
Abstract
Achieving sufficient therapeutic payload delivery remains a significant challenge in gene therapy, particularly for cancer immunotherapy, where payload thresholds are critical for efficacy. To address this, we developed a Cascade Amplification of Therapeutic Payloads (CATP) system, leveraging lipid nanoparticles (LNPs) to co-deliver self-amplifying mRNA (SamRNA) and modified mRNA encoding alphavirus capsids and envelopes. The CATP system initiates a dual-amplification process: SamRNA amplifies therapeutic payloads within transfected cells, while capsid and envelope proteins package SamRNA into defective viral particles to infect neighboring cells, enabling secondary payload amplification. This single-cycle infection ensures enhanced efficacy while maintaining safety. In vitro and in vivo studies demonstrated the CATP system’s superiority over conventional SamRNA delivery. In a B16F10 melanoma model, CATP achieved a 525-fold increase in intratumoral IL-12 levels, resulting in tumor regression and long-term immune memory. The platform also showed broad applicability, effectively treating MC38 colorectal cancer, CT26 colon cancer, and P53null KrasG12D pancreatic ductal adenocarcinoma. Additionally, optimization of therapeutic payloads with mutant IL-18 further enhanced anti-tumor efficacy. The CATP system represents a transformative approach to gene therapy, providing a scalable, safe, and potent platform for cancer immunotherapy. Its dual-amplification strategy offers new opportunities for overcoming payload limitations across diverse malignancies.
Similar content being viewed by others


Introduction
Messenger RNA (mRNA) has emerged as a transformative platform for therapeutic applications, leveraging its potential for transient, tunable protein expression. Among its various forms, modified mRNA, circular mRNA, and self-amplifying mRNA (SamRNA), SamRNA has garnered significant attention due to its capacity for intracellular amplifications to increase protein expression, enabling therapeutic efficacy at substantially lower initial doses compared to conventional mRNA1,2. However, mRNA inherently act as a pathogen-associated molecular patterns (PAMP) that can be recognized by endosomal sensors, such as Toll-like receptors 3 (TLR3) or 7 (TLR7), as well as cytosol sensors, including MDA5 or RIG1. Activation of these receptors and/or sensors triggers the release of interferons resulting in fevers, local and systemic inflammation as well as other side effects3,4,5,6,7. Though various strategies have been developed such as modification of uridine and other nucleotides8, as well as mimicking native mRNA caps9, to reduce the stimulations of interferon responses, the side effects, including fever, remain a common challenge. For example, the side effects during the clinical applications of COVID 19 vaccines are highly relevant to the dosage levels and the extent of interferon induction10.
Thus, there is an urgent need for a novel modality that maintains low mRNA dosage while dramatically increasing therapeutic payloads to reduce the mRNA-associated cytotoxicity and broaden its therapeutic application11. To address these challenges, we introduce the Cascade Amplification of Therapeutic Payloads (CATP) system, a groundbreaking platform that leverages SamRNA to encode therapeutic payloads, initiating a first wave of therapeutic payload expression. Simultaneously, the system co-delivers modified mRNA encoding viral capsid and envelope proteins which generate defective viral particles to achieve a second wave of therapeutic payload expression. This dual-amplification mechanism offers a highly effective solution for gene therapies and cancer immunotherapies. As a proof of concept, we employed IL-12 as the initial therapeutic payload, given its extensive evaluation in preclinical studies and advancement into clinical trials12,13,14, thereby enhancing the translational potential of the CATP system for clinical applications.
Results
In vitro and in vivo validation of cascade amplification of therapeutic payloads (CATP)
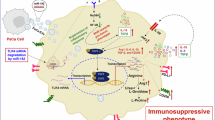
To overcome the limitations of existing mRNA and SamRNA approaches, which often require increased dosages or frequent administrations, we introduce a novel strategy to amplify therapeutic payloads in vivo without escalating the dosage. By co-delivering SamRNA from Venezuelan equine encephalitis (VEE) encoding therapeutic payloads and modified mRNA encoding optimized viral capsids/envelop using lipid nanoparticles, the inherent amplification of SamRNA generated the first wave of therapeutic payloads produced in transfected cells. Meanwhile, the translated capsid and envelop proteins in transfected cells package SamRNA and produce defective yet infectious viral particles15 to transduce adjacent cells with SamRNA, further amplifying the therapeutic payloads and enhancing overall efficacy (Fig. 1a, b). We refer to this new technology as cascade amplification of therapeutic payloads (CATP) in this work. Importantly, since the mRNA encoding capsids/envelop is not encapsulated in the viral genome, the infection is restricted to a single cycle, thereby ensuring safety.

a Illustration of the cascade amplification of therapeutic payloads (CATP) was created in BioRender. Li, Y. (2025) https://BioRender.com/g50p020. b Illustrations of constructs for CATP. c Effects of CATP in HEK293 cells. HEK293 cells were treated by LNP encapsulated SamRNA mRNA encoding with eGFP and modified mRNA encoding with envelop and capsids from VEE or LNP encapsulated SamRNA mRNA encoding with eGFP and modified mRNA encoding firefly luciferase. Then the GFP percentages were determined by flow cytometer at days 1, 2, 3, and 4 post transfection. The shown are the changes of GFP versus days post transfection. d Scheme of tumor inoculation and treatment: Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with 1 million B16F10 melanoma cells. Seven days post-inoculation, mice were intratumorally treated with PBS (Control group of basal line), or LNP encapsulating 10 µg SamRNA encoding with mouse IL-12 (mIL-12) and 1 µg modified mRNA encoding with firefly luciferase (Control group of treated group) or LNP encapsulated 10 µg of SamRNA encoding mIL-1212 and 1 µg modified mRNA encoding capsids/envelops from VEE, following the principle that molar ratio of modified mRNA (1 µg) is smaller than it of samRNA (10 µg). e mIL-12 expression in sera and tumors. The P-Values labeled in e were determined by Tukey’s multiple comparisons test.
