits, such as peritoneal thickening, calcification, and organ adhesions (Figs. 1F-f to 1F-h). These similarities between the mouse model and human disease underscore the utility of this model for studying EPS pathogenesis and evaluating potential therapeutic interventions. In summary, the combination of PD, LPS, and SHS successfully accelerated the development of EPS in mice, with key anatomical and pathological features closely mimicking human EPS. Given its efficiency and reproducibility, this model will be used for further studies aimed at understanding the mechanisms of EPS and testing potential therapeutic strategies.
EPS mouse model exhibits inflammation, fibrosis, and increased vascular density in peritoneum
To further characterize the pathological changes in our EPS mouse model, we performed detailed histopathological analyses of the peritoneum. As shown in Fig. 2A, cross-sectional images of the abdominal cavity revealed widespread and diffuse adhesions throughout the peritoneum in EPS mice. These adhesions formed dense, clot-like structures that encompassed multiple abdominal organs, closely resembling advanced EPS pathology observed in humans. Histological staining provided insight into the structural and cellular changes associated with EPS. Hematoxylin and eosin (H&E) staining indicated substantial thickening of both the parietal and visceral peritoneum in the EPS group, with marked infiltration of inflammatory cells (Fig. 2B, C). This infiltration signifies a heightened inflammatory response within the peritoneal tissues, which is a key feature of EPS progression. In addition, Masson’s trichrome staining highlighted extensive fibrotic deposition, further confirming that fibrosis is a central component of EPS pathology (Fig. 2B, C). Quantitative analysis of peritoneal thickness revealed a significant increase in EPS mice compared to controls, as depicted in Fig. 2G, H, reflecting the overall fibrotic burden in the disease. To explore the molecular drivers of this fibrotic response, we conducted immunohistochemical analyses, which demonstrated the upregulation of extracellular matrix (ECM) markers, including collagen type I alpha 1 (COL1A1), fibronectin (FN) and α-smooth muscle actin (α-SMA), in the peritoneum of EPS mice (Fig. 2D, Supplemental Fig. 2A). These markers are indicative of active fibroblast proliferation and matrix remodeling, key processes in tissue fibrosis.

A Cross-sectional images of the abdominal cavity in EPS mice, showing widespread, diffuse adhesions forming clot-like structures throughout the peritoneal cavity. Hematoxylin and eosin (HE) and Masson’s trichrome staining of the parietal (B) and visceral (C) peritoneum, demonstrating significant peritoneal thickening and fibrotic deposition in EPS mice. D Immunohistochemical analysis of the parietal peritoneum showing increased expression of extracellular matrix (ECM) markers, including COL1A1, fibronectin (FN), and α-smooth muscle actin (α-SMA), indicative of active fibrosis. E Immunohistochemical analysis of inflammatory markers IL-1β, IL-6, and TNF-α in the parietal peritoneum, showing significant upregulation of these cytokines in EPS mice, highlighting the inflammatory component of the disease. F Immunofluorescent staining of CD31 in the visceral peritoneum (omentum) showing denser angiogenesis in EPS mice compared to controls. Quantification of peritoneal thickness in the parietal (G) and visceral (H) peritoneum, revealing significant thickening in EPS mice (n = 6 per group). I ELISA analysis of peritoneal lavage fluid, confirming an increase in IL-6 levels in EPS mice compared to controls (n = 6 per group). Data are presented as mean ± SEM.*** p < 0.001, two-tailed Student’s t-test.
Inflammation is known to play a critical role in EPS development21,22. In consistent with these observations, our immunohistochemical analysis showed significant upregulation of pro-inflammatory cytokines, including interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), within the peritoneal tissues of EPS mice (Fig. 2E, Supplemental Fig. 2B). Furthermore, enzyme-linked immunosorbent assay (ELISA) confirmed elevated expression of IL-6 in the peritoneal lavage fluid of EPS mice compared to controls (Fig. 2I), highlighting the systemic inflammatory milieu. Another critical aspect of EPS pathology is increased vascular density, often associated with chronic inflammation and fibrosis. In the EPS model, we observed a notable increase in blood vessel density in the visceral peritoneum, as demonstrated by CD31 staining (Fig. 2F, Supplemental Fig. 2C). Collectively, these results provide a comprehensive view of the severe inflammation, fibrosis, and angiogenesis occurring in the peritoneum of EPS mice. Importantly, these pathological changes closely mirror those reported in human EPS, validating the utility of this mouse model for studying disease mechanisms and potential therapeutic interventions.
Dynamic collagen deposition and inflammatory cell infiltration during EPS formation
To investigate the dynamic progression of intraperitoneal adhesion formation in EPS, we sacrificed mice at different time points and analyzed the histopathological changes in the peritoneum. On day 1, no noticeable infiltration of inflammatory cells or collagen fiber deposition was observed on the peritoneal surface. By day 7, inflammatory cells had begun infiltrating the peritoneum, accompanied by the initial deposition of collagen fibers (Fig. 3A). This early infiltration laid the groundwork for adhesion formation, with immune cells accumulating on the surface of fibers by day 14, and the fibrils progressively thickening and interconnecting to form adhesions. As the process advanced, collagen fibers continued to proliferate and gradually became the dominant structural component of the adhesions, while the infiltration of immune cells began to subside. By day 21, the adhesions had evolved into dense fibrous scar tissue, with minimal remaining immune cell infiltration (Fig. 3A). These findings closely mirror the pathological evolution observed in human EPS, where early inflammation gives way to excessive fibrotic deposition over time.

