
Abstract
Translation of mRNA into protein is a fundamental process and tightly controlled during development. Several mechanisms acting on the mRNA level regulate when and where an mRNA is expressed. To explore the effects of conditional and transient gene expression in a developing organism, it is vital to experimentally enable abrogation and restoration of translation. We recently developed the FlashCaps technology allowing preparation of translationally muted mRNAs and their controlled activation by light. Here, we validate its functionality in vivo. We demonstrate that translation of FlashCap-eGFP-mRNA can be triggered in zebrafish embryos with spatiotemporal control. The injected FlashCap-mRNA is stable for hours and remains muted. Light-mediated activation up to 24 h post fertilization produces visible amounts of eGFP and can be restricted to distinct parts of the embryo. This methodology extends the toolbox for vertebrate models by enabling researchers to locally activate mRNA translation at different timepoints during development.
Similar content being viewed by others


Introduction
Translation of mRNAs into proteins is a central and conserved process in eukaryotes. It is employed with endogenous as well as exogenous transcripts1, which can originate from viruses or biotechnological manufacturing. The ability to synthesize and administer mRNAs led to the development of the Corona vaccine and provides the basis for current developments in protein replacement therapy2,3. The ability to control when and where an mRNA becomes activated for translation bears potential for new applications in targeted therapies and regenerative medicine.
The 5′ cap is a key feature of mRNAs and is required for proper translation in eukaryotes4,5. It consists of an N7 methylated guanosine linked to the first transcribed nucleotide via a 5′-5′ triphosphate bridge and is formed co-transcriptionally. Viruses have evolved various ways to generate capped mRNAs underscoring the functional importance of this structure6. Similarly, mRNAs used for biological and medical studies require a 5′ cap. The 5′ cap protects the mRNA from exonucleolytic decay and coordinates additional maturation steps, such as splicing and polyadenylation7,8,9,10. Most importantly, direct binding of the 5′ cap to the eukaryotic translation initiation factor 4E (eIF4E) is the first and rate-limiting step in translation initiation11,12. Cap-dependent translation is responsible for most of the proteins produced in cells, although cap-independent translation exists and is relevant for certain viral RNAs13.
The control of gene expression at the mRNA level is vital for virtually all complex biological processes, such as cell differentiation, cell proliferation, and embryogenesis14,15,16. In particular, the early embryonic development of zebrafish, which refers to the period from fertilization to 3.5 h post fertilization (hpf) and the switch from maternal to zygotic transcription, is primarily driven by maternal mRNAs17,18. Zebrafish embryos constitute an important vertebrate model organism for biomedical research, particularly for organogenesis, cardiovascular, and neurobiological research19,20,21. The zebrafish offers an experimentally extremely accessible system due to the ex utero development of the transparent embryos22. However, further in-depth investigation and manipulation of many cellular processes during embryogenesis and beyond requires a tool that enables researchers to initiate gene expression with a high spatiotemporal resolution.
Several zebrafish lines for ectopic expression of a target gene in distinct tissues based on cell-specific promoters and enhancers are available and modular systems, such as Gal4-UAS, facilitate generation of novel zebrafish lines23,24. Alternatively, promoter systems that are responsive to heat25 or small molecules26 allow for temporal—and in some cases spatial—control of gene expression in zebrafish. However, the generation of zebrafish lines is tedious, and promoters/enhancers for the desired expression pattern are not always available. Conditional expression in response to an inducer requires time for signaling, transcription, and translation, preventing its use in studying immediate effects.
An alternative is the transient overexpression of exogenous mRNA, which in zebrafish is routinely performed by direct injection of mRNA into the fertilized egg. The mRNA can be conveniently injected into the yolk until the 32-cell stage and still becomes homogenously distributed and reliably translated in contrast to the injection of DNA, which leads to mosaic expression patterns27. This greatly facilitates expression studies, as genetically modified fish are not required if transient expression suffices. However, the injected mRNA is translated almost immediately by all cells that inherit it, and the ectopic expression is difficult to control in temporal and spatial terms.
Post-transcriptional regulation of gene expression is commonly investigated in zebrafish embryos by injecting miRNA or morpholino oligomers into zebrafish embryos directly after fertilization28,29. Especially morpholino oligomers were intensively used to globally knock-down certain gene functions in zebrafish embryos30,31. The combination of morpholino oligomers with photo-cleavable groups provided an additional layer of control to this gene inactivation approach. In the past few years, numerous endogenous and exogenous genes were successfully downregulated by using caged morpholino oligomers32,33,34. Morpholinos, like all approaches relying on hybridization (e.g., siRNA or antisense oligonucleotides), are, by definition, indirect. A comprehensive independent comparison of zebrafish phenotypes caused by either gene knock-out via mutation or knock-down of the same gene with a morpholino oligomer revealed that morpholino knock-downs poorly correlate with mutant phenotypes in zebrafish embryos35,36,37.
