
Abstract
Luminescent lanthanide complexes can exhibit temperature-sensitive metal-centered emission due to energy transfer quenching from the lanthanide to the ligand triplet states, which have been promising application in emission lifetime-based thermometers. However, the long-lived ligand triplet state limits the temperature sensitivity of lanthanide emission. This study demonstrates an enhancement in the temperature sensitivity of Tb(III) emission by introducing an energy escape pathway from the ligand triplet state. A dinuclear Tb(III)–Nd(III) complex containing hexafluoroacetylacetonate (hfa) and triphenylene bridging ligands was prepared, which exhibits temperature-dependent energy transfer from the Tb(III)-emitting state to the hfa triplet state. The triplet level of the hfa ligand is similar to that of the triphenylene ligand, inducing effective energy transfer from hfa to Nd(III) via the triphenylene ligands. This energy transfer pathway provides a short-lived excited state of hfa ligands, resulting in the highest temperature sensitivity (4.4% K−1) among emission lifetime-based thermometers of lanthanide complexes.

Similar content being viewed by others

Introduction
Molecular luminescent sensors have attracted considerable attention due to their structural versatility and high processability, which have led to significant interest in the fields of chemical biology and engineering1,2,3,4. The sensing parameters (i.e., emission intensity, lifetime, and wavelength) can be tuned through interactions with various chemical species (e.g., metal ions and biomolecules)5,6 and the surrounding environment (e.g., temperature and pressure changes)7,8. Among these emission parameters, the emission lifetime is independent of the excitation intensity, the scattering of light and concentration of the emitter, thereby rendering it an attractive parameter2,9.
Emission lifetime-based molecular sensors, such as organic compounds10,11 and metal complexes12, have been reported for the development of advanced biosensors and environmental sensing systems13,14. Among them, trivalent europium (Eu(III)) and terbium (Tb(III)) complexes exhibit long-lived emissions (> 0.1 ms) with narrow emission bands based on 4f-4f transitions15, which are promising candidates for emission lifetime-based molecular sensors14,16,17,18,19. In such systems, the emission intensity is enhanced by energy transfer from the lowest excited triplet (T1) of the organic ligand to the lanthanide emitting state (Ln*). The Tb(III) and Eu(III) complexes with small energy gaps (ΔE(T1−Ln*) < 2000 cm−1) can cause emission quenching due to energy transfer from the Ln* state to the T1 state at ambient temperature20,21. The energy transfer rate depends on both the temperature and the energy gap (ΔE(T1−Ln*)), which has been utilized for emission lifetime-based thermometers with high thermal sensitivities (> 1% K−1)22,23,24,25,26,27.
Previously, Gálico et al. demonstrated that Eu(III) complexes with 1-phenyl-1,3-butanedionate ligand (ΔE(T1 − 5D1) = 1000 cm−1) exhibit emission lifetime-based thermometer with high thermal sensitivity (1.35% K−1)26. Lapaev et al. also reported a high thermal sensitivity (1.4% K−1) for the Tb(III) emission lifetime based on energy transfer from Tb(III) to 1-(4-(4-propylcyclohexyl)phenyl)decane-1,3-dionate ligand (ΔE(T1 − 5D4) = 1340 cm−1)24. However, the Tb(III) and Eu(III) complexes often exhibit lanthanide emission in the presence of a thermally populated ligand T1 state, which is caused by rapid back energy transfer and a long-lived T1 state (μs–ms at room temperature, Fig. 1a)28,29,30,31,32,33,34. The characteristic excited-state dynamics limit the thermal sensitivity of the lanthanide emission lifetime. Therefore, a strategy for enhancing thermal sensitivity is crucial for the further development of emission lifetime-based thermometers.

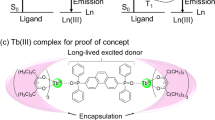
Schematic images of excited state dynamics for emission with thermal sensitivity in lanthanide complexes (a) previous study, (b) this study). c Molecular design for proof of concept. d The chemical structures of Tb(III), Nd(III) and Gd(III) complexes.
In this study, we demonstrate a strategy to enhance the thermal sensitivity based on lanthanide emission quenching, which was achieved by the introduction of an energy escape pathway from the ligand T1 state (Fig. 1b). To demonstrate this concept, a Tb(III)–Nd(III) dinuclear complex composed of [Tb(hfa)3] (hfa: hexafluoroacetylacetonate) and [Nd(hfa)3] units bridged by two bidentate triphenylene ligands (dptp: 2,7-bis(diphenylphosphoryl)triphenylene oxide) was designed ([TbNd(hfa)6(dptp)2], Fig. 1c). The Tb(III) was selected as the emitting metal center. The T1 level of the dptp ligand (T1 = 22,500 cm−1) is comparable to that of the hfa ligand (T1 = 21,700 cm−1), thereby inducing energy transfer between hfa and dptp ligands. The Nd(III) was chosen as the energy acceptor because its 4G11/2 level (~21,700 cm−1)35,36 is well matched with the triplet level of hfa ligands, allowing efficient energy transfer from hfa to Nd(III). The energy escape provides a short-lived hfa T1 state, resulting in enhanced thermal sensitivity. Three types of dinuclear trivalent lanthanide complexes ([Ln2(hfa)6(dptp)2], Ln = Tb, Nd, or Gd) were also prepared to clarify the excited state dynamics of [TbNd(hfa)6(dptp)2] (Fig. 1d). While many studies have reported heteronuclear lanthanide complexes employed as ratiometric luminescent thermometers based on lanthanide emission intensity ratios37,38,39, the present work offers a complementary approach that utilizes their emission lifetime for temperature sensing.