A Representative images of H&E and Masson’s trichrome staining showing the dynamic pathological progression of adhesion formation in EPS mice. Mice were sacrificed at days 1, 7, 14, and 21 to capture different stages of adhesion development (AW: abdominal wall; BW: bowel wall). B Immunohistochemistry staining of adhesion cross-sections showing the distribution of key inflammatory cell types involved in adhesion formation. CD3 was used to identify lymphocytes, Ly6G for neutrophils, and F4/80 for macrophages. C Quantitative analysis of immune cell infiltrations in Fig. 3B (n = 6 per group). D A graphical diagram depicting the dynamic formation of interorgan adhesions, illustrating the transition from early inflammatory cell infiltration to fibrous scar tissue formation over time. Data are presented as mean ± SEM. *** p < 0.001, two-tailed Student’s t-test.
In addition to collagen depositions, H&E and Masson’s trichrome staining revealed significant infiltration of inflammatory cells within the adhesion regions (Fig. 3A). To determine the types of immune cells involved, we conducted immunohistochemical staining with markers for specific cell types at the conclusion of EPS molding (day 21). The markers used were F4/80 for macrophages, CD3 for T lymphocytes, and Ly6G for neutrophils (Fig. 3B). Among these, macrophages were found to be the predominant immune cells present within the adhesions, suggesting their key role in driving the fibrotic response (Fig. 3C). This was further supported by the extensive infiltration of macrophages in the parietal peritoneum (Supplemental Fig. 3), reinforcing the critical involvement of macrophages in the adhesion formation process. The graphical diagram in Fig. 3D illustrates the dynamic formation process of interorgan adhesions, depicting the transition from early inflammatory infiltration to the eventual deposition of fibrous scar tissue. The findings indicate the role of macrophage recruitment in the development of EPS, offering additional understanding of the cellular processes involved in adhesion formation within this disease model.
Transcriptomic alterations in peritoneal tissues of EPS mouse models
To gain insights into the transcriptomic alterations in the peritoneal tissues of EPS mice, we performed bulk RNA sequencing on the visceral peritoneum, using three biological replicates from both the Control and EPS groups (Fig. 4A). Our analysis identified a total of 3,900 upregulated genes and 4,397 downregulated genes in the EPS group compared to controls (adjusted p-value < 0.05). Notably, macrophage-associated genes such as Marco and Csf3r, along with chemokine-related genes, exhibited significant upregulation in the EPS group, underscoring the inflammatory nature of the disease (Supplemental Fig. 4).

A Heatmap of RNA-seq data comparing gene expression in the visceral peritoneum of Control (n = 3) and EPS (n = 3) mice. B Gene Ontology (GO) biological process enrichment analysis of differentially expressed genes (DEGs) significantly upregulated in EPS mice compared to controls. C GSEA) showing enhanced myeloid leukocyte activation and regulation of leukocyte adhesion in EPS mice. D UMAP plot illustrating cell clusters from two publicly available single-cell RNA sequencing (scRNA-seq) datasets of mouse omentum. Mes: mesothelial cells; Endo: Endothelial cells; Fib: Fibroblasts; Neutro: Neutrophils; Macro: Macrophages; T: T cells. E Venn diagram showing the overlap between cell type-specific marker genes from scRNA-seq datasets and DEGs identified in our bulk RNA-seq data. F Deconvolution analysis using scRNA-seq datasets to compare cellular proportions in the Control and EPS groups, highlighting an increased proportion of fibroblasts among parenchymal cells and macrophages among immune cells in EPS mice (n = 3 per group). G Flow cytometry analysis of visceral peritoneum from intestine, confirming a significant increase in immune cell infiltration, particularly macrophages, in EPS mice compared to controls (n = 5 per group). H Correlation analysis between COL1A1 expression and the infiltration of various inflammatory cell types, showing a strong positive correlation between macrophage infiltration and extracellular matrix (ECM) progression. I Correlation analysis between the macrophages proportion detected by flow cytometry with COL1A1 expression in the peritoneum by immunohistochemistry and peritoneal thickness. Data are presented as mean ± SEM. * p < 0.05, *** p < 0.001, two-tailed Student’s t-test.
To better understand the functional implications of these differentially expressed genes (DEGs), we performed Gene Ontology (GO) enrichment analysis. As shown in Fig. 4B, the upregulated genes in the EPS group were predominantly enriched in biological processes associated with immune activation, such as leukocyte migration, myeloid leukocyte activation, and interleukin production. These findings suggest that immune cell activation, particularly the infiltration of mononuclear macrophages, plays a central role in the progression of EPS. In addition, Gene Set Variation Analysis (GSVA) further supported this, with a marked increase in myeloid leukocyte activation scores in the EPS mice (Fig. 4C). This aligns with the observed increase in macrophage infiltration, indicating that these immune cells are key contributors to the ongoing inflammatory and fibrotic processes.
