
Abstract
Ejaculated mammalian sperm must undergo a series of biochemical changes called capacitation to gain fertilizing competence. Mounting evidence on protein synthesis during capacitation contradicts the widely accepted dogma of translational quiescence in sperm. However, mechanisms regulating mRNA translation in sperm is ambiguous, necessitating elucidation and understanding its role in enabling fertilizing competence. Here, we perform proteome analysis from bovine sperm and identify proteins involved in translation, encompassing initiation and elongation factors, ribosomal proteins, tRNA synthetase, ligase, and RNA-binding proteins (RBPs) involved in mRNA export, degradation, and binding. We further explore the mRNA-binding activity of RBPs during capacitation, identifying 48 RBPs; 13 and 8 RBPs were exclusive to fresh-uncapacitated and capacitation groups, respectively, with an overlap of 27 RBPs present in both groups. Interestingly, cytoskeletal proteins and metabolic enzymes associate differentially with mRNAs during capacitation. Since phosphorylation is a known regulatory mechanism dynamically modulating RBPs’ interactions with mRNAs, we performed sperm phosphoproteome analysis, revealing few RBPs to phosphorylate during capacitation. These observations suggest that RNA-binding functions of these proteins are coupled with capacitation-associated phosphorylation events, enabling concomitant protein synthesis and fertilizing competence in sperm. These findings will assist in elucidating translational regulation of mRNA in sperm and advancing our knowledge in regulation of male fertility.
Similar content being viewed by others

Introduction
The past decade has seen a surge of interest in improving male fertility across species. Male factors contribute to approximately half of infertility cases in humans1, with unexplained infertility affecting one out of three couples, primarily attributed to these factors2. The clinical values of conventional semen analysis, such as sperm motility, concentration, and morphology, are inadequate in diagnosing male infertility as they do not account for submicroscopic or molecular-level differences in sperm. For an improved evidence-based diagnosis and infertility treatment, a better understanding of molecular mechanisms governing male fertility is necessary. Similarly, male fertility is crucial in animal breeding systems as cryopreserved semen from an elite male is distributed worldwide to breed numerous females through artificial insemination. Propagation of germplasm from sub-fertile bulls can reduce conception rates and calf birth and result in economic loss. To understand the factors enabling sperm fertilizing potential, we use a bovine model that enables the use of semen from bulls with known fertility status (availability of fertility data from thousands of inseminations). It allows correlation analysis between sperm characteristics and fertility with potential implications across different species.
A long-standing belief in translational quiescence in sperm has impeded investigating de novo protein synthesis in sperm and its potential impact on male fertility. Ejaculated mammalian sperm were considered translationally quiescent due to inherently fragmented 28S/18S ribosomal RNA across species3 and the exclusion of most cytoplasm during spermatogenesis4. Consequently, existing sperm proteins and their post-translational modifications (PTMs) were believed to regulate sperm functions during fertilization. Contrary to the dogma of translational quiescence in sperm, our previous work demonstrated de novo protein synthesis in bull sperm5 during capacitation5. We also demonstrated an increase in the content and activity of testis-specific isoform of Na+/K+-ATPase (ATP1A4) during capacitation, which was inhibited in the presence of chloramphenicol, implicating mitochondrial translation5. Other studies also demonstrated protein synthesis in sperm across species through the incorporation of [3H]-amino acids6,7, [35S]-methionine and [35S]-cysteine8, and puromycin (SUnSET assay)9 into newly synthesized sperm proteins. However, none of the studies have explored post-transcriptional gene regulation in sperm, an area that could revolutionize the diagnosis and management of male-factor infertility.
RNA-binding proteins (RBPs) and their PTMs are well known to modulate the translation of stage-specific mRNAs and control the differentiation of primordial germ cells to sperm during spermatogenesis10. Since protein phosphorylation is a hallmark of sperm capacitation across species11, we hypothesized that capacitation alters the interaction of RBPs with mRNAs in a phosphorylation-associated manner, releasing them from translational repression and resuming de novo protein synthesis. We report here, for the first time, a comprehensive assessment of the bovine sperm proteome in identifying translational machinery and investigating the dynamic interaction of RBPs and mRNAs during bovine sperm capacitation. Sperm proteomics identified several RBPs known for their role in translational regulation and their physiological interaction with mRNAs was further investigated during capacitation. Several metabolic enzymes and cytoskeletal proteins were found interacting with mRNAs in a phosphorylation-associated manner during capacitation, revealing their moonlighting effects as RBPs. Several of these proteins were previously reported to correlate with bull fertility. Altogether, our study comprehensively investigated the sperm proteome associated with mRNA translation and demonstrated the phosphorylation-associated interaction of RBPs with mRNAs during bovine sperm capacitation.
Results
Confirmation of capacitation status based on sperm kinematics and protein tyrosine phosphorylation
Total motile and progressively motile sperm did not differ between the control (C4) and CAP (heparin as a capacitation-inducing agent) groups after 4 h of incubation. However, the amplitude of lateral head displacement (ALH) and curvilinear velocity (VCL) increased (p ≤ 0.05) in CAP compared to the control group indicating sperm hyperactivation (Table 1). Sperm capacitation status was further confirmed by immunoblotting for protein tyrosine phosphorylation. The signal intensity of 4 tyrosine phosphorylated protein bands (indicated by arrows in Fig. 1) increased (p ≤ 0.05) in the CAP group (heparin) compared to the control group, indicating successful induction of capacitation in vitro.

A A representative immunoblot to detect tyrosine phosphorylation in sperm proteins following an in-vitro capacitation assay using heparin (10 µg/ml) as capacitation (CAP) inducing agent. Ouabain (50 nM) was only used here as another capacitation-inducing agent to compare its effects with heparin. All the following experiments in the manuscript strictly use heparin (10 µg/ml) to induce capacitation in vitro. Total sperm protein extracts (from three Holstein bulls) were immunoblotted with an anti-phosphotyrosine antibody (1:10,000; Millipore, clone 4G10®) and reprobed with β-tubulin (1:10,000; lower panel) for equal protein loading. B For signal quantification (data shown for band 1), each lane was normalized to its β-tubulin band intensity. Western blot analysis revealed increased band density following heparin-induced capacitation (mean ± SEM, n = 3, p ≤ 0.05). Numerical data corresponding to signal quantification for band 1 to detect tyrosine phosphorylation are presented in Supplementary Table 3. A similar trend was observed for other bands (marked 2–4).
Identification of protein candidates involved in translation and its regulation during sperm capacitation
To identify the proteins involved in translational machinery in sperm, we performed proteomic analysis of fresh-uncapacitated sperm in four bulls where Scaffold 5 identified a total of 1319 sperm proteins, after excluding contaminants (keratin and albumin). We identified peptide fragments of proteins involved in translation, including initiation and elongation factors, ribosomal proteins, and enzymes such as tRNA synthetase, ligase, deacylase, and hydrolase involved in tRNA aminoacylation (Supplementary Data S1, Sheet 1). Several sperm proteins discovered by mass spectrometry are known to regulate translation in somatic cells through mRNA processing, export, degradation, and binding, which suggests they could regulate mRNA translation during sperm capacitation (Supplementary Data S1, Sheet 2). We further investigated the abundance of these RBPs through the Exponentially Modified Protein Abundance Index (emPAI), which estimates the absolute protein amount in a complex mixture using the number of sequenced peptides per protein12. We concluded these proteins are lowly abundant in sperm proteome with a molecular weight <150 kDa. In our discovery-based approach to acquiring preliminary data, we also chose to report all proteins of interest with the “minimum one peptide” criterion as some of the peptides were likely legitimate and could be missed with the “minimum two peptides” criterion (Supplementary Data S1, Sheet 1). Most of the identified proteins were detected and validated in at least 2 of 4 biological replicates. Next, we aimed to determine if these proteins are just remnants from spermatogenesis or physiologically interact with sperm RNAs. Therefore, we captured mRNA interactome using RNA antisense purification coupled with mass spectrometry (RAP-MS) (workflow in Fig. 2).

