
Abstract
Hydrogen bond cooperativity (HBC) is the phenomenon where the collective strengthening of hydrogen bonds, by mutual polarization and non-additive electrostatic effects, in a network exceeds the sum of individual interactions. HBC has been well-studied in α-helices and β-sheets, where it critically stabilizes amyloid structures. During the last 20 years, several natural proteins were characterized which contain multiple aligned and hydrogen-bonded polyproline II (PPII) helices. HBC was recently reported for these PPII helices when arranged along one parallel layer, but its existence in the more abundant antiparallel or mixed parallel/antiparallel PPII assemblies is still unknown. Utilizing computational approaches validated through experimental observables, we report that both canonical CO···HN and non-canonical CO···HαCα hydrogen bonds exhibit mutual reinforcement, revealing a complex hydrogen bonding scheme that manifests HBC in antiparallel PPII helices and in mixed parallel/antiparallel orientations. These findings have fundamental relevance for understanding protein conformational stability and implications for PPII helices as a structural building block for protein design.

Similar content being viewed by others
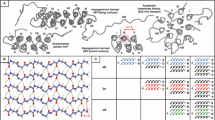

Introduction
The study of protein secondary structures has traditionally focussed on α-helices and β-sheets, the main secondary structures in globular proteins. These structures have been instrumental in pioneering advancements in protein design and enzymatic functionality, shaping our broader understanding of biological systems1. However, the other main class of protein secondary structure, the polyproline II (PPII) helix, has been less characterized. PPII helices are the basis of the collagen triple helix, and there are several proteins and protein domains that are composed of bundles of Gly-rich PPII helices2. In previous studies, we, among others, employed the short isoform of the Hypogastrura harveyi “snow flea” antifreeze protein (HhAFP) as a model system to investigate Gly-rich PPII helical bundles3,4. The short isoform of the HhAFP is composed by six PPII helices arranged in parallel on two faces. One face is rich in polar amino acid residues, and the other has more hydrophobic residues, and the two faces and their PPII helices are arranged antiparallel to each other. Intriguingly, despite a high glycine content and a lack of a stable hydrophobic core, these bundles display remarkable conformational stability3,5. This stability was ascribed to contributions from canonical CO···HN and non-canonical CO···HαCα H-bonds, together with other energetic factors4,5. The unfolded ensemble of the HhAFP short isoform is compact, containing nonlocal electrostatic interactions that may initiate its folding process6, which has been established to be cooperative through kinetic studies5.
The remarkable conformational stability and abundant H-bonds observed in Gly-rich PPII helical bundles, exemplified by the HhAFP structure, bear a striking resemblance to amyloids, which consist of nearly endless β-sheets featuring aligned networks of H-bonds. In both PPII helical bundles and amyloids, the extended configuration of the polypeptide chain promotes self-assembly. One of the key factors contributing to the extraordinary stability of amyloids is the cooperative nature of H-bonding or H-bond cooperativity (HBC), where the alignment of CO···HN backbone or side-chain (i.e., Asn and Gln) H-bonds results in enhanced polarization and bond strength, as we and others previously reported7,8,9,10. Our prior research examined this underexplored property for PPII helical bundles, particularly those rich in glycine residues, revealing for the first time unique HBC between both canonical CO···HN and CO···HαCα H-bonds between parallel PPII helices11. We hypothesize that HBC across both faces of the short isoform of HhAFP could be one of the reasons for its conformational stability. This cooperative effect could also stabilize other larger antifreeze proteins, such as the Granisotoma rainieri “springtail” antifreeze protein12 or the long isoform of HhAFP13, made out of nine and thirteen PPII helices, respectively. These PPII helices are also arranged in parallel on a polar and a hydrophobic face, and the two faces are antiparallel to each other (Fig. 1). Despite these advances, the H-bonding patterns specific to Gly-rich antiparallel and mixed parallel/antiparallel PPII helical topologies remain unexplored, like those present in Bacillus subtilis Obg GTPase14, Aromatoleum aromaticum acetophenone carboxylase15, Salmonella phage S16 tail fiber adhesin16, and anaplastic lymphoma kinase17 with six, eight, ten, and fourteen PPII helices, respectively (Fig. 1).

Experimentally determined structures from PDB for Hypogastrura harveyi AFP short isoform (PDB ID: 3BOG), Salmonella phage S16 tail fiber adhesin Gly-rich domain (PDB ID: 6F45), ALK Gly-rich domain (PDB ID: 7LS0/7LRZ), Aromatoleum aromaticum acetophenone carboxylase Gly-rich domain (PDB ID: 5L9W), Bacillus subtilis Obg GTPase Gly-rich domain (PDB ID: 1LNZ) and Granisotoma ranieri AFP (PDB ID: 7JJV), and predicted structure from Hypogastrura harveyi AFP long isoform (AF2 ID: D7PBP2). Roman numerals denote the distinct helices in each protein. From the reader’s perspective, the yellow PPII helices run from the N-terminus (bottom) to the C-terminus (top) and are parallel to each other; the gray PPII helices run from the C-terminus (bottom) to the N-terminus (top) and are parallel to each other, so the yellow and gray helices are antiparallel to each other.
The ability of polyGly to adopt a PPII helical structure was proposed by Crick and Rich18 on the basis of X-ray diffraction patterns. However, they could not distinguish whether the orientation of the helices was parallel or antiparallel. In the 1980’s, Krimm and co-workers pointed out that in parallel polyGly in PPII helices, all -CH2- groups can form H-bonds whereas in antiparallel orientations, only every third -CH2- group does so19. This means that there is one kind of -CH2- IR signal for the parallel orientation, but two kinds of peaks for the antiparallel orientation. In the IR spectra, two types of -CH2- signals are seen, and the position of the peaks agrees with normal mode calculations19. This is good evidence that the antiparallel orientation is dominant. More recently, a survey of PPII helices in folded proteins revealed that the predominant orientation is antiparallel20. Nevertheless, the bases of this preference is still not clear. Some possibilities are that it could be due to a stronger H-bonding network, more favorable helix macrodipole interactions, and/or less unfavorable entropic effects due to shorter connecting loops. By studying the H-bonding network, we aim to test the role of H-bonding for the preference for the antiparallel PPII helix orientation.
To bridge this gap in our understanding of PPII helical bundle stability, the current study employs a multifaceted methodology, integrating Dispersion-corrected Density Functional Theory (DFT-D) calculations with Natural Bonding Orbital (NBO) and Quantum Theory of Atoms in Molecules (QTAIM) analyzes, together with Symmetry-Adapted Perturbation Theory (SAPT) assessments. We present evidence for the existence of HBC between antiparallel PPII helices in one and multiple, multidirectional layers, and when considering them together with parallel PPII helices, with particular attention to the role of non-canonical CO···HαCα H-bonds, especially in Gly-rich regions. This finding not only improves our understanding of PPII helix assemblies but also reveals a more complex H-bonding network, broadening the scope of known H-bonding dynamics in protein chemistry.
Results
In the following sections, we begin by describing the construction of representative 2D and 3D PPII helical bundles and the theoretical framework used to analyze HBC in these models. We then examine H-bond patterns and strengths in antiparallel 2D systems, followed by an in-depth analysis of 3D models with various helical arrangements. Finally, we summarize key trends and validate the models through NMR chemical shift comparisons.
Model systems and theoretical framework to study PPII helical bundles
Construction of 2D and 3D computational models
To explore the origin of HBC in PPII helical bundles, we constructed theoretical models that reproduce the most representative arrangements observed in natural Gly-rich PPII helical domains. We selected two major topological schemes: a 2D antiparallel bundle consisting of aligned helices growing in one direction and 3D bundles composed of helices arranged both parallel and antiparallel oriented in two directions and stacked in two layers. This setup mimics proteins in which the organization of the PPII helices adopts alternating orientations within and between faces of the bundle. The overall goal was to test whether HBC could manifest in different architectural motifs, especially those lacking a hydrophobic core.
The 2D antiparallel computational model consisted of replicating five times, at 4.7 Å separation, the nine residues (31-39, i.e., three turns) of the central PPII helix from the polar face of the short isoform of HhAFP (Fig. 1). To emulate the continuity of the natural sequence, the N and C termini of each PPII helix were capped with acetyl and methylamine groups, respectively. To extend the applicability of this model to every Gly-rich antiparallel PPII helical bundle, every non-glycine residue was replaced by glycine (i.e., H32, N35, and N38). The second and fourth helices were rotated 180°, yielding the final 2D antiparallel PPII helical computational model (Fig. 2A).