To validate our platform in vitro, HEK293 cells were co-transfected with SamRNA encoding eGFP and modified mRNA encoding VEE capsids/envelop (CATP: Sam(eGFP)_mod(VEE)) and were compared to cells transfected with SamRNA encoding eGFP (Contr: Sam(eGFP)_mod(Luci). As anticipated, the percentages of eGFP-positive cells were initially lower in the CATP group than the control group within the first three days post transfection. However, eGFP-positive cells in the CATP group markedly expanded on day 3 and surpassed those in the control group after day 4. In contrast, the control group started to decay after day 2 (Fig. 1c). To track the function of the defective viruses, the naive HEK293 cells were incubated with the supernatants of the cells treated with CATP: Sam(eGFP)_mod(VEE), or Contr: Sam(eGFP)_mod(Luci), or PBS. A day post incubation, the CATP group showed clear GFP populations, indicating the formations of defective VEE viral particles (Supplementary Fig. 1a). This observation underscored the key feature of CATP in terms of amplifying reporter genes in vitro.
Further validation of the amplification capacity of CATP was performed in a highly immune resistant mouse B16F10 melanoma model16 via a single intratumoral17 injection of lipid nanoparticles encapsulating CATP encoding mouse cytokine IL-1212,18. Delivery of recombinant IL-12 via different technologies has been shown to exhibit potent anti-tumor immunity by stimulating CD4, CD8 T cells, NK cells, and B cells, while its prolonged systemic exposure is associated with host toxicity. To track the function of the defective viruses, the naive HEK293 cells were incubated with the supernatants of the cells treated with CATP: Sam(IL-12)_mod(VEE), or Contr: Sam(IL-12)_mod(Luci), or PBS. Two days post incubation, the CATP group showed 64% more of IL-12 expression, indicating the formations of defective VEE viral particles encoding mouse IL-12 and enhancing the IL-12 expression (Supplementary Fig. 1b). Compared to the control group containing SamRNA encoding mouse IL-12 (Contr: Sam(IL-12)_mod(Luci)), the CATP group containing SamRNA-IL12 and VEE capsids/envelop (CATP: Sam(IL-12)_mod(VEE)) displayed five-fold increase in mouse serum IL-12 levels on day 1, remained detectable on day 3, and became undetectable by day 7. Notably, the mouse IL-12 levels in tumor sites increased by 525 times in CATP group relative to that of control group (Fig. 1d, e). These results demonstrated that the CATP system outperformed the conventional SamRNA platforms in the expression of transgenes both in vitro and in vivo, though the outperformances by CATP are much faster in vivo.
Enhanced therapeutic efficacy of CATP-mIL-12 in the B16F10 melanoma model
We further evaluate whether the increased therapeutic levels achieved through a single dose of CATP-mIL-12 improves therapeutic efficacies in the B16F10 melanoma model that’s a poorly immunogenic and highly aggressive model for cancer immunotherapy19. On day 7 post-inoculation of cancer cells when tumor areas reached approximately 50 mm2, mice were intratumorally treated with a single dose of CATP or corresponding controls as described in Fig. 1d. Consistent with the increased therapeutic levels observed in Fig. 1e, tumors in the CATP-IL-12 group showed improved regression (Fig. 2a), resulting in 40% of fully tumor regressed mice (p = 0.0357, Fig. 2b). Notably, body weight changes were minimal at 3% (Fig. 2c), aligning with the undetectable serum IL-12 levels on day 7 post injections of LNP-mRNA (Fig. 1e). In summary, the CATP not only increases the expression of therapeutic payloads but also enhances therapeutic efficacy while maintaining the same dosage and safety profiles, demonstrating its potential as a safe and effective cancer immunotherapy strategy.

Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with 1 million B16F10 melanoma cells. Seven days post-inoculation, mice were intratumorally treated with PBS (Control group of basal line), or LNP encapsulating 10 µg SamRNA encoding with mouse IL-12 (IL-12) and 1 µg modified mRNA encoding with firefly luciferase (Control group of treated group) or LNP encapsulated 10 µg of SamRNA encoding IL-12 and 1 µg modified mRNA encoding capsids/envelops from VEE. Results are shown as: Tumor areas (Y-axis), Survival rates (Y-axis), and Body weight changes (Y-axis) versus days post-B16F10 melanoma cell inoculation (X-axis) a, b, and c, respectively. The P-Values label in b were determined by a Comparison of Survival Curves (Kaper-myer) test.
Comparative analysis of capsids and envelopes for optimizing CATP therapeutic efficacy
To investigate the roles of different capsids/envelops in determining the amplification efficiency for CATP, we evaluated capsids/envelops derived from three commonly used alphaviruses, including VEE, Sindbis virus (SIN), and Semliki Forest virus (SFV4)20. Interestingly, the SFV4-derived capsids/envelops exhibited the strongest regression of tumor growth (Fig. 3a), resulting in a 60% fully tumor regressed rate with minimal weight changes (Fig. 3b, c). These findings suggest that CATP leveraging SFV4 capsids/envelops may generate oncolytic virus-like particles (Fig. 3d), in agreement with other researchers’ observations of oncolytic effects associated with SFV4 viruses21. The oncolytic viral effects will be helpful to release the tumor associated antigens (TAA) and trigger the inflammasomes along with the LNP-mRNA, which are critical prime immune responses against tumor as in the Fig. 3b, d.

Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with 1 million B16F10 melanoma cells. Seven days post-inoculation, mice were intratumorally treated with PBS (Control group of basal line), or LNP encapsulated 10 µg of SamRNA encoding mIL-12 and 1 µg modified mRNA encoding capsids/envelops from VEE (Control group of treated group), SIN, and SFV4 as indicated. Results are shown as: Tumor areas (Y-axis), Survival rates (Y-axis), and Body weight changes (Y-axis) versus days post-B16F10 cell inoculation (X-axis) (a, b, c), respectively. d Illustrations of CATP with oncolytic effects by the capsids/envelop such as SFV4 was created in BioRender. Li, Y. (2025) https://BioRender.com/bguaszo. The SamRNA, regular mRNA, therapeutic payloads, capsids/envelop, defective viruses, tumor associated antigens (TAA), and inflammasomes triggered by defective viruses and LNP-mRNA are indicated. The P-Values labeled in a and c were determined by a two-way ANOVA test and Comparison of Survival Curves (Kaper-myer) test.
Optimization of combinatorial cytokines for CATP therapy in multiple tumor models
To further enhance therapeutic efficacy, we optimized therapeutic payloads by combining mouse IL-12 with mouse IL-18, as IL-18 enhances polarizations towards Type 1 inflammation in the presence of IL-1222,23. However, IL-18 binding protein (IL-18BP) has been reported to be upregulated in solid tumors thereby inhibiting IL-18 activity24. Therefore, we compared wildtype mouse IL-18 to a mutant form, which was designed to disrupt interactions with IL-18BP24,25.
Following a single dosing in tumors with a starting size of ~50 mm2, the combination of mIL-12 with mutant mIL-18 in the CATP system significantly inhibited the tumor growth and increased the fully tumor regressed rates to 80% in the B16F10 melanoma model. This outcome markedly surpassed the 40% fully tumor regressed rates achieved by mIL-12 plus wild-type mIL-18, or mIL-12 alone (Fig. 4a, b). While the CATP with mouse mutant IL-18 alone effectively inhibited tumor growth, it failed to fully suppress any tumor-bearing mouse (Fig. 4a, b), indicating that the anti-tumor effects of mutant IL-18 were dependent on mouse IL-12. Although the combination of mIL-12 with mutant mIL-18 was initially associated with body weight loss of > 6% after CATP administration, they returned to normal body weights similar to the control group (PBS) one week after the dosing (14 days post tumor inoculation, p = 0.8421) (Fig. 4c). These findings suggest that the CATP system combining mIL-12 and mutant IL-18 displayed superior efficacy without compromising the safety following a single administration in tumors.

Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with 1 million B16F10 melanoma cells. Seven days post-inoculation, mice were intratumorally treated with PBS (Control group of basal line), or LNP encapsulating 5 µg SamRNA encoding with mouse IL-12 (IL-12), 5 µg SamRNA encoding with firefly Luciferase, and 1 µg modified mRNA encoding capsids/envelops from SFV4 (Control group of treated group); or LNP encapsulating 5 µg SamRNA encoding with mouse wild type IL-18 (wtIL-18) or mutant IL-18 (mtIL-18), 5 µg SamRNA encoding with firefly Luciferase, and 1 µg modified mRNA encoding capsids/envelops from SFV4; or LNP encapsulating 5 µg SamRNA encoding with mouse IL-12 (IL-12), 5 µg SamRNA encoding with wild type IL-18 (wtIL-18) or mutant IL-18 (mtIL-18), and 1 µg modified mRNA encoding capsids/envelops from SFV4 as indicated. Results are shown as: Tumor areas (Y-axis), Survival rates (Y-axis), and Body weight changes (Y-axis) versus days post B16F10 melanoma cell inoculation (X-axis) (a–c), respectively. The P-Values labeled was determined by a two-way ANOVA test or Comparison of Survival Curves (Kaper-myer) test.
To generalize the CATP system to other syngeneic mouse cancer models, we next evaluated the MC38 colorectal cancer model and demonstrated that the CATP therapy combing mIL-12 and mutant mIL-18 fully suppressed all the tumors (Supplementary Fig. 2a, b). Furthermore, in the CT26 colon cancer model, the CATP therapy with mIL-12 and mutant mIL18 achieved 80% fully tumor regressed rates, outperforming 20% fully tumor regressed rates with CATP mIL-12 alone (Supplementary Fig. 3a, b). There is no significant difference between the body weights of CATP therapy combing mIL-12 and mutant mIL-18 group and the control group (PBS) one week after the dosing (Supplementary Figs. 2c, 3c).
CATP-mediated delivery of mIL-12 and mutant IL-18 in P53 null Kras G12D pancreatic cancer
To assess the efficacy of this approach in genetically driven cancers, mice were subcutaneously inoculated with P53null KrasG12D pancreatic duct cancer26 cells and treated intratumorally. Consistent with previous findings, the CATP combination therapy with mIL-12 and mutant mIL-18 resulted in better inhibition of tumor growth and 80% of mice being fully tumor regressed, compared to 60% with IL-12 alone (Fig. 5a–c). In summary, CATP combining IL-12 and mutant IL-18 demonstrates superior efficacy across multiple cancer models. Moreover, no overt body weight differences were detected between the combination and the vehicle control group (PBS) one week after dosing, although the combination treatment was associated with an initial decrease in body weight within 2–3 days after dosing (Supplementary Figs. 2c, 3c and Fig. 5c).

Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with cancer cells as indicated. Seven days post inoculation, the tumor was intratumorally treated with the LNP encapsulated with mRNA as indicated, with PBS (Control group of basal line), or LNP encapsulating 5 µg SamRNA encoding with mouse IL-12 (IL-12), 5 µg SamRNA encoding with firefly Luciferase, and 1 µg modified mRNA encoding capsids/envelops from SFV4 (Control group of treated group); or LNP encapsulating 5 µg SamRNA encoding with mouse IL-12 (IL-12), 5 µg SamRNA encoding with mutant IL-18 (mtIL-18), and 1 µg modified mRNA encoding capsids/envelops from SFV4. Results are shown as: Tumor areas (Y-axis), Survival rates (Y-axis), and Body weight changes (Y-axis) versus days post KPC (P53null KRasG12D) pancreatic duct cancer cell inoculation (X-axis) (a–c), respectively. The P-Values labeled in a and b were determined by a two-way ANOVA test or Comparison of Survival Curves (Kaper-myer) test.
Induction of long-term immune memory by CATP therapy against tumor recurrence
To determine whether fully tumor regressed mice treated with the CATP combination therapy developed long-term immune memory in B16F10 and MC38 models, we rechallenged the treated mice with original tumor cells, which more than 75 days. Of note, all fully tumor regressed mice were able to reject the tumor cells, in contrast to the age- and sex-matched naïve mice that developed tumors (Fig. 6a, b and Supplementary Fig. 4a, b). These results suggested that the CATP system induces robust long-term immune memory. To investigate the mechanisms underlying this immune memory, we intratumorally treated B16F10 melanoma with the CATP therapy using mouse IL-12 alone (Fig. 6c). Flow cytometry analysis of tumor draining lymph nodes (TDLNs) and spleen showed that the CATP group significantly (p = 0.0006) increased the number of memory precursor CD8 T cells characterized by CD62L+ CD122+ over the control group in TDLNs despite no significant difference between the two groups in spleens27,28 (Fig. 6d, e and Supplementary Fig. 5). Additionally, no significant differences were observed in CD62L- KLRG1+ cytotoxic CD8 T cells either in TDLN or in spleen (Fig. 6e)29. These findings suggested that the CATP therapy is likely to expand the population of memory precursor CD8 T cells in TDLNs, contributing to long term protection against tumor recurrence.

a, b Re-challenges of fully tumor regressed mice in the group LNP encapsulated 5 µg of SamRNA encoding with mouse mutant IL-18 plus 5 µg of SamRNA encoding mouse IL-12 and 1 µg of modified mRNA encoding with SFV4 capsids/envelop proteins. The treated mice (n = 10) and naïve mice (n = 7, Control group) matched with age and sex and were challenged with 0.1 million B16F10 cells. Results are shown as: Tumor areas (Y-axis) and Survival rates (Y-axis) versus days post B16F10 melanoma cancer cell inoculation (X-axis). c Scheme of tumor inoculation and treatment. d, e Phenotyping of memory precursor of CD8 T cells and cytotoxic CD8 T cells in spleen. Six- to eight-week-old C57BL/6 mice (n = 5 per group, a cage of animal) were subcutaneously inoculated with 1 million B16F10 cells. Seven days post-inoculation, mice received a single intratumorally injection of PBS (Control group of basal line), LNP encapsulated 10 µg of SamRNA encoding with mIL-12 and 1 µg of modified mRNA encoding with luciferase (Control group of treated group) or LNP encapsulated 10 µg SamRNA encoding with mIL-12 and 1 µg of modified mRNA encoding VEE capsids and envelop proteins. On day 7 injections of LNP-mRNA, the mice were sacrificed. Shown are the numerates of the CD8+ CD62L+ CD122+ and CD8+ CD62L- KLRG1+ in draining lymph nodes (DLN) (d) and spleens (e), respectively. The P-Values labeled in a, b, d were determined by a two-way ANOVA test for a Comparison of Survival Curves (Kaper-myer) test for b or a two-way ANOVA test for d.
Discussion
The Cascade Amplification of Therapeutic Payloads (CATP) system presented in this study introduces a transformative approach to addressing the persistent challenge of insufficient therapeutic payload delivery in cancer therapy. Utilizing a dual-amplification mechanism that combines SamRNA with modified mRNA, the CATP system demonstrates remarkable efficiency in amplifying therapeutic proteins both in vitro and in vivo. This innovative strategy holds significant promise for cancer immunotherapy, enabling higher therapeutic efficacy at a single dosage, thereby mitigating toxicity concerns commonly associated with conventional mRNA and SamRNA therapies.
The B16F10 melanoma mouse model results demonstrated the CATP system’s exceptional capacity to achieve a 525-fold increase in intratumoral IL-12 levels compared to controls. This remarkable amplification underscores the robustness of the CATP platform and its potential to overcome critical limitations of existing gene therapy approaches, which often rely on escalating dosages to achieve therapeutic efficacy. Crucially, the transient nature of CATP-driven IL-12 expression ensures minimal systemic exposure, thereby substantially mitigating the risk of off-target effects and toxicity. This represents a notable advancement over current IL-12 delivery strategies, which have historically been hindered due to systemic toxicity in clinical settings30.
