
Abstract
The absence of intrinsic protons in proton-conducting oxides (PCO) is a significant challenge that limits the proton conductivity of proton-conducting perovskites, such as Y-doped BaMO3 (M = Zr, Ce), in proton ceramic fuel cells exhibit low conductivity (10-3 to 10-2 S cm-1 at 600 °C). Herein, we introduce a photo-assisted synthesis method for incorporating protons into Al-doped ceria (AlxCe1-xO2-δ, x = 0.2; M-ACO), leveraging the open cubic fluorite structure and photo-activated radical reactions. Specifically, photon-generated hydroxyl reactive \(\left({{{\rm{OH}}}}^{{{\bullet }}}\right)\) and superoxide (\({{{\rm{O}}}}_{2}^{{{\bullet }}-}\)) Radicals are generated and interact with the ACO crystal lattice, facilitating proton incorporation and resulting in the synthesis of native-proton-type PCO. This process results in a protonated (H-ACO) with a high proton conductivity of 0.14 S cm-1 and exceptional power density of 922 mW cm-2 at 500 °C. This versatile synthesis methodology offers broader development of advanced PCO for energy-related applications.
Similar content being viewed by others


Introduction
Protonic ceramics hold promise for various electrochemical processes, including hydrogen production, energy storage/conversion, ammonia synthesis, and carbon dioxide utilization, making them highly versatile and applicable across a wide range of technologies1,2,3,4. PCOs play a pivotal role as electrolytes in electrochemical devices and as substrates for electrocatalysts. These materials are fundamental to the operation of proton ceramic fuel cells (PCFCs), protonic ceramic electrolysis cells (PCECs), and reversible proton ceramic cells, all of which are at the cutting edge of energy conversion and storage technologies5,6,7,8. Their ability to transport protons efficiently, with low migration barriers and high proton mobility, enables exceptional ionic conductivity across a broad temperature spectrum, which is crucial for optimizing device performance9,10,11. Commonly used PCOs include proton-conducting perovskite oxides such as Y-doped barium zirconate (BZY) and barium cerate (BCY). Non-native proton containment limits the conductivity of BZY/BCY, typically ranging from 10−3 to 10−2 S cm−1 at 600 °C, posing significant obstacles to the advancement of PCFCs12,13,14. The absence of intrinsic proton carriers in these materials leads to reliance on external sources for proton conduction, which severely hinders performance. Traditional synthesis approaches, such as high-temperature solid-state reactions and wet chemical methods, often exclude protons from the bulk lattice15, typically due to the high sintering temperatures exceeding 1000 °C. Moreover, proton incorporation for conductivity relies solely on external proton sources, like water vapor or hydrogen. This constraint underscores the urgent need for novel synthesis techniques capable of increasing native proton concentration within the oxide lattice while maintaining structural integrity.
Recent advances in the development of proton-conducting perovskites for PCFCs have introduced novel synthesis techniques. One such synthesis approach is microwave sintering, which uses microwave radiation to heat materials through dielectric loss, resulting in efficient and uniform volumetric heating. This process offers several advantages over traditional sintering, including faster heating rates, greater energy efficiency, and improved microstructural control. By minimizing grain growth and coarsening, microwave sintering helps preserve the fine microstructures necessary for enhancing proton conductivity in ceramic oxides16. As a result, it may improve ionic conductivity and structural stability at lower processing temperatures. Another method, spark plasma sintering (SPS), utilizes high-energy, low-voltage pulsed currents to generate localized plasma, and enables rapid heating under uniaxial pressure at much lower temperatures (400–500 °C) compared to conventional methods. This unique technique combines Joule heating with plastic deformation and reduces grain growth during densification while increasing proton mobility in the resultant oxides17. Despite the advances in both microwave sintering and SPS, the challenge of non-native proton containment and insufficient proton conductivity remains unresolved.
In this work, we present a paradigm-changing synthesis technique “photo-assisted protonation”. First, we utilize microwave synthesis to prepare M-ACO with an open cubic fluorite structure. This initial step creates a framework that is well-suited for subsequent proton incorporation due to its open lattice and active redox couple (Ce3+/Ce4+). Next, UV light irradiation facilitates protonation by utilizing photon energy to generate radicals, which splits water molecules to produce hydroxyl radicals \(\left({{{\rm{OH}}}}^{{{\bullet }}}\right)\), protons \(({{{\rm{H}}}}^{+})\), and superoxide (\({{{\rm{O}}}}_{2}^{{{\bullet }}-}\)) radicals. These radicals interact with the ACO lattice, enabling the incorporation of protons into the material. This radical-mediated process facilitates substantial native proton incorporation, enhancing proton mobility throughout the structure. As a result, the protonated H-ACO exhibits excellent proton conductivity of 0.14 S cm−1 at 500 °C. The combination of microwave synthesis for structural preparation and photon-induced radical reactions for protonation produces materials with superior proton conductivity and fuel cell performance. Our synthesis offers a transformative solution to the challenges faced by conventional methods that rely on elevated temperatures and chemical doping. The photo-assisted process can directly facilitate proton incorporation, leading to high proton conductivity, and can be extended to a wider range of proton-conducting oxide systems, creating a versatile platform for developing advanced PCOs for next-generation 300–500 °C PCFC and beyond. Exploring the use of other proton donors, such as organic acids, could improve proton incorporation and open new avenues for future research. This technique broadens the potential uses of photo-assisted synthesis technology, opening the door to more diverse and controllable proton-conducting oxide systems.
Experimental methodology
Figure 1 presents a scientific comparison of conventional synthesis techniques and the photo-assisted synthesis approach, emphasizing their mechanistic discrepancies and significant outcomes. Traditional methods comprehend multiple phases, such as heating and sintering, which demand substantial energy input and yield limited structural alterations. These approaches are marked by sluggish reaction kinetics and the lack of a protonation process, resulting in the absence of proton incorporation. As a result, materials produced via this route often display restricted functionalities, particularly ionic conductivity, due to the lack of structural elements that promote proton transport. Whereas, the photo-assisted synthesis approach introduces a revolutionary step involving UV irradiation, which generates radicals that drive the protonation process. This method facilitates the integration of protons onto the material’s surface, leading to improved functionality. The utilization of UV-induced radical formation not only enables more effective protonation but also introduces structural modifications that enhance proton diffusion. Additionally, the photo-assisted technique employs microwave sintering, which is more energy-efficient compared to traditional sintering processes, while preserving the integrity of the protonated structure. The comparison in the figure underscores the scientific advancements of the photo-assisted synthesis methodology. It illustrates how the introduction of UV irradiation and radical-mediated processes overcomes the limitations of conventional methods, enabling faster reaction kinetics, improved proton incorporation, and enhanced structural integration. These characteristics collectively contribute to the superior performance of the material synthesized using this method, demonstrating its potential for applications requiring high ionic conductivity and structural stability. This mechanistic differentiation highlights the unique advantages of the photo-assisted synthesis methodology in advancing the field of material science.