Taken together, a tool providing transient overexpression of ectopic genes with high spatiotemporal precision, easy application, and no side and off-target effects would be very desirable. However, the approaches for controlled activation of protein production in a local manner are limited thus far and often depend on, e.g., the use of heat shock promoters or tissue-specific promotor constructs. While spatiotemporal knock-down approaches, e.g., caged morpholinos, are used to assess whether the local inactivation of the expression of a given target gene would have an effect on the developmental process, there are few attempts to achieve transient overexpression with spatiotemporal control in zebrafish as an in vivo model. In a seminal study, photo-cleavable groups were randomly installed throughout the mRNA and could activate its translation in zebrafish38. However, the chemical treatment of mRNA with 6-bromo-4-diazomethyl-7-hydroxycoumarin caused significant mRNA degradation. The resulting mRNA preparation contained a statistical distribution of a varying number of photo-cleavable groups, and multiple photo-cleavable groups needed to be removed to activate translation, compromising the efficient release of an unprotected mRNA38. More recently, mRNAs with a photo-switchable 5′ cap were reported to be activated and deactivated for translation in zebrafish39. This approach is limited by the photochemical properties of diazo-based photo-switches, which restrict the cis-to-trans isomerization to 80%39. We recently developed a highly efficient optochemical approach to activate mRNA translation by light40. Synthetic 5′ caps with a photo-cleavable group (FlashCaps) prevent binding to eIF4E40,41. An mRNA equipped with a FlashCap is thus translationally muted. Irradiation induces photolysis, which liberates the natural 5′ cap (cap 0), allowing light-mediated activation of translation. We demonstrated the activation of translation by light for different FlashCap-mRNAs in vitro and in different cell lines.
We investigated the functionality and implementation of this concept in a developing vertebrate organism and, thus, in an in vivo setting. For this purpose, we prepared FlashCap-mRNA and injected zebrafish embryos at the 1-cell stage. The modified mRNAs were found translationally muted. Upon irradiation with light at 365 nm at different timepoints during embryonic development, protein production became activated in vivo. This study, therefore, provides a proof-of-concept for the activation of translation in a spatially and temporally controlled manner in a developing embryo.
Results and discussion
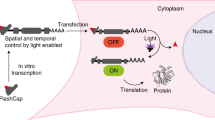
We recently reported photo-protected 5′ cap analogs, termed FlashCaps, as a tool to control the translation of mRNAs by light in mammalian cell lines (Fig. 1A). FlashCaps can be incorporated at the 5′ end of any given mRNA by in vitro transcription to yield FlashCap-mRNAs that are translationally muted in vitro and in mammalian cell lines40. Short irradiation of cells at 6 h post-transfection removed the photo-cleavable group and released mRNA with a cap 0 (Fig. 1A). This procedure restored binding to eIF4E and hence activated translation of FlashCap-mRNAs, resulting in a remarkable 27-fold increase of the respective reporter protein35. In mammalian cell lines, FlashCap-mRNAs exhibited similar stability as the corresponding transcript with the naturally occurring cap 0 and thus did not alter the half-life40,41. However, it was not clear, how long after transfection the translationally muted FlashCap-mRNAs could still be activated by light to provide a detectable output. Limited stability of mRNA and the intracellular localization could hamper such a later activation, as mRNAs that are not translated can be sequestered to P-bodies14.

A Photolysis reaction of the FlashCap-mRNA. B Schematic illustration of light-mediated activation of translation. Irradiation was performed at different timepoints. C Fluorescence confocal microscopy images of HeLa cells transfected with FlashCap-eGFP-mRNA. Cells were either kept in the dark or irradiated (365 nm, 15 s) at indicated timepoints (6 h or 12 h) after transfection. Cap0-eGFP-mRNA serves as a positive control and was kept in the dark. Samples were imaged for eGFP fluorescence 24 h after transfection. Shown is one representative set of n = 3 independent replicates.
To elucidate whether FlashCap-mRNA could be stable enough and available for later activation of translation, we produced FlashCap-eGFP-mRNA with the widely used α-globin 3′ UTR for cell studies. The synthetic 5′ cap (FlashCap or m7GpppG as control) was incorporated during in vitro transcription (IVT) by transcriptional priming using T7 RNA polymerase, resulting in capped and uncapped RNA. For our experiments, we routinely remove uncapped mRNA by enzymatic digestion with polyphosphatase and exoribonuclease XRN-140,42.
To assess the translation of FlashCap-mRNA after light-mediated activation at 6 h and 12 h post-transfection, we transfected HeLa cells and performed the light-activation procedure using an LED (Fig. 1B). As expected, cells transfected with FlashCap-capped eGFP-mRNA developed green fluorescence when irradiated with 365 nm for 15 s at 6 h post-transfection (Fig. 1C). When we performed the irradiation 12 h post-transfection, cells transfected with FlashCap-eGFP-mRNA still developed green fluorescence, albeit with lower signal intensity. Without irradiation, almost no fluorescence was observed when FlashCap-eGFP-mRNA was used. As expected, the positive control, i.e., cells transfected with cap0-eGFP-mRNA but without irradiation, resulted in the highest fluorescence signal of the four samples tested (Fig. 1C).