A 2D, Gly-rich antiparallel PPII helical bundle computational model: red spheres represent oxygen; blue, nitrogen; gray, carbon, and white, hydrogen. B Graphical representation of a PPII helix as a triangle and of the three-fold symmetry axis as a dashed line. C Graphical side-view representation of the 3D, Gly-rich PDPR configuration. D Graphical side-view representation of the 3D, Gly-rich PDAR configuration. E Graphical side-view representation of the 3D, Gly-rich ADPR configuration. F Graphical side-view representation of the 3D, Gly-rich ADAR configuration. The gray PPII helices are parallel to each other and the yellow PPII helices are parallel to each other, so the yellow and gray helices are antiparallel to each other. PDPR: parallel PPII helices along both the direct growth direction (from the carbonyl oxygen of B and the reverse (from the amide hydrogen of B); PDAR: parallel PPII helices along the direct growth direction and antiparallel along the reverse; ADPR: antiparallel PPII helices along the direct growth direction and parallel along the reverse, and ADAR: antiparallel PPII helices along both the direct and the reverse growth directions.
Considering a Gly-rich PPII helix, bundle growth can occur along two distinct axes: either from the carbonyl oxygens (C=O axis) or from the amide hydrogens (N-H axis) (Fig. 2B). Given the three-fold symmetry of PPII helices, adding helices along either axis is equivalent across their respective symmetry-related positions. The growth direction along the C=O axis will be referred to as the direct direction, used in the 2D antiparallel model and the previously reported 2D parallel model11, while growth along the N-H axis will be termed the reverse direction.
The 3D computational models comprised four distinct topologies defined by the relative orientations of their constituent PPII helices: PDPR (parallel in direct and reverse directions), PDAR (parallel in direct, antiparallel in reverse), ADPR (antiparallel in direct, parallel in reverse), and ADAR (antiparallel in both directions). The same nine-Gly PPII helix was duplicated twice in both direct and reverse directions, again separated by 4.7 Å, forming the PDPR model (Fig. 2C). To obtain the PDAR model (Fig. 2D), the central reverse-direction PPII helix was rotated 180°; in the ADPR model (Fig. 2E), the central direct-direction PPII helix was rotated 180°; finally, the ADAR model (Fig. 2F) had both central direct and reverse PPII helices rotated 180°. For instance, bundles like those in Salmonella phage S16 TFA, ALK, and B. subtilis Obg GTPase feature several parallel and antiparallel PPII helices distributed across layered architectures (Fig. 1).
H-bonding strength and other parameters as a metric of cooperativity
We define H-bonding strength (HBS) as the pairwise interaction energy between neighboring PPII helices, computed through DFT-D and SAPT calculations. In bundles, if HBS increases (i.e., becomes more negative) as more PPII helices are added, it would reflect the reinforcement of individual H-bond interactions through the network. This additive behavior (i.e., more favorable per PPII helix with increasing bundle size) is a hallmark of HBC. Furthermore, the variation of other computational parameters, such as those of NBO and QTAIM analysis, derived from the growth of the PPII helical bundle, can be used to describe HBC in detail. Figure 3 schematically represents the configurations for every calculation or analysis in each model, as well as the labeling convention for interfaces between PPII helices, which is maintained throughout this article.

The different interfaces of the 2D antiparallel computational model of Fig. 2A and between the PPII helices of the four 3D configurations depicted in Figs. 2C, D, E, and F are shown. Blue PPII helices give rise to the a···b interface; the red ones, to the b···c interface, and the green ones, to the c···d interface of the 2D antiparallel computational model. The gray PPII helices are those that give rise to the a···bc interface of the 3D computational model, that is, where the interaction of helix a with the neighboring helices b and c takes place. In both cases, the black PPII helices represent the ones that were already part of the bundle or the added ones when growing the bundle, i.e., those that do not form the interface where the calculations take place.
H-bonding cooperativity in 2D antiparallel PPII helical bundles
To investigate the energetic signatures of HBC, we focused first on the 2D model containing five aligned, antiparallel Gly-rich PPII helices (a, b, c, d and e) (Fig. 2A). Through DFT-D calculations, we systematically computed the HBS across the different interfaces of all its subunits in order of increasing size, i.e., dimer (ab), trimer (abc), tetramer (abcd) and pentamer (abcde), which represents the full system (Fig. 3). Figure 4 illustrates how the interaction energy evolves as more PPII helices are added to the antiparallel array. Notably, the energy becomes increasingly favorable (i.e., more negative) with each added PPII helix: HBS increases progressively from interface a···b, to b···c and c···d, with values shifting from −52.62 to −57.43 and −57.60 kcal/mol, respectively, representing an overall HBS gain of approximately 9.5%. The only interface not considered is d···e since it represents the limit of the model, obstructing the assessment of how adding more PPII helices affects the interaction energy.

The monotonic trend in HBS clearly demonstrates the presence of cooperativity in the 2D antiparallel configuration. The increase in interaction energy per PPII helix as more chains are added cannot be explained by pairwise additivity alone, indicating that a collective reinforcement mechanism, which is typical of HBC, is operative. Table 1 quantifies these results, confirming that HBC is not exclusive to parallel PPII helical arrangements, as shown previously11, but also applies to antiparallel configurations.
It is particularly noteworthy that this behavior arises in the absence of hydrophobic stabilization. These Gly-rich PPII helical bundles lack the hydrophobic cores that often contribute to bundle stability in other secondary structures, such as α-helical coiled coils21. Instead, the observed stabilization originates entirely from polar interactions and electrostatic contributions. The results indicate that aligned arrays of H-bonds formed by backbone atoms in antiparallel PPII helical geometry are sufficient to yield cooperative stabilization.
These findings broaden our understanding of HBC beyond better-known examples such as amyloids7,8,9,10. They also provide a strong rationale for examining more complex 3D PPII helical assemblies, where additional orientational effects may influence the magnitude and nature of HBC.
H-bonding patterns in 2D antiparallel PPII helical bundles
To understand the physical basis of HBC in antiparallel bundles, we examined the specific types of H-bonds formed between PPII helices. In antiparallel arrangements, carbonyl groups from one PPII helix can form H-bonds with either the backbone amide hydrogen (CO···HN) or with the alpha hydrogen of glycine residues (CO···HαCα) from an adjacent chain. These CO···HαCα contacts can be subdivided into two categories based on their geometric proximity: proximal, when the interacting carbonyl and HαCα groups are in adjacent turns, and distal, when they are offset by one turn, resulting in a longer donor-acceptor distance and a bond angle that deviates more from the optimal, which is 180°. These H-bonds collectively form a repetitive zigzag pattern along the interface of two antiparallel helices (Fig. 5A). The zigzag topology is unique to antiparallel geometry and is distinct from the more linear H-bond chains found in parallel arrays11. This arrangement allows alternating CO···HN and CO···HαCα interactions to occur along the interface, potentially enhancing structural stability through dense polar contacts.