The figure compares the traditional synthesis methods with the photo-assisted approach, showcasing the unique advantages of the photo methodology, including enhanced proton incorporation, radical generation, and improved structural integration.
Building on the comparison presented above, the detailed mechanism of the novel “photo-assisted protonation” process is presented, utilizing UV light to stimulate radical generation and facilitate proton incorporation into metal oxide structures. Unlike traditional protonation methods that rely on heat or chemical activation, this method uses UV irradiation to generate reactive oxygen species and oxygen vacancies, which act as active sites for proton incorporation. This technology offers a significant improvement in the field of PCOs since it allows for precise regulation over the protonation process while also increasing proton conductivity. UV light’s unique ability to activate specific sites without the need for high temperatures or hazardous chemicals represents a significant advancement in material modification and proton conduction optimization.
We adopted this novel approach to introduce proton defects (such as \({{{\rm{OH}}}}_{{{\rm{o}}}}^{{{\bullet }}}\)) In pristine M-ACO utilizing the vacant oxygen vacancy sites, enhanced the protonic conductivity in the material. Initially, M-ACO was prepared using the sol-gel method, and microwave sintering (200–4000 W at a frequency of 2.45 GHz ± 50 MHz) as shown in Fig. 2. The sample was further treated under UV irradiation to yield H-ACO. The basic mechanism includes a photo-redox reaction in M-ACO in water under the action of UV light. When it is exposed to high energy light of wavelength 280 nm, it generates highly reactive oxygen species (ROS) on the surface (Fig. 2), which further interact with oxygen and water molecules adsorbed on the surface of the material, allowing protons to be incorporated into the material. This interaction generates proton defects, which considerably increase the proton conductivity of H-ACO as compared to M-ACO. This “photo-assisted protonation” process not only enhances proton incorporation but also creates active sites that increase the material’s overall performance in proton-conducting applications. Using this methodology, we can achieve a more efficient and effective modification of ACO, resulting in promising advances in fuel cell technology. The novel nature of this method has the potential to influence future research, propelling additional advances in energy materials and technology.

UV irradiation generates electron–hole pairs, promoting radical formation (\({{\rm{OH}}}^{{{\bullet }}}\) and \({{{\rm{O}}}}_{2}^{{{\bullet }}-}\)) and oxygen vacancies, which facilitate proton incorporation and enhance proton conductivity by modifying the material’s surface chemistry.
Design of protonic oxide conductor
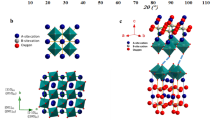
For an effective protonic conductor, the presence of protons and pathways for proton diffusion is crucial. These pathways must be accompanied by a sufficient concentration of charge carriers to achieve high protonic conductivity. Figure 3 shows a schematic illustration of the conduction pathways in ceria oxide. In doped ceria-based oxides, it is well established that oxygen vacancies and the above photo-protonation resulting proton defects act as the primary pathways for proton conduction. In the context of ACO, cerium oxide (CeO2) serves as the parent phase, a portion of Ce at the cationic sites is replaced by Al. As a result, positively charged oxygen vacancies (\({{{\rm{V}}}}_{{{\rm{O}}}}^{{{\bullet }}{{\bullet }}}\)) are generated to maintain charge balance, whereas negatively charged Al´Ce defects occur simultaneously. This can be described using Kröger–Vink notation:

a The structure of CeO2 pure phase, b the schematic structure of Al-doped CeO2, a part of Ce at the B-site is replaced by Al then oxygen vacancies \(({{{\rm{V}}}}_{{{\rm{O}}}}^{{{\bullet \bullet }}})\) are formed, c the schematic structure of H-ACO. Proton defects \(({{{\rm{OH}}}}_{{{\rm{o}}}}^{{{\bullet }}})\) are formed in H-ACO after integration of UV irradiation in water, d the schematic diagram conduction pathways in H-ACO.
When Al-doped ceria, interacts with water under UV irradiation, proton defects (\({{{\rm{OH}}}}_{{{\rm{o}}}}^{{{\bullet }}}\)) are formed at oxygen vacancy sites:
The presence of proton defects (\({{{\rm{OH}}}}_{{{\rm{o}}}}^{{{\bullet }}}\)) are essential to achieve high protonic conductivity in doped ceria structures. The mobility of protons within the material rises with the concentration of these defects. Proton conduction occurs predominantly via a hopping mechanism in which protons migrate between adjacent oxygen ions, aided by the presence of oxygen vacancies and hydroxyl groups. These defects coexist in the lattice and influence proton mobility by providing conduction pathways. The total protonic conductivity of the material is mostly determined by the presence and concentration of proton defects as well as how they move across the lattice18,19. Incorporated protons bond with lattice oxygen, creating OH defects that act as stable sites within the material. These OH defects reduce the energy barriers for proton movement, leading to enhanced proton conductivity. As a result, the material demonstrates improved proton transport properties due to the stabilizing and shielding effects of the OH defects. Effective proton conduction at lower temperatures is a crucial prerequisite for applications involving PCFCs, where this mechanism is especially helpful. In conclusion, the formation of oxygen vacancies, the creation of proton defects, and the resulting enhancement in proton mobility are all outcomes of the UV-assisted protonation process in H-CAO, which together contribute to enhanced protonic conductivity.
Results and discussion
Radical reactions and protonation evidence
Understanding the fundamental principles of proton incorporation is critical for maximizing material performance while developing enhanced proton-conducting oxides (PCOs). In this study, we investigate the radical-driven reactions triggered by UV light in M-ACO, which play an important role in producing proton defects and increasing protonic conductivity. The photo-assisted protonation process uses UV-induced electron excitation to trigger a series of redox processes on the material’s surface, as described below. M-ACO absorbs photons, promoting electrons from the valence band (VB) to the conduction band (CB) as shown in Eq. (1):
Then, these photogenerated \({{{\rm{h}}}}_{{{\rm{VB}}}}^{+}\) will react with water molecules \(({{{\rm{H}}}}_{2}{{\rm{O}}})\) adsorbed on the surface of the material to form hydroxyl radicals (OH•) and the \({{{\rm{e}}}}_{{{\rm{CB}}}}^{-}\) (electrons at the CB) will reduce \({{{\rm{O}}}}_{2}\) as \({{{{\rm{O}}}}_{2}^{-}}^{{{\bullet }}}\) (Eqs. (4)–(6)).
These reactions are significant as they begin with the generation of hydroxyl radicals, which are required for the subsequent proton incorporation into the lattice, as seen in Fig. 1. Hydroxyl radicals are highly reactive species that aggressively interact with various chemical substances20,21. The strong oxidizing nature of these radicals is attributed to their high oxidation potential. Due to their nonselective reactivity, hydroxyl radicals can effectively degrade or modify a wide range of susceptible substances, including metal oxides. The short life span of hydroxyl radicals necessitates in-situ generation, typically achieved through the oxidation of water or hydroxide ions, as delineated in Eqs. (7)–(9) or by using a combination of various oxidation methods, namely H2O2 and UV light.