These data show that FlashCap-mRNAs can still be activated at later timepoints, suggesting that their stability and availability in cells is sufficient for experiments up to at least 12 h post-transfection. Processes such as mRNA degradation and sequestration are likely contributors to signal reduction over time.
Encouraged by these results we sought to utilize FlashCap-mRNAs in a living organism. Zebrafish is a prominent model organism for vertebrate development, and due to its optical transparency renders itself extremely useful for such studies. We, therefore, set out to test FlashCap-mRNAs in zebrafish embryos as an in vivo model organism43,44.
In order to induce ubiquitous expression of a target gene in the embryo, the respective mRNA is routinely injected into the yolk of the zebrafish zygote directly after fertilization (Fig. 2A). In the very early stages (up to the 32-cell stage, when the embryo is still syncytial), the mRNA is taken up by all cells of the embryo via a process called cytoplasmic streaming, leading to homogenous distribution and expression of mRNA in all cells. However, neither the effect nor the fate of translationally muted mRNAs with a photo-protected 5′ cap had been tested in a developing embryo. Furthermore, the consequences of irradiation required for photo-deprotection of FlashCap-mRNAs on the development of zebrafish embryos and the translation of the injected mRNA had to be investigated for in vivo use.

A Scheme illustrating time-dependent activation of translation using FlashCap-mRNA in zebrafish embryos. B Effect of irradiation on the translation of cap0-eGFP-mRNA. Differential interference contrast (DIC) and fluorescence images of zebrafish embryos injected with 70 pg cap0-eGFP-mRNA with or without irradiation (365 nm, 20 s). C Activation of translation of FlashCap-eGFP-mRNA in developing zebrafish embryos by light. Time course of irradiation to activate translation in different stages of developing embryos. DIC and fluorescence images of zebrafish embryos injected with 70 pg FlashCap-eGFP-mRNA with and without irradiation (365 nm, 20 s). B, C Shown is one representative experiment of n = 3 independent replicates.
First, we assessed the effect of irradiation on the translation of injected eGFP-mRNA with a regular cap 0. To this end, cap0-eGFP-mRNA was injected into the yolk of zebrafish embryos directly after fertilization (0 h post fertilization (hpf)). The mRNA for zebrafish experiments was prepared by in vitro transcription (IVT) using a pCS2+ vector encoding eGFP. After transcription and removal of uncapped mRNA, a poly(A) tail was added post-transcriptionally using poly(A) polymerase. We found that poly(A) tailing improved the amount of protein produced in zebrafish embryos, despite the fact that the pCS2+ vector itself contains a poly(A) signal (Supplementary Fig. 1).
The injected embryos looked normal without developmental defects. Embryos were kept in the dark or irradiated at 2 hpf with an LED at 365 nm for 20 s. After irradiation, the embryos were kept in a dark incubator for 24 h at 28.5 °C and then imaged under a fluorescence microscope (Fig. 2A). As expected, embryos injected with cap0-eGFP-mRNA showed bright green fluorescence (Fig. 2B). Embryos that had been injected with cap0-eGFP-mRNA and irradiated also showed strong green fluorescence, albeit slightly less than in case of the non-irradiated control. Control embryos that had not been injected did not exhibit green fluorescence, confirming that the background fluorescence was very low. The slight reduction of translational output upon irradiation was also observed in cell culture (Supplementary Fig. 2) and is most likely caused by ribotoxic stress45.
Next, we asked whether FlashCap-eGFP-mRNAs would remain translationally silent in zebrafish embryos and whether they could be activated for translation by light. For mRNA preparation, we used the pCS2+ vector and confirmed that FlashCaps are incorporated during in vitro transcription (IVT) with SP6 polymerase. The FlashCap-eGFP-mRNA was prepared in the same way as the cap0-eGFP-mRNA, including the removal of uncapped RNA and poly(A)-tailing. To remove double-stranded RNA that can elicit an immunological response, the mRNAs were HPLC-purified. Integrity of FlashCap-eGFP-mRNA was confirmed by polyacrylamide gel electrophoresis (Supplementary Fig. 3). We determined capping efficiencies of 40–50% and obtained ~10 µg of the desired capped mRNA (both for cap 0 and FlashCap-mRNA) from a 20 µL IVT reaction (Supplementary Table 1).
After injection of FlashCap-eGFP-mRNA at 0 hpf, zebrafish embryos developed only weak green fluorescence when kept in the dark. This low basal level of eGFP indicates that FlashCap-mRNA remained translationally muted within the 24 h of observation (Fig. 2C). However, when zebrafish embryos injected with FlashCap-eGFP-mRNA were irradiated at 2 hpf with 365 nm for 20 s, they developed strong green fluorescence, almost at the level of the positive control (Fig. 2C vs. 2B). These data show that translation of FlashCap-mRNA is suppressed and can be efficiently activated by irradiation with light in living embryos.