A H-bonds between antiparallel PPII helices in a 2D antiparallel PPII helical bundle: CO···HN, CO···HαCα proximal and CO···HαCα distal. B Simplified structures for CO···HN (C, formamide), CO···HαCα proximal (P, N-methylformamide), CO···HαCα distal (D, acetaldehyde), and (CO)2···(Hα)2Cα proximal+distal (P + D, N-(2-oxoethyl)formamide) H-bonds.
CO···HN and CO···HαCα proximal contacts were consistently observed across all modeled interfaces, with short donor-acceptor distances and favorable angles, suggesting strong directional H-bonding. In contrast, CO···HαCα distal contacts were less frequent, forming only in approximately one-third of the possible positions (see below). To assess the prevalence of each interaction type, we examined in detail the nature and distribution of H-bonds in 2D antiparallel bundles through NBO analysis, whose results are summarized in Tables 2, 3 and Fig. 6.

A Mean occupancy (q) of the corresponding σ*(NH/CαHα) orbitals and B mean stabilization energy (E(2)) of the corresponding n(O)/σ(CO)/π(CO)→σ*(NH/CαHα) electron delocalization for each CO···HN (squares), CO···HαCα proximal (right triangles) and CO···HαCα distal (left triangles) H-bond between the antiparallel PPII helices of the 2D computational model. The ab interface for the different systems is indicated in blue; the bc interface, in red, and the cd interface, in green.
As new PPII helices are added to the bundle, according to the scheme of Fig. 3, we observe progressive reinforcement of CO···HN H-bonds. This is reflected in several electronic indicators inherent to the NBO methodology. In particular, the occupancy of the σ* orbitals in N-H bonds, q(σNH), increases systematically at each added interface, indicating enhanced electron donation as the H-bond network becomes more extended. Likewise, the stabilization energy corresponding to the delocalization of not only the lone pairs (n) from the oxygen in those orbitals, but also of the σ and π orbitals of the C=O bonds, E(2)n(O)/σ(CO)/π(CO)→σ*(NH), becomes more extended. For CO···HαCα proximal H-bonds, we also observe the same trends for both the q(σ*CαHα) and E(2)n(O)/σ(CO)/π(CO)→σ(CαHα), but within a smaller range. On the other hand, the CO···HαCα distal H-bonds show small values of E(2)n(O)→σ(CαHα) and constant values of q(σ*CαHα), and only involve the lone pairs of the oxygen. This electron delocalization into the σ* orbitals in Cα-Hα bonds only occurs in the middle turn of these PPII helices, which contain three turns.
The same conclusion can be drawn from the QTAIM analysis, extensively detailed in the supplementary material. Its results are reported in Tables S1, S2, S5, S6, and Figs. S1, S6. The magnitude and variation of parameters specific to the QTAIM methodology, like the electron density and its Laplacian at the critical points corresponding to the CO···HN and CO···HαCα proximal H-bonds, are consistent with strong and cooperative interactions, while evincing that CO···HαCα distal H-bonds are weak and non-cooperative. The same holds true for the relative positions of the critical points. These results confirm that cooperativity, or the lack of, is not only detectable at the energetic level but also manifests in the electron distribution within the H-bonding network.
Strength of individual H-bonds in 2D antiparallel PPII helical bundles
To isolate the contribution of each type of H-bond to the total interaction energy, we designed minimal molecular systems representing individual CO···HN, CO···HαCα proximal and CO···HαCα distal H-bonds, following previous procedures employed by us and others11,22,23,24. These systems were built using simple amide or aldehyde derivatives such as formamide, N-methylformamide, acetaldehyde, and N-(2-oxoethyl)formamide. Each model was constructed in layers (from dimers to pentamers), mimicking the stacking of antiparallel PPII helices and subjected to DFT-D energy calculations. Figure 5B illustrates these simplified mimetic models.
The calculated interaction energies, according to the scheme depicted by Fig. 3, showed clear differences in strength and cooperativity across H-bond types. CO···HN dimers exhibited the highest interaction energy and the strongest increase with added layers, consistent with marked cooperativity. CO···HαCα proximal H-bonds displayed moderate energies and cooperative gains, albeit saturating more quickly. CO···HαCα distal H-bonds showed the weakest and least cooperative behavior. Additionally, when both proximal and distal interactions were combined in a bifunctional model, the resulting interaction energy exceeded the additive sum of the individual components, suggesting synergistic effects. When simply summing all these individual interaction energies, they initially yield lower values than the total HBS calculated directly from complete PPII helices. However, as additional layers are considered, the summed contributions surpass the global HBS values, highlighting again the global HBC of the antiparallel arrangement. Table 4 and Fig. 7 summarize and show the HBS trends.

Mean HBS between the molecules that reproduce each CO···HN (formamide, squares), CO···HαCα proximal (N-methylformamide, right triangles), CO···HαCα distal (acetaldehyde, left triangles), and (CO)2···(Hα)2Cα proximal+distal (N-(2-oxoethyl)formamide, rhombuses and dashed line) H-bond of the 2D antiparallel computational model. As in the Fig. 3 color scheme, the ab interface for the different systems is indicated in blue; the bc interface, in red, and the cd interface, in green.
To better understand the physical origin of these interactions, we performed SAPT energy decomposition calculations at a high level of theory, which are extensively detailed in the supplementary material. The results are reported in Tables S9, S10, S11, S12, S13 and Figs. S11, S12, S13, S14, S15, S16. For CO···HN and CO···HαCα proximal H-bonds, the primary contributors to the SAPT interaction energy were electrostatics and induction, consistent with strong polar character and cooperative stabilization. For distal H-bonds, dispersion and exchange dominated, reflecting weaker and less directional bonding.
H-bonding cooperativity in 3D PPII helical bundles
We next evaluated HBC across the four 3D topologies (PDPR, PDAR, ADPR, and ADAR) to assess how different PPII helix orientations influence cooperative stabilization. As with the 2D bundles, each system was grown by adding PPII helices, and the pairwise interaction energy between them was computed and tracked as a function of bundle size. We only focused on the a···bc interface of all the 3D models when considering three and five PPII helices (Fig. 3). We did not analyze b···d or c···e interfaces because they represent the limit of our systems and the HBS increases at these interfaces cannot be computed since no more PPII helices are added to the four different 3D bundles.
In all cases, we observed that the total interaction energy became increasingly negative as the number of PPII helices increased (Fig. 8), confirming that HBC operates not only in 2D bundles but also across more complex 3D architectures. In particular, an increase in HBS values of ~9.3–9.8% is observed when three-helical bundles are expanded into five-helical bundles. However, the extent of this cooperative energy gain differed among topologies. PDPR bundles, which are composed entirely of parallel helices, showed the strongest cooperativity, with the largest incremental increase in stabilization per PPII helix. PDAR and ADPR, which combine parallel and antiparallel orientations in different directions, exhibited intermediate levels of cooperativity. In contrast, ADAR bundles, which are those composed exclusively of antiparallel PPII helices in both growth directions, displayed the weakest energetic reinforcement. These differences are quantitatively summarized in Table 5.

HBS between the parallel and/or antiparallel PPII helices of the four different 3D computational models. The PDPR arrangement is indicated in purple; the PDAR arrangement, in magenta, the ADPR arrangement, in orange, and the ADAR arrangement, in gray.
Altogether, these results establish that HBC is a general phenomenon across PPII helical bundles of varying orientations but that its magnitude depends strongly on the specific spatial organization of the PPII helices. This suggests that the presence of competing orientations, which is common in layered 3D topologies, influences the cooperativity of H-bonds in the assembly. Consequently, we examined in detail the nature and distribution of H-bonds that form in these arrangements.
H-bonding patterns in 3D PDPR PPII helical bundles
We began our electronic analysis with the PDPR configuration (Fig. 2C). This topology provides a clean reference framework to assess both canonical and non-canonical HBC, since the PPII helices interact predominantly through CO···HN, but also through the greater number of CO···HαCα H-bonds seen in these systems, and avoid the geometric variability introduced by antiparallel pairings. To assess the prevalence of each interaction type, we examined in detail the nature and distribution of these H-bonds through NBO analysis, whose results are summarized in Tables 6 and 7 and Fig. 9.