Results and discussion
Preparation and coordination structures
The dptp ligand was prepared from 2,7-dibromotriphenylene via a two-step reaction and was identified using proton (1H), carbon (13C), and phosphorus (31P) nuclear magnetic resonance (NMR) spectroscopy (Supplementary Figs. 1–3). The Tb(III), Nd(III), and Gd(III) complexes were prepared by the complexation of [Ln(hfa)3(H2O)2] (Ln = Tb, Nd, or Gd) with dptp. Single crystals of the Tb(III) complex were obtained via recrystallization from a chloroform/methanol solution. Single-crystal X-ray diffraction (SCXRD) analysis revealed the formation of a dinuclear complex with stacked dptp bridging ligands ([Tb2(hfa)6(dptp)2], as presented in Fig. 2 (crystallographic data are shown in Supplementary Table 1)). The distance between the dptp ligands was measured to be 3.6 Å, representing the presence of a polyaromatic π–π interaction in the Tb(III) complex. The distance between the two Tb(III) ions in [Tb2(hfa)6(dptp)2] was determined to be 14.6 Å. According to the SCXRD analysis, the Gd(III) complex also formed a dinuclear structure, [Gd2(hfa)6(dptp)2] (Supplementary Fig. 4). The powder X-ray diffraction (PXRD) patterns of the Tb(III) and Gd(III) complexes were similar to the simulated pattern of [Tb2(hfa)6(dptp)2] (Supplementary Fig. 6). The SCXRD analysis revealed that the Nd(III) complex formed a coordination polymer, [Nd(hfa)3dptp]n (Supplementary Fig. 5). The PXRD pattern of Nd(III) complex was similar to the simulated pattern obtained from SCXRD analysis (Supplementary Fig. 6). Electrospray ionization mass spectrometry (ESI-MS) analyses of [Tb2(hfa)6(dptp)2], [Nd(hfa)3dptp]n, and [Gd2(hfa)6(dptp)2] in methanol showed strong fragment signals corresponding to [Tb2(hfa)5(dptp)2]+, [Nd2(hfa)5(dptp)2]+, and [Gd2(hfa)5(dptp)2]+, which indicate the formation of dinuclear complexes under solution conditions (Supplementary Fig. 7). The major luminescent species in the Nd(III) complex under solution conditions is [Nd2(hfa)6(dptp)2], which was further supported by dynamic light scattering (DLS) measurements, the comparison of Nd(III) emission spectral shapes and the single-exponential Nd(III) emission decay curve (Supplementary Note 1).

ORTEP drawing (ellipsoids set at 50% probability) of [Tb2(hfa)6(dptp)2] without hydrogen atoms. Carbon, oxygen, fluorine, phosphorus, and terbium atoms are represented by gray, red, light green, orange, and green spheres, respectively.
Tb-Nd mixed crystal was also prepared by the complexation of [Tb(hfa)3(H2O)2] and [Nd(hfa)3(H2O)2] with dptp (Tb/Nd/dptp molar ratio = 0.1:0.9:1). This stoichiometric ratio was chosen to favor the formation of the heteronuclear [TbNd(hfa)6(dptp)2] species over the homonuclear [Tb2(hfa)6(dptp)2] species, allowing us to distinguish the Tb(III) emission lifetime of [TbNd(hfa)6(dptp)2] from that of [Tb2(hfa)6(dptp)2] in the mixed system. Powder X-ray diffraction pattern of Tb-Nd mixed crystal was comparable to that of [Tb2(hfa)6(dptp)2], which suggests the formation of dinuclear structure in the mixed crystal (Supplementary Fig. 6). ESI-MS analysis of the Tb-Nd mixed crystal in chloroform showed fragment peaks corresponding to [Tb2(hfa)5(dptp)2]+, [TbNd(hfa)5(dptp)2]+, and [Nd2(hfa)5(dptp)2]+, indicating that the Tb-Nd mixed crystal contains three types of dinuclear complexes with different lanthanide compositions under solution condition (Supplementary Fig. 8).
Photophysical properties and performance for luminescent thermometers
Photophysical measurements were conducted for the [Ln2(hfa)6(dptp)2] (Ln = Tb, Nd, or Gd) in 2-methyltetrahydrofuran (2-MeTHF) solution (0.1 mM). The solution of 2-MeTHF forms a transparent glass below 137 K, which is suitable for photophysical measurements under solution condition at low temperature. Absorption bands were observed at 266 and 363 nm for [Ln2(hfa)6(dptp)2], which were assigned to the π–π* transitions of the dptp ligands (Supplementary Fig. 12). The absorption bands observed at 363 nm were red-shifted compared to that of the free dptp ligand in 2-MeTHF (355 nm), which was attributed to the presence of polyaromatic π–π interaction29. Broad absorption bands at around 300 nm were observed, which mainly originate from the π–π* transitions of the hfa ligands40. These absorption bands were also observed for the Tb-Nd mixed crystals dissolved in 2-MeTHF (Tb-NdMeTHF, which contains three types of dinuclear complexes, [Tb2(hfa)6(dptp)2], [Nd2(hfa)6(dptp)2], and the heteronuclear complex [TbNd(hfa)6(dptp)2]).
The emission spectra of [Tb2(hfa)6(dptp)2] and [Nd2(hfa)6(dptp)2] are shown in Fig. 3 (293 K, 0.1 mM, 2-MeTHF). The [Tb2(hfa)6(dptp)2] showed sharp emission bands at 484, 542, 585, 620, and 650 nm, which were assigned to the 5D4 → 7FJ (J = 6, 5, 4, 3, and 2, respectively) transitions of Tb(III) ions (Fig. 3a). The [Nd2(hfa)6(dptp)2] showed emission bands at 880, 1060, and 1330 nm, which were assigned to the 4F3/2 → 4IJ (J = 9/2, 11/2, and 13/2, respectively) transitions of Nd(III) ions (Fig. 3b). Both [Tb2(hfa)6(dptp)2] and [Nd2(hfa)6(dptp)2] also exhibited additional broad emission bands at 390 nm, which were attributed to the fluorescence of dptp ligands in the dinuclear complexes (Supplementary Fig. 13). In the excitation spectra of [Tb2(hfa)6(dptp)2] and [Nd2(hfa)6(dptp)2], excitation bands were observed in the ultraviolet region, indicating the occurrence of energy transfer from the ligands to the lanthanide ions (Supplementary Fig. 14). The energy transfer pathway for Tb(III) sensitization is further discussed in the Supplementary Information (Supplementary Note 5). The emission decay curves for the Tb(III) and Nd(III) complexes in 2-MeTHF were analyzed using single exponential functions (Fig. 4 and Supplementary Fig. 37). The emission lifetimes for [Tb2(hfa)6(dptp)2] and [Nd2(hfa)6(dptp)2] were determined to be 150 and 1.5 μs, respectively (Table 1 and Supplementary Table 16).