Intrigued by the functionality of FlashCaps in vivo, we wanted to test for how long mRNA translation can be efficiently activated. Starting at 2 hpf, we briefly irradiated several sets of embryos at different timepoints post fertilization in 3 h intervals until 14 hpf (Fig. 2A/C). The timepoint of analysis was at 28 hpf in all cases (Fig. 2A). Importantly, we found that the irradiation of embryos injected with FlashCap-eGFP-mRNA even at later timepoints, such as 14 hpf, consistently led to a strong fluorescent signal. The signal was comparable to the positive control, in which cap0-eGFP-mRNA was injected and the zebrafish embryo irradiated (Fig. 2B).
Taken together, FlashCap can efficiently be incorporated during IVT with various RNA polymerases and the resulting FlashCap-mRNAs remain translationally muted in zebrafish embryos until uncaged by light. Irradiation until at least 14 hpf allows for efficient light-mediated activation of translation in zebrafish embryos.
Having in mind possible applications in the field of developmental biology, we wanted to determine the time gap between FlashCap-mRNA activation and the earliest possible detection of eGFP protein expression. We, therefore, irradiated embryos at the earliest possible stage following injection and imaged them in 2 h intervals. At 23 and 28 h post irradiation (hpi), images served as a control for comparison to previous experiments. The earliest feasible detection timepoint of photo-deprotected FlashCap-eGFP-mRNA is 2–4 h after irradiation (Fig. 3, Supplementary Figs. 4–6). We further wondered whether this time window would also be sufficient to give rise to detectable eGFP expression on a single-cell level. Therefore, embryos were injected with FlashCap-eGFP-mRNA, immediately irradiated, and subsequently dissociated into single-cell suspensions 3 and 5 hpi. Subsequent imaging of the cells under the confocal microscope showed detectable levels of eGFP expression already after 3 h, which were further increased at 5 hpi, indicating that also on a single-cell level, about 3–5 h are sufficient to detect a robust eGFP signal after activation of the FlashCap-eGFP-mRNA (Supplementary Fig. 9).

Zebrafish were irradiated directly after injection (0 h, 365 nm, 20 s) or left in the dark. Uninjected zebrafish embryos serve as the negative control. Images were taken at 0 hpi, 2 hpi, 4 hpi, 6 hpi. Shown is one representative experiment of n = 3 independent replicates. For comparability, images were also shown after 23 hpi and 28 hpi.
In addition to temporal control of translation, we were curious about the possibility of spatial control of translation. As locally restricted translation is an important feature of eukaryotes per se, but also highly desirable in an experimental setting, we asked whether FlashCap-mRNA can be activated in specific regions of a zebrafish embryo. To this end, zebrafish embryos were injected at 0 hpf with FlashCap-eGFP-mRNA as described above and kept in the dark until the following day. After manual removal of the chorion, 24 hpf embryos were embedded in a glass-bottom dish using 0.8% low melting point agarose, and a defined region within the trunk of the embryo was irradiated using a CLSM (Fig. 4A). To achieve local irradiation, embryos were embedded laterally, and a z-stack spanning the full depth of the embryo at the indicated position (white square in Fig. 4B, approximately 0.054 mm2 or 4 somites in width) was irradiated in 4 µm steps using the 405 nm laser (2x scanning with 100% laser power per z-plane). Embryos were subsequently transferred into the E3 medium again and kept in a dark incubator. Analysis of induced eGFP production was performed on the following day at 48 hpf. We observed a prominent fluorescent signal in the irradiated region (Fig. 4B), indicating an obvious induction of eGFP production after local uncaging of the FlashCap-eGFP-mRNA. The directly adjacent somites, which were also partially included in the irradiated area due to the V-shape of the somites, showed a slightly reduced induction of eGFP expression (see false-color version depicting pixel intensities in Fig. 4B). However, we consistently also noted a weaker eGFP expression outside of the target region, reflecting a baseline production of eGFP even without laser-induced photo-deprotection of the mRNA. Whether this basal eGFP signal reflects a general leakiness in the FlashCap system or whether this expression results from an unintended photo-deprotection of FlashCap-mRNA by exposure of the embryos to light during injection and subsequent handling or from other environmental effects is currently unclear. Although we have tried to minimize the exposure of embryos to any kind of light, it cannot be eliminated completely.

A Schematic illustration of light-mediated local activation of translation in zebrafish embryos. FlashCap-mRNA is injected at the one-cell stage. Local irradiation in the trunk area is initiated at 24 hpf using the confocal laser scanning microscope and a 405 nm laser. The zebrafish embryo is then imaged at 48 hpf. B Z-projection of the GFP channel in the trunk region of an embryo at 48 hpf. Left: After local irradiation of the indicated area in the trunk (boxed area) of a FlashCap-eGFP-mRNA injected zebrafish embryo at 24 hpf, a strong induction of eGFP expression is evident at 48 hpf. Right: False-colored version of the GFP channel depicting different pixel intensities in different colors and highlighting the strong induction of eGFP expression in the irradiated area. Shown is one representative experiment of n = 3 independent replicates.