A Mean occupancy (q) of the corresponding σ*(NH/CαHα) orbitals and B mean stabilization energy (E(2)) of the corresponding n(O)/σ(CO)/π(CO)→σ*(NH/CαHα) electron delocalization for each CO···HN (squares), CO···HαCα front (circles), CO···HαCα inner (triangles) and CO···HαCα outer (inverted triangles) H-bond/interaction between parallel PPII helices of the 3D PDPR configuration. Dark purple for the direct growth direction and light purple for the reverse.
Following the scheme of Fig. 3, we find that the same H-bonds that occur in a 2D parallel arrangement (Fig. 10) exist and show similar behavior in this 3D parallel arrangement as PPII helices are added to the bundle11. For canonical CO···HN bonds, the bundle growth in both the direct and the reverse direction involves an increase in both q(σNH) and E(2)n(O)/σ(CO)/π(CO)→σ*(NH). For CO···HαCα inner H-bonds, we also observe the same trends for both q(σ*CαHα) and E(2)n(O)/σ(CO)/π(CO)→σ(CαHα), but within a smaller range. Regarding the other type of non-canonical H-bond, the CO···HαCα front, while E(2)n(O)→σ*(CαHα) slightly increases as PPII helices are added to the bundle in both growth directions, q(σ*CαHα) slightly decreases. Finally, the potential CO···HαCα outer interactions show q(σ*CαHα) values that remain virtually constant and negligible E(2)n(O)→σ*(CαHα) as the bundle grows in both directions, suggesting their non-existence.

A H-bonds/interactions between parallel PPII helices in a 2D parallel PPII helical bundle: CO···HN, CO···HαCα front, CO···HαCα inner, and CO···HαCα outer. B The three H-bond types sharing carbonyl oxygen in the 3D PDAR model system. C The two H-bond types sharing carbonyl oxygen in the 3D ADAR model system.
These findings are corroborated by the QTAIM analysis, which is thoroughly detailed in the supplementary material in Tables S3, S4, S7, S8 and Figs. S2, S7. The magnitude and variation of the electron density, its Laplacian, and the relative position of the critical points corresponding to the CO···HN and CO···HαCα inner H-bonds are consistent with strong and cooperative interactions; with limitedly strong and non-cooperative for CO···HαCα inner H-bonds, and with non-existent for CO···HαCα outer contacts. The behavior of these interactions is also clearly manifested in the electron distribution across the H-bonding network of the PDPR model, beyond its energetic implications.
H-bonding patterns in 3D PDAR PPII helical bundles
We next examined the PDAR configuration (Fig. 2D). This mixed arrangement is an informative intermediate case to evaluate how the previously described interactions respond to geometric asymmetry. Continuing with our previous approach, we again used NBO methodology to analyze in depth the nature and distribution of the H-bonds and to assess how prevalent each interaction type is. The results can be found in Tables 6, 7, and Fig. 11.

A Mean occupancy (q) of the corresponding σ*(NH/CαHα) orbitals and B mean stabilization energy (E(2)) of the corresponding n(O)/σ(CO)/π(CO)→σ*(NH/CαHα) electron delocalization for each CO···HN (squares), CO···HαCα front (circles), CO···HαCα inner (triangles) and CO···HαCα outer (inverted triangles) H-bond/interaction between parallel PPII helices and for each CO···HN (squares), CO···HαCα proximal (right triangles) and CO···HαCα distal (left triangles) H-bond between antiparallel PPII helices of the 3D PDAR configuration. Dark magenta for the direct growth direction and light magenta for the reverse.
For the parallel direction of this system, we observe the same H-bonding pattern and behavior along both directions of the previous 3D parallel model. However, in the reverse direction, where PPII helices are antiparallel, the H-bonding patterns and behaviors seen in the previous 2D antiparallel model hold true only for canonical interactions. This time, CO···HαCα proximal H-bonds reveal little to no gain in q(σ*CαHα) when adding PPII helices to the bundle and E(2)n(O)/σ(CO)/π(CO)→σ*(CαHα) is found at the energetic minimum to consider an existing interaction. Meanwhile, CO···HαCα distal H-bonds go from occurring one out of three times with constant q(σ*CαHα) and small E(2)n(O)→σ*(CαHα) to decreasing q(σ*CαHα) and negligible E(2)n(O)→σ*(CαHα) all the time.
The supplementary material contains a detailed QTAIM analysis that confirms these findings. The magnitude and variation of the electron density, its Laplacian, and the relative position of the critical points corresponding to the CO···HN and CO···HαCα H-bonds are reported in Tables S3, S4, S7, S8 and Figs. S3, S8. Despite being geometrically well-placed, the CO···HαCα proximal H-bonds along the reverse direction of the PDAR reveal modest energetic and electronic reinforcements, which suggests that cooperativity is absent in these interactions, despite their structural potential. In the same way, the poor geometry of the CO···HαCα distal contacts presents negligible energetic and electronic parameters, so they are classified as non-existent.
H-bonding patterns in 3D ADPR PPII helical bundles
The ADPR configuration presents a compelling contrast to the PDAR model. While both are mixed topologies, their orientation of key interactions leads to markedly different cooperative behavior as the NBO analysis showed. Its results are summarized in Tables 6, 7 and Fig. 12.

A Mean occupancy (q) of the corresponding σ*(NH/CαHα) orbitals and B mean stabilization energy (E(2)) of the corresponding n(O)/σ(CO)/π(CO)→σ*(NH/CαHα) electron delocalization for each CO···HN (squares), CO···HαCα proximal (right triangles) and CO···HαCα distal (left triangles) H-bond between antiparallel PPII helices and for each CO···HN (squares), CO···HαCα front (circles), CO···HαCα inner (triangles) and CO···HαCα outer (inverted triangles) H-bond/interaction between parallel PPII helices of the 3D ADPR configuration. Dark orange for the direct growth direction and light orange for the reverse.
The type of H-bonds and their behavior in the reverse direction of this configuration are the same as in the direct direction of the previous one, both with parallel PPII helices. The same holds true for the canonical H-bonds and the lack of the non-canonical distal ones in the direct direction of this configuration and the reverse of the previous one, both with antiparallel PPII helices. However, the CO···HαCα proximal H-bonds show a modest but consistent recovery of electronic cooperativity compared to the PDAR model, as indicated by the increasing values of E(2)n(O)/σ(CO)/π(CO)→σ*(CαHα) and q(σ*CαHα) across interfaces as the number of PPII helices grows. Although the magnitude of these increases is lower than that observed for CO···HN H-bonds, the trend is robust, indicating that these interactions are not electronically inert.
The supplementary material provides a comprehensive QTAIM analysis supporting the previous results. The electron density values, their Laplacians, and the spatial arrangement of critical points associated with the canonical and non-canonical H-bonds are detailed in Tables S3, S4, S7, S8 and Figs. S4, S9. The corresponding values of CO···HαCα proximal interactions rise slightly with bundle expansion, suggesting that the structural context of ADPR favors cooperative enhancement, even in weaker bonds. Compared to PDAR, where competition between canonical and non-canonical interactions hampers delocalization, ADPR seems to alleviate electronic conflict by distributing donor-acceptor roles more evenly.
H-bonding patterns in 3D ADAR PPII helical bundles
The ADAR configuration represents the most geometrically diverse yet electronically balanced topology among the bundles analyzed. In contrast to mixed configurations, ADAR avoids the directional asymmetry of the PDPR model, allowing for a clear view of how non-canonical interactions behave when unconstrained. The corresponding NBO findings are summarized in Tables 6, 7 and Fig. 13.