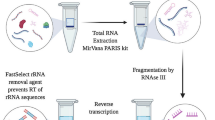
(1) Briefly, the physiological RBP-mRNA interaction was stabilized in vivo using UV crosslinking. (2) Sperm were lysed to release mRNA-RBP complex in the lysate (3) which was pulled down using streptavidin bead-conjugated biotinylated oligo (dT) probes hybridizing to poly A RNAs. (4) mRNA-RBP complex was eluted from beads by heat treatment. (5) The mRNA-RBP complex was treated with proteinase K to facilitate the recovery of mRNAs, which were then analyzed for nucleotide size distribution. The results were compared to total sperm RNA using an electrophoretic profile in a Bioanalyzer. Benzonase treatment digested mRNAs and recovered RBPs were visualized on a polyacrylamide gel using silver staining and identified using LC-MS/MS. Created in BioRender. Tiwari, S. (2025) https://BioRender.com/i9n9awi.
Considerations while adapting mRNA interactome capture in bovine sperm
We performed RAP-MS and investigated RBPs physiologically interacting with mRNAs.
To secure in vivo mRNA-RBP interaction, we crosslinked sperm with UV irradiation and paraformaldehyde (PFA) parallelly. PFA-treated sperm (various times and concentrations) failed to release adequate total RNA in sperm lysate. Owing to the PFA-mediated promiscuous crosslinking and challenges, UV irradiation was used for further experiments. The known low-crosslinking efficiency of UV irradiation was compensated by using high sperm quantity (5–6 million sperm).
High UV dosages can result in a general loss of proteins13. Therefore, we compared protein and RNA profiles of sperm irradiated with UV doses of 150–600 mJ/cm2 (treatment) to those without irradiation (control). Both groups exhibited similar protein and RNA profiles as indicated by Coomassie Blue staining, immunoblotting (Fig. 3A, B), and electrophoretic bioanalyzer analysis (Fig. 3C), respectively, suggesting no UV-induced degradation. Since the sperm head contains condensed paternal DNA, we avoided sperm head decondensation and therefore co-purification of DNA-binding proteins by performing sperm lysis at 4 °C for 1 h. Lysis beyond 1 h of incubation did not retrieve more sperm RNA. Sperm head intactness, confirmed by microscopic examination following the lysis, suggested an absence of genomic DNA (Fig. 3D). This was confirmed by the amplification of sperm cDNA (synthesized without prior DNase treatment) using intron-spanning PRM1 primer yielding a 285 bp PCR product (sperm genomic DNA yields a 376 bp PCR product, Fig. 3E). PCR using somatic-cell-specific genes observed no amplification, confirming the absence of somatic-cell contamination (Fig. 3F). Furthermore, bioanalyzer profile of sperm RNA had no additional peaks running above 6000 nt, suggesting an absence of genomic DNA (Fig. 3G). Bead elution at 80 °C for 3 min yielded maximum recovery in the supernatant while maintaining RNA integrity.

A Sperm irradiated with a range of UV dosages had similar band intensity on Coomassie Blue staining or B immunoblotting for β-tubulin compared to control (no UV irradiation; equal protein loading in all groups); C Similarly, the Bioanalyzer profile of RNA was similar across UV dosages (representative image, upper panel) and control group (lower panel). The dissimilar area under the curve between the treatment and control groups corresponds to different RNA inputs in the Bioanalyzer system. D During sperm lysis, the microscopic evaluation revealed that the sperm head maintained a condensed state, indicating no gDNA release. E To confirm, cDNA (synthesized without DNase treatment) amplified intron-spanning PRM1 primer yielding a 285 bp PCR product (sperm genomic DNA yields 376 bp PCR product) with no amplification in No-RT control, concluding the absence of DNA contamination (original uncropped/unedited agarose gel images). F Total RNA (in sperm lysate) did not amplify any somatic-cell-specific genes confirming the absence of somatic cell contamination (original uncropped/unedited agarose gel images). G Total sperm RNA following lysis did not reveal any peaks >6000 nucleotides in the Bioanalyzer, which also indicates no gDNA contamination during mRNA interactome capture.
Metabolic enzymes and cytoskeletal proteins associate with mRNAs during bovine sperm capacitation
The mRNAs and associated RBPs were enriched in fresh-uncapacitated (C0) and heparin-induced capacitated (CAP) sperm with RAP-MS using biotinylated oligo (dT) probes in 3 independent biological replicates. The oligo (dT) conjugated streptavidin beads enriched poly-A RNAs 500–6000 nucleotides in size (Fig. 4A) from total sperm RNA (Fig. 4B) in the C0 and CAP group whereas bead control (BC; unconjugated streptavidin beads) did not associate with any RNAs (Fig. 4C). Enrichment of proteins in the C0 and CAP over bead control group was confirmed using silver-stained SDS-PAGE gels (Fig. 4D) and validated for hexokinase-1 using immunoblotting (Fig. 4E). A very faint RBP profile in the C0 group compared to the CAP group (Fig. 4D and Supplementary Fig. 1) in silver-stained gels could indicate a lesser abundance of RBPs bound to the poly-A RNAs or may also indicate the differences in RBPs’ extractability between fresh-uncapacitated (C0) and capacitated (CAP) sperm cohorts.

A Oligo (dT) beads enriched poly-A RNAs ranging from 500 to 6000 nucleotides in size from total RNA. B Total RNA isolated from bovine sperm before enrichment. C No RNA was associated with unconjugated streptavidin beads (bead control, BC). The peak at 25 nt corresponds to the internal standard (lower marker) to align the ladder data with samples to determine sizing. D Poly-A RNA-associated proteins run on a 3–10% gradient gel and visualized by silver staining showed distinct band patterns in the CAP group compared to C0 and BC groups (E). Western blot analysis (n = 3) validated the presence of one of the RBPs, hexokinase 1 (~103 kDa), in poly-A–associated proteins using an anti-hexokinase 1 antibody.
Through Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS), altogether 48 RBPs were identified across three biological replicates, where 13 and 8 RBPs were exclusive to the C0 and CAP groups, respectively, with an overlap of 27 RBPs present in both groups (Fig. 5A; Supplementary Data S2, Sheet 1). Minimum two peptides were identified for these RBPs, in at least two out of three biological replicates, excluding proteins that were non-specifically bound to the beads. Several proteins did not pass these selection parameters but were exclusively present in C0 or CAP groups or both. As these could be important RBPs with a role in translational regulation during bovine sperm capacitation, they were classified into another set called the “candidate RBPs” (Supplementary Data S2, Sheet 2). emPAI analysis for RBPs revealed their moderate abundance in the sperm proteome and <100 kDa molecular weight (Fig. 5B).

A Venn diagram representing C0- and CAP-group (heparin) specific and overlapping RBPs (Supplementary Data S2). B Scatter dot blot representing RBP’s absolute concentration (magenta color dots) in sperm proteome (black color dots excluding RBPs). Representative top 10 pathway enrichment analysis (KEGG; C) and functional enrichment analysis for RBPs (D: Biological processes; E: Cellular components and F: Molecular functions).
The top 20 enriched functional analyses of RBPs revealed various biological processes (Fig. 5D), mainly glycerol-3-phosphate metabolism, tricarboxylic acid cycle, flagellated sperm motility, and reproductive functions such as sperm-oocyte binding and fertilization. The identified RBPs were a component of sperm mid-piece, principal piece, actin cytoskeleton, and acrosomal matrix (Fig. 5E). The RBPs were involved in molecular functions (Fig. 5F) such as glycerol kinase activity, actin filament binding, calcium ion binding, and nucleotide binding. Pathway enrichment analysis identified RBPs involved in the citrate cycle (TCA cycle), oxidative phosphorylation, PPAR signaling pathway, complement and coagulation cascades and muscle contraction (Fig. 5C).
