
Abstract
Using Pd homogeneous catalysts for direct C–H arylation is an attractive approach for synthesizing natural products and organic functional materials, but limitations in terms of cost and catalyst recovery could be alleviated by alternative heterogeneous catalysts. Here we demonstrated that a type of Pd single atoms/clusters cocatalyst localized on non-enzymatic browning glucose acted as a highly active and stable heterogeneous catalyst for direct coupling of a variety of inert C-H bonds with aryl iodides. Oriented to either C(sp3)-H or C(sp2)-H bond activation, designing targeted heterogeneous Pd catalyst was achieved by tailoring the structure of support in our catalyst. Multi-technique characterizations confirmed that the strong metal-support interaction (SMSI) between Pd single atoms/clusters with various coordination groups, especially imino (C = N), of support cans stabilize high valent (d electron-deficient) active Pd sites on support surface in reaction process and break the activity-stability trade-off in chelation-assisted C(sp3)-H arylation reaction via PdII/PdIV catalytic cycle.
Similar content being viewed by others

Introduction
Compared via with traditional cross-coupling reactions, the direct coupling of inert C-H bonds with aryl halides has emerged as the most attractive C-C bond-forming method enroute to a wide range of natural products, synthetic drugs, and organic functional materials (Fig. 1a)1,2. To data, a range of homogeneous transition-metal catalysts, particularly palladium catalysts, have been developed for its use in various inert C(sp2)-H and C(sp3)-H arylation reactions1,2,3,4,5,6. However, the obvious limitations of homogeneous catalysts are the technical challenge and quite high cost of catalyst-product separating before the precious metal recycled7. Several heterogeneous palladium nanoparticles (NPs) catalysts such as Pd/C (commercially available)8,9,10,11, Pd/ODDMA-MP12, HCP-NHC-Pd13, CB[6]-PdNPs14 and Pd-NPs (biogenically synthesized)15 have been used for C(sp2)-H arylation using aryl halides or [Ph2I]+ salts as coupling partner (Fig. 1b)16. However, these Pd NPs catalysts still have challenges for lower catalytic turnover numbers (TONs), Pd aggregates/leaching, and limited reaction scope17. Our previous work achieved the supported Pd NPs catalyzed benzylic C(sp3)-H bonds arylation, but stoichiometric AgI precipitate generated from the reaction process led to the hard recovery of the catalyst18. To the best of our knowledge, there is no previous report on aliphatic C(sp3)-H arylation using a heterogeneous catalyst. The poor reactivity of C(sp3)-H bonds is often attributed to its higher bond energy (~110 kcal/mol) and the complexity of precise controlling the site-selectivity1.

a C-H arylation for the synthesis of drug molecules; b Challenges of heterogeneous catalyzed C-H arylation; c Non-enzymatic browning glucose supported Pd single atoms/clusters cocatalyzed C-H arylation.
The chelation-assisted strategy was a general method in homogeneous catalysis to overcome the difficulty of controlling selectivity1,2,3,4,5,6. But the chelation-assisted C-H activation step needed adding three coordination bonds on Pd active center and the coordination number added up to >5 after oxidative addition of aryl iodide6. So low-coordinated Pd active sites or convenient ligand exchange on Pd active sites were required to achieve heterogeneous catalyzed chelation-assisted C-H arylation. However, both low-coordination or convenient ligand exchange would lead to the loss of stability of supported catalyst. And meanwhile, the oxidative addition of aryl iodide would weaken the Pd-support coordination bonds. Therefore, how to break the activity-stability trade-off is the key challenge for designing a high-efficient heterogeneous Pd catalyst for chelation-assisted C-H arylation. Moreover, different from Pd catalyzed C(sp2)-H arylation, the widely approved mechanism of the direct coupling reaction of C(sp3)-H bonds with aryl halides is PdII/PdIV catalytic mechanism as proposed by Sanford6 and Yu1 (Fig. 1b), therefore, how to stabilize high valent (d electron-deficient) active Pd sites on the support surface in reaction process remains an unsolved problem in the heterogeneous catalysis field.
Recently, the atomically dispersed palladium catalysts have been widely used in organic synthesis reactions, including hydrogenation and reduction19,20,21,22, oxidation23,24, C-C coupling7,25,26,27,28,29, C-N coupling30, and light-driven organic synthesis31. In spite of the vast potential for applications of single-atomic catalysis, single atoms (SAs) catalysts encounter issues due to their straightforward structure and the absence of synergistic active sites required to exceed the inherent performance limitation in more intricate reactions, such as site-selective C(sp3)-H activation reaction32. Integrating SAs with clusters (Cs) or NPs into a unified catalyst is an effective way to address these drawbacks via synergistic catalysis33,34,35,36.