A Mean occupancy (q) of the corresponding σ*(NH/CαHα) orbitals and B mean stabilization energy (E(2)) of the corresponding n(O)/σ(CO)/π(CO)→σ*(NH/CαHα) electron delocalization for each CO···HN (squares), CO···HαCα proximal (right triangles) and CO···HαCα distal (left triangles) H-bond between antiparallel PPII helices of the 3D ADAR model system. Dark gray for the direct growth direction and light gray for the reverse.
The antiparallel arrangement promotes a zigzag network of CO···HN and CO···HαCα proximal H-bonds in both directions. In this context, both H-bond types exhibit cooperative behavior as indicated by a steady increase in the occupancy of the σ*(NH) and σ*(CαHα) orbitals, as well as in the stabilization energies for the electronic donation of the corresponding carboxylic oxygens. In addition, CO···HαCα distal H-bonds are not present like in the PDAR and ADPR configurations, as indicated by the decreasing q(σ*CαHα) and negligible E(2)n(O)→σ*(CαHα) values.
QTAIM descriptors also indicate enhanced electron density, favorable Laplacian, and appropriate positions for the critical points corresponding to CO···HN and CO···HαCα proximal H-bonds; while the opposite for CO···HαCα distal contacts, evincing their absence. These results are widely discussed in the supplementary material and reported in Tables S3, S4, S7, S8 and Figs. S5, S10.
Summary of interactions and corroboration of the computational models
Energetic and geometric summary of H-bonds
Table S14 consolidates the computational metrics of the H-bonds examined in this study, i.e., between antiparallel PPII helices, thus collecting the same information already existing between parallel ones11. This average variation should be understood as the mean difference between the last and first values for the interfaces ab (Xabcde – Xab), bc (Xabcde – Xabc), and cd (Xabcde – Xabcd) of the 2D computational model, calculated for parameter X. Our NBO and QTAIM analyzes, as well as the DFT-D and SAPT calculations of the interaction energy or HBS, point to the existence and cooperativity of CO···HN and CO···HαCα proximal H-bonds. The case of CO···HαCα distal H-bonds is more complex, since they are only found one-third as often as the previous two, and when they are present, they are uncooperative and weak. In fact, when more than one growth direction of the bundle is considered, i.e., in the 3D computational models, the distal H-bonds never exist. The distances between H-bond acceptors (carbonyl oxygen) and donors (HN or Hα) mirror the observed strengthening, or lack thereof, of these proposed H-bond types between antiparallel PPII helices (Table S15 and Fig. S17). Specifically, the longest distances are associated with CO···HαCα distal H-bonds, which are the weakest. Conversely, shorter distances are observed for the cooperative CO···HN and CO···HαCα proximal H-bonds, which indicate stronger interactions. Figure 14 offers a graphical comparison of interactions according to the strength and cooperativity of the different existing H-bonds between parallel and antiparallel PPII helices.