This notwithstanding, these results provide a proof-of-concept that ubiquitously distributed muted mRNA can be activated for translation with temporal and spatial control in developing zebrafish embryos. After injection at 0 hpf, the FlashCap-eGFP-mRNA is most likely uniformly distributed in the developing embryo but muted. Generic irradiation of the zebrafish embryo at different timepoints leads to uniform distribution of strong eGFP fluorescence (Fig. 2). Importantly, the FlashCap-eGFP-mRNA is sufficiently stable to allow for activation at later developmental stages, in which certain regions of the embryo can be targeted by light.
In summary, we present an optochemical approach for activating mRNA translation by light in vivo. We used zebrafish as a model organism and found that translationally muted FlashCap-mRNA remains remarkably stable (>24 h). This feature enables light-activation of protein production in zebrafish embryos, enabling researchers to, e.g., mark defined cell populations or tissues at various developmental stages. Irradiating several segments within the trunk at 24 hpf led to a marked increase in eGFP production that could be detected 24 h later. The technique is simple to use by all researchers familiar with mRNA preparation. It requires the FlashCap, which is compatible with all steps of mRNA preparation. We anticipate the technique to facilitate in vivo studies in developmental biology.
We note, however, that in the current study, we used the baseline eGFP production of unirradiated FlashCap-eGFP-mRNAs as a reference set to zero. Further technical improvements dampening the observed baseline expression of FlashCap-mRNAs in zebrafish tissues will be important to allow for the controlled expression of more critical FlashCap-mRNAs encoding, e.g., dominant negative constructs, where even weaker background expression in non-irradiated tissues could affect normal development. For such biological studies, reducing the basal expression levels might be as important as solely focusing on further improving the activation per se, which already provides robust protein production in its current form.
This is the first demonstration of activating FlashCap-mRNA for translation in a living organism. It shows that mRNA technology bears potential in targeted therapies by activating its translation in selected regions.
Methods
FlashCaps were synthesized as previously described40. Figures were created with Biorender.
In vitro T7-transcription and purification of RNA (used for cell studies)
A linearized pmRNA plasmid containing the sequence coding for eGFP and a T7 promoter was used for the preparation of mRNA for cell studies (Supplementary Table 2). Plasmid DNA (3 µg) was incubated with 1× FastDigest buffer and 3 µL of PacI FastDigest enzyme (Thermo Fisher) for 10 min at 37 °C, followed by inactivation at 65 °C for 10 min for linearization. Dephosphorylation of the ends was achieved by adding 3 µL of FastAP and incubation at 37 °C for 15 min, followed by inactivation at 65 °C for 5 min. The DNA was purified via NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). The concentration was measured at 260 nm with a Tecan Infinite M1000 PRO instrument. The resulting linear dsDNA was used as a template (400 ng). The in vitro transcription was performed with T7 polymerase (Thermo Scientific) in transcription buffer (40 mM Tris/HCl, 25 mM NaCl, 8 mM MgCl2, 2 mM spermidine (HCl)3) by adding A/m5C/m1ΨTP mix (0.5 mM), GTP (0.25 mM), the respective 5′ cap analog (1 mM) (FlashCap was previously synthesized40 and m7GpppG (Jena Bioscience)), T7 RNA polymerase (50 U; Thermo Scientific) and pyrophosphatase (0.1 U; Thermo Scientific) for 4 h at 37 °C. 2 U of DNase I was added for 1 h at 37 °C to remove the DNA. The mRNA was purified using the RNA Clean & Concentrator™-5 kit (Zymo Research). Non-capped RNAs were digested by 10 U of RNA 5′-polyphosphatase (Epicenter). After an incubation period of 30 min at 37 °C, 0.5 U of the 5′–3′ exoribonuclease XRN-1 (NEB) and MgCl2 (5 mM) were added. The reaction mixture was incubated for 1 h at 37 °C. Subsequently, capped mRNAs were purified using the RNA Clean & Concentrator™-5 kit (Zymo Research). RNA integrity and length were confirmed via 7.5% polyacrylamide gel electrophoresis. For this purpose, 100 ng mRNA was loaded onto PAA gel, and electrophoresis was performed for 1 h in 1 × Tris/Borate/EDTA (TBE) buffer).