The comprehensive mechanism describes the UV light-induced excitation of electrons, instigating intricate redox reactions within ACO and it can generate two distinct forms of hydrogen within the oxide lattice: one as protons bonded to oxygen ions (forming O–H bonds) and the other as highly mobile and reactive hydroxyl radicals (\({{\rm{OH}}}^{{{\bullet }}}\)). These radicals can further oxidize the ceria surface and modify its structure22,23. The formation of \({{{\rm{OH}}}}^{-}\) hydroxyl group could also play a role in modifying the crystal structure, which could impact the fuel cell’s operational characteristics. Understanding the behavior of these hydroxyl radicals and their interactions with the ceria lattice is crucial for optimizing the performance of ACO electrolytes in fuel cell applications. UV-induced radical reactions, as described in Eqs. (1) to (7), not only produce hydroxyl radicals and proton defects, but also cause major changes in the chemical bonding and structure of H-ACO. These changes can be well viewed and confirmed using several characterization techniques such as XRD, Raman, FTIR, XPS, and NMR, which provide information about changes in structure, bonding, defect formation, and proton incorporation within the materials.
Furthermore, the X-ray diffraction was performed for both samples, which shows a cubic fluorite structure like that of pure ceria (Fig. 4a). The peaks are analyzed using PDF# 75-0076 (which belongs to the Fm-3m space group)24. The XRD pattern for M-ACO shows the presence of a high-intensity peak (111) at 28.54°, confirming its cubic fluorite structure, other lower intensity peaks (200), (220), (311) was found at 33.11°, 47.54°, 56.40° were observed, all corresponding to the fluorite structure. For the H-ACO sample, all diffraction peaks show a slight shift toward higher angles: (111), (200), (220), and (311) at 28.59°, 33.17°, 47.57°, and 56.47°, respectively. The shift indicates a change in lattice parameters, most likely caused by the incorporation of protons into the crystal structure25. The crystallite size was calculated by the Scherrer formula to be 29.66 nm for M-ACO and 27 nm for H-ACO. This XRD data, supported by Rietveld refinement, gives compelling evidence for the proposed radical-driven protonation mechanism, as demonstrated by changes in lattice parameters, peak shifting, and reduced crystallite size all confirming proton incorporation into the ceria lattice. These structural changes maintain the cubic fluorite structure and prevent the secondary phase from being beneficial in the improvement of proton mobility and conductivity.

a XRD pattern, b Raman analysis, c FTIR analysis, d, e XPS spectra of M-ACO and H-ACO, highlighting their physical and chemical properties.
Figure 4b displays Raman spectra for both M-ACO and H-ACO in a comparative study. A sharp peak at 460 cm−1 and lower intensity peaks at 943 and 1054 cm−1, all associated with the fluorite typical F2g band reflecting the vibrational modes of ceria26,27,28, are notable for both samples. Two weaker second-order peaks can be seen around 261 cm−1, ascribed to double degenerate transverse acoustic (TA) mode and non-degenerate longitudinal optical (LO) mode at 615 cm−1, also observed in Fig. 4b. Intriguingly, these secondary peaks are found exclusively in the H-ACO sample, signifying the generation of defects that are created during the protonation process, specifically oxygen vacancies29,30, also see XPS results discussed later (Fig. 4e). When protons (H+ ions) are integrated, they generate hydroxyl groups (O–H) that can participate in hydrogen bonding with neighboring oxygen atoms (O–H–O bonds). Furthermore, hydroxyl groups interact with nearby vacancies (O–H vacancy interactions), providing a diverse bonding environment that influences the material’s property31,32. The Raman analysis reveals the presence of these defects associated with the photo radicals and protonation reaction of the material.
FTIR analysis (Fig. 4c) provides further pieces of evidence. A prominent band in the 3400–3430 cm−1 range, corresponding to O–H stretching vibrations, is present in H-ACO, which is significantly stronger compared to M-ACO. The increased intensity of the O–H band in the H-ACO sample demonstrates that UV-induced radical processes have successfully introduced proton defects and hydroxyl groups into the ceria lattice. This discovery emphasizes the importance of hydroxyl radicals (\({{\rm{OH}}}^{{{\bullet }}}\)) produced during UV irradiation in promoting proton incorporation into the material. Additionally, a sharp band at 1629 cm−1 is associated with the O–H bending vibrations of water molecules, indicating a higher amount of surface hydroxyl groups33. The presence of these hydroxyl groups is important to assist proton transport inside the material, contributing to the increased proton conductivity shown in the H-ACO samples34. Two distinctive bands at 1344 cm−1 and 1458 cm−1 in H-ACO emerge, which correspond to the stretching vibrations of oxygen ions and the bending modes of O–H groups, respectively35,36. These bands encounter vibrational frequency shifts as a result of the interaction between introduced protons and oxygen ions within the lattice, leading the original double peaks to degenerate into a single peak with a shoulder37. This spectrum shift is an obvious consequence of protonation, emphasizing the structural changes. The H-ACO samples show a band at 1540 cm−1, which corresponds to the bending mode of H–OH adsorbed on the surface38. The absence of 1344 cm−1 and 1458 cm−1 peaks in M-ACO samples demonstrates that the photo-assisted protonation process made significant structural modifications (Fig. 4c). The FTIR spectra show that the UV-assisted process successfully incorporates proton defects, as demonstrated by the creation of O–H bonds and surface hydroxyl groups (Eqs. (7)–(9)), as well as other distinctive peaks at 1344 cm−1 and 1458 cm−1.
XPS (Fig. 4d) was deployed to investigate the surface chemical state of Ce 3d, and O1s of M-ACO and H-ACO, also support the material design methodology, revealing the impacts of photo-assisted protonation. The relative percentages of Ce and O species were obtained based on the peak area ratio of the XPS spectra. The Ce 3d spectra show eight distinct peaks that correspond to the spin-orbit pairs of Ce 3d5/2 (v) and Ce 3d3/2 (u), deconvoluted using a mixed Gaussian-Lorentzian function. The binding energies (BE) and relative area ratios of the different peaks are listed in Supplementary Table. 1. The peaks v1(884.65 eV) and u1(902.40 eV) are assigned to Ce3+, while others correspond to Ce4+39,40. A quantitative analysis shows that H-ACO has a greater relative Ce3+ concentration (19.3%) on the ceria surface than that of M-ACO (17.9%), indicating enhanced Ce⁴⁺ reduction, due to photo-activated H+ incorporation into the lattice. As a result of receiving one H+ into the lattice, Ce4+ must be reduced to Ce3+ for charge neutrality (Fig. 4e).