RBPs’ association with mRNAs during capacitation is phosphorylation-associated
The PTMs of RBPs are well-reported to regulate translation in somatic cells. Since phosphorylation is a hallmark of sperm capacitation across species, we hypothesized that RBPs differentially bind to mRNAs during capacitation in a phosphorylation-dependent manner. We performed phosphopeptide enrichment and identified phosphopeptides for a total of 304 proteins in the C0 and CAP (heparin) group (Supplementary Data S3, Sheet 1). A total of 10 RBPs (among 48 RBPs) were phosphorylated meeting the “minimum two phosphopeptides” criteria with the presence in two of three biological replicates (peptide threshold—95%, protein threshold—99%, Table 2). Interestingly, one RBP was exclusively phosphorylated in the CAP group and four RBPs were differentially phosphorylated during capacitation (fold change cutoff: ≤0.8 or ≥1.2, p ≤ 0.05, false detection rate ≤0.01). Due to technical limitations with LC-MS/MS, certain RBPs (among 48 RBPs) that do not meet the specified criteria (minimum two phosphopeptides) may be excluded. As a result, we have included these RBPs in our supplementary data for comprehensive reporting (Supplementary Data S3, Sheet 2).
Few RBPs are differentially expressed during capacitation
In an independent experiment, we explored whether RBPs in mRNA interactome show differential expression during heparin-induced bovine sperm capacitation. A similar band pattern of total sperm protein extracts in the C0 and CAP groups on a Coomassie blue-stained gel suggested that capacitation does not affect sperm protein extractability, which is within the detection limit of this method (Supplementary Fig. 2). LC-MS/MS identified differential expression of 70 proteins, where 60 proteins were upregulated, and 10 proteins were downregulated in the CAP group compared to the C0 (minimum two peptides’ criteria with the presence in two of three biological replicates, fold change cutoff: ≤0.8 or ≥1.2, p ≤ 0.05, false detection rate ≤0.01) (Supplementary Data S4). Interestingly, one RBP (Fibrous Sheath CABYR-binding Protein) was upregulated while three (Hexokinase, Zonadhesin, and NADH-cytochrome b5 reductase) were downregulated during capacitation. However, it is possible that during sperm capacitation, protein-dense structures such as the perinuclear theca, mitochondrial and fibrous sheath, and outer dense fibers undergo morphological changes as a preparative step for post-fertilization decondensation. It could alter the solubility and extractability of sperm proteins and could contribute to their differential expression during capacitation.
Discussion
Sperm are highly specialized cells delivering paternal genes, epigenetic signatures14, coding and non-coding RNAs15, centrioles, and proteins such as PLCζ16,17 to an oocyte during fertilization.
Surface-associated decapacitation factors modulate the fertilizing ability of ejaculated sperm. To gain fertilizing potential, they must undergo a species-specific time-dependent series of biophysical and biochemical events in the female reproductive tract called capacitation18. These events include the removal of decapacitation factors and several biochemical changes which lead to phosphorylation of tyrosine residues in several proteins, inducing changes in sperm motility from linear progressive (moving in a relatively straight line) to irregular trajectory with high-amplitude asymmetrical flagellar beating (hyperactivation)18, facilitating sperm-oocyte interaction. Subsequently, sperm undergo an acrosome reaction to penetrate the oocyte vestments before fertilization. In the present study, heparin-induced capacitation (in vitro) was confirmed by increased tyrosine phosphorylation and a change in kinematic parameters (increased ALH and VCL).
Our previous study5, including others6,7,8,9,19,20, provided strong evidence that spermatozoa synthesize proteins de novo. However, it is unknown which factors of translational machinery in sperm are excluded in residual bodies or are retained in ejaculated spermatozoa. The translational machinery comprises the ribosomes, transfer RNAs (tRNAs), aminoacyl-tRNA synthetases, and translation factors21. Additionally, it constitutes other proteins called RNA-binding proteins21 that serve as essential factors in post-transcriptional gene regulation. They regulate RNA splicing, mRNA polyadenylation, subcellular compartmentalization, translation22, and their stabilization or destabilization in response to stress or extracellular signals23. Our proteomic analysis of bovine sperm inferred that ejaculated sperm harbor translational machinery that could be regulated by RBPs for de novo protein synthesis, supporting sperm functions. RBPs identified in sperm, such as YBX2, DAZAP1, CARHSP1, and ANGEL2, are known to regulate spermatid differentiation through mRNA translation24,25 during spermatogenesis. However, other germ cell-specific RBPs (e.g., ADAD1, CSTF2T, TSN aka TB-RBP) were not detected in ejaculated bovine sperm.
We further aimed to investigate the mRNA-binding activity of RBPs during sperm capacitation. We initially performed the orthogonal organic phase separation26; however, we could not adapt this protocol to bovine sperm. Therefore, we used the oligo (dT) affinity purification approach to investigate RBPs physiologically interacting with mRNAs.
RBPs form a firm non-covalent complex with RNA nucleotides using hydrogen bonds, Van der Waals, and hydrophobic interactions27. On UV (254 nm) irradiation of these complexes, RNA nucleotides generate reactive intermediates, creating new covalent bonds with amino acid residues of the bound RBPs28,29. We stalled the in vivo-specific RNA-RBP interaction dynamics with UV crosslinking (without promoting protein-protein crosslinks as in formaldehyde-mediated crosslinking)30. Sperm lysis with SDS yielded total sperm RNA, where biotinylated oligo (dT) probes conjugated with streptavidin beads specifically hybridized to poly A RNAs (500–6000 nucleotides in size) and pulled down the RNA-RBP complex. This approach allows capturing RNA-RBP complexes under stringent denaturing conditions and eliminates noncovalent binders, providing RBPs with high confidence31. We expect that our lysis buffer extracts most sperm proteins, excluding nuclear proteins (absence of head decondensation) and perinuclear theca proteins that require stringent extraction32. Capturing mRNA interactome in sperm is challenging due to low mRNA concentration compared to somatic cells33. UV irradiation crosslinks only 1–5% of RBPs and is biased towards certain amino acid residues, pyrimidines, and single-stranded RNA13, which poses a technical limitation; however, we compensated for these limitations using high (500–600 million) sperm count. Sperm lysis in our protocol maintained the nucleus in a condensed state and it was unclear if any sperm RBPs or poly-A RNAs were associated with it, which could limit detection of RBPs. Furthermore, RBPs associated with histone transcripts were excluded as they lack a poly-A tail34 and could not be captured using oligo (dT) probes. Since sperm are abundant in transcript fragments33,35,36, our study could not differentiate between RBPs associated with full-length and fragmented transcripts, as we did not conduct RNA sequencing of poly-A RNAs. However, we confirmed the presence of intact transcripts for candidate mRNAs in RNA-RBP complexes through PCR (Supplementary Note 1, Supplementary Table 2 and Supplementary Figs. 3 and 4).
Sperm RBPs enriched using an affinity protocol revealed moonlighting effects of metabolic enzymes and cytoskeletal proteins, indicating their multiple functions beyond their primary catalytic and structural role, respectively. Interestingly, RBPs regulating mRNA translation during spermatogenesis (e.g., YBX2, DAZ, TSNAXIP1, etc.) were not detected to associate with sperm mRNAs during capacitation. Perhaps these proteins are remnants from spermatogenesis with no roles in translational regulation in sperm. GO-enrichment analysis revealed that sperm RBPs predominantly participate in metabolic activities with nucleotide-binding and calcium-binding activity during capacitation.