The CATP platform was successfully extended beyond IL-12 to incorporate a combination of IL-12 and mutant IL-18, achieving synergistic anti-tumor effects across multiple cancer models, including B16F10 melanoma, MC38 colorectal cancer, CT26 colon cancer, and P53null KRasG12D pancreatic ductal cancer. The incorporation of mutant IL-18, which resists inhibition by IL-18BP, exemplifies the platform’s adaptability and its potential for rational therapeutic payload design to maximize efficacy. Notably, the 80% fully tumor regressed survival rate observed across diverse mouse tumor models highlights the broad applicability of the CATP platform, positioning it as a versatile and powerful tool in cancer immunotherapy.
The findings further indicate that the therapeutic efficacy of the CATP system is significantly influenced by the selection of viral capsid/envelop protein. For instance, constructs utilizing capsids derived from SFV4 exhibited superior tumor regression and survival outcomes compared to those derived employing capsids from VEE or Sindbis virus. These results underscore the importance of optimizing viral components to enhance the efficiency and therapeutic potential of the CATP system, offering a promising avenue for further development.
An additional compelling aspect of the CATP system is its capacity to induce robust long-term immune memory, evidenced by the successful rejection of tumor rechallenges in treated mice. This phenomenon is attributed to the expansion of memory precursor CD8 T cells in tumor-draining lymph nodes, a key feature that may provide durable protection against tumor recurrence. This immunological advantage further strengthens the therapeutic potential of CATP system, positioning it as a long-term prevention of relapse.
The further compelling aspect of the CATP system lies in its capability to achieve potent therapeutic efficacies through the combination of IL-12 and mutant IL-18 without the need for immune checkpoint blockade (ICB) therapies, such as anti-PD1 or anti-PD-L1, as utilized in clinical trials NCT03946800 and NCT03871348. This distinction is critical, as ICB therapies may limit the capacity of rejuvenated cells to generate long-lived antitumor immunity following tumor clearance31. Additionally, CATP treatment requires only a single administration, unlike alternative approaches such as those in NCT0387134814, which involve multiple injections. These repeated treatments risk continuous antigen stimulations and subsequent exhaustion of tumor specific CD8 T cells32.
In this work, the CATP system was administrated intratumorally.The advantages of intratumourally injections with therapeutic agents (e.g., CATP, LNP-mRNA encoding with therapeutic) could enhance antitumour immune responses, reduce the risk of systemic toxicities, and achieve higher local bioactive drug concentrations, which was widely validated in preclinical and clinical practices12,17. The intatumorally injection is powerful to be used for those tumors easier to reach, such as melanoma, breast cancer, head-neck cancer, colon cancers et al. However, intatumorally injection is very difficult to touch those deeper tumors, limiting its applications.
Interestingly, the CD62L+ CD122+ population in tumor draining lymph nodes (TDLN) were increased in the CATP group (Fig. 6d). The CD62L is highly expressed on the naïve CD8 T cells and memory CD8 T cells and CD62L on most of the CD8 T cells are decreased after CD8 T cells activations. The CD122+ CD8 T cells are considered as a new regulator with central memory phenotypes highly expressing CD62L28. The increased CD62L+ CD122+ populations in the CATP group in TDLN are likely the memory precursors against the tumor antigens. Other CD62L+ CD122+ populations in the PBS group in TDLN are likely the memory precursors against various other antigens. In consistent with such kind of hypothesis, the CD62L+ CD122+ populations in various groups in spleen are comparable (Fig. 6e). The further evidence is the activated CD8 T cells are comparable in different groups either in TDLN or spleen ((Fig. 6d, e).
In conclusion, the CATP system represents a groundbreaking advancement in cancer therapy, effectively addressing critical challenges in payload delivery and therapeutic efficacy. Its innovative dual-amplification strategy, combined with its safety and adaptability, establishes a transformative platform for the development of next-generation cancer immunotherapies. By enabling robustly higher payload expression without exacerbating systemic toxicity, the CATP platform holds significant potential to reshape clinical practice, revolutionize the treatment landscape across a wide range of malignancies, and promote durable immune memory for sustained therapeutic benefits.
Materials and methods
Constructs, in vitro transcription, capping/methylating for self-amplifying mRNA (SamRNA) and modified mRNA
VEE SamRNA plasmid DNA was de novo synthesized based on the viral sequences of the VEE-TC-83 strain (Genebank ID: L01443.1) and prepared based on the constructs previously developed in the WO2023220693A1, “Synthetic SamRNA Molecules with Secretion Antigen and Immunomodulator”. Modified mRNA plasmid DNA was prepared based on the constructs previously developed in the PCT/US2023/085919, “Compositions and Methods for Delivering Molecules”.
eGFP, firefly luciferase, mouse IL-12, and mouse IL-18 were cloned into XbaI and ClaI after the subgenomic promoter of VEE SamRNA plasmid DNA. Capsids/envelops from Venezuelan equine encephalitis virus (VEE), Sindbis virus, and Semliki Forest virus (SFV) 4 were cloned after T7 promoter using seamless cloning technology.
SamRNA were in vitro transcribed (IVT) from the templates of linearized VEE DNA constructs using the NEB RNA synthesize kit (Catalog No. E2050S). Then the synthesized mRNAs were capped and methylated by Cellscript kits (Catalog No. C-SCCS1710). Modified mRNAs were in vitro transcribed (IVT) from the templates of linearized VEE DNA constructs using the NEB RNA synthesize kit (Catalog No. E2040S) plus pseudouridine (Catalog No. N-1019) and Cleancap (Catalog No. N-7413) from Trilink. The quantity and purity of SamRNA and modified mRNA were assessed by Nanodrop and gel electrophoresis.