The O 1s spectra (Fig. 4f) further support these findings. The prominent peak around 528.94 eV (Olat) corresponds to lattice oxygen, while the peak around 529.83 eV (Ovac) reflects O-vacancies, and the peak at 531.29 eV (Osur) indicated surface hydroxyl-groups and the ratio of oxygen species, specifically the Ovac/Ovac + Olat + Osur, indicates the relative abundance of surface oxygen vacancies41,42. The quantitative analysis shows that in M-ACO the oxygen lattice (Olat) predominates (54.9%), while surface oxygen species (Osur) and oxygen vacancies (Ovac) are relatively low at 40.37% and 5.04%, respectively. While photo-assisted protonation in H-ACO, the surface hydroxyl content (Osur) increases to 43.38%, and Ovac% rises to 6.5%, indicating a significant enhancement in oxygen vacancies, which facilitates defect creation and proton incorporation.
The detailed surface morphology of the powders was analyzed via scanning electron microscopy (SEM). The SEM analysis reveals distinct morphological differences between M-ACO and H-ACO. M-ACO exhibits a porous structure with higher surface area, typically favorable for surface-mediated ionic conduction in our case (Fig. 5a). In contrast, H-ACO displays a denser, tightly packed morphology, not favor for surface ionic transport. However, the observed higher ionic conductivity in H-ACO contradicts the morphology-driven expectations, confirming that the enhancement is not due to microstructural factors, as shown in Fig. 5b. Instead, this improvement is attributed to the photo-protonation process. The photo-assisted synthesis enhances intrinsic proton concentration and creates favorable conduction pathways, overriding the impact of reduced porosity.

a SEM image of M-ACO, b SEM image of H-ACO, c–f elemental mapping images of M-ACO elements (Al, Ce, O), g M-ACO interface h, EDS analysis of M-ACO.
The morphological characteristics of the M-ACO sample were analyzed using high-resolution transmission electron microscopy (HR-TEM) and energy-dispersive X-ray spectroscopy (EDS) (Fig. 5c–f). The HR-TEM images reveal distinct interfaces between individual grains in the M-ACO structure. The lattice planes and interplanar spacing (d-spacing) were measured using both the line intercept method and Digital Micrograph software. The measured d-spacing of 0.315 nm is consistent with the expected crystallographic values, confirming the successful formation of the material’s structure (Fig. 5g). The elemental distribution within the M-ACO sample was verified through EDS mapping analysis, which shows the uniform distribution of Al, Ce, and O elements throughout the sample (Fig. 5h). These results demonstrate the successful synthesis of the M-ACO structure with well-defined morphological features.
Figure 6a shows electron spin resonance (ESR) spectra of the ACO and H-ACO samples, highlighting the differences in oxygen vacancy concentrations and radical species between the two materials. The M-ACO sample exhibits a low-intensity paramagnetic resonance signal at g = 1.9626 due to unpaired electrons associated with the oxygen vacancy defects, i.e., the M-ACO sample has a sizable fraction of oxygen vacancy defects. This signal indicates that even in the non-protonated state, M-ACO possesses some oxygen vacancies, which are fundamental to the material’s intrinsic properties. Conversely, the EPR spectrum for H-ACO shows two distinct paramagnetic resonance signals notably with higher intensity. This observation indicates a substantial increase in oxygen vacancies as a result of the protonation process. The stronger ESR signals in H-ACO suggest that the protonation not only increases the number of oxygen vacancies but also contributes to the formation of Hydroxyl radicals \(\left({{{\rm{OH}}}}^{{{\bullet }}}\right)\) associated with these vacancies. This enhanced presence of oxygen vacancies in the protonated sample underscores their critical role in facilitating radical stabilization and potentially improving the material’s performance by providing effective pathways for proton diffusion and interaction. This direct characterization of oxygen vacancies in H-ACO demonstrated through increased intensity and multiple peaks in the ESR spectrum, provides vigorous evidence supporting the transformative effect of the protonation process on the material’s structural and electronic properties.

a ESR spectra of M-ACO and H-ACO, highlighting oxygen vacancy formation and radical presence upon protonation, b TGA analysis of M-ACO and H-ACO, highlighting thermal stability and weight changes upon protonation.
Thermogravimetric analysis (TGA) was conducted to evaluate the thermal stability and volatile content of M-ACO and H-ACO samples. The results from TGA are presented in Fig. 6b, depicting two distinct thermal behaviors indicative of differences in composition and volatile retention between the samples. Thermogravimetric analysis (TGA) of M-ACO and H-ACO samples reveals distinct differences in thermal behavior and stability. M-ACO shows a significant initial weight loss of 7.94% between 150 and 450 °C, indicating a high content of volatile components that are readily lost, likely due to the evaporation or decomposition of water. This rapid loss suggests a less stable matrix with a high proportion of easily decomposable components. A further weight loss of 0.84% from 450 to 550 °C in M-ACO indicates crystal-absorbed water and moisture resulting in no decomposition reaction. Conversely, H-ACO exhibits a more controlled and lower total weight loss of 5.76% in the 150–450 °C range, reflecting its enhanced stability and ability to retain structural integrity due to protonation. The minimal weight loss of 0.36% between 450 and 550 °C further underscores the strength of H-ACO, suggesting a tightly bound matrix that is less disposed to thermal degradation compared to M-ACO. The gradual mass loss rate in H-ACO indicates that the material structure retains its volatiles more effectively, pointing to enhanced thermal stability and possibly a higher efficacy in proton retention. In contrast, M-ACO, lacking such treatment, demonstrates a lesser capacity for volatile retention, indicative of a simpler matrix. The differences in thermal degradation contours between M-ACO and H-ACO highlight the impact of protonation on material properties, notably affecting their thermal stability and volatile handling characteristics.
Magic angle spinning (MAS) NMR is used to identify structural changes, particularly as it undergoes the process of photo-assisted protonation. The spectra are shown in Fig. 7. The cerium cations in CeO2 are all eight-coordinated Ce (VIII)). When Ce atoms were substituted by Al atoms, the latter atoms will exist as six coordination (AlO6, Al (VI)), five (AlO5, Al (VI)), and four (AlO4, Al (IV)) coordination with possible formation of oxygen vacancies. The spectra (Fig. 4a) display two main peaks at 4 and 30 ppm and minor signals at 70 ppm, corresponding to Al in six (VI)-, five (V)-, and four (IV)-coordinated environments, respectively43,44. After protonation in H-ACO, the relative amount of Al (V) increases, suggesting enhanced oxygen vacancy to maintain charge neutrality. The slight upfield shift in the 27Al (VI) NMR signal (maximum position from 4 ppm to near 0 ppm) following photo-activation could be associated with aluminum species coordinated with protons or hydroxyl groups (Al3+–OH environment)45, introduced through the protonation process, as depicted in Fig. 7a. This shift in 27Al NMR spectrum between M-ACO and H-ACO reflects structural changes around aluminum atoms due proton incorporation, indicating successful protonation and restructuring environments.

a 27Al and b 1H NMR spectra of M-ACO and H-ACO before and after proton injection. The chemical shifts are noted with purple numbers.