To gain further insight into the cellular composition of the peritoneal tissues, we utilized two publicly available single-cell RNA sequencing (scRNA-seq) datasets from mouse omentum and classified them into seven distinct clusters based on canonical marker genes (Fig. 4D) (GSE 136636, GSE176254). Differential expression analysis between the Control and EPS groups revealed that 63% of the marker genes identified from scRNA-seq overlapped with those from our bulk RNA-seq data (Fig. 4E). Using this dataset, we applied deconvolution analysis with the R package MuSiC to assess the relative proportions of different cell types. Our analysis showed a marked decrease in the proportion of mesothelial cells in the EPS group, suggesting a disruption in the mesothelial layer, which is a known contributor to adhesion formation by exposing adhesive fibrin clots to surrounding tissues23. Conversely, we observed a significant increase in the proportion of fibroblasts in the EPS mice, which likely contributes to the excessive fibrous deposition seen in the disease. In terms of immune cell populations, both macrophages and T cells were significantly elevated in the EPS group, with macrophages being the most predominant immune cell type (Fig. 4F). To validate these findings, we performed flow cytometry on peritoneal tissues, confirming that the percentage of macrophages was significantly higher in EPS mice, corroborating the RNA-seq results (Fig. 4G). Furthermore, we examined the correlation between different immune cell types and the expression of the fibrosis marker Col1a1. Among the immune cell markers evaluated, F4/80, a macrophage marker, demonstrated the strongest positive correlation with Col1a1 expression (R2 = 0.471, p = 0.042) (Fig. 4H). Concurrently, the proportion of macrophages detected by flow cytometry showed a significant positive correlation with COL1A1 expression in the peritoneum as measured by immunohistochemistry (R2 = 0.9311, p < 0.0001), as presented in Fig. 4I, suggesting that macrophages are the main immune cell type involved in fibrotic deposition in EPS. These findings, when combined with our previous immunohistochemistry data, emphasize the pivotal role of macrophage infiltration in the formation of EPS and further highlight the synergistic relationship between immune cell infiltration and fibrotic deposition in this disease model.
Activation of the cGAS-STING pathway in mesothelial cells regulates macrophage chemotaxis
To investigate the mechanism underlying the significant macrophage infiltration observed in the EPS peritoneum, we reanalyzed our bulk RNA sequencing data. KEGG analysis revealed that genes upregulated in EPS mice were enriched in pathways related to cell chemotaxis and cytoplasmic DNA sensing, particularly the cGAS–STING pathway (Supplemental Fig. 5A–B), highlighting a potential link between immune cell recruitment and intracellular immune surveillance mechanisms (Fig. 5A). Additionally, GSVA demonstrated a strong positive correlation between the cytoplasmic DNA-sensing pathway and cytokine chemotaxis and inflammatory response pathways, further supporting the role of intracellular surveillance system in EPS progression (Fig. 5B, C).

A KEGG analysis showing significant enrichment of upregulated genes in cell chemotaxis and cytoplasmic DNA-sensing pathways in EPS mice. GSVA scores demonstrating a positive correlation between the cytoplasmic DNA-sensing pathway and cytokine chemotaxis (B) and inflammatory response pathways (C). D ELISA results showing elevated CCL2 levels in the peritoneal lavage fluid of EPS mice (n = 6 per group). E Immunofluorescence indicating increased CCL2 expression in the EPS parietal peritoneum, primarily co-localized with Cytokeratin 7+ mesothelial cells. F Immunofluorescence showing increased STING expression in the EPS parietal peritoneum, also co-localized with Cytokeratin 7+ mesothelial cells. G Western blot confirming the activation of cGAS-STING and downstream proteins in the EPS peritoneum (n = 6 per group). H Cytoplasmic DNA leakage observed in mesothelial cells stimulated with LPS + SHS. I, J Western blot and immunofluorescence showing STING activation and downstream signaling in mesothelial cells under EPS stimulation, with H151 reducing this activation (n = 4 independent experiments). K qPCR showing upregulation of inflammatory-related genes in mesothelial cells under EPS stimulation, with or without H151 pretreatment (n = 3 independent experiments). L qPCR demonstrating increased CCL2 gene expression in mesothelial cells under EPS stimulation, with H151 reducing the effect (n = 3 independent experiments). M Immunofluorescent staining of CCL2 in EPS-stimulated mesothelial cells. N ELISA showing elevated CCL2 levels in mesothelial cell supernatant under EPS stimulation, with H151 reducing CCL2 secretion (n = 3 independent experiments). O Schematic of the trans-well assay to assess macrophage migration. Mesothelial cells were seeded in the lower chamber, stimulated with EPS, and co-cultured with macrophages from the upper chamber (Created in BioRender. Sun, J. (2025) https://BioRender.com/vbajaol). P Bright-field image of macrophage migration after co-culturing with EPS-stimulated mesothelial cells. Q Quantification of macrophage migration under different conditions, with H151 alleviating macrophage migration (n = 3 independent experiments). R, S Representative image and quantification of macrophage migration under anti-CCL2 treatment, (n = 3 independent experiments). Data are presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, two-tailed Student’s t-test.
Given that CCL2 is a well-established chemokine involved in macrophage recruitment24,25, our qPCR validation confirmed that CCL2 exhibited the most pronounced expression change among chemokines in the peritoneal tissues of EPS mice (Supplemental Fig. 6A). ELISA analysis of the peritoneal lavage fluid revealed significantly higher levels of CCL2 in the EPS group compared to controls (Fig. 5D). Immunofluorescence also showed a marked increase in CCL2 expression in the parietal peritoneum, primarily co-localizing with Cytokeratin 7+ mesothelial cells (Fig. 5E, Supplemental Fig. 6B). These findings suggest that mesothelial cells are the primary source of CCL2 secretion in EPS, driving the chemotaxis of macrophages.