Metabolic enzymes such as glyceraldehyde-3-phosphate dehydrogenase37, pyruvate kinase 138, enolase 139, etc., are well-known for their moonlighting effects, regulating cross-talk between cellular metabolism and gene expression, forming the basis of the RNA-enzyme-metabolite (REM) hypothesis40. We identified the RNA-binding activity of hexokinase 1 during sperm capacitation as previously reported in somatic cells41. The energy production in sperm undergoing capacitation is fueled by the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS), plus glycolysis42. Bull sperm undergoing heparin-induced capacitation efficiently use pyruvate to generate ATP through the TCA cycle43, which provides a phosphate group for capacitation-associated phosphorylation events44,45. Interestingly, protein synthesis during sperm capacitation reportedly depended on mitochondrial-synthesized ATP and mitochondrial membrane potential8. It would be interesting to investigate whether the mRNA binding activity of metabolic enzymes during sperm capacitation is coupled with capacitation-induced phosphorylation events and metabolism and could determine the fertilizing potential of sperm. Evidence also suggests sperm metabolism is closely related to bull fertility. High-fertile bulls have prominent oxidative phosphorylation and pyruvate metabolism pathways46 while low-fertility bulls have an altered expression of molecules associated with sperm metabolism47.
A few identified RBPs are involved in the binding of cytoskeletal proteins and exhibit calcium-binding activity. A strong association of the cytoskeleton with total poly (A) RNAs is well documented in neurons, oligodendrocytes, and oocytes (reviewed by Jansen)48. During spermatogenesis, TB-RBP, a phosphoprotein, mediates the association of translationally repressed testicular mRNAs to microtubules through highly conserved sequences (Y and H elements) in the 3’ UTRs49. Capacitation involves dynamic cytoskeletal remodeling where PKA-dependent phosphorylation events activate phospholipase D, stimulating filamentous (F)-actin polymerization and sperm hyperactivation18. For example, FSCB, an RBP, is located in the proximity of fibrous sheath proteins and gets phosphorylated during capacitation to bind calcium and contribute to hyperactivation50. The role of cytoskeletal proteins has been reported in mRNA transport, localization to the translation site, and protein synthesis48. Previously, Gur and Breitbart demonstrated localization of several sperm mRNAs and their translated proteins both inside and outside mitochondria51. Therefore, mRNA translation in sperm may occur either in the mitochondria and/or by mitochondrial-type ribosomes located outside the mitochondria51. Sperm RBPs, being a component of the mid-piece and principal piece, should be investigated to determine if they facilitate the transport of transcripts to the site of translation, most likely in the midpiece8,51 or sperm head where extra-mitochondrial rRNA is localized52. However, these arguments arise from indirect evidence and need further investigation.
The PTMs of testicular-RBPs (especially phosphorylation events) are known to regulate spermatogenesis53,54. Electrostatic charges (e.g., negative charge by phosphorylation) introduced by PTMs of RBPs could alter their interactions with RNA, other proteins and affect the structural stability of RNA-binding domains (RBDs)55,56. We hypothesized that capacitation alters the RBPs’ phosphorylation state with a change in the RNA-binding profile of translationally repressed mRNAs, initiating translation under capacitating conditions. RBP-binding to 5’ UTR could sterically hinder translation57 and their phosphorylation state could determine transcript localization58 or sequester translational factors from mRNA targets59,60, thereby repressing translation. A few RBPs undergoing phosphorylation during sperm capacitation were common to C0 and CAP group, which could modulate their affinity or steric hindrance and serve as a mechanism to resume mRNA translation (Fig. 6). These phosphorylated RBPs were a component of the principal piece of sperm tail and dynactin complex involved in cilium or flagellum-dependent cell motility. It restates the significance of understanding the possible role of cytoskeletal proteins in translational regulation through mRNA transport and localization to the translation site. A few RBPs were specific to associate with mRNAs in C0 and CAP groups, which could be due to their specificity towards RNA sequence motifs, and capacitation-specific conditions modulating cooperative or competitive interactions with other RBPs or miRNAs, as in other cells61,62.

Several sperm proteins undergo phosphorylation events during capacitation. Phosphoproteome (Supplementary Data S3) and differential protein expression (Supplementary Data S4) analysis revealed 10 and 4 RBPs to be phosphorylated and differentially expressed, respectively. This could influence mRNA processing, export, degradation, and binding to regulate translation during capacitation. Created in BioRender. Tiwari, S. (2025) https://BioRender.com/gmecv67.
Our observations on phosphorylation-associated RBP-mRNA interaction during capacitation further strengthen the significance of sperm protein phosphorylation in regulating sperm functions and enabling fertilizing competence. In contrast, mouse sperm could fertilize cumulus-intact oocytes when the cAMP pathway was inactivated63, indicating sperm fertility could be independent of capacitation-associated signaling pathways and phosphorylation events. However, it is important to consider species-specific differences, e.g., AKAP4, in mouse sperm is phosphorylated at serine or threonine residues than at tyrosine residues as in humans64,65. Since AKAPs can bind protein kinases, protein phosphatases, ion channels, and small GTP-binding proteins, they may assist in integrating cAMP and other signaling pathways66,67. Individual sperm vary in their ability to undergo capacitation within and across ejaculates in bulls68 and humans69, which could affect fertility70. Defects in sperm capacitation are highly prevalent in men seeking fertility assistance71,72,73 and therefore necessitate further investigation of its molecular mechanisms governing fertilizing competence.
Altogether, our study demonstrated that ejaculated bovine sperm harbor translational machinery where metabolic enzymes and cytoskeleton proteins serve as RBPs beyond their primary catalytic and structural role, respectively. Furthermore, capacitation altered the phosphorylation state of a few RBPs interacting with poly-A RNAs, suggesting it could be a potential mechanism to regulate translation in sperm similar to spermatogenesis53,54. These findings will assist in elucidating the translational regulation of mRNA in sperm. Few sperm proteins reported in the literature correlate with bull fertility and were identified as RBPs in our study. Therefore, it will be interesting to study their role in mRNA translation, which could advance our knowledge in regulating male fertility.
Future directions
Future studies should focus on elucidating the molecular mechanisms governing translational regulation during sperm capacitation and its association with sperm functions and their fertilization competence. A better understanding of how specific mRNAs associate with RBPs and are selectively translated in physiological and clinical conditions, for example, environmental stressors such as heat-induced testicular dysfunction, nutrition, and lifestyle factors, could provide insights into the etiology of sperm defects and male infertility. Extending these investigations to animal reproduction, particularly through comparative analyses of low- and high-fertility bulls, could facilitate the identification of biomarkers for improved fertility prediction and contribute to enhanced breeding efficiency and reproductive performance in livestock.
Materials and methods
Semen collection and processing
Fresh semen ejaculates collected from mature Holstein bulls (3–4 years of age) were retrieved from a commercial artificial insemination center, diluted (1:1) with TALPH and transported to the laboratory in a thermos (~35 °C). The diluted semen was laid on the top of a Percoll gradient (45–90%) in a 15 mL tube and centrifuged at 800 × g for 30 min (25 °C). The sperm pellet at the bottom was suspended in TALPH and centrifuged at 400 × g for 10 min (25 °C) to remove Percoll. The pellet was resuspended in TALPH, sperm motility was assessed with a microscope and sperm concentration was determined using a hemocytometer.
In-vitro capacitation of bovine sperm using heparin
An in-vitro capacitation assay was performed using 200 × 106 sperm in a 1 mL volume. Two control groups were used: a) control 0 h (C0): fresh sperm suspended in capacitating medium i.e., Sp-TALP medium [containing 1 mM pyruvate, 25 mM NaHCO3, 2 mM Ca2+ and 0.6% (w/v) Bovine Serum Albumin (BSA)] and maintained on ice for 4 h; and b) control 4 h (C4): sperm incubated in Sp-TALP medium for 4 h at 39 °C under high humidity. In the treatment group (capacitation; CAP), sperm were incubated in Sp-TALP supplemented with the capacitation-inducing agent heparin (Sigma, Catalog no. H3393-10KU; final concentration of 10 µg/ml) for 4 h at 39 °C under high humidity. Sperm motility was evaluated on an hourly basis during a 4-h incubation in C4 and CAP groups. Following incubation, sperm were pelleted by centrifugation at 10,000 × g for 5 min. at 4 °C. The supernatant was removed, and TALPH was added to the tube (without resuspending sperm pellet) to remove residual albumin, followed by centrifugation at 10,000 × g for 5 min at 4 °C. The sperm pellets were either snap-frozen and stored at −80 °C until protein extraction and precipitation for LC-MS/MS or processed for validating capacitation through immunoblotting for protein tyrosine phosphorylation.