1H NMR spectra of M-ACO and H-ACO (Fig. 7b) exhibit signals of double-bridged hydroxyl groups at 0.8–2.0 ppm, with an extra-framework Al-OH signal at 2.1 ppm46, which is consistent with the results of 27Al spectra, confirming protons generation around Al. A tailed signal at ~8 ppm corresponds to surface hydroxyls, while the main signal at ~5.4 ppm is assigned to lattice -OH, i.e., protons in oxide lattice47,48. Compared to M-ACO, the increased signal of double-bridged hydroxyls in H-ACO suggests the generation of reactive oxygen species and surface defects during the photo-activation process. H-ACO shows significant peak intensities at 5.4 ppm, 2.1 ppm, 1.4 ppm, and 0.9 ppm, indicating the successful incorporation of protons into various environments within the lattice. This difference highlights the effectiveness of our photo-assisted synthesis method in introducing intrinsic, mobile protons, fundamentally differentiating H-ACO from M-ACO, which lacks lattice-bound protons. Additionally, the spectrum of M-ACO shows minimal peak intensity, indicating an almost negligible proton presence. This confirms that M-ACO has no substantial proton concentration before undergoing the novel synthesis process. These findings demonstrate that the photo-assisted protonation process not only modifies aluminum coordination but also creates a new proton environment, enhancing proton conductivity in H-ACO. All the above structural characterizations and analyses suggest that photo-assisted protonation with UV-induced radical reactions and protonation optimize the material for proton conduction, making it ideal for PCOs and fuel cell applications.
The electrochemical performances of M-ACO and H-ACO-based PCFC across 500–300 °C are presented in Fig. 8. The peak power outputs are summarized in the Supplementary Table 2. PCFC based on H-ACO consistently outperforms that with M-ACO, with a peak power density of 922 mW cm−2 at 500 °C vs. 719 mW cm−2 for M-ACO. Even at 380 °C H-ACO maintain a high-power density of 484 mW cm−2, while drops to 125 mW cm−2 for M-ACO. Below 350 °C, only the H-ACO-based cells remain operational, delivering power densities of 344, 203, and 78 mW cm-2 at 350, 320, and 300 °C, respectively (Fig. 8a, b). These results highlight the superior performance of H-ACO across the entire temperature range, significantly outperforming previous works49,50. The superior performance of H-ACO is attributed to proton incorporation facilitated by UV-induced radical reactions, which enhance protonic conduction via the protonation of the material. As the temperature decreases, the performance gap between M-ACO and H-ACO becomes more noticeable. At lower temperatures, H-ACO exhibits even better conductivity than M-ACO due to the stabilization of proton conduction channels established by the photo-assisted protonation process.

a, b IV-IP characteristics of M-ACO and H-ACO operated at various temperatures, showcasing their respective efficiency and output characteristics, c, d Nyquist plot obtained under an OCV condition for M-ACO and H-ACO, providing insights into their electrochemical behavior, e, f activation energy measurements obtained from IV-IP curves, and from the Nyquist plot for M-ACO and H-ACO, highlighting their energy conversion efficiency.
The Nyquist plots, under OCV conditions for both M-ACO and H-ACO-based PCFC in an Air/H2 atmosphere (300–500 °C) were analyzed and fitted by the equivalent circuit in ZView2 software (Fig. 8c, d). In this equivalent circuit, Rb, Rgb, and Rct signify the bulk, grain-boundary, and electrode polarization resistance, respectively, and were subsequently converted into corresponding conductivities for temperature dependence analysis (Supplementary Table. 3). The H-ACO sample shows significantly lower bulk and grain boundary resistances, 0.083 and 0.054 Ω cm2 compared to M-ACO, 0.15 and 0.09 Ω cm2 at 500 °C, respectively, indicating enhanced ionic conductivity due to photo-assisted protonation process. Notably, at temperatures of 500 °C to 300 °C, the H-ACO showcased lower Ro values of 0.137–0.981 Ω cm2 (Fig. 8d and Supplementary Fig. 1c, d) compared to the M-ACO’s values of 0.245-99.51 Ω cm2 (Fig. 8c and Supplementary Fig. 1a, b). This reduction in Ro for H-CAO is associated with protons incorporation which enhances ionic conduction. Thus, proton incorporation via radical reactions boosts charge transfer efficiency and stabilizes electrochemical performance over a broad temperature range, making H-ACO more efficient for PCFC applications.
The temperature-dependent conductivity measurements confirm H-ACO’s superiority with conductivity of 0.36 S cm−1 (at 500 °C) and lower activation energy Ea = 0.36 eV, compared to M-ACO’s conductivity of 0.20 S cm−1 (at 500 °C) and higher activation energy Ea1 = 0.63 and Ea2 = 1.14 eV which shows transition effect (Fig. 8f). Similarly, ionic conductivity from the IV-IP curves shows H-ACO with 0.14 S cm−1 (at 500 °C) and exhibited lower activation Ea1 = 0.30 and Ea2 = 0.95 eV compared to M-ACO which has 0.11 S cm−1 ionic conductivity (at 500 °C) and higher activation energy Ea1 = 0.32 and Ea2 = 1.52e as depicted in Fig. 8e. This higher conductivity in H-ACO attributed to the “photo-assisted protonation” process which enhance protons and oxygen ion transport within the lattice, ultimately enhances fuel cell performance under Air/H2 conditions. These findings highlight the significant advantages of H-ACO for PCFC applications, due to its higher ionic conductivity and lower activation energy when compared to M-ACO.
Following the successful protonation of H-ACO, its DC conductivity was evaluated at 500 °C using the Hebb–Wagner method under N2 atmosphere to quantify the material’s ionic conductivity, as shown in Fig. 9a. Given the negligible electronic conductivity of H-ACO, the measurement primarily reflects proton transport facilitated by the protonation process. The prepared cell was subjected to a constant potential of 1 V for approximately 30 min, and the DC conductivity (σ) was determined using the formula:
where L is the cell thickness, I is the current, A is the cross-sectional area, and V is the applied voltage.

a DC polarization conductivity in N2 atmosphere, b, c proton conductivity as a function of water partial pressure at 500 °C and 400 °C, d stability under constant current conditions, confirming long-term durability.
The electronic conductivity of the H-ACO material at 500 °C was measured to be 7.85 × 10−5 S cm−1, confirming the dominant ionic conduction mechanism. The negligible electronic contribution aligns with the material’s protonated nature, emphasizing its role as a pure proton conductor. This result further validates the success of the protonation process, which enhances ionic pathways and facilitates proton transport across the membrane. The DC polarization study solidifies the material’s potential as a high-performance proton-conducting electrolyte for advanced applications.