In vitro SP6-transcription and purification of RNA used for in vivo studies
For zebrafish experiments, a linearized pCS2+ vector containing the sequence coding for eGFP and the SP6 promoter was used (Supplementary Table 2 and Supplementary Fig. 7). This construct (3 µg pDNA) was linearized in 1x FastDigest buffer by incubation with 3 µL of FastDigest enzyme Not I (Thermo Fisher Scientific) for 30 min at 37 °C and inactivated at 80 °C for 5 min. The generation of eGFP-mRNA was carried out with the Invitrogen™ MEGAscript™ SP6 Transcription Kit (Invitrogen) according to the manufacturer´s instructions. In a 20 µL reaction, 1 µg of linearized plasmid, 2 µL of ATP, CTP, and UTP, and 0.5 µL of GTP from the kit’s stock solutions were mixed according to the manufacturer’s instructions. Moreover, 2 µL of 10x reaction buffer, 1 µL of RiboLock, and 4 µL of 5′ cap analog were added. Finally, 2 µL of the enzyme mix was added to the mixture. The IVT reaction was incubated for 4 h at 37 °C, followed by incubation with Turbo DNase I for 15 min at 37 °C, and purification using the RNA Clean & Concentrator™-5 Kit (Zymo Research) according to the manufacturer´s instructions. The concentration was determined via absorbance at 260 nm. Using 5′-polyphosphatase (20 U) in 1x polyphosphatase buffer (Biozym Scientific GmbH) for 30 min at 37 °C, followed by MgCl2 (5.3 mM) and XRN-1 (1 U) (New England Biolabs GmbH) treatment for 1 h at 37 °C uncapped mRNA was digested. Again, purification was performed using the RNA Clean & Concentrator™-5 Kit (Zymo Research). Yeast poly(A) polymerase was used for post-transcriptional poly(A) addition. In a total volume of 20 µL, 3–4 µg mRNA, ATP (1 mM), and poly(A) polymerase (600 U) were incubated in 1x poly(A) polymerase-reaction buffer at 37 °C for 1 h. Afterwards, the mRNA was precipitated overnight at –20 °C using isopropanol, glycogen, and sodium acetate (NaOAc, final concentration: 0.3 M), followed by washing with 70% ethanol. HPLC purification of mRNA was performed using RiboSep RNA Column (ConciseSeparations), 0.1 M Triethylammonium acetate (TEAA) in water, and elution buffer 0.1 M TEAA in 25% acetonitrile (55 °C, 0.9 mL/min). RNA was collected based on the absorption at 260 nm. To the isolated mRNA 3 M NaOAc (final concentration: 0.3 M), 0.5 µL glycogen and 1.2 vol.-% isopropanol were added for precipitation. After flash freezing in liquid nitrogen and centrifugation for 1 h, at 4 °C and 21.000 × g, the mRNA was washed with 70% ethanol. After an additional 30 min centrifugation, the supernatant was removed, and the mRNA was dried and dissolved in 15 µL ddH2O. The concentration was determined via absorbance at 260 nm. RNA integrity and length were confirmed via 7.5% polyacrylamide gel electrophoresis. For this purpose, 100 ng mRNA was loaded onto PAA gel, and electrophoresis was performed for 1 h in 1× Tris/Borate/ EDTA (TBE) buffer).
Complete irradiation and stereo microscopy of zebrafish embryos
Wildtype zebrafish embryos were injected with mRNA with indicated 5′ caps. One nanoliter, equaling 70 pg of mRNA, was injected into the yolk of zebrafish embryos directly after egg deposition. Zebrafish embryos were subsequently kept in the dark (no direct LED irradiation) until irradiation. Embryos were cultivated in an incubator at 28.5 °C. Non-fertilized eggs and dead embryos were removed after 5 hpf and 10 hpf. Generic irradiation of the zebrafish embryos was performed at indicated timepoints (2 hpf, 5 hpf, 8 hpf, 11 hpf, 14 hpf) in a 96-well plate at 365 nm (460 mW/cm2) for 20 s using an LED with a collimator lens (Supplementary Fig. 8). The UV–A–LED (λmax = 365 nm) was operated with 1700 mA (for details, see ref. 40). After irradiation, the embryos were placed in a petri dish containing E3 medium. Before the images were taken, the chorion was removed with tweezers. The eGFP fluorescence was imaged at 24 hpf. For image acquisition, the gain was set as 17x, and for DIC images at 1.5x, the LED power was set to 70%, and the exposure time for the eGFP excitation was set to 200 ms (filter block: EX: 480/40, DM: 510, BA: 510). During image acquisition, the zebrafish embryos were sedated using a final concentration of 0.014% Tricaine Methanesulfonate (MS-222). For all microscopy images, contrast, and brightness adjustments were performed with ImageJ.
Dissociation of zebrafish embryos into single-cell suspensions
In total, 40 embryos per condition were dechorionized with 200 µL Pronase (30 mg/mL) at 3 and 5 h post irradiation. The remaining E3 medium was replaced with 600ul 1xPBS and incubated at RT for 1 min. For the dissociation, the embryos were incubated in 1 mL 0.5%trypsin-EDTA (Gilbco) at 30 °C for 10 min. After the addition of 300 µL 1xPBS, the mixture was centrifuged at RT for 5 min at 900 rpm. The supernatant was discarded, whereby care was taken to remove lipid droplets accumulated at the surface of the supernatant and the pellet was resuspended in 200 µL 4% BSA in PBS. Cell suspensions were subsequently imaged on a Leica SP8 confocal microscope using a 40x/1.10 water objective.