Assessment of sperm kinematics under capacitating conditions
Sperm kinematics were evaluated with a computer-assisted sperm analyzer (CASA; Sperm Vision; Minitube, Ingersoll, ON, Canada). The sample (3 μL) was loaded into a chamber of prewarmed (37 °C) Leija slide (IMV Technologies, Ref. 025107-025108) and 7 fields per sample were analyzed using the bovine sperm kinematics program (Sperm Vision® SAR). An array of sperm kinematic parameters was analyzed, including the ALH (Amplitude of Lateral Head Displacement), Linearity (LIN), Curvilinear Velocity (VCL), and Straight-Line Velocity (VSL).
Evaluation of protein tyrosine phosphorylation using immunoblotting
For evaluation of sperm protein tyrosine phosphorylation, capacitated (heparin-induced) and control sperm samples were re-suspended in 100 μL TALPH supplemented with 25 μL of 5× sample buffer (Tris base: 0.25 M, DTT: 0.5 M, SDS: 10%, Glycerol: 50% (v/v), Bromophenol blue: 0.3%, pH = 6.8) and phosphatase inhibitor (1 mM Na3VO4). Another group of ouabain-induced (50 nM) capacitation was used to compare the protein tyrosine phosphorylation with heparin. However, the manuscript strictly uses heparin for all the following experiments to induce capacitation. Samples were boiled for 5 min at 95 °C followed by centrifugation (13,000 × g for 5 min) at room temperature (RT). The supernatant was loaded on a 3% stacking polyacrylamide gel and proteins were separated on a 10% resolving polyacrylamide gel and electro-transferred to 0.45 μM nitrocellulose membranes. These membranes were blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (TTBS), followed by overnight incubation with Anti-Phosphotyrosine Antibody, clone 4G10® (1:10,000; Millipore, Billerica, MA, USA) at 4 °C. The next day, membranes were washed thrice in TTBS for 10 min. followed by 1 h of incubation in HRP-conjugated goat anti-mouse IgG (1:10,000; Millipore, Catalog no. 12-349) at RT. Membranes were washed thrice in TTBS for 10 min, followed by the detection of immunoreactive bands in the control (C0 and C4) and CAP groups using chemiluminescence in ChemiDoc XRS+ System (BioRad). The membranes were stripped and probed with monoclonal anti-β-tubulin antibody (1:10,000; Sigma, Catalog no. T4026) for loading control. The signal intensity ratios were compared for tyrosine phosphorylated protein bands following normalization with β-tubulin intensity ratios using Image J software.
Investigating sperm proteome for translational machinery
The Percoll-washed sperm (n = 4 bulls) were diluted to 100 × 106/mL using TALPH and centrifuged at 400 × g for 10 min. The sperm pellets were snap-frozen and stored at −80 °C until protein extraction and precipitation for LC-MS/MS. The Percoll-washed sperm were checked for the presence of somatic cell contamination by PCR using somatic cell-specific genes (PCR details in Supplementary Table 1).
Investigating sperm proteome for differential protein expression and phosphoproteins during capacitation
The Percoll-washed sperm (n = 3 bulls) were in vitro capacitated using heparin and samples from C0 (control 0 h) and CAP (Capacitation) groups were processed for protein extraction and precipitation as described below.
Protein extraction and precipitation for LC-MS/MS
Sperm pellets (200 × 106 sperm) were thawed on ice and suspended in 100 μL TALPH with the addition of 25 μL of 5× sample buffer (Tris base: 0.25 M, DTT: 0.5 M, SDS: 10% without glycerol and bromophenol blue) and phosphatase inhibitor (1 mM Na3VO4). Samples were boiled for 5 min at 95 °C, followed by centrifugation (13,000 × g; 5 min) at RT. The supernatant was collected and diluted to 400 μL with TALPH for protein quantification using DC Protein Assay (BioRad, catalog no. 500-0112) as per the manufacturer’s protocol. An aliquot equivalent of 100 μg total protein was used for protein precipitation using the methanol chloroform water (MCW) extraction method. Due to the high SDS concentration (2%) in our sample buffer, the MCW extraction method was used for MS sample preparation to efficiently eliminate SDS74. Briefly, a 100 µL aliquot was added to 400 µL methanol in a 1.5 mL microcentrifuge tube and thoroughly vortexed, followed by addition of 100 µL chloroform and vortexing (all steps performed at room temperature). Finally, 300 µL MilliQ water was added, vortexed (the solution became cloudy) and incubated on ice for 3 min. The solution was centrifuged for 10 min at 13,000 × g (4 °C) and separated into 3 layers: a large aqueous layer on top, a circular flake of protein in the interphase, and a smaller chloroform layer at the bottom. The top aqueous layer was carefully removed without disturbing the interphase. Then, 400 µL methanol was added to the microcentrifuge tube followed by gentle mixing and centrifugation at 13,000 × g for 5 min (4 °C). The protein appeared as a very fine precipitate against the tube wall. The supernatant was removed without allowing the pellet to dry and fresh methanol was added (4 °C) until further processing.
Sample preparation for LC-MS/MS
Protocols for protein digestion, peptide extraction, nano LC-MS/MS analysis of the digest, and Mascot database search were performed by the Southern Alberta Mass Spectrometry Facility (SAMS), University of Calgary. The sample preparation was performed with FASP (Filter Aided Sample Preparation). Briefly, the precipitated protein was resuspended in Urea/Tris (8 M Urea in 100 mM Tris-HCl, pH 8), added with 10 mM of Dithiothreitol and incubated at 37 °C for 30 min. The reduced proteins were transferred into Microcon YM-30 (Millipore MRCF0R030) and centrifuged at 14,000 × g for 15 min. 100 μl of iodoacetamide (Sigma I6125-5g, 50 mM) prepared in Urea-Tris was added and incubated in the dark at room temperature for 20 min. Samples were centrifuged at 14,000 × g for 10 min, added with 100 μl of Urea-Tris and centrifuged at 14,000 × g for 15 min. 100 μl of 50 mM ammonium bicarbonate was added and centrifuged at 14,000 × g for 15 min. Forty microliters of trypsin (0.22 µg/µl) in 50 mM ammonium bicarbonate was added to YM-30 and incubated overnight at 37 °C. The next day, peptides were collected from YM-30 and resuspended in 40 μl of 1% Formic acid for desalting on UPLC or 40 μl of 200 mM of ammonium formate for high pH reverse phase fractionation. The peptides were then cleaned up using Pierce™ Peptide Desalting Spin Columns (Catalog no. 89852, ThermoFisher). For phosphopeptide enrichment, these clean peptides were processed with the SMOAC kit (Catalog nos. A32993 and A32992, ThermoFisher).
LC-MS/MS analysis for sperm proteomics and phosphopeptide detection
Tryptic peptides were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) operated with Xcalibur (Version 4.4.16.14) and coupled to a Thermo Scientific Easy-nLC (Nanoflow Liquid Chromatography) 1200 system. Tryptic peptides were loaded onto a C18 trap (75 µm × 2 cm; Acclaim PepMap 100, P/N 164946; Thermo Scientific) at a flow rate of 2 µL/min of solvent A (0.1% formic acid in LC-MS grade water). Peptides were eluted using a 120 min gradient from 5 to 40% (5–28% in 105 min followed by an increase to 40% B in 15 min) of solvent B (0.1% formic acid in 80% LC-MS grade acetonitrile) at a flow rate of 0.3 μL/min and separated on a C18 analytical column (75 µm × 50 cm; PepMap RSLC C18; P/N ES803; ThermoScientific). Peptides were then electrosprayed using 2.1 kV voltage into the ion transfer tube (300 °C) of the Orbitrap Lumos operating in positive mode. The Orbitrap first performed a full MS scan at a resolution of 120000 FWHM to detect the precursor ion having a m/z between 375 and 1575 and a +2 to +7 charge. The Orbitrap AGC (Auto Gain Control) and the maximum injection time were set at 4e5 and 50 ms, respectively. The Orbitrap was operated using the top speed mode with a 3 s cycle time for precursor selection. The most intense precursor ions presenting a peptidic isotopic profile and having an intensity threshold of at least 5000 were isolated using the quadrupole and fragmented with HCD (30% collision energy) in the ion routing multipole. Fragment ions (MS2) were analyzed in the ion trap at a rapid scan rate. The AGC and the maximum injection time were set at 1e4 and 35 ms, respectively, for the ion trap.