To analyze the behavior of the H-ACO electrolyte under varying water partial pressures, electrochemical impedance spectroscopy (EIS) was conducted at 500 °C and 400 °C. Initially, measurements were carried out in an air–air atmosphere to establish a baseline for comparison. Subsequently, one gas feed was replaced with water vapor, and its partial pressure was systematically varied to investigate its influence on the electrochemical properties. As shown in Supplementary Fig. 8a, b, increasing water partial pressure resulted in a noticeable reduction in both Ohmic and polarization resistances at both temperatures. This indicates enhanced proton (H+) transport and increased mobility of hydroxyl (OH−) ions across the H-ACO membrane. At 500 °C, the conductivity in air was calculated to be approximately 5.88 × 10−6 S cm−1. Upon introducing water vapor, the conductivity progressively increased, stabilizing at 6.28 × 10−1 S cm−1 at the highest water vapor flow rate. A similar trend was observed at 400 °C, where the conductivity initially measured 2.38 × 10−6 S cm−1 in air and improved significantly with the addition of water vapor, reaching a stable value of 4.33 × 10−1 S cm−1 (Fig. 9b, c). The observed enhancement in conductivity under water vapor conditions is attributed to the higher availability of protons and hydroxyl ions, which facilitate ion transport through the membrane. Water molecules create additional pathways for proton conduction, reducing resistances and significantly improving overall ionic conductivity. This demonstrates that water vapor serves as a superior medium to air for enhancing proton conduction in H-ACO membranes.
Furthermore, to evaluate the stability and feasibility of the material under operational conditions, long-term stability tests were conducted using a fuel cell configuration incorporating the synthesized H-ACO material as the electrolyte. The tests were performed at 500 °C under H₂/air conditions. Under a constant current density of 110 mA cm−2, stable operation was achieved for 250 h, maintaining an operational voltage above 0.80 V, as shown in Fig. 9d. Minor fluctuations in the operational voltage were observed, attributed to variations in experimental setup and environmental conditions, such as slight changes in gas flow or humidity. The stability of H-ACO is due to the enhanced surface interactions facilitated by protonation, which creates a dynamic equilibrium between oxygen vacancies and proton carriers, preventing structural degradation and maintaining consistent performance under operational conditions. These results confirm that the H-ACO material demonstrates good stability and functionality as an electrolyte under fuel cell operating conditions. Its durability could be further enhanced with optimized engineering efforts and better compatibility between electrodes and the electrolyte material.
More experiment validation of proton conduction was performed for M-ACO and H-ACO. Firstly, fuel cell experiments with a well-known proton conducting BaZr0.1Ce0.7Y0.2O3-δ (BZCY) filter verified both materials in proton conduction, although H-ACO achieved significantly higher power densities. Impedance spectroscopy further demonstrates the increased conductivity and improved proton diffusion in H-ACO over M-ACO (Supplementary Fig. 2). Secondly, the isotopic effect by comparing the proton conduction in H2 and D2 environments. The findings indicated a notable increase in conductivity when transitioning from D2 to H2, particularly for H-ACO, emphasizing its superior proton mobility (Supplementary Fig. 5). These results demonstrate the superior proton conduction properties and electrochemical performance of H-ACO.
Conclusion
In this pioneering work, we have developed a novel photo-assisted synthesis method for protonated Al-doped ceria (H-ACO) that utilizes UV-activated radical reactions to drive protonation, generating hydroxyl (\({{\rm{OH}}}^{{{\bullet }}}\)) and superoxide ion (\({{{\rm{O}}}}_{2}^{{{\bullet }}-}\)) radicals. These radicals enhance vacancy creation and incorporate protons directly into the oxide lattice, resulting in intrinsic mobile protons in the material, i.e., protonated H-ACO. This innovative synthesis method sets a new direction for the development of PCOs, with intrinsic protons, unlike conventional materials e.g., BZY and BZCY. The synthesized H-ACO exhibits remarkable proton conductivity of 0.14 S cm−1, lower activation energy of 0.36 eV, and high-power density of 922 mW cm−2 at 500 °C. These properties validate the effectiveness of the “photo-assisted protonation” method which outperforms conventional techniques by delivering higher intrinsic proton conductivity and significantly reducing activation energy. When directly compared to widely studied perovskite proton conductors like BZY and BCY, it not only offers superior performance but also establishes a more efficient and scalable pathway for advancing proton-conducting oxide systems. The impact of this research, the photo-assisted synthesis method, presents a novel approach for producing advanced PCOs with greatly improved performance characteristics. Consequently, this technique has the potential to shape future research platforms in materials science and technology, particularly in the realm of sustainable energy applications.
Methods
Step 1: Preparation of aluminum doped ceria (ACO)
A sol–gel method was used to synthesize Al0.2Ce0.8O2−δ (ACO) powders. The precursors employed in the synthesis were Al (NO3)3·9H2O (Sigma Aldrich, 99.5%), and Ce (NO3)3·6H2O (Sigma Aldrich, 99%). To summarize, Al (NO3)3·9H2O and Ce (NO3)3·6H2O were dissolved in deionized water according to their stoichiometric ratios while continuously heating and stirring. The solution was then treated with citric acid in the mole quantities of metal cations. The solution was then agitated for 2 h, followed by 4 h of heating to evaporate the water, yielding a gel. The solution was kept stirred and heated until a suitable gel formed. The gel was dried at 120 °C, then heated at 180 °C for 2 h, ground into, and calcined at 300 °C for 4 h before being sintered in a microwave oven. The microwave oven (WBMW-JS4) operates at 220 ± 10 V and 50 Hz, with an output power of 200–4000 W at a frequency of 2.45 GHz ± 50 MHz. A vacuum tube called a magnetron generates microwave radiation at 2.45 GHz in a microwave oven by converting electrical energy through electric and magnetic field interaction, followed by an adequate grind to obtain fine M-ACO material. Figure 1 shows a schematic diagram of the synthesis of M-ACO.
Step 2: Preparation of protonated ACO through the photo-assisted process
The prepared M-ACO is then poured into 50 ml water (H2O) and ACO is formed by the irradiation of a precursor solution with UV light (280 nm wavelength) after a continuous stirring of 5 h. When the protonation process is completed, the H-ACO is subjected to washing to remove impurities or residual reactants and dried at 180 °C for 2 h to get H-ACO. Then H-ACO oxide material was subjected to a microwave sintering and then again ground in agate mortar and pestle to get the final powder of H-ACO. The microwave sintering ensures the consolidation of the H-ACO powders while maintaining the desired protonation state achieved through photo-activated radicals’ reaction.