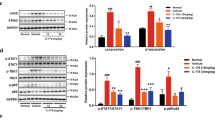
We then investigated the activation of the cGAS-STING pathway, which is known to mediate responses to cytoplasmic DNA. Both Western blot and immunofluorescence confirmed significant activation of the cGAS-STING pathway in EPS peritoneum, with STING co-localizing primarily with mesothelial cells (Fig. 5F, G, Supplement Supplemental Fig. 6C, D), suggesting that activation of the cGAS-STING pathway in mesothelial cells under EPS conditions may be responsible for promoting the chemotaxis of inflammatory cells, such as macrophages. After initial experiments to set up the in vitro EPS model (Supplemental Fig. 6E-H), we simulated the EPS environment by treating human peritoneal mesothelial cells (HPMCs) with LPS and SHS. Under these conditions, mesothelial cells exhibited cytoplasmic DNA leakage (Fig. 5H), consistent with cGAS-STING pathway activation. Western blotting and immunofluorescence confirmed the activation of STING and its downstream proteins in mesothelial cells upon EPS stimulation, while treatment with the STING inhibitor H151 partially mitigated this effect (Fig. 5I, J). qPCR analysis further demonstrated that the expression of inflammatory-related genes was significantly upregulated in mesothelial cells under EPS stimulation, and this effect was also suppressed by H151 (Fig. 5K). We next examined the role of CCL2 in this process. RT-qPCR and cellular immunofluorescence showed that both CCL2 gene expression and CCL2 protein levels were significantly elevated in mesothelial cells under EPS stimulation, and these increases were attenuated by H151 (Fig. 5L, M). The secretion of CCL2 in the cell supernatant was also significantly higher in EPS-stimulated mesothelial cells (Fig. 5N). STING agonists ADU-S100 also significantly induced CCL2 expression (Supplemental Fig. 6I, J), verifying the role of STING activation in promoting CCL2 secretion.
To directly assess the impact of mesothelial cell activation on macrophage migration, we performed a transwell co-culture assay, where mesothelial cells were co-cultured with macrophages (Fig. 5O). EPS-stimulated mesothelial cells significantly promoted macrophage migration (Fig. 5P, Q), while treatment with H151 partially alleviated this effect. These findings demonstrate that activation of the cGAS-STING pathway in mesothelial cells leads to the secretion of CCL2, which in turn induces macrophage chemotaxis and migration. Collectively, our results highlight the critical role of mesothelial cells in sensing EPS-related stimuli and activating the cGAS-STING pathway, thereby driving macrophage recruitment through CCL2 secretion. Inhibition of STING effectively reduces both CCL2 production and macrophage migration, offering a potential therapeutic approach for mitigating macrophage-driven inflammation in EPS. Additionally, we observed that anti-CCL2 intervention also significantly attenuated in vitro EPS-induced macrophage migration, again indicating that CCL2 acts as the predominant cytokine driving macrophage chemotaxis in this microenvironment (Fig. 5R, S).
Inhibition of cGAS-STING activation ameliorates EPS formation in mice
To investigate whether inhibiting the cGAS-STING pathway could reduce the severity of EPS, we administered the STING inhibitor H151 via intraperitoneal injection in mice (Fig. 6A). Western blot and immunofluorescence analyses confirmed that H151 effectively suppressed the activation of STING and downstream signaling proteins in the peritoneal tissues (Fig. 6B), validating its ability to block STING signaling in vivo. In parallel, H151 treatment significantly lowered the expression of CCL2 in peritoneal tissue (Fig. 6C, Supplemental Fig. 7A) and in the peritoneal lavage fluid (Fig. 6D). This reduction in CCL2 was associated with a decrease in macrophage infiltration on the peritoneal surface, observed through immunohistochemistry (Fig. 6E, F). Correlation analysis between the CCL2 concentration in peritoneal lavage fluid and the F4/80 infiltration area showed a significant positive correlation (Fig. 6G). These results indicate that STING-mediated chemotaxis, driven by CCL2, plays a central role in recruiting macrophages during EPS progression. Therapeutically, H151 treatment markedly improved the pathological features of EPS. Mice treated with H151 showed a significant reduction in adhesion scores during gross assessments (Fig. 6H, I), reflecting decreased intra-abdominal adhesions. Histological staining also revealed a notable reduction in collagen fiber deposition in the adhesion regions (Fig. 6Ja-b and 6K). The thickness of the parietal peritoneum, a hallmark of fibrosis in EPS, was significantly decreased in H151-treated mice (Fig. 6J-c and L), indicating a direct impact on fibrotic progression. Immunohistochemistry showed that ECM-related proteins (COL1A1, fibronectin, α-SMA) (Fig. 6d-f), inflammatory markers (IL-1β, IL-6, TNF-α), and pro-angiogenic VEGF were significantly reduced in the H151-treated group (Fig. 6M), supporting the inhibitor’s anti-adhesion effect. In summary, these findings suggest that inhibiting cGAS-STING activation with H151 reduces CCL2 secretion and macrophage recruitment, ultimately mitigating the excessive collagen deposition and adhesion formation characteristic of EPS. This highlights the potential of targeting the STING pathway as a therapeutic strategy for EPS.