Formulations of lipid nanoparticles and encapsulation of SamRNA and modified mRNA
P6 ionizable lipid was designed and synthesized as in the patent (WO2025010420A2). Delivery efficiency of P6-LNP-SamRNA (EGFP) was evaluated by 293 cells and the formulations were optimized using an orthogonal experimental design. As illustrated and data in Supplementary Fig. 6a–f, four concentration levels were evaluated for each of the four key components: P6, DOPE, cholesterol, and DMG-PEG2000. This approach enabled systematic assessment of individual and interactive effects using only 16 experimental conditions, representing a significant reduction from the 256 possible combinations in a full factorial design.The predicted optimal formulation was identified as P6 /DOPE /Chol /DMG -PEG2000 = 30/15/50/1.5 (referred to as “Opt”). The Opt formulation was subsequently evaluated based on its EGFP expression level in HEK293 cells (Supplementary Fig. 6b), demonstrating the highest delivery efficiency among all tested conditions.
An ethanol phase was prepared by dissolving P6 (Supplementary Fig. 7a), DOPE (Catalog No. 870341, Avanti Research), cholesterol (Catalog No. 7001, Avanti Research), and DMG-PEG2000 (Catalog No. 880151, Avanti Research) at a predetermined molar ratio of 30:15:50:1.5 for P6-LNPs and SM102 (Catalog No. HH119909, Arctom Scientific), DSPC (Catalog No. 850333, Avanti Research), cholesterol (Catalog No. 7001, Avanti Research) and DMG-PEG2000 (Catalog No. 880151, Avanti Research) at a molar ratio of 50:10:38.5:1.5 for SM102-LNPs at finally total lipids concentration is at 10 mg/ml. The aqueous phase was prepared in 50 mM citrate buffer (pH 4.5, AAJ60024AK, Fisher Scientific) containing SamRNA and/or mRNA. All mRNA samples were stored at −80 °C and thawed on ice prior to use. The aqueous and ethanol phases were combined at a 3:1 ratio with a lipid-to-RNA (N/P) ratio of 4.2 for P6-LNPs and 6 for SM102-LNPs, respectively, using a microfluidic chip device (INano™ L system, Micro & Nano) at a flow rate of 12 mL/min. The resulting lipid nanoparticles (LNPs) were dialyzed against 1X PBS (MT21040CMX, Fisher Scientific) using a Slide-A-Lyzer™ MINI Dialysis Devices, 20 K MWCO (Catalog No. 88405 Fisher Scientific) at 25 °C for 80 min and stored at 4 °C prior to injection.
Characterization of ionizable lipid N-(2-(cyclohex-1-en-1-ylamino)-1-(1-ethylpiperidin-4-yl)-2-oxoethyl)-N-(heptadecan-9-yl)palmitamide (P6)
QTOF MS (ESI): m/z calculated for C48H92N3O2+ (M + H+), 742.7185; found, 742.7156. 1H NMR (400 MHz, CDCl3) δ 9.47 (s, 1H), 6.07 (s, 1H), 3.55 (s, 1H), 3.15 (d, J = 8.0 Hz, 1H), 3.08 (s, 2H), 2.73 (s, 1H), 2.52 (s, 2H), 2.36–2.31 (m, 2H), 2.07–2.04 (m, 6H), 1.83–1.79 (m, 2H), 1.66–1.15 (m, 60H), 0.89–0.79 (m, 12H). 13C NMR (400 MHz, CDCl3) δ 175.97, 170.26, 132.75, 112.15, 60.44, 52.81, 52.67, 52.48, 35.05, 34.06, 32.29, 32.01, 31.90, 29.85, 29.79, 29.78, 29.74, 29.69, 29.57, 29.53, 29.44, 29.37, 29.28, 28.16, 27.29, 27.10, 25.45, 24.05, 22.77, 22.74, 22.72, 22.61, 22.18, 14.19, 14.18.
LNP characterization
The size, polydispersity index and zeta potentials of LNPs were measured using dynamic light scattering (Z-100-Z2(MTS), HORIBA, Ltd.). Diameters are reported as the intensity mean peak average. To calculate the nucleic acid encapsulation efficiency, a modified Quant-iT RiboGreen RNA assay (Invitrogen) was used. Results of encapsulation efficiency can be found in the Supplementary Fig. 7b.
Stability evaluation of P6-LNPs
The formulated LNPs (P6-LNP-SamRNA (eGFP) and SM102-LNP-SamRNA (eGFP)) were buffer changed with Tris HCl buffer (20 mM, pH 7.5, 8.5% sucrose w/w) and stored at 4 °C. The in vitro transfection efficiency of the LNPs were evaluated using HEK293 after 2, 6, 10 and 20 weeks (Supplementary Fig. 8).
To address the in vivo stability and the resistances of RNase, the formulated LNPs, P6-LNP-SamRNA (Luciferase) and P6-LNP-modified mRNA (Luciferase) were administrated by intramuscularly or intravenously. The images of bioluminescence were measured by IVIS (In vitro imaging system) (Supplementary Fig. 9).
To address the effects of the serum to the formulated LNPs, fetal bovine serum was filtered through 0.22 mm filter and the flowthrough was incubated with LNPs for two hours. Then the nanoparticles size was measured (Supplementary Fig. 10).
Transfections of lipid nanoparticles in vitro and in vivo
For in vitro transfection, the cells were plated at 30% confluence at day 0. The formulated nanoparticles containing 100 ng mRNA were added to 100 μl of medium in a well of a 96-well plate at day 1. Then the transfected cells were followed with analysis accordingly.