Local irradiation and confocal laser scanning microscopy of zebrafish embryos
Zebrafish embryos were injected and thereafter incubated at 28.5 °C as described above. At 24 hpf, zebrafish embryos were manually dechorionated, embedded in a lateral orientation in 0.8% low melting point agarose (LPA) supplied with 0.0168% Tricaine Methanesulfonate (MS-222) in a glass-bottom imaging dish. Zebrafish embryos were irradiated using a Leica SP8 confocal laser scanning microscope (CLSM) with a 405 nm laser, which was set to 100% power. In the target area, a z-stack spanning the whole depth of the embryo was defined with a step size of 4 µm. Each plane of the stack was scanned twice with the 405 laser line. After the local irradiation, the zebrafish embryos were placed back in normal embryo medium (E3) and kept in a dark incubator at 28.5 °C for 24 h. For image acquisition, zebrafish embryos were embedded again in 0.8% LPA with 0.0168% Tricaine Methanesulfonate. Images were taken on a Leica SP8 CLSM using a 20x/0.75 dry objective. Z-projections and image processing were done employing Fiji [(Fiji Is Just)ImageJ 2.14.0 /1.54 f].
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the results reported in this paper are available in the Supporting Information.
References
Eisenstein, R. & Harper, A. Characterization of a protein synthesis system from rat liver. Translation of endogenous and exogenous messenger RNA. J. Biol. Chem. 259, 9922–9928 (1984).
Hogan, M. J. & Pardi, N. mRNA vaccines in the COVID-19 pandemic and beyond. Annu. Rev. Med. 73, 17–39 (2022).
Vavilis, T. et al. mRNA in the context of protein replacement therapy. Pharmaceutics 15, 166 (2023).
Shatkin, A. Capping of eucaryotic mRNAs. Cell 9, 645–653 (1976).
Izaurralde, E., Stepinski, J., Darzynkiewicz, E. & Mattaj, I. W. A cap binding protein that may mediate nuclear export of RNA polymerase II-transcribed RNAs. J. Cell Biol. 118, 1287–1295 (1992).
Decroly, E., Ferron, F., Lescar, J. & Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 10, 51–65 (2012).
Izaurralde, E. et al. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell 78, 657–668 (1994).
Jiao, X., Chang, J. H., Kilic, T., Tong, L. & Kiledjian, M. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol. Cell 50, 104–115 (2013).
Flaherty, S. M., Fortes, P., Izaurralde, E., Mattaj, I. W. & Gilmartin, G. M. Participation of the nuclear cap binding complex in pre-mRNA 3′ processing. Proc. Natl Acad. Sci. USA 94, 11893–11898 (1997).
Furuichi, Y., LaFiandra, A. & Shatkin, A. J. 5’-Terminal structure and mRNA stability. Nature 266, 235–239 (1977).
Topisirovic, I., Svitkin, Y. V., Sonenberg, N. & Shatkin, A. J. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip. Rev. RNA 2, 277–298 (2011).
Borden, K. L. B. & Volpon, L. The diversity, plasticity, and adaptability of cap-dependent translation initiation and the associated machinery. RNA Biol. 17, 1239–1251 (2020).
Merrick, W. C. Cap-dependent and cap-independent translation in eukaryotic systems. Gene 332, 1–11 (2004).
Besse, F. & Ephrussi, A. Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat. Rev. Mol. Cell Biol. 9, 971–980 (2008).
Gingold, H. et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 158, 1281–1292 (2014).
Holler, K. et al. Spatio-temporal mRNA tracking in the early zebrafish embryo. Nat. Commun. 12, 3358 (2021).
Tadros, W. & Lipshitz, H. D. The maternal-to-zygotic transition: a play in two acts. Development 136, 3033–3042 (2009).
Winata, C. L. et al. Cytoplasmic polyadenylation-mediated translational control of maternal mRNAs directs maternal-to-zygotic transition. Development 145, dev159566 (2018).
Feitsma, H. & Cuppen, E. Zebrafish as a cancer model. Mol. Cancer Res. 6, 685–694 (2008).
Steenbergen, P. J., Richardson, M. K. & Champagne, D. L. The use of the zebrafish model in stress research. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 35, 1432–1451 (2011).
Adams, M. M. & Kafaligonul, H. Zebrafish—a model organism for studying the neurobiological mechanisms underlying cognitive brain aging and use of potential interventions. Front. Cell Dev. Biol. 6, 135 (2018).
Dawid, I. B. Developmental biology of zebrafish. Ann. N. Y. Acad. Sci. 1038, 88–93 (2004).
Basu, S. & Sachidanandan, C. Zebrafish: a multifaceted tool for chemical biologists. Chem. Rev. 113, 7952–7980 (2013).
Davison, J. M. et al. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev. Biol. 304, 811–824 (2007).
Halloran, M. C. et al. Laser-induced gene expression in specific cells of transgenic zebrafish. Development 127, 1953–1960 (2000).
Knopf, F. et al. Dually inducible TetON systems for tissue-specific conditional gene expression in zebrafish. Proc. Natl Acad. Sci. USA 107, 19933–19938 (2010).
Gilmour, D., Jessen, J. R. & Lin, S. In Zebrafish, A Practical Approach. Vol. 261, Ch. 5 (Oxford Academic, 2002).