Herein, we successfully prepared the non-enzymatic browning glucose supported Pd catalyst by the Maillard reaction of glucose with silane coupling agents following impregnation method (Fig. 1c). Compared with silane coupling agents modified cellulose37, the non-enzymatic browning glucose contains various structure motifs, especially imino (C = N) which can form strong coordination bond with Pd species resulting stable Pd SAs/Cs coexisted catalyst. Two Pd SAs/Cs catalysts were prepared for direct coupling of either C(sp3)-H or C(sp2)-H bonds with aryl iodides, respectively. By means of the systematic comparative study on homogeneous Pd catalyst, Pd NPs and Pd SAs/Cs, we verify that this type of Pd SAs/Cs cocatalyst is the best active, stable and general heterogeneous catalyst for direct coupling of a variety of inert C(sp3)-H or C(sp2)-H bonds with aryl iodides.
Results and discussions
Intrinsic structural characterization of non-enzymatic browning glucose
Non-enzymatic browning between reducing sugars and amino acids or proteins, also known as Maillard reaction, has received much attention in food science where it significantly contributes to taste, aroma, and color38,39. The Maillard reaction is initialized by the condensation of reducing sugars and amino compounds and a subsequent Amadori rearrangement and a variety of parallel and consecutive reactions resulting high-molecular-weight polymers40. The non-enzymatic browning sugars may contains a variety of structure motifs such as carboxyl (COOH), double-bond carbon (C = C), acrylamide, imino (C = N), pyridone or pyranone, and behave as anionic material to form stable complexes with metal cations41,42,43,44,45,46. However, as far as we know, because of the colloidal character and uncertain structure of non-enzymatic browning sugars, its application in catalytic chemistry field remains very rare.
The non-enzymatic browning glucose support was prepared according to Bäckvall’ work by changing cellulose to glucose37. To unveil the structure features of support, non-enzymatic browning glucoses and catalysts were analyzed by Fourier Transform Infrared spectrometer (FT-IR) and solid state NMR (Supplementary Methods). As shown as Fig. 2a, the FT-IR spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose showed the same broad band around 3300 cm−1, corresponding to the stretching vibration of the -OH and -NH2 groups, whereas glucose showed two peaks at 3409 and 3315 cm−1. The new peak at 1661 cm−1 in FT-IR spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose attributed to the stretching vibration of C = N bond47,48, indicating that the condensation reaction between glucose with the -NH2 groups happened, which is the first step of Maillard reaction. The peak at 1573 cm−1 in FT-IR spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose, and Pd/APTE-glucose corresponding to the N-H scissoring bending vibration49. The two strong absorption peaks at 1132 and 1034 cm-1 in FT-IR spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose representing the symmetric and asymmetric stretching vibrations of Si-O-Si and Si-O-C bridge49,50. By contrast, only a new obvious peak at 1697 cm−1 and 1699 cm−1 was observed in FT-IR spectra of APTE-BTMO-cellulose and Pd/APTE-BTMO-cellulose, respectively, corresponding to the stretching vibration of C = O bond (amide group) (Fig. 2b). The Si-O-Si and Si-O-C asymmetric stretching vibrations signal was overlapped by C-O-C signal ( ~ 1058 cm−1) in cellulose chain.

a FTIR spectra of glucose, APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose; b FTIR spectra of cellulose, APTE-BTMO-cellulose and fresh Pd/APTE-BTMO-cellulose; c 13C NMR of glucose, APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose; d 13C NMR of cellulose, APTE-BTMO-cellulose and Pd/APTE-BTMO-cellulose.
The solid state 13C NMR spectra illustrated that the typical peaks of glucose in the 119-63 ppm51 range (C1-C5) were disappeared in the spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose, and Pd/APTE-glucose (Fig. 2c). Instead, two new peaks at 164 ppm (C = N) and 177 ppm (C = O of amide group) were observed in the spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose and Pd/APTE-glucose. In addition, a new peaks 130 ppm (benzene ring of BTMO) was observed in both the spectra of APTE-BTMO-glucose, Pd/APTE-BTMO-glucose. Three new peaks at around 10, 23, and 43 ppm were assigned to the α-CH2, β-CH2, and γ-CH2 resonances of the aminopropyl groups of APTE and BTMO. The peak at ~50 ppm may be attributed to C6 of glucose or C-O of APTE. On the contrary, there is only a new peak at 179 ppm (C = O of amide group) in the 13C NMR spectra of APTE-BTMO-cellulose and Pd/APTE-BTMO-cellulose except the carbon resonances of the cellulose chain, APTE (aminopropyl groups, 10-43 ppm) and BTMO (benzene ring, 127, 135 ppm) (Fig. 2d).
Given the FT-IR and NMR analysis, it could be concluded that the Millard reaction of glucose with APTE or both APTE and BTMO was completely proceed catalyzed by L-lysine. APTE-glucose and APTE-BTMO-glucose are a new type of support containing a variety of coordination groups, especially a unique imino (C = N) group, compared with usual silane coupling agents modified supports.