For phosphopeptide detection, the most intense precursor ions presenting a peptidic isotopic profile and having an intensity threshold of at least 2e4 were isolated using the quadrupole (Isolation window (m/z) of 1.2) and fragmented using HCD (30% collision energy) in the ion routing multipole. The fragment ions (MS2) were analyzed in the Orbitrap at a resolution of 30000. The AGC and the maximum injection time were set at 5e4 and 60 ms, respectively.
Dynamic exclusion was enabled for 45 s to avoid acquisition of the same precursor ion having a similar m/z (±10 ppm).
Database searching and criteria for protein and phosphopeptide identification
The Lumos raw data files (*.raw) were converted into Mascot Generic Format (MGF) using RawConverter (v1.1.0.18; The Scripps Research Institute) operating in a data-dependent mode. Monoisotopic precursors having a charge state of +2 to +7 were selected for conversion. The MGF file was used to search the database for Bos taurus taxonomy (139498 protein entries) using the Mascot algorithm (Matrix Sciences; Version 2.7) assuming the digestion enzyme trypsin. Search parameters for the MS data included a maximum number of missed cleavages of 1, with a fragment ion mass tolerance of 0.020 Da and a parent ion tolerance of 10.0 ppm. Carbamidomethyl of cysteine was specified in Mascot as a fixed modification. Oxidation of methionine (+16) and phosphorylation (+80) of serine, threonine and tyrosine were specified in Mascot as variable modifications. The correspondent % decoy false discovery rate (FDR) was 0.0% for proteins and 0.8% for peptides. Common contaminants (e.g., keratins and albumin) were manually excluded.
For phosphopeptide identification, Mascot results obtained from the target and decoy databases searches were subjected to Scaffold software (Version Scaffold_5.1.2, Proteome Software Inc., Portland, OR) to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability by the Peptide Prophet algorithm with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at >95.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Identifying mRNA interactome during bovine sperm capacitation using RAP-MS
As mRNA interactome capture has been described only in somatic cells, the RAP-MS protocol41 in sperm was adapted through several optimizations. A crosslinking and oligo (dT) affinity purification approach was used to capture mRNA-interacting RBPs during bovine sperm capacitation. Buffers were prepared in DEPC-treated water, and all practices were followed to ensure RNase-free conditions.
Optimizing UV dosage for in vivo crosslinking of mRNA-RBP complex in sperm
UV irradiation (254 nm) induces the formation of covalent bonds between RBPs directly bound to mRNAs; however, it can degrade proteins and RNA in high dosages. Therefore, the UV dosage for crosslinking was optimized by irradiating sperm (100 × 106) with a total UV (254 nm) dose of 150–600 mJ/cm2 in a UV Crosslinker (VWR, Catalog no. 89131-484). Sperm with no UV exposure served as a control. Effects of various dosages of irradiation on proteins were investigated in triplicate through SDS-PAGE of similar protein loading on gel and Coomassie blue staining, followed by immunoblotting for β-tubulin. Effects on RNA integrity were determined (triplicate) with a bioanalyzer.
Sperm lysis and capture of mRNA-RBP complex
Approximately 500–600 million sperm from C0 and CAP (heparin) groups were thawed on ice and suspended in 1.5 mL lysis buffer [(50 mM Tris (pH 7.5), 1 mM EDTA, 500 mM LiCl, 0.1% LiDS, 1% ethylene carbonate, 10 mM DTT, cOmplete™ protease inhibitor cocktail (Roche, 11697498001), and 1 mM Na3VO4] in a microcentrifuge tube. The sperm were sonicated on ice (thrice for ~10 s in pulses) and transferred to a 100 mm petri dish for irradiation with a UV (254 nm) dose of 400 mJ/cm2. The petri dish was kept on ice over a rotor for 1 h and the sperm suspension in lysis buffer was transferred to a microcentrifuge tube and centrifuged (12,000 × g, 5 min, 4 °C) to collect supernatant (sperm lysate). An aliquot of lysates was collected to validate RBPs through immunoblotting. RNA isolated from sperm lysate was used to generate a bioanalyzer profile and was later converted to cDNA (without DNase digestion) to check genomic DNA contamination. cDNA was amplified using intron-specific primer PRM1 and somatic cell-specific genes (PCR details in Supplementary Table 1).
In parallel, 200 μL streptavidin agarose beads (Sigma, Catalog no. S1638) were washed and resuspended in lysis buffer, without or with 3.5 μL biotinylated oligo (dT) probes (Promega, Catalog no. Z5261) on ice for 1 h over a rotor. The beads conjugated with probes were collected, and an equal volume of beads was added to the C0 and CAP lysates. Agarose beads (without probe conjugation) were added to C0 and CAP lysates (bead control, BC) to identify non-specific protein binding. Beads were incubated on ice for 1 h over a rotator and retrieved by centrifugation (12,000 × g, 5 min, 4 °C) followed by sequential washing in wash buffers (WB) WB-I [50 mM Tris (pH 7.5), 1 mM EDTA, 500 mM LiCl, 0.1% LiDS], WB-II [50 mM Tris (pH 7.5), 1 mM EDTA, 500 mM LiCl] and WB-III [50 mM Tris (pH 7.5), 1 mM EDTA, 200 mM LiCl].
After the last washing, beads were suspended in 30 μL elution buffer [50 mM Tris (pH 7.5), 1 mM EDTA] and incubated at 80 °C for 3 min. The supernatant over the beads was collected, transferred on ice, and partitioned for RNA extraction or proteomics. Briefly, the supernatant was treated with Proteinase K for 30 min at 55 °C and RNA was extracted using TRIzol, followed by generating an electrophoretic profile in Bioanalyzer. The RBPs were identified by digesting RNA in mRNA-RBP complexes using Benzonase (Millipore, Catalog no. 70664; 37 °C, 30 min), followed by boiling in Lamelli buffer at 95 °C for 5 min and loading on a 3–10% gradient gel for SDS-PAGE. Gels were silver-stained using Pierce™ Silver Stain (ThermoFisher, Catalog no. 24600).
Proteins run in 10% polyacrylamide gel (~1 mm3 pieces) were excised, placed in Milli-Q water, and sent to the SAMS facility at the University of Calgary for LC-MS/MS analysis.
Functional and pathway enrichment analysis of mRNA interactome and validation using immunoblotting
Sperm RBPs were subjected to ShinyGO (0.80), a graphical gene-set enrichment tool, for related cell components, molecular function, and biological process terms. The FDR ≤ 0.05 was set as the threshold for significant enrichment. Using the application program interface access to KEGG, functional pathway analysis was performed. The proportional abundance of a protein in a protein mixture can be estimated with emPAI (Exponentially Modified Protein Abundance Index) from LC-MS/MS data. Proteins related to translational machinery and mRNA binding (RBPs) were plotted on a scatter dot plot with log 10 (emPAI) against molecular weight to check their abundance and distribution among total sperm proteins. Immunoblotting was performed (as described in Section 2.2.3) to validate LC-MS/MS results for one of the RBPs, hexokinase-1, using an anti-hexokinase 1 antibody (1:2,500; NBP-1-31600, Novus Biologicals) in 3 biological replicates. The immunoreactive bands were detected using SuperSignal™ West Femto Maximum Sensitivity Substrate (Catalog no. 34094, ThermoFisher) in the ChemiDoc XRS+ System.