Westerich, K. J. et al. Bioorthogonal mRNA labeling at the poly(A) tail for imaging localization and dynamics in live zebrafish embryos. Chem. Sci. 11, 3089–3095 (2020).
Nadarajah, N. et al. A novel splice-site mutation in VEGFC is associated with congenital primary lymphoedema of gordon. Int. J. Mol. Sci. 19, 2259 (2018).
Nasevicius, A. & Ekker, S. C. Effective targeted gene ‘knockdown’in zebrafish. Nat. Genet. 26, 216–220 (2000).
Draper, B. W., Morcos, P. A. & Kimmel, C. B. Inhibition of zebrafish fgf8 pre‐mRNA splicing with morpholino oligos: A quantifiable method for gene knockdown. Genesis 30, 154–156 (2001).
Deiters, A. et al. Photocaged morpholino oligomers for the light-regulation of gene function in zebrafish and Xenopus embryos. J. Am. Chem. Soc. 132, 15644–15650 (2010).
Wang, Y. et al. Manipulation of gene expression in zebrafish using caged circular morpholino oligomers. Nucleic Acids Res. 40, 11155–11162 (2012).
Shestopalov, I. A., Sinha, S. & Chen, J. K. Light-controlled gene silencing in zebrafish embryos. Nat. Chem. Biol. 3, 650–651 (2007).
Kok, F. O. et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97–108 (2015).
Eve, A. M., Place, E. S. & Smith, J. C. Comparison of Zebrafish tmem88a mutant and morpholino knockdown phenotypes. PLoS ONE 12, e0172227 (2017).
Place, E. S. & Smith, J. C. Zebrafish atoh8 mutants do not recapitulate morpholino phenotypes. PLoS ONE 12, e0171143 (2017).
Ando, H., Furuta, T., Tsien, R. Y. & Okamoto, H. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat. Genet. 28, 317–325 (2001).
Ogasawara, S. Duration control of protein expression in vivo by light-mediated reversible activation of translation. ACS Chem. Biol. 12, 351–356 (2017).
Klöcker, N. et al. Photocaged 5′ cap analogues for optical control of mRNA translation in cells. Nat. Chem. 14, 905–913 (2022).
Bollu, A. et al. Light‐activated translation of different mRNAs in cells via wavelength‐dependent photouncaging. Angew. Chem. Int. Ed. 62, e202209975 (2023).
Cornelissen, N. et al. Post-synthetic benzylation of the mRNA 5′ cap via enzymatic cascade reactions. Chem. Sci. 14, 10962–10970 (2023).
White, R. J. et al. A high-resolution mRNA expression time course of embryonic development in zebrafish. Elife 6, e30860 (2017).
Howley, C. & Ho, R. K. mRNA localization patterns in zebrafish oocytes. Mech. Dev. 92, 305–309 (2000).
Andrade, T. S. et al. Zebrafish embryo tolerance to environmental stress factors-concentration-dose response analysis of oxygen limitation, pH, and UV-light irradiation. Environ. Toxicol. Chem. 36, 682–690 (2017).
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 772280; A.R.). We thank M. Hußmann for assistance with zebrafish handling and for providing us with injection needles (University of Münster). Further, we thank Thomas Zobel and Sarah Weischer for their excellent support and technical explanations of the CLSM (Multiscale Imaging Center, University Münster).
Author information
Authors and Affiliations
Contributions
A.R. conceived the study. All authors (A.R., S.S.M., F.P.W., M.P., H.S., S.H., N.K., S.H., and A.v.I.) contributed to the design of experiments. F.P.W., M.P., H.S., S.H., N.K., S.H., and A.v.I. performed the experiments and collected the data. All authors (A.R., S.S.M., F.P.W., M.P., H.S., S.H., N.K., S.H., and A.v.I.) discussed the results. F.W. and A.R. wrote the first draft of the manuscript with contributions from all authors (A.R., S.S.M., F.P.W., M.P., H.S., S.H., N.K., S.H., and A.v.I.)). All authors (A.R., S.S.M., F.P.W., M.P., H.S., S.H., N.K., S.H., and A. v. I.) have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Andrea Rentmeister is a Guest Editor for Communications Chemistry’s Nucleic Acid Chemistry Collection but was not involved in the editorial review of or the decision to publish this article. N.K., F.W., and A.R. are the inventors on a European patent application (EP1184349.5, pending) of the University of Münster covering the synthesis and use of photo-cleavable 5′ cap analogs as well as RNA molecules comprising photo-cleavable 5′-cap analogs. All other authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Weissenboeck, F.P., Pieper, M., Schepers, H. et al. Spatiotemporal control of translation in live zebrafish embryos via photoprotected mRNAs. Commun Chem 8, 16 (2025). https://doi.org/10.1038/s42004-025-01411-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01411-7
This article is cited by
-
Optochemical control over mRNA translation by photocaged phosphorodiamidate morpholino oligonucleotides in vivo
Nature Communications (2025)