The gray PPII helices are parallel to each other and antiparallel to the yellow one. Five stars for strength are assigned to the CO···HN H-bond of a parallel arrangement, three stars to the CO···HαCα inner H-bond, and the one to the CO···HαCα front H-bond based on the second presenting half the HBS of the first and three times that of the third11. Regarding cooperativity, five stars are assigned to the CO···HN H-bond for showing the maximum increase when completing the parallel PPII helical bundle, three stars to CO···HαCα inner H-bond for showing half of that increase, and none to CO···HαCα front H-bond for not showing cooperativity11. Likewise, for the H-bonds of an antiparallel arrangement: five stars for strength and cooperativity for CO···HN H-bonds for presenting more than twice the HBS and interaction energy increases as CO···HαCα proximal H-bonds (Table 4 and Fig. 7), which have been awarded two stars for each category. Meanwhile, only one star of strength for CO···HαCα distal H-bonds for having half the HBS of their proximal counterparts and zero stars of cooperativity for not presenting HBC (Table 4 and Fig. 7). Note that non-canonical distal H-bonds are only present in 2D bundles, but not the 3D bundles.
NMR validation of theoretical parameters
In order to validate our findings, we explored both experimental and computational changes in NMR chemical shifts induced by H-bond formation. Gly-rich peptides in solution are generally inclined to assume an extended PPII helical structure rather than a random coil or other conformations25,26,27. The two 1Hα NMR signals are typically degenerate in disordered proteins. Interestingly, we previously found that in PPII helical bundles, these two 1Hα shifts are distinct, offering the first set of conformational chemical shifts, Δδ, specific to Gly-rich PPII helical bundles4. To further validate our observations, we predicted conformational chemical shifts for 13Cα, 13CO, and 1Hα nuclei involved in CO···HαCα H-bonds of the 2D and 3D computational models. The experimental and these theoretical conformational shifts are shown in Table 8. In every case, the computational values fall within the range of the empirical ones, lending additional credence to the presence of the H-bonds analyzed here.
Discussion
The stability of proteins and protein domains featuring Gly-rich PPII helical bundles has long been a subject of intrigue, given the inherently high flexibility of glycine residues4,5,6,11,20,25. Various factors have been identified as contributing to the stable folded state of these proteins, like a moderate conformational entropy in the denatured state6 or a significant number of CO···HαCα H-bonds4,5. Previously, we reported the HBC as a possible stabilizing effect in Gly-rich parallel PPII arrangements like those displayed by both layers of HhAFP or GrAFP11. In this study, we have employed five carefully designed computational models, corroborated by NMR observables, to probe the potential stabilizing forces at play in between Gly-rich antiparallel PPII helices when considering them alone and in combination with parallel ones. Our investigation addresses the different types of H-bonds between antiparallel PPII helices, which display a unique zigzag pattern (Fig. 5A), as well as the effect of introducing into the H-bonding network the aligned interactions between parallel PPII helices (Fig. 10).
First, by means of NBO and QTAIM methodology, as well as DFT-D and SAPT calculations, our computational analyzes have unveiled the existence of HBC between antiparallel PPII helices when considering a bundle that grows only in one direction. This revelation significantly enhances our understanding of the stabilizing forces in Gly-rich PPII helical bundles. We have confirmed the existence and cooperativity of CO···HN H-bonds and those that we call CO···HαCα proximal H-bonds, while our results strongly suggest that those that we call CO···HαCα distal H-bonds do not always form, and when they do, they are weak and noncooperative. Thus, they do not contribute as much as the others to the stability of the antiparallel arrangement along one plane. This reconciles with experimental evidence where through NMR two different chemical shifts are obtained for the Hαs of the glycines in PPII bundles containing both parallel and antiparallel helices, which is consistent with only one of them being involved in H-bonds, or with one being involved in a stronger bond and the other in a weaker one4.
While the major contribution to the antiparallel arrangement comes from canonical H-bonds, the non-canonical ones also make significant contributions (Table 4 and Fig. 7). In the interaction between two antiparallel PPII helices in our 2D model before experiencing cooperativity, i.e., helices a and b without considering c, d, and e, each CO···HN H-bond represented by two formamide molecules contributes ≈5.5 kcal/mol. If we additionally consider the ≈3.5 kcal/mol arising from the CO···HαCα proximal+distal H-bonds, adequately represented by a pair of N-(2-oxoethyl)formamide molecules, we reach an HBS of ≈9.0 kcal/mol per PPII helical turn, as opposed to the initial ≈5.5 kcal/mol corresponding only to the canonical H-bonds. In this way, the important role of non-canonical H-bonds in the stability of antiparallel PPII bundles is highlighted.
These observations suggest that while CO···HN and CO···HαCα proximal interactions between antiparallel PPII helices are robust contributors to bundle stability, the distal ones play a more auxiliary role. The relative contribution of each interaction type likely depends on sequence composition and steric constraints, particularly in Gly-rich regions that favor flexibility and close packing. Interestingly, the total energy of complete bundles was found to be less than the sum of their isolated parts, indicating that long-range effects and macrodipole interactions may regulate the overall energy landscape.
When comparing the hydrogen-bonding patterns of parallel and antiparallel arrangements more closely, both exhibit cooperativity between canonical and one type of non-canonical H-bonds: inner for the former and proximal for the later. However, parallel PPII helices consistently display an additional non-cooperative interaction: the CO···HαCα front H-bond11. In contrast, the corresponding non-cooperative interaction in antiparallel PPII helical bundles, the CO···HαCα distal H-bond, is not always observed. This could be the reason why parallel PPII helices show somewhat higher interaction energies (about 1 kcal/mol per turn of PPII helix) compared to the antiparallel ones. This may also be one of the reasons why Gly-rich sequences adopting parallel arrangements, moderately more stable, give rise to layers with some larger number of PPII helices12,13 compared to the number of helices that can be found in the layers of antiparallel arrangements14,15,16,17, moderately less stable. Nevertheless, a long turn or linker would be needed to connect PPII helices if they were to be all parallel. Long connecting turns or linkers are entropically destabilizing and are also expected to lead to slower folding28,29.
The relevant formula from polymer theory is:
where L is the loop length in number of residues, Lref is a reference loop length, c is a constant = 1.63 (taking into account excluded volume effects of a random coil) or 2.4 (taking into account excluded volume effects of a random coil and end effects). In our opinion, the value of 2.4 may well have a stronger theoretical basis as it also takes into account end effects. Nevertheless, previous publications have used both the 2.428 and the 1.6329 values. Here, we decided to present the results obtained using both options so that the reader can see that even with the lower c value, 1.63, the free energy cost of the linker is substantial. Therefore, for the case of PPII helices with about three turns, about ten residues are required to make a connection between parallel helices, but just two for antiparallel ones. Taking T = 298.2 K, we get
or
for c = 1.63 or c = 2.4, respectively. This is a substantial value and could explain why antiparallel orientations are favored in model peptides19 and natural proteins with PPII helical domains20.
Furthermore, PPII helix macrodipole interactions are destabilizing in the parallel configuration but stabilizing for antiparallel PPII helices30. In α-helices, a \(\varDelta\)G of approximately 0.6 kcal/mol for altering an unfavorable interaction to a favorable one at one end can be calculated based on previously reported data31. Therefore, if two PPII helices in parallel reverse their orientation, the stabilization could amount to 1.2 kcal/mol, accounting for two interactions. Considering the macrodipoles in PPII helices are weaker than in α-helices, a contribution of one kcal/mol can be considered an approximate upper limit32. In our recent study of parallel PPII helices11, the HBS is reported to be 10 kcal/mol per helical turn: 6 kcal/mol from canonical H-bonds, 3 kcal/mol from inner, and 1 kcal/mol from front non-canonical H-bonds. This is 1 kcal/mol per turn more than for the antiparallel helices. Thus, for PPII helices with less than three turns, which are the most common in naturally occurring PPII helical bundle domains20, the contributions of the loop length (2 kcal/mol) and macrodipoles (1 kcal/mol) would be large enough to favor the antiparallel orientation over the parallel one. Interestingly, on the basis of these calculations, it is possible to predict that PPII helices with four or more turns would favor the parallel orientation.
Then, using once again both energetic and electronic descriptors, we have also established that when considering multiple planes, every possible configuration of parallel and antiparallel PPII helices leads to bundles that gain stability with size. However, it is important to note that, while the H-bonding patterns and cooperativity occurring along a single plane of parallel PPII helices hold when a multiplane bundle is considered, the H-bonding patterns and cooperativity occurring along a single plane of antiparallel PPII helices vary.
The symmetry and homogeneity of the PDPR model make it a robust baseline to distinguish cooperative versus competitive effects in more complex topologies. The CO···HN and the CO···HαCα inner H-bonds between parallel PPII helices always show cooperativity, more noticeable for the former than for the latter, while the CO···HαCα front H-bonds never cooperate due to the competition with the more favored CO···HN H-bonds, with which they share carbonyl oxygen11 (Fig. 10A). The same statements hold true for the parallel layers of the PDAR and ADPR configurations, along with the CO···HαCα outer interactions never existing.