Emission spectra of [Tb2(hfa)6(dptp)2] (black line) and [Nd2(hfa)6(dptp)2] (blue line) in 2-MeTHF (0.1 mM) and Tb-NdMeTHF (0.1 mM, red broken line) at 293 K (λex = 360 nm). The spectra in visible (a) and near infrared regions (b) are normalized at 542 nm and 1060 nm, respectively.

Tb(III) emission decay curves of [Tb2(hfa)6(dptp)2] in 2-MeTHF (0.5 mM, black line) and Tb-NdMeTHF (0.5 mM, red line) at 300 K (λex = 360 nm, λem = 542 nm). The emission decay curves are normalized at their maximum intensity.
Herein, we evaluated the photophysical properties of the Tb-NdMeTHF. Emission spectral shape for Tb-NdMeTHF is comparable to those of [Tb2(hfa)6(dptp)2] and [Nd2(hfa)6(dptp)2] (Fig. 3), which suggests a similar coordination geometry between [TbNd(hfa)6(dptp)2] and [Ln2(hfa)6(dptp)2] (Ln = Tb and Nd). Time-resolved emission spectroscopy (300 K) revealed a double-exponential decay of Tb(III) emission in Tb-NdMeTHF (Fig. 4). The Tb(III) emission lifetimes for Tb-NdMeTHF were determined to be 151 μs (12%) and 6.5 μs (88%) (Table 1). The long emission lifetime component was attributed to [Tb2(hfa)6(dptp)2] species in Tb-NdMeTHF. The short emission lifetime component can be assigned to the Tb(III) emission in [TbNd(hfa)6(dptp)2], which is caused by the presence of the additional emission quenching pathway induced by Nd(III). This quenching pathway does not correspond to the direct energy transfer from Tb(III) to Nd(III) since the rate constant of the energy transfer (< 10 s−1) was estimated to be three times smaller than the radiative rate constant of Tb(III) ion (~103 s−1, see Supplementary Note 2).
To perform a comparative study for evaluating the luminescent thermometer properties of [Tb2(hfa)6(dptp)2] and [TbNd(hfa)6(dptp)2], the temperature-dependent Tb(III) emission lifetimes of [Tb2(hfa)6(dptp)2] dissolved in 2-MeTHF and Tb-NdMeTHF were measured (0.5 mM, Fig. 5). The emission lifetime of [Tb2(hfa)6(dptp)2] increased upon lowering the temperature from 300 to 125 K and remained constant between 125 and 100 K (Supplementary Figs. 15–17 and Supplementary Table 6). The Tb-NdMeTHF exhibited two Tb(III) emission lifetime components within the temperature range of 100–300 K (Supplementary Figs. 18–20 and Supplementary Table 6). The longer emission lifetime component was comparable to that of [Tb2(hfa)6(dptp)2], indicating that the shorter emission lifetime component originated from [TbNd(hfa)6(dptp)2]. The temperature-dependent Tb(III) emission lifetimes for [Tb2(hfa)6(dptp)2] and [TbNd(hfa)6(dptp)2] were also analyzed using the Mott–Seitz model41,42,43 which is expressed as follows:
where \({\tau }_{{{{\rm{rad}}}}}\) is the radiative lifetime of Tb(III), \({A}_{1}\) and \({A}_{2}\) are the pre-exponential factors, \({E}_{{{{\rm{a}}}}1}\) and \({E}_{{{{\rm{a}}}}2}\) are the activation energies, \({k}_{{{{\rm{B}}}}}\) is the Boltzmann constant, and \(T\) is temperature. The fitting parameters are listed in Table 2. Fitting with a single activation term failed to reproduce the experimental data for both [Tb2(hfa)6(dptp)2] and [TbNd(hfa)6(dptp)2], necessitating the use of the two-component Mott–Seitz model (Supplementary Note 3). The radiative lifetime was approximated as the emission lifetime of [Tb2(hfa)6(dptp)2] measured at 100 K (764 μs) where nonradiative deactivation is significantly suppressed. This approximation is supported by the temperature independence of the emission lifetime at 100–125 K44,45,46. Subsequently, the emission lifetime-based relative thermal sensitivities were evaluated as follows:
where \(\tau\) is the Tb(III) emission lifetimes of [Tb2(hfa)6(dptp)2] or [TbNd(hfa)6(dptp)2].

Temperature-dependent Tb(III) emission lifetimes (λex = 360 nm, λem = 542 nm) of [Tb2(hfa)6(dptp)2] in 2-MeTHF (0.5 mM, green dots) and Tb-NdMeTHF (0.5 mM, black and red dots). The solid lines are obtained by fitting with the Mott-Seitz model (Eq. 1). Error bar represents standard deviation.
Relative thermal sensitivity is a useful parameter that allows direct comparison with other luminescent thermometers, compared to the absolute thermal sensitivity defined as
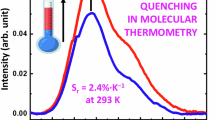
As presented in Fig. 6, the calculated relative thermal sensitivity of [TbNd(hfa)6(dptp)2] was determined to be higher than that of [Tb2(hfa)6(dptp)2]. The maximum relative thermal sensitivities of [Tb2(hfa)6(dptp)2] and [TbNd(hfa)6(dptp)2] were estimated to be 2.5% K−1 (300 K) and 4.4% K−1 (256 K), respectively. The sensitivity calculated for [TbNd(hfa)6(dptp)2] represents the highest value among lifetime-based thermometers of lanthanide complexes (see Supplementary Table 8 for comparison with previously reported systems).

Relative thermal sensitivities of [Tb2(hfa)6(dptp)2] (black line) and [TbNd(hfa)6(dptp)2] (red line) derived by their temperature-dependent Tb(III) emission lifetimes.