A The schematic diagram of administering the STING inhibitor H151 (Created in BioRender. Sun, J. (2025) https://BioRender.com/vbajaol). Western blot (B) and immunofluorescence (C) demonstrating that H151 effectively reduced the activation of STING and its downstream proteins in peritoneal tissues (n = 6 per group). D ELISA exhibited that H151 partially lowered the CCL2 concentration in peritoneal lavage fluids (n = 6 per group). E–F Immunohistochemistry (IHC) showed H151 effectively reduced macrophage infiltration (n = 6 per group). G Correlation analysis between the CCL2 concentration in peritoneal lavage fluid and the F4/80 infiltration from (F) among the 3 groups. Gross macroscopic viewing of abdomen (H) and the macroscopic adhesion score (I) demonstrated from a macro perspective that H151 effectively alleviated abdominal adhesion (n = 6 per group). J–L Histopathological assessment of intra-abdominal adhesions and fibrous deposition on peritoneal surface. a MASSON staining of the abdominal cross-sections presented the adhesion condition in different groups, and the MASSON+ area (b) was calculated and the statistic diagram is shown in Figure K (n = 6 per group). c Masson’s trichrome staining illustrates the thickness of the parietal peritoneum among the three groups, with (L) showing the corresponding statistical bar graph (n = 6 per group). d–f Immunohistochemical images presenting the expression of three classic ECM-related proteins in parietal peritoneum: COL1A1, Fibronectin, and α-SMA. M Immunohistochemical analysis of inflammatory markers IL-1β, IL-6, and TNF-α in the parietal peritoneum. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed Student’s t-test.
IRF3 as the key transcription factor promoting CCL2 secretion in mesothelial cells
Cyclic GMP-AMP synthase (cGAS) detects cytosolic DNA during cellular stress and activates the adaptor protein STING, which triggers immune responses by activating the downstream transcription factor IRF326,27,28. We investigated whether the activation of STING in mesothelial cells under EPS condition regulates CCL2 secretion via IRF3. Using the Univariate Linear Model (ULM) method in the DecoupleR package, we analyzed our bulk RNA-seq data and found that IRF3 transcriptional activity was significantly elevated in the EPS group (Fig. 7A). This increase in IRF3 activity aligns with our observation of increased phospho-IRF3 levels in mesothelial cells under EPS condition (Fig. 5I). Among the genes predicted to be regulated by IRF3, CCL2 and CCL5 were the most upregulated in the EPS group (Fig. 7B), with CCL2 had the higher p-value (p < 10-6). To further investigate how IRF3 promotes CCl2 expression, we used the JASPAR database to analyze the transcription factor binding sites within the CCL2 promoter and identified a high-scoring potential IRF3 binding site (Fig. 7C). This suggested a direct interaction between IRF3 and the CCL2 promoter region (Fig. 7D). To test this possibility, we conducted a chromatin immunoprecipitation (ChIP) assay, which validated that IRF3 indeed binds to the promoter of the CCL2 gene (Fig. 7E, F, Supplemental Fig. 8). These findings demonstrate that under EPS condition, IRF3 binding to the CCL2 promoter facilitates increased CCL2 production and secretion. Thus, IRF3 serves as a key transcription factor driving CCL2 secretion in mesothelial cells in response to EPS. IRF3 knockdown significantly reduced both the transcription and secretion of CCL2 in response to stimulation with a STING agonist (Supplemental Fig. 6I, J) and EPS-conditioned media (Fig. 7G, H). Furthermore, this genetic intervention markedly inhibited macrophage migration, as shown by transwell assays (Fig. 7I, J). These results provide functional evidence that IRF3 is a key transcriptional regulator of CCL2 in EPS-associated mesothelial responses.

A Bar plot showing the activity scores of differentially expressed transcription factors in Saline and EPS group, based on bulk RNA-seq data. B Volcano plot displaying target genes of IRF3 that are differentially expressed between Saline and EPS group. Red dots indicate genes activated by IRF3, while blue dots represent genes inhibited by IRF3. C Predicted IRF3 binding motifs identified using the JASPAR database. D Schematic illustration showing the IRF3 binding motif within the promoter region of the CCL2 gene. E–F ChIP assays and ChIP-qPCR analysis of IRF3 binding to CCL2 in mesothelial cells treated with or without EPS (SHS + LPS) and H151 (n = 3 independent experiments). Gene silencing of IRF3 significantly reduced both the transcriptional expression (G) and secretion (H) of CCL2 induced by SHS + LPS. I–J Trans-well experiments demonstrating IRF3 silencing suppressed EPS-induced macrophage migration. Data are presented as mean ± SEM. ***p < 0.001, two-tailed Student’s t-test.
Clinical relevance of STING activation and EPS
For a clinical investigation of the cGAS-STING pathway, we collected peritoneal tissues, including adhesion tissues, from patients with EPS and healthy controls. Immunofluorescence staining revealed significant activation of the cGAS-STING pathway in mesothelial cells from the peritoneal surface of EPS patients (Fig. 8A), along with a marked increase in CCL2 expression (Fig. 8B). Given that a significant number of mesothelial cells are shed into the peritoneal dialysis fluid under EPS condition, we also prepared smears of the shed cells from the peritoneal dialysis effluent for immunofluorescence staining, which yielded results consistent with the peritoneal tissue (Fig. 8C–H). Additionally, ELISA measurements confirmed that the concentration of CCL2 in the peritoneal dialysis effluent was significantly higher in EPS patients compared to controls (Fig. 8I), so as the cGAMP, which serves as an indicator of STING activation (Fig. 8J). Furthermore, there is a positive correlation between CCL2 and cGAMP concentrations (Fig. 8K). These clinical findings corroborate the link between STING activation and increased CCL2 levels in EPS, further supporting the role of the cGAS-STING pathway in driving macrophage chemotaxis and inflammation.