Material characterizations
The X-ray diffraction (XRD) was conducted using an X-ray diffractometer, Bruker, Germany with a Cu Kα radiation (1.5419 Å) to identify the crystalline phases in the samples. The diffraction scans were collected in a 2θ range from 5° to 90° with a step size of 0.013° with a counting time of 110 s per step. The data were analyzed using the Jade software. Rietveld refinement for representative fluorite oxides was performed by FullProf software. The morphology of the samples was examined using a field emission scanning electron microscope (FE-SEM, JEOL JSM7100F, Germany). Additionally, the microstructure of the samples specimens was analyzed with a transmission electron microscope (TEM, JEOL JEM-2100F), operated at an accelerating voltage of 200 kV. Raman spectroscopy was conducted by a LabRAM Soleil Raman spectrometer (Jobin Yvon, France), utilizing a laser wavelength of 532 nm, and the scanning range was 100–2000 cm−1. Fourier transform infrared spectroscopy (FTIR) was carried out with a Vertex 70 time-resolved infrared spectroscopy (Bruker, German), and calibration of the results followed the binding energy referencing method. The surface properties of both Al-doped ceria samples were analyzed by X-ray photoelectron spectroscopy (XPS, AXIS ULtrabld, Kratos, England). The electron paramagnetic resonance (EPR) spectra were acquired using a Bruker EMX-PLUS spectrometer, operating at a central magnetic field of 6900G and a frequency of 9.74 GHz, and differential scanning calorimetry/thermogravimetric analysis (DSC/TG) for thermal properties. Additionally, 1H magic angle spinning nuclear magnetic resonance (NMR) measurements were performed at a Larmor frequency of 400.33 MHz using a Bruker Avance Neo 400WB spectrometer with an H13925_0025 probe.
Fuel cell and electrochemical characterizations
PCFC with ACO composite electrolytes were fabricated via a dry pressing procedure. The ACO electrolyte powder was compacted between two pieces of Ni0.8Co0.15Al0.05LiO2−δ-pasted Ni foam (NCAL-Ni) electrodes uniaxially under a load of 250 MPa into one pellet, followed by applying Ni foam as the current collector. The assembled PCFC devices have a structure of NCAL-Ni/ACO/NCAL-Ni, with a thickness of approximately 1.5 mm, an active area of 0.64 cm2, and an electrolyte layer thickness of 0.76 cm. Before fuel cell measurements, the constructed cell with the configuration (Ni-NCAL/ACO/NCAL-Ni) was sintered at 500 °C for 1 h. The electrodes were made from commercial NCAL powder. Initially, the NCAL powder was ground, and then an adequate amount of terpineol was poured into it and combined to form the NCAL slurry. In comparison, the nickel foam was cut into button shapes, and a prepared NCAL slurry was applied to the surface. The NCAL electrodes were then dried at 120 °C for 20 min. The pellet (Ni-NCAL/ACO/NCAL-Ni) was then placed in the testing instrument to evaluate fuel cell performance and perform electrochemical impedance spectroscopy (EIS). The electrochemical reaction was facilitated by supplying hydrogen to the anode side at 80–120 mL min−1 and purging air to the cathode side at 150–200 mL min−1. The programmable electronic load (IT8511A) was used to evaluate fuel cell performance via current and voltage measurements. The acquired data was then plotted using Origin software. EIS analysis was performed using a Solartron Analytical instrument (UK) in an Air/H2 atmosphere at temperatures at different temperatures (500–300 °C).
Data availability
The data used for our analyses are available from the corresponding author upon request
Change history
03 July 2025
In the version of the Supplementary information initially published, Supplementary Fig. 1 was incorrect and has now been amended. The updated Supplementary information file is available online.
References
Azad, A. K. et al. Improved mechanical strength, proton conductivity, and power density in an all-protonic ceramic fuel cell at intermediate temperature. Nature 11, 19382 (2021).
Saini, D. S. et al. A promising proton conducting electrolyte BaZr1-xHoxO3-δ(0.05≤x≤ 0.20) ceramics for intermediate temperature solid oxide fuel cells. Nature 10, 3461 (2020).
Zheng, S., Bian, W. & Ding, H. A robust protonic ceramic fuel cell with a triple conducting oxygen electrode under accelerated stress tests. Mater. Adv. 5, 2296–2305 (2024).
Duan, C. et al. Highly durable, coking and sulfur tolerant, fuel-flexible protonic ceramic fuel cells. Nature 557, 217–222 (2018).
Li, Y. et al. Mutual conversion of CO–CO2 on a perovskite fuel electrode with endogenous alloy nanoparticles for reversible solid oxide cells. ACS Appl. Mater. Interfaces 14, 9138–9150 (2022).
Yang, H. et al. A power-to-hydrogen nearby consumption system based on a flat-tube rSOC coupled with local photovoltaics and Yellow River water. Int. J. Hydrog. Energy 57, 1111–1117 (2024).
Benghanem, M., Almohamadi, H., Haddad, S., Mellit, A. & Chettibi, N. The effect of voltage and electrode types on hydrogen production powered by photovoltaic system using alkaline and PEM electrolyzers. Int. J. Hydrog. Energy 57, 625–636 (2024).
Jing, Y. et al. Synergistic coupling among Mg2B2O5, polycarbonate and N, N-dimethylformamide enhances the electrochemical performance of PVDF-HFP-based solid electrolyte. J. Energy Chem. 94, 158–168 (2024).
Yamate, S. & Otomo, J. Design of cost-effective and highly efficient systems for protonic ceramic fuel cells based on techno-economic analysis. Energy Conv. Manag. 301, 118016 (2024).
Antunes, I. et al. Structure and electrical-transport relations in Ba(Zr, Pr)O3−δ perovskites. Inorg. Chem. 56, 9120–9131 (2017).
Sherafat, Z. et al. Modeling of electrical conductivity in the proton conductor Ba0. 85K0.15ZrO3−δ. Electrochim. Acta 165, 443–449 (2015).
Chen, G. et al. Advanced fuel cell based on perovskite La-SrTiO3 semiconductor as the electrolyte with superoxide-ion conduction. ACS Appl. Mater. Interfaces 10, 33179–33186 (2018).
Le, L. Q. et al. Proton-conducting ceramic fuel cells: scale up and stack integration. J. Power Sources 482, 228868 (2021).
Bibi, B. et al. Emerging semiconductor ionic materials tailored by mixed ionic-electronic conductors for advanced fuel cells. APM 3, 100231 (2024).
Zhou, J. et al. Wet-chemical synthesis of Li7P3S11 with tailored particle size for solid state electrolytes. Chem. Eng. J. 429, 132334 (2022).
Wang, R. et al. Densification behavior and microstructure evolution of Mo manocrystals by microwave sintering. ES Mater. Manuf. 13, 97–105 (2021).
Simonenko, T. L. et al. Synthesis of BaCe0.9-xZrxY0.1O3-δ nanopowders and the study of proton conductors fabricated on their basis by low-temperature spark plasma sintering. Int. J. Hydrog. Energy 44, 20345–20354 (2019).
Zou, P. et al. A fast ceramic mixed OH−/H+ ionic conductor for low-temperature fuel cells. Nat. Commun. 15, 909 (2024).
Yang, X. et al. Mechanism of proton conduction in doped barium cerates: a first-principles study. J. Phys. Chem. C 124, 8024–8033 (2020).
Hayashi, T. et al. OH, radical formation by the photocatalytic reduction reaction of H2O2 on the surface of plasmonic excited Au-TiO2 photocatalyts. Chem. Phys. Lett. 739, 136958 (2020).
Medel, A., Treviño-Reséndez, J., Brillas, E., Meas, Y. & Sirés, I. Contribution of cathodic hydroxyl radical generation to the enhancement of electro-oxidation process for water decontamination. Electrochim. Acta 331, 135382 (2020).