Mechanistic study 1: Determination of the organic ligand energy levels
To determine the excited triplet energy levels of the hfa and dptp ligands, time-resolved emission spectra were measured for [Gd2(hfa)6(dptp)2]. The time dependence of the emission spectral shape was observed (Fig. 7 and Supplementary Fig. 22), and the corresponding emission decay curve was analyzed using a double exponential function (Supplementary Fig. 23 and Supplementary Table 9), indicating the existence of two luminescent centers in [Gd2(hfa)6(dptp)2]. The emission spectrum of [Gd2(hfa)6(dptp)2] with a delay of 10 ms after photoexcitation was similar to that of [Gd(hfa)3(tppo)2] (tppo: triphenylphosphine oxide, Supplementary Fig. 24). This result suggests that the main component of the emission band is the phosphorescence of the hfa ligand in [Gd2(hfa)6(dptp)2]. By band deconvolution, the lowest excited triplet level of the hfa ligand was estimated to be 21,700 cm−1. The phosphorescence spectrum of [Gd2(hfa)6(dptp)2] with a delay of 200 ms after photoexcitation was similar to that of the free dptp ligand (Supplementary Fig. 25), suggesting that the additional emission band of [Gd2(hfa)6(dptp)2] could be attributed to dptp phosphorescence. This interpretation was also supported by the observation of comparable phosphorescence decay curves for free dptp ligand and [Gd2(hfa)6(dptp)2] within the 1–10 s range (Supplementary Fig. 23). The lowest excited triplet level of dptp in [Gd2(hfa)6(dptp)2] was determined to be 22,500 cm⁻1 based on the band deconvolution of the phosphorescence spectrum.

Phosphorescence spectra (red lines, λex = 360 nm) of [Gd2(hfa)6(dptp)2] in 2-MeTHF (0.5 mM) at 80 K with different delay times ((a) 10 ms and (b) 200 ms). Blue lines are obtained by band deconvolution analyses with Gaussian functions. Black broken lines are the summations of the (a) five and (b) seven blue lines.
Mechanistic study 2: Determination of the Tb(III) emission quenching pathway
To determine the Tb(III) emission quenching pathways in [Tb2(hfa)6(dptp)2] and [TbNd(hfa)6(dptp)2], the calculated activation energies (\({E}_{{{{\rm{a}}}}1}\) and \({E}_{{{{\rm{a}}}}2}\)) and pre-exponential factors (\({A}_{1}\) and \({A}_{2}\)) from Eq. 1 were considered (Table 2). Similar \({E}_{{{{\rm{a}}}}1}\) values were estimated for [Tb2(hfa)6(dptp)2] (2160 cm−1) and [TbNd(hfa)6(dptp)2] (2380 cm−1), which are consistent with previously reported values for Tb(hfa)3-typed complexes39,44. These results indicate that the quenching pathways originate from endothermic energy transfer from the Tb(III) emitting level (5D4) to the localized excited triplet level of hfa. The additional quenching pathway related to \({E}_{{{{\rm{a}}}}2}\) is discussed in the Supplementary Information (Supplementary Note 4). The pre-exponential factor \({A}_{1}\) for [TbNd(hfa)6(dptp)2] (1.3 × 1010 s−1) was found to be two orders of magnitude higher than that for [Tb2(hfa)6(dptp)2] (1.35 × 108 s−1), indicating the stronger emission quenching process in [TbNd(hfa)6(dptp)2] than in [Tb2(hfa)6(dptp)2], which occurs via energy transfer from the 5D4 level of Tb(III) to the triplet level of hfa. To reveal the origin of the difference, the oxygen concentration-dependent emission lifetimes of the dinuclear complexes were measured, which is a useful method for revealing the triplet population during Tb(III) emission44,47. The emission lifetime for [Tb2(hfa)6(dptp)2] depends on the oxygen concentration (N2 bubblingː τ = 141 μs, O2 bubbling: τ = 28 μs). In contrast, that for [TbNd(hfa)6(dptp)2] is almost independent of the oxygen concentration (N2 bubblingː τ = 7.1 μs, O2 bubblingː τ = 6.2 μs, Supplementary Table 10). The less populated triplet (hfa) state during the Tb(III) emission in [TbNd(hfa)6(dptp)2] indicates the existence of triplet energy escape (hfa) upon the incorporation of Nd(III).
To discuss the detailed energy escape pathway from hfa (coordinated to Tb) to Nd, we showed the excited-state diagram of [TbNd(hfa)6(dptp)2] (Fig. 8). Herein, the hfa ligands coordinated to Tb and Nd are denoted as hfaTb and hfaNd, respectively. The possible quenching processes were classified into two pathways (Tb → hfaTb → dptp → (hfaNd → ) Nd and Tb → hfaTb → hfaNd → Nd). The possible Tb → dptp → Nd energy transfer pathway is considered negligible since the calculated energy transfer rate constant from hfaTb to Tb(III) is much larger than from dptp to Tb(III) (Supplementary Note 5). The localized triplet level (3LE) of the hfa ligand (21,700 cm−1) was determined to be close to that of dptp (22,500 cm−1), and the shortest distance between hfa and the dptp exciton center was measured to be 7.4 Å (Supplementary Fig. 26), which allows for effective triplet–triplet energy transfer between hfa and dptp48. The presence of this energy transfer pathway is suggested by nanosecond transient absorption spectroscopy (ns-TAS, Supplementary Note 6). On the other hand, the shortest distance between the hfaTb and hfaNd exciton centers was determined to be 15.8 Å, which suggests that triplet–triplet energy transfer between hfaTb and hfaNd represents a less effective quenching pathway. Energy matching between the donor (localized hfa/dptp triplet) and acceptor levels (Nd(III), 4G11/2: 21,700 cm−1)35,36 allows for effective energy transfer from the ligands to Nd(III). The energy transfer rate constant from the hfa triplet state to Nd(III) was calculated to be two-orders of magnitude faster than that from the dptp triplet state to Nd(III) (Supplementary Note 5). From these results, it was considered that the presence of a sequential energy transfer pathway (i.e., Tb → hfaTb → dptp → hfaNd → Nd) in [TbNd(hfa)6(dptp)2] shortens the 3LE lifetime of hfaTb compared to that in [Tb2(hfa)6(dptp)2], which is also supported by the Nd(III) emission lifetime analyses (Supplementary Note 7). The short-lived excited triplet state of hfa contributes to the high thermal sensitivity of the Tb(III) emission in [TbNd(hfa)6(dptp)2] compared to that in [Tb2(hfa)6(dptp)2].