Morphology and intrinsic structural characterization of catalyst
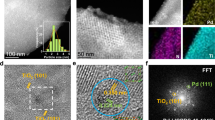
To explore the correlation between structure and catalytic performance, Pd/APTE-glucose and Pd/APTE-BTMO-glucose (fresh and used) were characterized comprehensively (Supplementary Methods). In order to investigate the distribution of Pd species at atomic scale, these catalysts were analyzed by High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The clear two-dimensional network structure was observed in the image of Pd/APTE-glucose (Fig. 3a). By contrast, the morphology feature of Pd/APTE-BTMO-glucose was similar to Pd/APTE-BTMO-cellulose (Supplementary Note 1, Fig. S1a). These results illustrated that the morphology features of the non-enzymatic browning glucose supported catalyst are affected by silane coupling agents. As shown as Fig. 3b, Pd SAs (red circle) and Pd Cs (yellow circle) coexisted on the support and some satellite Pd SAs closely distributed around Pd Cs in Pd/APTE-glucose. The size distribution of Cs in Pd/APTE-glucose was in the range of 0.5 nm to 1.5 nm, with average diameter of 0.99 nm (Fig. 3c). Similarly, subnanometric Pd Cs and SAs coexisted in fresh Pd/APTE-BTMO-glucose (Fig. 3d). Not only that, after 1st cycle, the Pd SAs/Cs coexistence state unchanged and the size of Pd Cs (from 0.96 to 1.17 nm) did not change obviously (Fig. 3e). It is notable that Si, N and O elements distributed uniformly and densely in the catalyst affording various coordination sites (Fig. 3f). Compared with Pd/APTE-BTMO-glucose, the uniformly distributed Pd NPs were observed in the TEM image of fresh Pd/APTE-BTMO-cellulose, but obvious Pd aggregation happened after the catalytic reaction (Supplementary Note 1, Fig. S1e, S1f). It is the most possible reason for the poor recyclability of Pd/APTE-BTMO-cellulose.

a–c AC-HAADF-STEM images of Pd/APTE-glucose; d AC-HAADF-STEM image of fresh Pd/APTE-BTMO-glucose; e AC-HAADF-STEM image of used Pd/APTE-BTMO-glucose; f The mapping of fresh Pd/APTE-BTMO-glucose.
In order to get information on the valence state of Pd SAs/Cs, Pd/APTE-glucose (fresh and used) and Pd/APTE-BTMO-glucose (fresh and used) were tested by X-ray photoelectron spectroscopy (XPS) analysis. As shown as Fig. 4a, three peaks attributed to amino groups (-NH2, 399.78 eV), hydrogen-bonded amines or quaternary ammonium nitrogen (-NH3+, 402.21 eV), and imino (C = N, 398.09 eV) were observed in the N 1 s spectra of fresh Pd/APTE-glucose. Compared with the APTE-glucose support, the peaks of amino groups shifted to higher binding energy (0.8-1.0 eV) in fresh and used Pd/APTM-glucose. It is noted that the peaks of imino groups significantly shifted to higher binding energy (2.33-2.39 eV), indicating the strong coordination of imine groups with Pd species. Likewise, the peak at ≈398 eV (C = N) was observed in the N 1 s spectra of fresh Pd/APTE-BTMO-glucose (Fig. 4b). By contrast, there were only two peaks attributed to amino groups and hydrogen-bonded amines or quaternary ammonium nitrogen in the N 1 s spectra of Pd/APTE-BTMO-cellulose (Fig. 4c)52. These results were consistent with the observation from FT-IR and NMR spectra. It can be concluded that the stronger coordination ability of imino (C = N) than amino (-NH2) in our catalyst is a key role of stabilizing Pd species. As shown in Fig. 4d, the Pd 3d5/2 peak was mainly located at 337.18 eV (PdII) along with a small shoulder at 335.66 eV (metallic state Pd) in fresh Pd/APTE-glucose, suggesting that almost all the Pd SAs and Pd atoms in Cs were coordinated with the N/O atoms. The new XPS signals at higher binding energy (3d5/2 peak at 338.59 eV) attributed to PdIV apart from PdII and Pd0 appeared in the XPS Pd 3 d spectrum of used Pd/APTE-glucose50,53. These results proved that a part of PdII was oxidized after the heterogeneous catalyzed C(sp3)-H arylation reaction and high valent (d electron-deficient) PdIV species can exist stably on the support surface. It is worth noting that the peak of Pd0 disappeared, but the peak of PdIV and PdII coexisted in the XPS Pd 3 d spectrum of used 1st and 5th Pd/APTM-BTMO-glucose after the heterogeneous catalyzed C(sp2)-H arylation reaction (Fig. 4e). Therefore, a small amount of Pd0 in fresh Pd/APTM-BTMO-glucose may not be the active species and the PdII/PdIV catalytic cycle could be concluded for the heterogeneous catalyzed C(sp2)-H arylation reaction. As same as Bäckvall’s work37, the XPS Pd 3 d spectrum of Pd/APTE-BTMO-cellulose showed the presence of two intense doublets at 335.73 and 340.86 eV related to Pd0 and peaks at 337.85 and 342.87 eV related to PdII corresponding to Pd 3d5/2 and Pd 3d3/2, respectively (Fig. 4f). But after used, the ratio of Pd0 obviously increased, and no obvious peak of PdIV was observed. Therefore, it can be concluded that the SMSI between Pd SAs/Cs with imino group in our catalysts may play a key role in generating stable high valent (d electron-deficient) PdIV species54.