Statistics and reproducibility
Sperm kinematic parameters were compared between the control and CAP groups with an unpaired Student’s t-test (p ≤ 0.05) using GraphPad Prism Version 10.00 (GraphPad Software, La Jolla, CA, USA). Western blot analysis for relative band intensity was performed through one-way ANOVA and Tukey’s multiple comparisons test. Data are represented as mean ± SEM. The LC-MS/MS data were visualized and validated using Scaffold 5. The total spectra were log-normalized for differential protein expression (n = 3) and phosphoproteome analysis (n = 3), and an F-test (p ≤ 0.05) was performed for differential protein expression between fresh-uncapacitated versus capacitated groups. FDR was controlled using the Benjamini–Hochberg correction (p ≤ 0.01). To ensure reproducibility, the protocols for in vitro capacitation of bull sperm and sample preparation were consistently used across all biological replicates, as detailed in the “Materials and Methods” section.
Ethical approval
We have complied with all relevant ethical regulations for animal use. This study was approved by the University of Calgary Institutional Animal Care and Use Committee (protocol number: AC25-0072).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All underlying data are available in the article itself and its supporting information. This article contains supplemental data, including original unedited/uncropped immunoblots, numerical source data for bar plot (Fig. 1B), stained SDS-PAGE gels, PCR data and Sanger sequencing data. The supplementary information file outlines the details of figures generated from the corresponding unedited/uncropped immunoblots and gel images. Numerical data for Fig. 1B corresponds to Supplementary Table 3. Numerical data in the Venn diagram in Fig. 5A corresponds to Supplementary Data S2. Numerical data for phosphoproteome and differential protein expression in the Venn diagram in Fig. 6 correspond to Supplementary Data S3 and S4, respectively. The LC-MS/MS raw and processed data can be accessed through PRISM Data: University of Calgary’s Data Repository (https://doi.org/10.5683/SP3/8DT2MU).
References
Winters, B. R. & Walsh, T. J. The epidemiology of male infertility. Urol. Clin. North Am. 41, 195–204 (2014).
Pandruvada, S. et al. Lack of trusted diagnostic tools for undetermined male infertility. J. Assist. Reprod. Genet. 38, 265–276 (2021).
Miller, D. Sperm RNA as a mediator of genomic plasticity. Adv. Biol. 2014, 179701 (2014).
O’Donnell, L. Mechanisms of spermiogenesis and spermiation and how they are disturbed. Spermatogenesis 4, e979623 (2014).
Rajamanickam, G. D., Kastelic, J. P. & Thundathil, J. C. Content of testis-specific isoform of Na/K-ATPase (ATP1A4) is increased during bovine sperm capacitation through translation in mitochondrial ribosomes. Cell Tissue Res. 368, 187–200 (2017).
Bragg, P. W. & Handel, M. A. Protein synthesis in mouse spermatozoa. Biol. Reprod. 20, 333–337 (1979).
Ahmed, N. A., Salem, M. H., El-Oksh, H. A. & Pursel, V. G. Effect of incubation conditions, inhibitors and seminal plasma on protein synthesis in ram spermatozoa. Reproduction 71, 213–219 (1984).
Gur, Y. & Breitbart, H. Mammalian sperm translate nuclear-encoded proteins by mitochondrial-type ribosomes. Genes Dev. 20, 411–416 (2006).
Corda, P. O., Silva, J. V., Almeida, C. R., Pierre, P. & Fardilha, M. De novo protein synthesis occurs through the cytoplasmic translation machinery in mammalian spermatozoa. J. Cell. Physiol. 240, e70038 (2025).
Sutherland, J., Siddall, N., Hime, G. & McLaughlin, E. RNA binding proteins in spermatogenesis: an in depth focus on the Musashi family. Asian J. Androl. 17, 529 (2015).
Ickowicz, D., Finkelstein, M. & Breitbart, H. Mechanism of sperm capacitation and the acrosome reaction: role of protein kinases. Asian J. Androl. 14, 816–821 (2012).
Ishihama, Y. et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 4, 1265–1272 (2005).
Urdaneta, E. C. & Beckmann, B. M. Fast and unbiased purification of RNA-protein complexes after UV cross-linking. Methods 178, 72–82 (2020).
Hosseini, M. et al. Sperm epigenetics and male infertility: unraveling the molecular puzzle. Hum. Genom. 18, 57 (2024).
Gross, N., Strillacci, M. G., Peñagaricano, F. & Khatib, H. Characterization and functional roles of paternal RNAs in 2–4 cell bovine embryos. Sci. Rep. 9, 20347 (2019).
Saunders, C. M. et al. PLCζ: a sperm-specific trigger of Ca2+ oscillations in eggs and embryo development. Development 129, 3533–3544 (2002).
Fujimoto, S. et al. Mammalian phospholipase Cζ induces oocyte activation from the sperm perinuclear matrix. Dev. Biol. 274, 370–383 (2004).
Yanagimachi, R. The movement of golden hamster spermatozoa before and after capacitation. Reproduction 23, 193–196 (1970).
Bhargava, P. M., Bishop, M. W. H. & Work, T. S. Incorporation of [14C]amino acids into the proteins of bull spermatozoa. Biochem. J. 73, 247–256 (1959).
Premkumar, E. & Bhargava, P. M. Isolation and characterization of newly synthesized RNA and protein in mature bovine spermatozoa and effect of inhibitors on these syntheses. Indian J. Biochem. Biophys. 10, 239–253 (1973).
Farabaugh, P. J. The translational machinery. in Programmed Alternative Reading of the Genetic Code 5–27 (Springer US, 1997).
Gerstberger, S., Hafner, M. & Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 15, 829–845 (2014).
Alves, L. R. RNA-binding proteins related to stress response and differentiation in protozoa. WJBC 7, 78 (2016).
Kleene, K. C. Poly(A) shortening accompanies the activation of translation of five mRNAs during spermiogenesis in the mouse. Development 106, 367–373 (1989).
Smith, R. W. P. et al. DAZAP1, an RNA-binding protein required for development and spermatogenesis, can regulate mRNA translation. RNA 17, 1282–1295 (2011).
Queiroz, R. M. L. et al. Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 37, 169–178 (2019).
Corley, M., Burns, M. C. & Yeo, G. W. How RNA-binding proteins interact with RNA: molecules and mechanisms. Mol. Cell 78, 9–29 (2020).
Saito, I. & Matsuura, T. Chemical aspects of UV-induced crosslinking of proteins to nucleic acids. Photoreactions with lysine and tryptophan. Acc. Chem. Res. 18, 134–141 (1985).
Wagenmakers, A. J. M., Reinders, R. J. & Van Venrooij, W. J. Cross-linking of mRNA to proteins by irradiation of intact cells with ultraviolet light. Eur. J. Biochem. 112, 323–330 (1980).
Pashev, I. G., Dimitrov, S. I. & Angelov, D. Crosslinking proteins to nucleic acids by ultraviolet laser irradiation. Trends Biochem. Sci. 16, 323–326 (1991).
Esteban-Serna, S., McCaughan, H. & Granneman, S. Advantages and limitations of UV cross-linking analysis of protein– RNA interactomes in microbes. Mol. Microbiol. 120, 477–489 (2023).
Tovich, P. R. & Oko, R. J. Somatic histones are components of the perinuclear theca in bovine spermatozoa. J. Biol. Chem. 278, 32431–32438 (2003).
Sellem, E. et al. A comprehensive overview of bull sperm-borne small non-coding RNAs and their diversity across breeds. Epigenetics Chromatin 13, 19 (2020).
Marzluff, W. F. Metazoan replication-dependent histone mRNAs: a distinct set of RNA polymerase II transcripts. Curr. Opin. Cell Biol. 17, 274–280 (2005).
Sendler, E. et al. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Res. 41, 4104–4117 (2013).
Jodar, M. et al. The presence, role and clinical use of spermatozoal RNAs. Hum. Reprod. Update 19, 604–624 (2013).