The energy diagram and triplet energy escape for [TbNd(hfa)6(dptp)2]. The hfaTb and hfaNd represent the hfa ligands coordinated to Tb(III) and Nd(III) ion, respectively. The 3LE denotes the locally excited triplet state.
Conclusion
In this study, we demonstrate an enhancement in the temperature sensitivity of Tb(III) emissions with a thermally populated ligand triplet state. This enhancement was achieved by introducing an energy-escape pathway from the triplet state. This energy escape pathway induces the formation of a short-lived ligand-excited triplet state, which promotes the quenching of Tb(III) emission. The maximum relative thermal sensitivity of the Tb(III) complex was enhanced to 4.4% K−1, which represents the highest value among emission lifetime-based thermometers of lanthanide complexes. The short-lived ligand excited triplet state can be also accomplished through the inclusion of certain functional groups with non-bonding lone pairs and/or heavy atom effect49. While lanthanide ions themselves exhibit a heavy atom effect50, incorporation of heavy atoms directly into the ligand structure can further promote intersystem crossing independently of the metal center51,52. These findings provide a new design principle for the development of highly sensitive emission lifetime-based thermometers using lanthanide complexes and are expected to promote the development of advanced non-contact temperature sensing applications in chemical biology and engineering.
Methods
Materials
2,7-Dibromotriphenylene (>98.0%), chlorodiphenylphosphine (>97.0%), and n-butyllithium (ca. 15% in hexane, ca. 1.6 M, n-BuLi) were purchased from Tokyo Chemical Industry Co., Ltd. Hydrogen peroxide (30%), terbium(III) acetate tetrahydrate (Tb(OAc)3·4H2O, 99.9%), and neodymium(III) acetate monohydrate (Nd(OAc)3·H2O, 99.9%) were purchased from Wako Pure Chemical Industries, Ltd. 2-Methyltetrahydrofuran (biorenewable, anhydrous, ≥99%, inhibitor-free, 2-MeTHF) and gadolinium acetate tetrahydrate (Gd(OAc)3·4H2O, 99.9%) were purchased from Sigma-Aldrich. Co. LLC. All the other chemicals were reagent grade and used without further purification.
General methods
1H-NMR, 13C-NMR, and 31P-NMR spectra were recorded on JEOL ECS400 instrument at 298 K. Tetramethylsilane (TMS, for 1H-NMR and 13C-NMR spectroscopies) and phosphoric acid (H3PO4 for 31P-NMR spectroscopy) were used as internal references. ESI-MS was performed by JEOL JMS-T100LP instrument. Elemental analyses were measured by Exeter Analytical CE440 instrument. DLS measurements were performed with a Microtrac NANOTRAC Wave II-UT151. Inductively coupled plasma optical emission spectrometry (ICP-OES) was carried out using Agilent 5900.
Crystallography
Crystallographic data for dinuclear complexes were measured by Rigaku XtaLAB Synergy with graphite monochromatic Mo-Kα radiation (λ = 0.71073 nm). These data were processed by direct methods and expanded using Fourier techniques. The atoms except for hydrogen atoms were refined anisotropically by the SHELX system53. Hydrogen atoms were refined based on the riding model. All calculations were carried out by Olex2 software package54. Powder X-ray diffraction patterns were measured by a Rigaku SmartLab diffractometer with Cu-Kα radiation and a D/teX Ultra detector at room temperature.
Optical measurements
UV-vis absorption spectra were measured by using a JASCO V-670 spectrometer. Emission and excitation spectra were recorded by using Edinburgh Instruments FLS1000 Photoluminescence Spectrometer. Emission decay curves of Tb(III) and Nd(III) emission were measured by HORIBA Fluorolog-3 spectrofluorometer with a cryostat (Oxford Instruments, OptistatDN) and a temperature controller (Oxford Instruments, ITC 502S). SpectraLED-355 (λex = 360 nm, FWHM = 17 nm) was used as an excitation power source for recording the emission decay curves. We used Decay Analysis Software v6.8 (HORIBA) for emission decay analysis. The emission decay curves of dinuclear complexes were analyzed by reconvolution fitting unless otherwise noted. The reported percentage contributions of the emission lifetimes were calculated based on their amplitude. The phosphorescence spectra of Gd(III) dinuclear complexes were recorded on JASCO FP-8550 with a cryostat (Oxford Instruments, OptistatDN). The phosphorescence decay curves were measured by using JASCO FP-6300. The phosphorescence decay curve of [Gd2(hfa)6(dptp)2] was analyzed by double-exponential tail fitting. The ns-TAS spectra were recorded with a UNISOKU TSP-2000 laser flash photolysis system.
Synthesis of 2,7-bis(diphenylphosphoryl)triphenylene oxide (dptp)
n-BuLi in hexane (1.6 M, 3.4 mL, 5.4 mmol) was added to the solution of 2,7-dibromotriphenylene (1.00 g, 2.59 mmol) in anhydrous THF at −80°C under argon atmosphere. After stirring the solution for 1 h with increasing temperature from −90°C to −60°C, chlorodiphenylphosphine (1.04 mL, 5.70 mmol) was added. The resulting solution was stirred for 60 h at room temperature. After the reaction, distilled water was added to the solution. The product was extracted by dichloromethane, and then the organic layer was dried over anhydrous Na2SO4. To the organic solution, 30% H2O2 aqueous solution (12 mL) was added at 0°C. The reaction mixture was stirred for 1 h. The product was extracted by using dichloromethane, and the extract was dried over anhydrous Na2SO4. The product was purified by silica gel column chromatography (ethyl acetate/methanol = 97/3-97/5) to give a colorless solid (1.38 g, 2.20 mmol, 85%).
1H-NMR (392 MHz, CDCl3, 298 K, TMS): δ 9.13 (d, J = 13.5 Hz, 2H), 8.71 (dd, J = 8.5, 2.7 Hz, 2H), 8.56 (q, J = 3.1 Hz, 2H), 7.71-7.86 (m, 10H), 7.47-7.70 (m, 14H) ppm.