HE staining and immunofluorescence of peritoneal tissue from clinical patients showed the increased expression of STING (A) and CCL2 (B) in EPS patients compared to healthy control (n = 6 per group). C–H Immunofluorescence of peritoneal dialysis effluent smear showed the increased expression of CCL2 (C), STING (D), and p-IRF3 (E) in EPS patients compared to short-term PD patients (SPD) (n = 6 per group). With the corresponding quantitative statistics of fluorescence co-localization showing in F, G and H. I–J ELISA measurements revealed a significant increased CCL2 concentrations (E) and cGAMP concentrations (F) in the peritoneal dialysis effluent in EPS patients compared to control (n = 6 per group). K Correlation analysis presented a significant positive correlation between cGAMP and CCL2 (n = 12). Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001, two-tailed Student’s t-test.
Discussion
Encapsulating Peritoneal Sclerosis is a severe complication of peritoneal dialysis, diagnosed through clinical symptoms, radiological findings, and histological criteria29. EPS pathogenesis is linked to prolonged PD duration and recurrent peritonitis, while some patients may develop EPS following discontinuing PD13,30,31. Despite these known risk factors, the precise mechanisms underlying EPS remain unclear, highlighting the need for animal models that closely mimic the human disease. In this study, we developed a novel mouse model of EPS that replicates key pathophysiological features observed in patients, such as inflammatory cell infiltration, extracellular matrix (ECM) deposition, and adhesion formation. We demonstrated that the cGAS-STING pathway plays a crucial role in EPS pathology by activating mesothelial cells, which secrete CCL2 and promote macrophage chemotaxis and inflammation. We also explored the therapeutic potential of targeting STING signaling, showing that the STING inhibitor H151 significantly reduces macrophage infiltration, collagen deposition, and adhesion formation. Lastly, we elucidated the cellular and molecular mechanisms of macrophage recruitment, confirming that mesothelial cells are the primary source of CCL2, driving macrophage migration via cGAS-STING activation. These findings offer fresh insights into EPS pathogenesis and suggest promising therapeutic interventions.
Previous EPS models relied on intraperitoneal injection of surgical hygiene solution (SHS)13,32, requiring 8 weeks to establish. Our modified model integrates both PD and lipopolysaccharide (LPS), leading to the formation of stable adhesions in just 3 weeks. This model better simulates the pathophysiological progression of EPS by reflecting more clinically relevant risk factors, such as PD and peritonitis, and captures key features like inflammatory infiltration and extracellular matrix (ECM) deposition.
While our model successfully recapitulates key features of PD-induced EPS, several limitations should be acknowledged. First, the model is specific to peritoneal dialysis–associated EPS and may not capture the full spectrum of etiologies underlying this condition. Future models incorporating alternative triggers could broaden translational relevance. Second, the relatively short duration of our experiments may not reflect the chronic nature of EPS or fully capture long-term therapeutic outcomes, including potential resistance or recurrence. Third, systemic inhibition of STING—a pathway critical for antiviral and antitumor immunity33—raises concerns about unintended immunosuppressive effects. Although our findings highlight the therapeutic potential of STING inhibition, we did not assess its safety profile, which remains an essential consideration for clinical translation.
Previous studies have identified multiple factors contributing to the initiation of abdominal adhesions, with a common emphasis on ECM deposition between the peritoneum and adjacent organs34. Using lineage tracing, Yuval Rinkevich’s team demonstrated that migration and infiltration of neutrophils and mesothelial cells, following mesothelial-to-mesenchymal transition, to the surgical adhesion site were key drivers of adhesion formation and ECM accumulation31,35. In contrast, Ito et al. proposed that adhesions arise from the loss of the mesothelial barrier, which is replaced by non-polarized macrophages forming a compensatory barrier23. Zindel et al. further advanced this concept by using intravital microscopy to show that peritoneal macrophages physically bridge intra-abdominal organs, such as the omentum, during the early stages of adhesion formation. These macrophage bridges subsequently serve as scaffolds for the recruitment of mesothelial cells, immune cells, and ECM components36. Consistent with prior studies on EPS, our findings highlight inflammatory infiltration, particularly by macrophages, as a central mechanism in disease progression37,38. The most significantly enriched pathways in our transcriptomic analysis were related to leukocyte migration and myeloid cell activation (primarily macrophages), consistent with sustained immune cell recruitment to the peritoneum. These macrophages may serve as a structural scaffold for subsequent adhesion bridge formation, as previously described by Zindel et al.36. In our model, early stages of EPS were dominated by immune cell infiltration, followed by extensive collagen deposition, which mirrors the progression seen in patients. RNA-seq analysis further confirmed upregulation of pathways involved in immune responses, particularly the cGAS-STING pathway, which mediates inflammation and fibrosis in multiple organ systems10,33. Activation of STING in mesothelial cells triggered CCL2 secretion, promoting macrophage chemotaxis and adhesion formation, consistent with findings in other fibrotic diseases39,40.