In terms of antiparallel PPII helices, the strong CO···HN H-bonds are always cooperative, and the CO···HαCα distal H-bonds go from existing weakly, non-cooperatively one out of three times in a single plane to never existing when considering more than one plane in the bundle. On the other hand, the case of the non-canonical CO···HαCα proximal H-bonds is the most curious. They exist in a noticeable and cooperative manner when all the planes of the bundle are antiparallel (ADAR), just as they do along a single plane, but they become weak and non-cooperative when parallel arrangements are also present, though only in certain configurations (PDAR), not all (ADPR). This imparts a somewhat random character to the strength and cooperativity of these interactions, even considering that the overall trend of the multiplane bundles, with parallel or antiparallel arrangements, or combination of both, is to display HBC.
In bundles like PDAR, the mixture of directionalities imposes steric and electrostatic constraints that affect how H-bonds form and interact across multiple planes. The carbonyl oxygens responsible for forming the CO···HαCα proximal H-bonds in the antiparallel direction are the same that form the CO···HN and CO···HαCα front H-bonds in the parallel direction (Fig. 8B). Thus, the donors can not engage in such a strong and cooperative interaction due to the limited number of electrons they can share with the acceptors. In this way, the directional asymmetry of PDAR creates zones of energetic imbalance, where only one interaction per site benefits from electronic and resulting energetic reinforcement. PDAR thus represents a case in which HBC is partially frustrated due to the interplay between canonical and non-canonical H-bonding within an asymmetrical scaffold.
On the other hand, in the ADAR configuration the non-canonical proximal H-bonds of one direction are not weakened or deprived of cooperativity by the more favored canonical H-bonds of the other, with which they also share a carbonyl oxygen, since this time there is not a third interaction to share electrons with (Fig. 8C). Meanwhile, in the other mixed model, this situation of electronic competition directly does not occur, showing that the spatial distribution of donor and acceptor orbital plays a defining role. By placing weaker interactions in regions of minimal competition, the ADPR configuration allows both canonical and non-canonical H-bonds to function synergistically, each reinforcing the other across the bundle’s growth. This implies that orthogonal complementarity, rather than competition, may allow both types of H-bonds to significantly contribute to the overall stabilization of the bundle.
Therefore, when involving antiparallel PPII helices, ADAR and ADPR stand out due to their ability to support cooperative reinforcement within a mixed H-bonding network, where even non-canonical interactions make significant contributions. Unlike PDAR, where some CO···HαCα interactions were electronically suppressed by competition with stronger CO···HN H-bonds, ADAR and ADPR provide an environment where each interaction can operate with minimal interference. This balanced interplay allows weaker H-bonds to actively contribute to the cooperative framework, echoing the behavior observed for parallel PPII helices in the PDPR configuration.
Taken together, these differences suggest that there is a limit to the cooperativity that can be achieved in complex layered arrangements, likely due to constraints in donor-acceptor alignment, competition among nearby H-bonds, and possible interference from long-range dipolar effects. While symmetry does not impact HBC interaction strength, it does affect spatial opportunity and electronic independence. This highlights the importance of geometric context in modulating HBC.
Thus, we can propose that the HBC in Gly-rich antiparallel PPII helical bundles, and in those in combination with parallel arrangements, could be a key factor contributing to the enhanced stability of structures like the ones displayed by natural protein domains Salmonella phage S16 TFA, ALK, and B. subtilis Obg GTPase (Fig. 1). The presence of HBC between antiparallel PPII helices is similar to the behavior observed in amyloids7,8,9,10 and between parallel PPII helices11. This suggests a universal principle affecting the stability of protein secondary structures with extensive H-bonding networks. Indeed, HBC is also found along α-helices and even 310-helices33,34,35,36.
Conclusions
In conclusion, our enhanced understanding of the stabilizing factors in PPII helical bundles, particularly the recently discovered HBC in antiparallel arrangements, when considered alone and in combination with parallel PPII helices, opens avenues for the design of Gly-rich PPII helical proteins and raises two important questions for future research. Firstly, the dipoles of HN, CαHα, and CO in isolated PPII helices align to form a macrodipole30,37, and future experimental studies could provide quantitative measurements of the free energy contribution of these macrodipoles in parallel versus antiparallel orientations. This approach could provide insights into the stability of proteins or protein domains composed of PPII helical bundles. Secondly, we suggest testing our prediction that longer PPII helices would adopt a parallel orientation. Designing experiments or computational models to investigate how the length of PPII helices influences their alignment could provide valuable insights not only to deepen our understanding of the stability of these bundles but also to offer a foundation for innovative applications in protein design.
Methods
Dispersion-corrected Density Functional Theory (DFT-D)
Density Functional Theory (DFT) calculations were performed using the high non-locality M06-2X functional38, which has been demonstrated to effectively characterize non-covalent interactions and is notably proficient for biological systems with extensive H-bonding9,11,39,40. We selected the 6-31 + G(d) basis set, considering its balance of precision and computational efficiency for large molecular systems. Drawing upon established literature methodologies tailored for expansive biomolecular structures characterized by multiple H-bonding interactions11,41,42,43, we incorporated the D3 dispersion correction scheme44 to all our calculations. Interaction energies were subsequently counterpoise (CP)-corrected, adhering to the Boys and Bernardi method, to account for the basis set superposition error (BSSE)45. It is important to note that all quantum mechanical calculations were conducted without the inclusion of any polarizable continuum model since the objective of this work is to know the intrinsic energy of the systems under study.
The geometry of the PPII helices in the 2D and 3D bundles was optimized by keeping the Cαs frozen, while allowing amide group atoms and hydrogens to undergo energy minimization to optimize the H-bonding distances without steric clashes, emulating the dense packing observed in experimental structures. The energies of the monomes were determined using their structure from the optimized bundle, ensuring the interaction energy directly relates to the formed H-bonds. In this way, the H-Bonding Strength or HBS for the XY interface can be expressed as:
As an example, the HBS of the cd interface in the case of four PPII helices of the 2D antiparallel model is determined by:
The remaining computational parameters from NBO and QTAIM analyzes can be directly sourced at the required interface for the specific number of PPII helices in question, while in SAPT calculations, the interaction energy is determined without computing the total energy of the monomers or n-mers. Each data point in the graphs of this study aligns with the images shown in Fig. 3.
Natural Bonding Orbital (NBO)
The NBO methodology46 was employed to scrutinize the H-bonding character of interactions among the Gly residues within the Gly-rich PPII helical bundles under examination. Within the ambit of NBO approach, H-bonds can be described as an electron delocalization from a lone pair (n) of the H-acceptor atom (e.g., an oxygen atom) onto the sigma antibonding (σ*) orbital of the covalent bond between the H-donor atom and the hydrogen atom itself (e.g., an N-H or C-H bond). This interaction can be quantitatively characterized via the stabilization energy, E(2), which is calculated according to the Second Order Perturbation Theory:
where qi denotes the occupancy of the electron-donor orbital i, Ei and Ej represent the respective energies of the orbitals i and j, and Fij signifies the Fock matrix element between these two NBOs. Elevated values of qj and E(2) signify augmented electron delocalization from orbital i to j, resulting in increased stabilization of the molecular entity.
Quantum Theory of Atoms in Molecules (QTAIM)
Canonical and non-canonical H-bonds in the Gly-rich PPII bundles have been also analyzed using the QTAIM approach47,48 as described in detail in the supplementary material.
Symmetry-Adapted Perturbation Theory (SAPT)
Canonical and non-canonical H-bonds in the Gly-rich PPII bundles have been further explored using the SAPT methodology49,50 as described in detail in the supplementary material.
Theoretical chemical shift calculations
Nuclear Magnetic Resonance (NMR) shielding tensors for both isolated PPII helices and the PPII helical bundle were evaluated employing the gauge-including-atomic-orbital (GIAO) method51,52. The corresponding chemical shifts, δ, can be determined from the difference between isotropic chemical shieldings in the nucleus under study of the model, σiso, and that of the reference compound, σiso,ref:
In protein studies, the conformational chemical shifts, Δδ, represent the deviation of the experimental chemical shifts, δ1, from those predicted for statistical coil, δ2, so they can be obtained through the difference between two different chemical shifts of the same nucleus for which the same reference compound has been used:
Within the framework of the DFT-GIAO calculations, structures are treated as static rather than dynamic entities. Taking into account observations suggesting that in solution, Gly-rich sequences tend to adopt extended PPII helix conformations over random coil25,26,27, a random coil state was not utilized as a reference for the theoretical Δδ values. Instead, Δδ values were derived by contrasting the σiso of the PPII helix in the model bundle (σiso,2), geometrically optimized as described in the DFT-D calculations section, to that of the isolated PPII helix (σiso,1), keeping the backbone atom positions consistent with the crystallographic structure (PDB ID: 3BOG) and optimizing only the 1H nuclei missing in X-ray determined structures.