Cell lines and animals
Cell lines HEK293 (ATCC CRL-1573), B16F10 (ATCC CRL-6475), CT26 (ATCC CRL-2638), were cultured following vendor instructions. MC38 and KPC (P53null KRasG12D) cells were prepared in the Li lab at the University of Michigan. All the cells into mice were performed with pathogen tests by third party as Mispro Biotech Service Corporation requires. The identity of cells into mice were determined by morphological records and tumor growth rates which were described by providers. Female C57BL/6 (Charles River Lab), Balb/C (Charles River Lab) mice at 6–8 weeks of age were purchased and maintained in the animal facility at the Mispro Biotech Service Corporation, Alewife, Massachusetts, USA. We have complied with all relevant ethical regulations for animal use. All animal studies and procedures were carried out following federal, state and local guidelines under an institutional animal care and use committee (IACUC)-approved animal protocols (2022-SUN-01, and 2024-SUN-02) by the Committee of Animal Care at Mispro Biotech Service Corporation. The IACUC of the Mispro Biotech Service Corporation approved all animal experiments and all animal experiments were conducted at the Mispro Biotech Service Corporation.
Tumor inoculation, tumor therapy, and rechallenge. One million B16F10, MC38, CT26 or KPC (P53null KRasG12D) cells in 50 μl of sterile PBS were s.c. injected into the flank of mice. At 7 days later when tumors reached ~50 mm2 in size, animals were injected intratumorally with PBS (control) or LNPs as described in figure legends in 50 μl of PBS as indicated. For the rechallenge, 0.1 million B16F10 or 0.2 million MC38 cancer cells were subcutaneously (s.c.) injected into another side flank of fully tumor regressed mice or naïve mice.
The tumor areas were used for tumor growth regression as the statistic differences observed by tumor area was more reliable though with lower sensitivity than by tumor volume33. The tumor area was determined by the width x length which was measured by caliper. The maximal tumor volumes that were permitted by the IACUC of Mispro Biotech Service Corporation were not exceeded in any of the animal experiments. All the tumor mice were followed with the endpoints (e.g., tumor size no more than 20 mm in diameter, or body condition score (BCS < 2), or weight loss >20% etc.) which were described in the IACUC animal protocol approved (2022-SUN-01, and 2024-SUN-02).
Antibodies, staining and FACS analysis
Antibodies against mouse CD4 (Catalog No. 100412), CD8 (Catalog No. 100766), CD3e (Catalog No. 155612), CD62L (Catalog No. 104406), CD122 (Catalog No. 123216), KLRG1 (Catalog No. 138418), 7-AAD (Catalog No. 420404), and Zombie Aqua (Catalog No. 423102) were bought from Biolegend. All the antibodies were diluted 1:50. The live/dead dye Zombie Aqua was diluted 1:300. The single-cell suspensions were filtered by 70-μm nylon strainers and stained as described34. Stained samples were analyzed using a Symphony A5 FACS analyzer from BD Biosciences. All flow cytometry data were analyzed using FlowJo software (Flowjo LLC).
ELISA analysis
Tumors were collected and ground in tissue protein extraction reagent (T-PERTM, Thermo Fisher Scientific, cat. no. 78510) in the presence of 1% proteinase and phosphatase inhibitors (Thermo Fisher Scientific, cat. no. 78442). The lysates were incubated at 4 °C for 30 min with slow rotation then centrifuged to remove debris. The supernatants were transferred to a clean tube for ELISA or Luminex analysis. Mouse IL-12 in tumor tissue supernatants or in serum were measured by ELISA kits from R&D (Catalog No. DY419) following the manufacturer’s instructions.
Plots
Plots were drawn by Prism and the illustrations Figs. 1a and 3d were created in BioRender. Li, Y. (2025) https://BioRender.com/g50p020” and https://BioRender.com/bguaszo”, respectively.
Statistics and reproducibility
Data were statistically analyzed by one-way or two-way ANOVA or by Student’s t-test using GraphPad PRISM as indicated. Animals were randomized to treatment groups once the mean tumor area around 50 mm2 was reached by the tumor-inoculated cohort. No data were excluded from the analyses. The investigators were not blinded to allocation during experiments and outcome assessments. The samples sizes for in vitro analysis were three (triplicates) and for in vivo analysis are as annotated in figure legends. The details of statistical analysis for figures and Extended Data Figures are included in the Source Data files. The sample size in animal studies were determined by “E = Total number of animals—Total number of groups” which lies in 10–20 that’s enough for ANOVA analysis35. The allocations of experimental units to control and treatment groups are random but not blindly by selection of cages for treatments as indicated.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data behind the graphs in the paper can be found in Supplementary Data 1 and other data (e.g., synthesis of lipids and plasmids) are available from the corresponding authors upon reasonable request, maybe under non-disclosure agreement (NDA) or material transferring agreement (MTA).
References
Bloom, K., van den Berg, F. & Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 28, 117–129 (2021).
Comes, J. D. G., Pijlman, G. P. & Hick, T. A. H. Rise of the RNA machines—self-amplification in mRNA vaccine design. Trends Biotechnol. 41, 1417–1429 (2023).
Kariko, K., Ni, H., Capodici, J., Lamphier, M. & Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279, 12542–12550 (2004).
Teijaro, J. R. & Farber, D. L. COVID-19 vaccines: modes of immune activation and future challenges. Nat. Rev. Immunol. 21, 195–197 (2021).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Schuberth-Wagner, C. et al. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2’O-methylated self RNA. Immunity 43, 41–51 (2015).
Dias Junior, A. G., Sampaio, N. G. & Rehwinkel, J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol 27, 75–85 (2019).
Andries, O. et al. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Release 217, 337–344 (2015).
Wang, J. et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res. 47, e130 (2019).