Chang, C.-H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Kejiou, N. S. et al. Pyruvate kinase M (PKM) binds ribosomes in a poly-ADP ribosylation dependent manner to induce translational stalling. Nucleic Acids Res. 51, 6461–6478 (2023).
Huppertz, I. et al. Riboregulation of Enolase 1 activity controls glycolysis and embryonic stem cell differentiation. Mol. Cell 82, 2666–2680.e11 (2022).
Hentze, M. W. & Preiss, T. The REM phase of gene regulation. Trends Biochem. Sci. 35, 423–426 (2010).
Castello, A. et al. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc. 8, 491–500 (2013).
Mostek, A., Janta, A., Majewska, A. & Ciereszko, A. Bull sperm capacitation is accompanied by redox modifications of proteins. IJMS 22, 7903 (2021).
Córdoba, M., Pintos, L. N. & Beconi, M. T. Heparin and quercitin generate differential metabolic pathways that involve aminotransferases and LDH-X dehydrogenase in cryopreserved bovine spermatozoa. Theriogenology 67, 648–654 (2007).
O’Flaherty, C. M., Beorlegui, N. B. & Beconi, M. T. Lactate dehydrogenase-C4 is involved in heparin- and NADH-dependent bovine sperm capacitation. Andrologia 34, 91–97 (2002).
O’Flaherty, C., Beorlegui, N. & Beconi, M. T. Heparin- and superoxide anion-dependent capacitation of cryopreserved bovine spermatozoa: Requirement of dehydrogenases and protein kinases. Free Radic. Res. 40, 427–432 (2006).
Muhammad Aslam, M. K. et al. Identification of biomarker candidates for fertility in spermatozoa of crossbred bulls through comparative proteomics. Theriogenology 119, 43–51 (2018).
Talluri, T. R. et al. Integrated multi-omics analyses reveals molecules governing sperm metabolism potentially influence bull fertility. Sci. Rep. 12, 10692 (2022).
Jansen, R. P. RNA-cytoskeletal associations. FASEB J. 13, 455–466 (1999).
Han, J. R., Yiu, G. K. & Hecht, N. B. Testis/brain RNA-binding protein attaches translationally repressed and transported mRNAs to microtubules. Proc. Natl Acad. Sci. USA 92, 9550–9554 (1995).
Liu, S.-L. et al. FSCB phosphorylation in mouse spermatozoa capacitation. BMB Rep. 44, 541–546 (2011).
Gur, Y. & Breitbart, H. Protein synthesis in sperm: Dialog between mitochondria and cytoplasm. Mol. Cell. Endocrinol. 282, 45–55 (2008).
Villegas, J. Localization of the 16S mitochondrial rRNA in the nucleus of mammalian spermatogenic cells. Mol. Hum. Reprod. 8, 977–983 (2002).
Morales, C. R., Wu, X. Q. & Hecht, N. B. The DNA/RNA-binding protein, TB-RBP, moves from the nucleus to the cytoplasm and through intercellular bridges in male germ cells. Dev. Biol. 201, 113–123 (1998).
Steger, K. Transcriptional and translational regulation of gene expression in haploid spermatids. Anat. Embryol. 199, 471–487 (1999).
Law, L. M. J., Everitt, J. C., Beatch, M. D., Holmes, C. F. B. & Hobman, T. C. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J. Virol. 77, 1764–1771 (2003).
Lovci, M. T., Bengtson, M. H. & Massirer, K. B. Post-translational modifications and RNA-binding proteins. in RNA Processing (ed. Yeo, G. W.) Vol. 907 297–317 (Springer International Publishing, Cham, 2016).
Felder, S., Nelson, I. M., Hatfield, B. M. & Weeks, K. M. Protein binding in an mRNA 5′-UTR sterically hinders translation. RNA 31, 143–149 (2025).
Yao, P. et al. The HILDA complex coordinates a conditional switch in the 3′-untranslated region of the VEGFA mRNA. PLoS Biol. 11, e1001635 (2013).
Michlewski, G., Sanford, J. R. & Cáceres, J. F. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol. Cell 30, 179–189 (2008).
Zou, Z. et al. FMRP phosphorylation modulates neuronal translation through YTHDF1. Mol. Cell 83, 4304–4317.e8 (2023).
Nag, S., Goswami, B., Das Mandal, S. & Ray, P. S. Cooperation and competition by RNA-binding proteins in cancer. Semin. Cancer Biol. 86, 286–297 (2022).
Ho, J. J. D. & Marsden, P. A. Competition and collaboration between RNA-binding proteins and microRNAs. WIREs RNA 5, 69–86 (2014).
Tateno, H. et al. Ca2+ ionophore A23187 can make mouse spermatozoa capable of fertilizing in vitro without activation of cAMP-dependent phosphorylation pathways. Proc. Natl Acad. Sci. USA 110, 18543–18548 (2013).
Brito, M., Figueroa, J., Maldonado, E. U., Vera, J. C. & Burzio, L. O. The major component of the rat sperm fibrous sheath is a phosphoprotein. Gamete Res. 22, 205–217 (1989).
Johnson, L. R. et al. Assembly of AKAP82, a protein kinase A anchor protein, into the fibrous sheath of mouse sperm. Dev. Biol. 192, 340–350 (1997).
Michel, J. J. C. & Scott, J. D. AKAP mediated signal transduction. Annu. Rev. Pharmacol. Toxicol. 42, 235–257 (2002).
Welch, E. J., Jones, B. W. & Scott, J. D. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interv. 10, 86–97 (2010).
Zicarelli, L. et al. Effect of bull on in vitro sperm capacitation induced by different agents in buffalo species (Bubalus bubalis). Ital. J. Anim. Sci. 8, 177–179 (2009).
Ostermeier, G. C. et al. Timing of sperm capacitation varies reproducibly among men. Mol. Reprod. Dev. 85, 387–396 (2018).
Zoca, S. M. et al. Bull field fertility differences can be estimated with in vitro sperm capacitation and flow cytometry. Front. Anim. Sci. 4, 1180975 (2023).
Cardona, C. et al. Defects in sperm capacitation and fertilizing ability are highly prevalent in men undergoing fertility workups, even if normospermic. Fertil. Steril. 112, e281–e282 (2019).
Sharara, F. I., Ostermeier, G. C. & Travis, A. J. Defects in sperm capacitation/fertilizing ability are equally prevalent across ages in men seeking fertility assistance. Reprod. BioMed. Online 46, 179–187 (2023).
Sharara, F. et al. Multicentric, prospective observational data show sperm capacitation predicts male fertility, and cohort comparison reveals a high prevalence of impaired capacitation in men questioning their fertility. Reprod. BioMed. Online 41, 69–79 (2020).
Puchades, M., Westman, A., Blennow, K. & Davidsson, P. Removal of sodium dodecyl sulfate from protein samples prior to matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 13, 344–349 (1999).
Acknowledgements
This work was supported by funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Grant No. RGPIN-2020-04585) awarded to Jacob Thundathil and “Alberta Graduate Excellence Scholarship (AGES) – International,” “Alberta Innovates Graduate Scholarship” and “Alberta Children’s Hospital Research Institute Graduate Scholarship” awarded to Saurabh Tiwari. We thank Alta Genetics Inc., Calgary, Alberta, Canada, for providing fresh bovine semen for this study. We also thank the SAMS Facility at the University of Calgary for their support with LC-MS/MS on a fee-for-service basis.
Author information
Authors and Affiliations
Contributions
Writing—original draft, validation, formal analysis, investigation, data curation: S.T.; Methodology, visualization, supervision, project administration, conceptualization, J.T., N.T. and S.T.; Funding acquisition: J.T.; Writing—review and editing: J.T. and N.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Michal Zigo and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Frank Avila, David Favero, and Kaliya Georgieva.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tiwari, S., Thakor, N. & Thundathil, J. Systematic mRNA interactome analysis reconceptualizes translational quiescence in bovine sperm. Commun Biol 8, 1419 (2025). https://doi.org/10.1038/s42003-025-08919-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08919-7