Our results suggest that targeting the STING pathway could mitigate the inflammatory and fibrotic processes driving EPS. STING inhibitors, such as H151, effectively reduced macrophage infiltration, ECM deposition, and adhesion formation in our mouse model, making this pathway a promising therapeutic target for EPS. However, given the complex signaling interactions between STING, TGF-β, and other fibrosis-related pathways like JAK/STAT and MAPK41,42, further studies are needed to elucidate these relationships and optimize treatment strategies. Additionally, long-term studies are necessary to evaluate the safety, efficacy, and resistance potential of STING inhibitors in chronic EPS.
Currently, clinical applications of STING modulation are largely centered on the use of STING agonists for cancer immunotherapy43,44, while research on the therapeutic use of STING inhibitors remains limited. Although STING inhibition has shown potential in animal models to reduce macrophage infiltration and adhesion formation, systemic blockade of STING raises important concerns. Given STING’s essential role in antiviral and antitumor immunity, its inhibition could impair host immune defenses, posing safety risks that must be carefully considered in clinical translation. Moreover, our findings are based on short-term experimental models, and the long-term effects of STING inhibition—particularly in chronic EPS—remain unknown. Future studies should evaluate the safety, durability, and potential for immune resistance associated with prolonged STING inhibition to determine its feasibility in clinical settings.
In conclusion, we established a robust mouse model that faithfully recapitulates the key histological, molecular, and clinical features of EPS. Our findings demonstrate that STING activation in mesothelial cells promotes CCL2 secretion, triggering macrophage recruitment and adhesion formation. Targeting the STING pathway represents a promising therapeutic strategy to mitigate inflammation and fibrotic remodeling in EPS, with potential relevance for preventing or treating this debilitating complication in patients.
References
Cheong, Y. C. et al. Peritoneal healing and adhesion formation/reformation. Hum. Reprod. Update 7, 556–566 (2001).
Foster, D. S. et al. Elucidating the fundamental fibrotic processes driving abdominal adhesion formation. Nat. Commun. 11, 4061 (2020).
Singhal, M. et al. Encapsulating peritoneal sclerosis: the abdominal cocoon. Radiographics 39, 62–77 (2019).
Yu, Q. et al. A zwitterionic hydrogel with anti-oxidative and anti-inflammatory properties for the prevention of peritoneal adhesion by inhibiting mesothelial-mesenchymal transition. Adv. Health. Mater. 12, e2301696 (2023).
Fatehi Hassanabad, A., Zarzycki, A. N., Jeon, K., Deniset, J. F. & Fedak, P. W. M. Post-operative adhesions: a comprehensive review of mechanisms. Biomedicines 9, 867 (2021).
diZerega, G. S. & Campeau, J. D. Peritoneal repair and post-surgical adhesion formation. Hum. Reprod. Update 7, 547–555 (2001).
Carmichael, S. P. et al. Regenerative medicine therapies for prevention of abdominal adhesions: a scoping review. J. Surg. Res 275, 252–264 (2022).
Tang, X. & Sun, L. Encapsulating peritoneal sclerosis. N. Engl. J. Med 388, 833 (2023).
Alston, H., Fan, S. & Nakayama, M. Encapsulating peritoneal sclerosis. Semin Nephrol. 37, 93–102 (2017).
Zhang, J. et al. The role of cGAS-STING signaling in pulmonary fibrosis and its therapeutic potential. Front Immunol. 14, 1273248 (2023).
Zhang, Z. et al. Multifaceted functions of STING in human health and disease: from molecular mechanism to targeted strategy. Signal Transduct. Target Ther. 7, 394 (2022).
Kawaguchi, Y., Kawanishi, H., Mujais, S., Topley, N. & Oreopoulos, D. G. Encapsulating peritoneal sclerosis: definition, etiology, diagnosis, and treatment. International Society for Peritoneal Dialysis Ad Hoc Committee on Ultrafiltration Management in Peritoneal Dialysis. Perit. Dial. Int 20, S43–S55 (2000).
Ishii, Y. et al. An experimental sclerosing encapsulating peritonitis model in mice. Nephrol. Dial. Transpl. 16, 1262–1266 (2001).
Wang, Y. et al. In-situ growth of robust superlubricated nano-skin on electrospun nanofibers for post-operative adhesion prevention. Nat. Commun. 13, 5056 (2022).
Zhang, T. et al. A ROS-responsive and scavenging hydrogel for postoperative abdominal adhesion prevention. Acta Biomater. 184, 98–113 (2024).
Huang, Q. et al. Extracellular vesicle-packaged ILK from mesothelial cells promotes fibroblast activation in peritoneal fibrosis. J. Extracell. Vesicles 12, e12334 (2023).
Sun, Y. et al. Mucosal-Associated Invariant T (MAIT) cell-mediated immune mechanisms of peritoneal dialysis-induced peritoneal fibrosis and therapeutic targeting. J. Am. Soc. Nephrol. 36, 1072–1087 (2025).
Badia-I-Mompel, P. et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinform Adv. 2, vbac016 (2022).
Türei, D. et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 17, e9923 (2021).
Yavaşcan, Ö et al. The importance of ultrasonographic measurement of peritoneal wall thickness in pediatric chronic peritoneal dialysis patients. Ren. Fail 37, 381–386 (2015).
Lambie, M. R. et al. Peritoneal inflammation precedes encapsulating peritoneal sclerosis: results from the GLOBAL Fluid Study. Nephrol. Dial. Transpl. 31, 480–486 (2016).