13C-NMR (100 MHz, CDCl3, 298 K, TMS): δ 132.9, 132.6, 132.2, 132.1, 131.8, 131.5, 131.4, 130.5, 130.4, 129.7, 129.6, 129.4, 128.7, 128.6, 128.4, 128.3, 124.0, 123.9, 123.6 ppm.
31P-NMR(159 MHz, CDCl3, 298 K, H3PO4): δ 29.48 ppm.
ESI-MS (m/z): calcd. for C42H30NaO2P2 [M+Na]+: 651.16, found: 651.16.
Synthesis of [Ln(hfa)3(H2O)2] (Ln = Tb, Nd, Gd)
The [Ln(hfa)3(H2O)2] were prepared according to a previously reported method55. To the water solution of Ln(OAc)3·nH2O (Tb(OAc)3·4H2O: 2.50 g, 6.13 mmol; Nd(OAc)3·H2O: 2.00 g, 5.89 mmol; Gd(OAc)3·4H2O: 2.00 g, 4.92 mmol), hexafluoroacetylacetone (for [Tb(hfa)3(H2O)2]: 2.57 mL, 18.4 mmol, for [Nd(hfa)3(H2O)2]: 2.49 mL, 17.8 mmol, and for [Gd(hfa)3(H2O)2]: 2.92 mL, 20.9 mmol) was added dropwise at room temperature. After stirring the mixture for 6 h at room temperature, the resulting product was filtered and washed with distilled water and chloroform, and then dried under vacuum to give [Ln(hfa)3(H2O)2] ([Tb(hfa)3(H2O)2]: colorless solid, 3.12 g, 3.82 mmol, 62%; [Nd(hfa)3(H2O)2]: light purple solid, 2.95 g, 3.68 mmol, 62%; [Gd(hfa)3(H2O)2]: colorless solid, 2.26 g, 2.77 mmol, 46%).
Synthesis of [Tb2(hfa)6(dptp)2]
To the solution of [Tb(hfa)3(H2O)2] (100 mg, 0.123 mmol) in solvent of chloroform/methanol = 1/1 (2 mL), dptp (77 mg, 0.12 mmol) was added. The mixture was heated to be a transparent solution. The solution slowly evaporated at room temperature to give colorless crystal (113 mg, 0.0401 mmol, 65%).
ESI-MS (m/z): calcd. for C109H65F30O14P4Tb2 [M−hfa]+: 2610.14, found: 2610.16.
Elemental analysis (%): calcd. for C114H66F36O16P4Tb2: C, 48.60; H, 2.36; found: C, 48.14; H, 2.23.
Synthesis of [Nd(hfa)3dptp]n
To the solution of [Nd(hfa)3(H2O)2] (300 mg, 0.374 mmol) in solvent of chloroform/methanol = 1/1 (3 mL), dptp (235 mg, 0.374 mmol) was added. The mixture was heated to be a transparent solution. The solution was slowly evaporated at room temperature to give light blue crystal (307 mg, 0.110 mmol, 59%).
ESI-MS (m/z): calcd. for C109H65F30O14P4Nd2 [Nd2(hfa)5(dptp)2]+: 2579.11, found: 2579.11.
Elemental analysis (%): calcd. for C57H33F18NdO8P2: C, 49.11; H, 2.39; found: C, 49.11; H, 2.35.
Synthesis of [Gd2(hfa)6(dptp)2]
To the solution of [Gd(hfa)3(H2O)2] (100 mg, 0.123 mmol) in solvent of chloroform/methanol = 1/1 (3 mL), dptp (77 mg, 0.12 mmol) was added. The mixture was heated to be a transparent solution. The solution was slowly evaporated at room temperature to give colorless crystal (112 mg, 0.040 mmol, 65%).
ESI-MS (m/z): calcd. for C109H65F30O14P4Gd2 [M−hfa]+: 2607.14, found: 2607.10.
Elemental analysis (%): calcd. for C114H66F36O16P4Gd2: C, 48.66; H, 2.36; found: C, 48.63; H, 2.53.
Synthesis of Tb-Nd mixed crystal
To the solution of [Tb(hfa)3(H2O)2] (11 mg, 0.13 mmol) and [Nd(hfa)3(H2O)2] (100 mg, 0.125 mmol) in solvent of chloroform/methanol = 1/1 (3 mL), dptp (87 mg, 0.14 mmol) was added. The mixture was heated to be a transparent solution. The solution was slowly evaporated at room temperature to give light blue crystal (123 mg, 0.044 mmol, 63%). The chemicalcomposition (Tb3+/(Tb3++Nd3+)) of Tb-Nd mixed crystal was determined to be 5.2% from ICP-OES analysis.
ESI-MS (m/z): calcd. for C109H65F30O14P4Tb2 [[Tb2(hfa)6(dptp)2]−hfa]+: 2610.14, found: 2610.14; calcd. for C109H65F30O14P4TbNd [[TbNd(hfa)6(dptp)2]−hfa]+: 2594.12, found: 2594.13; calcd. for C109H65F30O14P4Nd2 [[Nd2(hfa)6(dptp)2]−hfa]+: 2579.11, found: 2579.11.
Elemental analysis (%): calcd. for C114H66F36O16P4Tb0.2Nd1.8: C, 49.06; H, 2.38; found: C, 49.00; H, 2.37.
Synthesis of [Gd(hfa)3(tppo)2]44
ESI-MS (m/z): calcd for C46H32F12GdO6P2 [M− hfa]+: 1128.07, found: 1128.06.
Elemental analysis (%): calcd. for C51H33F18GdO8P2: C, 45.89; H, 2.49; found: C, 45.69; H, 2.33.
Data availability
The single-crystal data obtained in this study are deposited in The Cambridge Crystallographic Data Center under accession code CCDC-2446137 for [Tb2(hfa)6(dptp)2] (Supplementary Data 1), CCDC-2475844 for [Gd2(hfa)6(dptp)2] (Supplementary Data 2), and CCDC-2475842 for [Nd(hfa)3dptp]n (Supplementary Data 3). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All the other data supporting the findings of this study are stored on our institutional server and are available from the corresponding author upon reasonable request.