For geometry optimizations, single-point calculations, and NMR spectroscopic shielding computations, the Quantum Chemistry package Gaussian 1653 was employed. Visualization and modeling of the PPII helical bundle were facilitated by GaussView 654, Pymol 2.3.055, and Jmol 1456. NBO analysis was executed with NBO Version 3.157 as integrated within Gaussian 16. QTAIM analysis was conducted using the Multiwfn 3.8 software package58. Lastly, SAPT calculations were performed using the Psi4 software59.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
References
Kuhlman, B. & Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 20, 681–697 (2019).
Mompeán, M., Oroz, J. & Laurents, D. V. Do polyproline II helix associations modulate biomolecular condensates? FEBS Open Bio. 11, 2390–2399 (2021).
Pentelute, B. L. et al. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J. Am. Chem. Soc. 130, 9695–9701 (2008).
Treviño, M. Á. et al. The singular NMR fingerprint of a polyproline II helical bundle. J. Am. Chem. Soc. 140, 16988–17000 (2018).
Gates, Z. P. et al. Perplexing cooperative folding and stability of a low-sequence complexity, polyproline 2 protein lacking a hydrophobic core. Proc. Natl. Acad. Sci. USA 114, 2241–2246 (2017).
Treviño, M. Á. et al. Insight into polyproline II helical bundle stability in an antifreeze protein denatured state. Biophys. J. 121, 4560–4568 (2022).
Tsemekhman, K., Goldschmidt, L., Eisenberg, D. & Baker, D. Cooperative hydrogen bonding in amyloid formation. Protein Sci. 16, 761–764 (2007).
Mompeán, M. et al. Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP-43. J. Phys. Chem. Lett. 6, 2608–2615 (2015).
Mompeán, M., Nogales, A., Ezquerra, T. A. & Laurents, D. V. Complex system assembly underlies a two-tiered model of highly delocalized electrons. J. Phys. Chem. Lett. 7, 1859–1864 (2016).
Mompeán, M., Baralle, M., Buratti, E. & Laurents, D. V. An amyloid-like pathological conformation of TDP-43 is stabilized by hypercooperative hydrogen bonds. Front. Mol. Neurosci. 9, 125 (2016).
López-Sánchez, R., Laurents, D. V. & Mompeán, M. Hydrogen bonding patterns and cooperativity in polyproline II helical bundles. Commun. Chem. 7, 1–11 (2024).
Scholl, C. L., Tsuda, S., Graham, L. A. & Davies, P. L. Crystal waters on the nine polyproline type II helical bundle springtail antifreeze protein from granisotoma rainieri match the ice lattice. FEBS J. 288, 4332–4347 (2021).
Mok, Y.-F. et al. Structural basis for the superior activity of the large isoform of snow flea antifreeze protein. Biochemistry 49, 2593–2603 (2010).
Buglino, J., Shen, V., Hakimian, P. & Lima, C. D. Structural and biochemical analysis of the Obg GTP binding protein. Structure 10, 1581–1592 (2002).
Warkentin, E. et al. A rare polyglycine type II-like helix motif in naturally occurring proteins. Proteins Struct. Funct. Bioinforma. 85, 2017–2023 (2017).
Dunne, M. et al. Salmonella phage S16 tail fiber adhesin features a rare polyglycine rich domain for host recognition. Structure 26, 1573–1582.e4 (2018).
Li, T. et al. Structural basis for ligand reception by anaplastic lymphoma kinase. Nature 600, 148–152 (2021).
Crick, F. H. C. & Rich, A. Structure of polyglycine II. Nature 176, 780781 (1955).
Dwivedi, A. M. & Krimm, S. Vibrational analysis of peptides, polypeptides, and proteins. XV. crystalline polyglycine II. Biopolymers 21, 2377–2397 (1982).
Rodríguez, C. S. & Laurents, D. V. Architectonic principles of polyproline II helix bundle protein domains. Arch. Biochem. Biophys. 756, 109981 (2024).
Dürr, E., Jelesarov, I. & Bosshard, H. R. Extremely fast folding of a very stable leucine zipper with a strengthened hydrophobic core and lacking electrostatic interactions between helices. Biochemistry 38, 870–880 (1999).
Scheiner, S. Contributions of NH···O and CH···O hydrogen bonds to the stability of β-sheets in proteins. J. Phys. Chem. B 110, 18670–18679 (2006).
Scheiner, S. Theoretical analysis of the contributions made by CH···OH bonds to protein structure. Curr. Org. Chem. 14, 106–128 (2010).
Scheiner, S. Dissection of the factors affecting formation of a CH ∙ ∙ ∙ O H-Bond. A case study. Crystals 5, 327–345 (2015).
Ohnishi, S., Kamikubo, H., Onitsuka, M., Kataoka, M. & Shortle, D. Conformational preference of polyglycine in solution to elongated structure. J. Am. Chem. Soc. 128, 16338–16344 (2006).
Bykov, S. & Asher, S. Raman studies of solution polyglycine conformations. J. Phys. Chem. B 114, 6636–6641 (2010).
Mompeán, M. et al. Glycine rich segments adopt polyproline II helices: implications for biomolecular condensate formation. Arch. Biochem. Biophys. 704, 108867 (2021).
Nagi, A. D. & Regan, L. An inverse correlation between loop length and stability in a four-helix-bundle protein. Fold. Des. 2, 67–75 (1997).
Linse, S., Thulin, E., Nilsson, H. & Stigler, J. Benefits and constrains of covalency: the role of loop length in protein stability and ligand binding. Sci. Rep. 10, 20108 (2020).
Kuemin, M., Schweizer, S., Ochsenfeld, C. & Wennemers, H. Effects of terminal functional groups on the stability of the polyproline II structure: a combined experimental and theoretical study. J. Am. Chem. Soc. 131, 15474–15482 (2009).
Shoemaker, K. R., Kim, P. & Baldwin, R. L. Tests of the helix dipole model for stabilization of alpha-helices. Nature 326, 563–567 (1987).
Lauz, M., Eckhardt, S., Fromm, M. & Giese, K. B. The influence of dipole moments on the mechanism of electron transfer through helical peptides. Phys. Chem. Chem. Phys. 14, 13785–13788 (2012).
Ireta, J., Neugebauer, J., Scheffler, M., Rojo, A. & Galván, M. Density functional theory study of the cooperativity of hydrogen bonds in finite and infinite α-helices. J. Phys. Chem. B 107, 1432–1437 (2003).
Wieczorek, R. & Dannenberg, J. J. H-bonding cooperativity and energetics of α-helix formation of five 17-amino acid peptides. J. Am. Chem. Soc. 125, 8124–8129 (2003).
Wu, Y.-D. & Zhao, Y.-L. A theoretical study on the origin of cooperativity in the formation of 310- and α-helices. J. Am. Chem. Soc. 123, 5313–5319 (2001).
Wieczorek, R. & Dannenberg, J. J. Hydrogen-bond cooperativity, vibrational coupling, and dependence of helix stability on changes in amino acid sequence in small 310-helical peptides. A density functional theory study. J. Am. Chem. Soc. 125, 14065–14071 (2003).
Huang, K.-Y. & Horng, J.-C. Impacts of the terminal charged residues on polyproline conformation. J. Phys. Chem. B 123, 138–147 (2019).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Mompeán, M. et al. Molecular mechanism of the inhibition of TDP-43 amyloidogenesis by QBP1. Arch. Biochem. Biophys. 675, 108113 (2019).
Martín-Pintado, N. et al. Dramatic effect of furanose C2′ substitution on structure and stability: directing the folding of the human telomeric quadruplex with a single fluorine atom. J. Am. Chem. Soc. 135, 5344–5347 (2013).
Arey, J. S., Aeberhard, P. C., Lin, I.-C. & Rothlisberger, U. Hydrogen bonding described using dispersion-corrected density functional theory. J. Phys. Chem. B 113, 4726–4732 (2009).
DiLabio, G. A., Johnson, E. R. & Otero-de-la-Roza, A. Performance of conventional and dispersion-corrected density-functional theory methods for hydrogen bonding interaction energies. Phys. Chem. Chem. Phys. 15, 12821–12828 (2013).
Boese, A. D. Density functional theory and hydrogen bonds: are we there yet? ChemPhysChem 16, 978–985 (2015).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Boys, S. F. & Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. some procedures with reduced errors. Mol. Phys. 19, 553–566 (1970).
Reed, A. E., Curtiss, L. A. & Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926 (1988).
Bader, R. F. W. Atoms in molecules. Acc. Chem. Res. 18, 9–15 (1985).
Bader, R. F. W. Atoms in Molecules: A Quantum Theory (Clarendon Press, 1990).
Jeziorski, B., Moszynski, R. & Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der waals complexes. Chem. Rev. 94, 1887–1930 (1994).
Hohenstein, E. G. & Sherrill, C. D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 133, 014101 (2010).
Wolinski, K., Hinton, J. F. & Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 112, 8251–8260 (1990).
Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 27, 789–807 (1974).
Gaussian 16, Revision C.01, Frisch, M. J. et al. Gaussian, Inc. (Wallingford, CT, 2016).
GaussView, Version 6, Dennington, R.; Keith, T. A., Millam, J. M. Semichem Inc. (Shawnee Mission, KS, 2016).
The PyMOL Molecular Graphics System, Version 2.3.0. Schrödinger, LLC.
Jmol Development Team. Jmol. Retrieved from http://jmol.sourceforge.net/ (2016).
NBO Version 3.1Glendening, E. D., Reed, A. E., Carpenter, J. E., Weinhold, F.
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Smith, D. G. A. et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 152, 184108 (2020).
Acknowledgements
This paper is dedicated to Prof. C. Nick Pace, a wonderful teacher and a pioneer in demonstrating the importance of hydrogen bonds to protein stability. This work has been funded by grants PID2019-109306RB-I00 and PID2022-137806OB-I00 (D.V.L.) from MCIN/AEI/10.13039/501100011033. Co-funded by the European Union (ERC, 101042403 – BiFOLDOME, M.M.). Views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The authors also gratefully acknowledge Antonio Díaz Pozuelo from IQF-CSIC and the SGAI-CSIC for the assistance and help while using the LADON and DRAGO supercomputers, respectively.
Author information
Authors and Affiliations
Contributions
Conceptualization: R.L.S., M.M. and D.V.L.; Calculations: R.L.S. and M.M.; Data Analyzes: R.L.S., M.M. and D.V.L.; Writing original draft: R.L.S.; Writing revised draft: R.L.S., M.M. and D.V.L.; Funding Acquisition: M.M. and D.V.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
The authors state that this research has been conducted following ethical guidelines consistent with those embraced by this journal. In particular, we declare no competing financial interests. Individual data points are shown. No images have been manipulated and this research did not involve animals, human subjects or human cells or tissues. Finally, a data availability statement has been included.
Peer review
Peer review information
Communications Chemistry thanks Fei Xu and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
López-Sánchez, R., Mompeán, M. & Laurents, D.V. Orientation affects hydrogen bonding cooperativity in polyproline II helical bundles. Commun Chem 8, 195 (2025). https://doi.org/10.1038/s42004-025-01576-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01576-1