Habib, S. M., Korte, M. R. & Betjes, M. G. H. Lower mortality and inflammation from post-transplantation encapsulating peritoneal sclerosis compared to the classical form. Am. J. Nephrol. 37, 223–230 (2013).
Ito, T. et al. Cell barrier function of resident peritoneal macrophages in post-operative adhesions. Nat. Commun. 12, 2232 (2021).
Nagarsheth, N., Wicha, M. S. & Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572 (2017).
Guan, M. et al. Chemokine systems in oncology: from microenvironment modulation to nanocarrier innovations. Int J. Biol. Macromol. 268, 131679 (2024).
Stavrou, S., Blouch, K., Kotla, S., Bass, A. & Ross, S. R. Nucleic acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe 17, 478–488 (2015).
Wang, F. et al. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat. Immunol. 17, 514–522 (2016).
Chakraborty, A. et al. Epigenetic induction of smooth muscle cell phenotypic alterations in aortic aneurysms and dissections. Circulation 148, 959–977 (2023).
Summers, A. M. et al. A collaborative approach to understanding EPS: the European perspective. Perit. Dial. Int. 31, 245–248 (2011).
Johnson, D. W. et al. Encapsulating peritoneal sclerosis: incidence, predictors, and outcomes. Kidney Int. 77, 904–912 (2010).
Lambie, M. et al. Estimating risk of encapsulating peritoneal sclerosis accounting for the competing risk of death. Nephrol. Dial. Transpl. 34, 1585–1591 (2019).
Lin, G.-J. et al. Adoptive transfer of DMSO-induced regulatory T cells exhibits a similar preventive effect compared to an in vivo DMSO treatment for chemical-induced experimental encapsulating peritoneal sclerosis in mice. Toxicol. Appl Pharm. 378, 114641 (2019).
Liu, Y. et al. The effect of the cyclic GMP-AMP synthase-stimulator of interferon genes signaling pathway on organ inflammatory injury and fibrosis. Front Pharm. 13, 1033982 (2022).
Tsai, J. M. et al. Surgical adhesions in mice are derived from mesothelial cells and can be targeted by antibodies against mesothelial markers. Sci. Transl. Med 10, eaan6735 (2018).
Fischer, A. et al. Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat. Immunol. 23, 518–531 (2022).
Zindel, J. et al. Primordial GATA6 macrophages function as extravascular platelets in sterile injury. Science 371, eabe0595 (2021).
Habib, S. M. et al. CD4-positive T cells and M2 macrophages dominate the peritoneal infiltrate of patients with encapsulating peritoneal sclerosis. PLoS One 10, e0120174 (2015).
Reimold, F. R. et al. Transcriptional patterns in peritoneal tissue of encapsulating peritoneal sclerosis, a complication of chronic peritoneal dialysis. PLoS One 8, e56389 (2013).
Brassington, K. et al. Crosstalk between cytotoxic CD8+ T cells and stressed cardiomyocytes triggers development of interstitial cardiac fibrosis in hypertensive mouse hearts. Front Immunol. 13, 1040233 (2022).
Gao, L. et al. STING/ACSL4 axis-dependent ferroptosis and inflammation promote hypertension-associated chronic kidney disease. Mol. Ther. 31, 3084–3103 (2023).
Zhang, Y. et al. Signaling pathways involved in diabetic renal fibrosis. Front Cell Dev. Biol. 9, 696542 (2021).
Ni, J. et al. STING signaling activation modulates macrophage polarization via CCL2 in radiation-induced lung injury. J. Transl. Med. 21, 590 (2023).
Luke, J. J. et al. Intratumoral or subcutaneous MK-2118, a noncyclic dinucleotide STING agonist, with or without pembrolizumab, for advanced or metastatic solid tumors or lymphomas. Clin. Cancer Res. 31, 1233–1242 (2025).
Luke, J. J. et al. Phase I dose-escalation and pharmacodynamic study of STING agonist E7766 in advanced solid tumors. J. Immunother. Cancer 13, e010511 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC82170762 to HP), the National Natural Science Foundation of Guangdong, China (2022A1515012637 to HP), “Three Major Construction” of Large Science Program of Sun Yat-sen University, China (82000-18843406 to HP). Model cartoons in Figs. 1A, 1F, 5A, 6O were constructed using the Biorender website (https://www.biorender.com/) and we’ve obtained a usage license (Created in BioRender. Sun, J. (2025) https://BioRender.com/vbajaol).
Author information
Authors and Affiliations
Contributions
J.S., Z.Y.H. and H.P. designed the experiments and wrote the manuscript. J.S., Y.X.S., D.D.G., H.L.Y., Q.H., H.Z., C.M.L., P.Z., Z.Y.H. and H.P. performed the experiments and data analysis. H.L.S. and D.X.W. assisted clinical sample collection. H.L.Y., M.L., Y.J.Y. helped to conduct the ultrasound examination. Prof. JW assisted to revise the manuscript. All authors edited and approved the final version. J.S., Y.X.S., D.D.G., and H.L.Y. contributed as co–first authors; authorship order is based on the amount of data contributed.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Raffaele Strippoli and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Joao Valente. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, J., Sun, Y., Guo, D. et al. Targeting STING to disrupt macrophage-mediated adhesion in encapsulating peritoneal sclerosis. Commun Biol 8, 1266 (2025). https://doi.org/10.1038/s42003-025-08662-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08662-z