Li, Y., Miller, C. J., Wu, L. & Waite, T. D. Hydroxyl radical production via a reaction of electrochemically generated hydrogen peroxide and atomic hydrogen: an effective process for contaminant oxidation. Environ. Sci. Technol. 56, 5820–5829 (2022).
Wang, Q. et al. Laboratory experiment on the nano-TiO2 photocatalytic degradation effect of road surface oil pollution. Nanotechnol. Rev. 9, 922–933 (2020).
Xing, Y. et al. Proton shuttles in CeO2/CeO2−δ core–shell structure. ACS Energy Lett. 4, 2601–2607 (2019).
Saito, K. & Yashima, M. High proton conductivity within the ‘Norby gap’ by stabilizing a perovskite with disordered intrinsic oxygen vacancies. Nat. Commun. 14, 7466 (2023).
Akbar, M. et al. High-performing and stable non-doped ceria electrolyte with amorphous carbonate coating layer for low-temperature solid oxide fuel cells. Electrochim. Acta 393, 139067 (2021).
Shah, M. A. K. Y. et al. Semiconductor Nb-doped SrTiO3−δ perovskite electrolyte for a ceramic fuel cell. ACS Appl. Energy Mater. 4, 365–375 (2021).
Villa‐Aleman, E., Houk, A., Dick, D. & Murph, S. E. H. Hyper‐Raman spectroscopy of CeO2. J. Raman Spectrosc. 51, 1260–1263 (2020).
Tiseanu, C. et al. Isolated centres versus defect associates in Sm3+-doped CeO2: a spectroscopic investigation. J. Phys. D Appl. Phys. 46, 275302 (2013).
Kosacki, I., Petrovsky, V., Anderson, H. U. & Colomban, P. Raman spectroscopy of nanocrystalline ceria and zirconia thin films. J. Am. Ceram. Soc. 85, 2646–2650 (2002).
Xu, C. Entanglement entropy of coupled conformal field theories and Fermi liquids. Phys. Rev. B 84, 125119 (2011).
Norby, T. Solid-state protonic conductors: principles, properties, progress and prospects. Solid State Ion-. 125, 1–11 (1999).
Selvam, N. C. S., Manikandan, A., Kennedy, L. J. & Vijaya, J. J. Comparative investigation of zirconium oxide (ZrO2) nano and microstructures for structural, optical and photocatalytic properties. J. Colloid Interface Sci. 389, 91–98 (2013).
Ullah, A. K. M. A. et al. Green synthesis of bio-molecule encapsulated magnetic silver nanoparticles and their antibacterial activity. RSC Adv. 8, 37176–37183 (2018).
Pathan, A. A., Desai, K. R. & Bhasin, C. P. Synthesis of La2O3 nanoparticles using glutaric acid and propylene glycol for future CMOS applications. IJNC 3, 21–25 (2017).
Mu, Q. & Wang, Y. Synthesis, characterization, shape-preserved transformation, and optical properties of La(OH)3, La2O2CO3, and La2O3 nanorods. J. Alloy. Compd. 509, 396–401 (2011).
Wong, K.-L. et al. Functionalized europium nanorods for in vitro imaging. Inorg. Chem. 47, 5190–5196 (2008).
Valechha, D. et al. Study of nano-structured ceria for catalyticCO oxidation. J. Mater. Chem. 21, 3718–3725 (2011).
Fernández, C., Sassoye, C., Debecker, D. P., Sanchez, C. & Ruiz, P. Effect of the size and distribution of supported Ru nanoparticles on their activity in ammonia synthesis under mild reaction conditions. Appl. Catal. A Gen. 474, 194–202 (2014).
Lin, B. et al. Morphology effect of ceria on the catalytic performances of Ru/CeO2 catalysts for ammonia synthesis. Ind. Eng. Chem. Res. 57, 9127–9135 (2018).
Liu, P., Niu, R., Li, W., Wang, S. & Li, J. Morphology effect of ceria on the ammonia synthesis activity of Ru/CeO2 catalysts. Catal. Lett. 149, 1007–1016 (2019).
Seo, J., Gowda, A. & Babu, S. V. Almost complete removal of ceria particles down to 10 nm size from silicon dioxide surfaces. ECS J. Solid State Sci. Technol. 7, 243–252 (2018).
Zhao, Z. et al. Nature of five-coordinated Al in γ-Al2O3 revealed by ultra-high-field solid-state NMR. ACS Cent. Sci. 8, 795–803 (2022).
Avadhut, Y. S., Weber, J., Hammarberg, E., Feldmann, C. & Schmedt auf der Gunne, J. Structural investigation of aluminium doped ZnO nanoparticles by solid-state NMR spectroscopy. Phys. Chem. Chem. Phys. 14, 11610–11625 (2012).
Zakharov, D. M. et al. Catalytic methane activation over La1−xSrxScO3−α proton-conducting oxide surface: a comprehensive study. J. Catal. 394, 67–82 (2021).
Koller, H., Uesbeck, T., Hansen, M. R. & Hunger, M. Characterizing the first and second 27Al neighbors of brønsted and lewis acid protons in zeolites and the distribution of 27Al quadrupolar couplings by 1H {27Al} offset REAPDOR. J. Phys. Chem. C 121, 25930–25940 (2017).
Maekawa, H., Kashii, N., Kawamura, J.-I., Hinatsu, Y. & Yamamura, T. High temperature 1H NMR study of proton conducting oxide SrCe0.95Y0.05H0.0O3-δ. Solid State Ion. 122, 231–236 (1999).
Miyoshi, S. et al. Low-temperature protonic conduction based on surface protonics: an example of nanostructured yttria-doped zirconia. Chem. Mater. 26, 5194–5200 (2014).
Choi, S. et al. Exceptional power density and stability at intermediate temperatures in protonic ceramic fuel cells. Nat. Energy 3, 202–210 (2018).
Rauf, S. et al. Alternative strategy for development of dielectric calcium copper titanate-based electrolytes for low-temperature solid oxide fuel cells. Nano-Micro Lett. 17, 13 (2025).
Acknowledgements
We acknowledge generous funding by the Science and Technology Department of Jiangsu Province under Grant (BE2022029) and Jiangsu Provincial Innovation and Entrepreneurship Talent Program (JSSCRC2021491) and US Department of Energy DE-EE0011325.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.Z., A.N., and B.B. Methodology: B.Z., A.N., and B.B. Validation: B.Z., L.F., and M.T. Formal analysis: C.L., R.R., M.Y., Y.J., and S.L. Resources and funding acquisition: B.Z. Writing—original draft: A.N. and B.B. Writing—review and editing: F.Y., F.Q., M.A., ānd K.N. Visualization: A.N. and B.B. Supervision: B.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nazar, A., Bibi, B., Lou, C. et al. Photo-assisted synthesis of protonated oxides for fuel cells. Commun Chem 8, 120 (2025). https://doi.org/10.1038/s42004-025-01488-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-025-01488-0