References
Schäferling, M. The art of fluorescence imaging with chemical sensors. Angew. Chem. Int. Ed. 51, 3532–3554 (2012).
Zhang, K. Y. et al. Long-lived emissive probes for time-resolved photoluminescence bioimaging and biosensing. Chem. Rev. 118, 1770–1839 (2018).
Zhou, J., del Rosal, B., Jaque, D., Uchiyama, S. & Jin, D. Advances and challenges for fluorescence nanothermometry. Nat. Methods 17, 967–980 (2020).
Wang, X. D. & Wolfbeis, O. S. Optical methods for sensing and imaging oxygen: materials, spectroscopies and applications. Chem. Soc. Rev. 43, 3666–3761 (2014).
Lee, M. H., Kim, J. S. & Sessler, J. L. Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules. Chem. Soc. Rev. 44, 4185–4191 (2015).
Park, S. H., Kwon, N., Lee, J. H., Yoon, J. & Shin, I. Synthetic ratiometric fluorescent probes for detection of ions. Chem. Soc. Rev. 49, 143–179 (2020).
Brites, C. D. S. et al. Spotlight on luminescence thermometry: basics, challenges, and cutting-edge applications. Adv. Mater. 35, 2302749 (2023).
Marciniak, L., Woźny, P., Szymczak, M. & Runowski, M. Optical pressure sensors for luminescence manometry: classification, development status, and challenges. Coord. Chem. Rev. 507, 215770 (2024).
Stich, M. I. J., Nagl, S., Wolfbeis, O. S., Henne, U. & Schaeferling, M. A dual luminescent sensor material for simultaneous imaging of pressure and temperature on surfaces. Adv. Funct. Mater. 18, 1399–1406 (2008).
Mazza, M. M. A. & Raymo, F. M. Structural designs for ratiometric temperature sensing with organic fluorophores. J. Mater. Chem. C. 7, 5333–5342 (2019).
Zhang, X., Yin, J. & Yoon, J. Recent advances in development of chiral fluorescent and colorimetric sensors. Chem. Rev. 114, 4918–4959 (2014).
Li, R., Xu, F. F., Gong, Z. L. & Zhong, Y. W. Thermo-responsive light-emitting metal complexes and related materials. Inorg. Chem. Front. 7, 3258–3281 (2020).
Li, F., Wang, M., Liu, S. & Zhao, Q. Halide-containing organic persistent luminescent materials for environmental sensing applications. Chem. Sci. 13, 2184–2201 (2022).
Parker, D., Fradgley, J. D. & Wong, K. L. The design of responsive luminescent lanthanide probes and sensors. Chem. Soc. Rev. 50, 8193–8213 (2021).
Bünzli, J. C. G. Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 110, 2729–2755 (2010).
Hasegawa, Y. & Kitagawa, Y. Thermo-sensitive luminescence of lanthanide complexes, clusters, coordination polymers and metal-organic frameworks with organic photosensitizers. J. Mater. Chem. C. 7, 7494–7511 (2019).
Poole, R. A., Kielar, F., Richardson, S. L., Stenson, P. A. & Parker, D. A ratiometric and non-enzymatic luminescence assay for uric acid: differential quenching of lanthanide excited states by anti-oxidants. Chem. Commun. 4084, 4086 (2006).
Kitagawa, Y., Nakai, T., Hosoya, S., Shoji, S. & Hasegawa, Y. Luminescent lanthanide complexes for effective oxygen-sensing and singlet oxygen generation. ChemPlusChem 88, e202200445 (2023).
Wang, X. D., Wolfbeis, O. S. & Meier, R. J. Luminescent probes and sensors for temperature. Chem. Soc. Rev. 42, 7834–7869 (2013).
Latva, M. et al. Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield. J. Lumin. 75, 149–169 (1997).
Sato, S. & Wada, M. Relations between intramolecular energy transfer efficiencies and triplet state energies in rare earth β-diketone chelates. Bull. Chem. Soc. Jpn. 43, 1955–1962 (1970).
Cabral, F. M., Gálico, D. A., Mazali, I. O. & Sigoli, F. A. Crystal structure and temperature dependence of the photophysical properties of the [Eu(tta)3(pyphen)] complex. Inorg. Chem. Commun. 98, 29–33 (2018).
Costa, A. I. et al. Intensity and lifetime ratiometric luminescent thermometer based on a Tb(III) coordination polymer. Dalton Trans. 53, 3994–4004 (2024).
Lapaev, D. V., Nikiforov, V. G., Lobkov, V. S., Knyazev, A. A. & Galyametdinov, Y. G. A photostable vitrified film based on a terbium(III) β-diketonate complex as a sensing element for reusable luminescent thermometers. J. Mater. Chem. C. 6, 9475–9481 (2018).
Bhuyan, M. & Koenig, B. Temperature responsive phosphorescent small unilamellar vesicles. Chem. Commun. 48, 7489–7491 (2012).
Gálico, D. A., Mazali, I. O. & Sigoli, F. A. Nanothermometer based on intensity variation and emission lifetime of europium(III) benzoylacetonate complex. J. Lumin. 192, 224–230 (2017).
Yu, J., Sun, L., Peng, H. & Stich, M. I. J. Luminescent terbium and europium probes for lifetime based sensing of temperature between 0 and 70. C. J. Mater. Chem. 20, 6975–6981 (2010).
Kitagawa, Y. et al. Thermally-assisted photosensitized emission in a trivalent terbium complex. Commun. Chem. 6, 122 (2023).
Kitagawa, Y. et al. Stacked nanocarbon photosensitizer for efficient blue light excited Eu(III) emission. Commun. Chem. 3, 3 (2020).
Kitagawa, Y. et al. Effective photosensitization in excited-state equilibrium: brilliant luminescence of TbIII coordination polymers through ancillary ligand modifications. ChemPlusChem 87, e202200151 (2022).