Amer, S. A. et al. Exploring the reported adverse effects of COVID-19 vaccines among vaccinated Arab populations: a multi-national survey study. Sci. Rep. 14, 4785 (2024).
Bitounis, D., Jacquinet, E., Rogers, M. A. & Amiji, M. M. Strategies to reduce the risks of mRNA drug and vaccine toxicity. Nat. Rev. Drug Discov. 23, 281–300 (2024).
Li, Y. et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 1, 882–893 (2020).
Hewitt, S. L. et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin. Cancer Res 26, 6284–6298 (2020).
Hotz, C. et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci. Transl. Med. 13, eabc7804 (2021).
Vignuzzi, M. & Lopez, C. B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol 4, 1075–1087 (2019).
Overwijk, W. W. & Restifo, N. P. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. Chapter 20, Unit 20 21 https://doi.org/10.1002/0471142735.im2001s39 (2001).
Melero, I., Castanon, E., Alvarez, M., Champiat, S. & Marabelle, A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 18, 558–576 (2021).
Lasek, W., Zagozdzon, R. & Jakobisiak, M. Interleukin 12: still a promising candidate for tumor immunotherapy. Cancer Immunol. Immunother. 63, 419–435 (2014).
Wang, J., Saffold, S., Cao, X., Krauss, J. & Chen, W. Eliciting T cell immunity against poorly immunogenic tumors by immunization with dendritic cell-tumor fusion vaccines. J. Immunol. 161, 5516–5524 (1998).
Jose, J., Snyder, J. E. & Kuhn, R. J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4, 837–856 (2009).
Ramachandran, M. et al. Safe and effective treatment of experimental neuroblastoma and glioblastoma using systemically delivered triple MicroRNA-detargeted oncolytic Semliki Forest virus. Clin. Cancer Res 23, 1519–1530 (2017).
Ihim, S. A. et al. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front Immunol. 13, 919973 (2022).
Lucey, D. R., Clerici, M. & Shearer, G. M. Type 1 and type 2 cytokine dysregulation in human infectious, neoplastic, and inflammatory diseases. Clin. Microbiol. Rev. 9, 532–562 (1996).
Zhou, T. et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614 (2020).
Kim, S. H. et al. Site-specific mutations in the mature form of human IL-18 with enhanced biological activity and decreased neutralization by IL-18 binding protein. Proc. Natl. Acad. Sci. USA 98, 3304–3309 (2001).
Hingorani, S. R. et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005).
Yang, S., Liu, F., Wang, Q. J., Rosenberg, S. A. & Morgan, R. A. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS One 6, e22560 (2011).
Liu, J., Chen, D., Nie, G. D. & Dai, Z. CD8(+)CD122(+) T-cells: a newly emerging regulator with central memory cell phenotypes. Front Immunol. 6, 494 (2015).
Herndler-Brandstetter, D. et al. KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective. Immun. Immun. 48, 716–729 e718 (2018).
Momin, N. et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med. 11 https://doi.org/10.1126/scitranslmed.aaw2614 (2019).
Ghoneim, H. E., Zamora, A. E., Thomas, P. G. & Youngblood, B. A. Cell-intrinsic barriers of T cell-based immunotherapy. Trends Mol. Med. 22, 1000–1011 (2016).
Yi, J. S., Cox, M. A. & Zajac, A. J. T-cell exhaustion: characteristics, causes and conversion. Immunology 129, 474–481 (2010).
Brough, D., Amos, H., Turley, K. & Murkin, J. Trends in subcutaneous tumour height and impact on measurement accuracy. Cancer Inf. 22, 11769351231165181 (2023).
Li, Y. et al. Persistent antigen and prolonged AKT-mTORC1 activation underlie memory CD8 T cell impairment in the absence of CD4 T cells. J. Immunol. 195, 1591–1598 (2015).
Charan, J. & Kantharia, N. D. How to calculate sample size in animal studies. J. Pharm. Pharmacother. 4, 303–306 (2013).
Acknowledgements
We’d like to thank lots of persons for their generous helps to make this work happened, especially to thank Claire Johnson and Ana Lopez from Mispro Biotech Service Corporation for their kindly helps on drafting animal protocols and mouse maintenance, respectively. We’d like to thank our seeding investors for their continuous funding support and many advice. We’d like to thank Tom Shea and his team from Xentria for many thoughtful discussions on this work.
Author information
Authors and Affiliations
Contributions
Y.L. conceptualized, designed the experiments and L.Z. designed the ionizable lipid and LNP formulation. Y.L., J.W., N.L., M.W., and L.Z., carried out the experiments. J.L. and Z.W. prepared MC38 colon cancer and P53null KRasG12D pancreatic duct cancer cell lines. Y.L., L.Z., and J.W. organized and drafted the manuscript. J.L. edited the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.L. and L.Z. are inventors patent of WO2025010420A2 “Compositions and methods for delivering molecules”, which invented the mRNA delivery ionizable lipid P6 for this study; patent of WO2023220693A1 “Synthetic self-amplifying mRNA molecules with secretion antigen and immunomodulator”, which invented one of the self-amplifying mRNA for this study; patent of Methods for cascade amplifications of PCT/US2025/019051 “Therapeutic payloads (CATP) & compositions for cancer immunotherapies and gene therapy” for this study. The other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Rakesh Kumar Tekade and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Huan Bao and Johannes Stortz. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Wu, J., Li, N. et al. Cascade amplification of therapeutic payloads for cancer immunotherapy. Commun Biol 8, 1277 (2025). https://doi.org/10.1038/s42003-025-08735-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08735-z