Nakai, H., Kitagawa, K., Seo, J., Matsumoto, T. & Ogo, S. A gadolinium(III) complex that shows room-temperature phosphorescence in the crystalline state. Dalton Trans. 45, 11620–11623 (2016).
Sun, B. et al. Highly efficient room-temperature phosphorescence achieved by gadolinium complexes. Dalton Trans. 48, 14958–14961 (2019).
Zang, L. et al. Twenty-fold enhancement of gadolinium-porphyrin phosphorescence at room temperature by free gadolinium ion in liquid phase. J. Phys. Chem. C. 119, 28111–28116 (2015).
Ying, L. et al. Excited state properties and intramolecular energy transfer of rare-earth acylpyrazolone complexes. J. Phys. Chem. 100, 18387–18391 (1996).
Thor, W. et al. Unearthing the real-time excited state dynamics from antenna to rare earth ions using ultrafast transient absorption. JACS Au. 4, 3813–3822 (2024).
Carnall, W. T., Fields, P. R. & Rajnak, K. Electronic energy levels in the trivalent lanthanide aquo ions. I. Pr3+, Nd3+, Pm3+, Sm3+, Dy3+, Ho3+, Er3+, and Tm3+. J. Chem. Phys. 49, 4424–4442 (1968).
Wang, J., Dong, J., Cui, M., Li, X. & Wang, F. High quantum yields and energy transfer efficiency of lanthanide-based coordination polymers as luminescent thermometer in low temperature range. Appl. Organomet. Chem. 39, e7810 (2025).
Aragon-Alberti, M. et al. Rethinking assumptions: assessing the impact of strong magnetic fields on luminescence thermometry. J. Am. Chem. Soc. 146, 33723–33731 (2024).
Miyata, K. et al. Chameleon luminophore for sensing temperatures: control of metal-to-metal and energy back transfer in lanthanide coordination polymers. Angew. Chem. Int. Ed. 52, 6413–6416 (2013).
Miyazaki, S. et al. Dual energy transfer pathways from an antenna ligand to lanthanide ion in trivalent europium complexes with phosphine-oxide bridges. J. Phys. Chem. A 124, 6601–6606 (2020).
Mott, N. F. On the absorption of light by crystals. Proc. R. Soc. Lond. A 167, 384–391 (1938).
Seitz, F. An interpretation of crystal luminescence. Trans. Faraday Soc. 35, 74–85 (1939).
Li, L. et al. Visible-light excited luminescent thermometer based on single lanthanide organic frameworks. Adv. Funct. Mater. 26, 8677–8684 (2016).
Inage, K., Wang, M., Hasegawa, Y. & Kitagawa, Y. Effective photosensitized emission of a Tb(III) complex using a β-diketonate photosensitizer and an oxygen barrier system in a thermally populated triplet state. Dalton Trans. 53, 8555–8562 (2024).
Omagari, S. et al. Effective photosensitized energy transfer of nonanuclear terbium clusters using methyl salicylate derivatives. J. Phys. Chem. A 119, 1943–1947 (2015).
Sivakumar, S., Reddy, M. L. P., Cowley, A. H. & Butorac, R. R. Lanthanide-based coordination polymers assembled from derivatives of 3,5-dihydroxy benzoates: syntheses, crystal structures, and photophysical properties. Inorg. Chem. 50, 4882–4891 (2011).
Sørensen, T. J., Kenwright, A. M. & Faulkner, S. Bimetallic lanthanide complexes that display a ratiometric response to oxygen concentrations. Chem. Sci. 6, 2054–2059 (2015).
Yanai, N. & Kimizuka, N. New triplet sensitization routes for photon upconversion: thermally activated delayed fluorescence molecules, inorganic nanocrystals, and singlet-to-triplet absorption. Acc. Chem. Res. 50, 2487–2495 (2017).
Park, I. S., Min, H. & Yasuda, T. Ultrafast triplet–singlet exciton interconversion in narrowband blue organoboron emitters doped with heavy chalcogens. Angew. Chem. Int. Ed. 61, e202205684 (2022).
Mara, M. W. et al. Energy transfer from antenna ligand to europium(III) followed using ultrafast optical and X-ray spectroscopy. J. Am. Chem. Soc. 141, 11071–11081 (2019).
Wu, L. et al. Excited-state dynamics of crossing-controlled energy transfer in europium complexes. JACS Au 2, 853–864 (2022).
Isaac, M. et al. Lanthanide luminescence modulation by cation–π interaction in a bioinspired scaffold: selective detection of copper(I). Angew. Chem. Int. Ed. 54, 11453–11456 (2015).
Sheldrick, G. M. A short history of SHELX. Acta Cryst. A64, 112–122 (2008).
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 42, 339–341 (2009).
Hasegawa, Y. et al. Enhanced emission of deuterated tris(hexafluoroacetylacetonato)neodymium(III) complex in solution by suppression of radiationless transition via vibrational excitation. J. Phys. Chem. 100, 10201–10205 (1996).
Acknowledgements
This work was partially supported by a Grants-in-Aid for Scientific Research (grant numbers JP24K21774, JP23H04863, and JP20H02748) and by Iketani Science and Technology Foundation. This work also supported by the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology Agency (JST), Japan (Grant Number JPMJTR23T5), JST SPRING (Grant Number JPMJSP2119), and the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Center Initiative (WPI) of MEXT, Japan. We are grateful to Dr. Tatsuo Nakagawa and Mr. Toshiyuki Kawabata (UNISOKU Co., Ltd., Japan) for supporting the transient absorption measurements.
Author information
Authors and Affiliations
Contributions
Y.K. designed research. Y.Y. performed syntheses. Y.Y. and T.N. performed X-ray crystal measurements. Y.Y. performed optical measurements. Y.Y., T.N., S.O., and Y.K. discussed the research. S.O. performed the estimation of energy transfer rate constant from Tb(III) to Nd(III). Y.Y., T.N., S.O., M.W., Y.H., and Y.K. wrote the paper. All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yamaguchi, Y., Nakai, T., Omagari, S. et al. Ligand triplet energy escape in lanthanide complexes for developing luminescent molecular thermometers. Commun Chem 8, 269 (2025). https://doi.org/10.1038/s42004-025-01673-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-025-01673-1