a N 1 s XPS spectra of APTE-glucose, fresh and used Pd/APTE-glucose; b N 1 s XPS spectra of fresh Pd/APTE-BTMO-glucose; c N 1 s XPS spectra of fresh Pd/APTE-BTMO-cellulose; d Pd 3 d XPS spectra of Pd/APTE-glucose (fresh and used); e Pd 3 d XPS spectra of Pd/APTE-BTMO-glucose (fresh and used); f Pd 3 d XPS spectra of Pd/APTE-BTMO-cellulose (fresh and used).
The valence states of Pd were further studied by the X-ray absorption near-edge structure (XANES) analysis. As shown in Fig. 5a, the absorption edge positions at the Pd K-edge display a right shift in the order of Pd foil (blue line), fresh Pd/APTE-glucose (red line), PdO (green line), fresh Pd/APTE-BTMO-glucose (orange line), and used Pd/APTE-BTMO-glucose (cyan line). The absorption edge energy of fresh Pd/APTE-glucose is close to that of PdO, indicating that the average oxidation state of Pd SAs/Cs is about +2. By contrast, the average oxidation state of Pd SAs/Cs in fresh Pd/APTE-BTMO-glucose is between +2 and δ (δ > +2) and the used Pd/APTE-BTMO-glucose is higher positive-charged than fresh Pd/APTE-BTMO-glucose55. These results further proved the existence of stable high valent (d electron-deficient) PdIV species in our catalysts.

a Palladium K-edge XANES spectra of Pd/APTE-glucose, Pd/APTE-BTMO-glucose (fresh and used), reference Pd-foil and PdO; b k2-weighted χ(k) function of FT-EXAFS spectra of Pd/APTE-glucose, Pd/APTE-BTMO-glucose (fresh and used), reference Pd-foil and PdO; c–g Wavelet transforms of the k2-weighted EXAFS of fresh Pd/APTE-glucose, reference Pd foil, reference PdO, fresh Pd/APTE-BTMO-glucose and used Pd/APTE-BTMO-glucose.
The Pd K-edge extended X-ray absorption fine structure spectra (EXAFS, Fig. 5b and Supplementary Note 3) proved the coexistence of Pd-N/O and Pd-Pd coordination in Pd/APTE-glucose, fresh Pd/APTE-BTMO-glucose, and used Pd/APTE-BTMO-glucose (red, orange and cyan lines) with reference to Pd foil (blue line) and bulk PdO sample (green line) (k-range used for Fourier transform is 2.5-12.35). The EXAFS of three samples showed predominant peaks at 1.54, 1.52, and 1.49 Å in R space, similar to the reference peak of PdO (1.53 Å), which cans be ascribed to Pd-O or Pd-N bond56,57. Additionally, a small peak indexed to the first shell Pd-Pd scattering at 2.39, 2.41, and 2.42 Å, similar to the reference peak of Pd foil (2.48 Å), could be observed in three samples, indicating the existence of subnanometric Pd Cs. It is noted that there is no Pd-O-Pd coordination (∼2.96 Å) peak compared to PdO reference in the EXAFS spectra of three samples, suggesting the Pd-N bonds may be the main coordination bonds of Pd Cs and Pd SAs56,57. The EXAFS fitting further cleared the coordination of Pd in three samples (Supplementary Note 4, Table S3). The coordination number (CN) of the Pd atom was calculated to be ≈4 in three samples, and the mean Pd-N bond length was approximately 2.03 Å (R factor of 0.008, 0.011, and 0.009), implying that each Pd atom was coordinated to four nitrogen atoms in average. The fitting results of Pd/APTE-glucose and fresh Pd/APTE-BTMO-glucose showed that the average CN of Pd-Pd is less than one. The relatively low Pd-Pd coordination number further illustrated the formation of subnanometric Pd Cs. Moreover, the wavelet transformed (WT) EXAFS oscillations of samples were obtained, providing more detailed information about the R-space and k-space resolutions of the scattering atoms (Fig. 5c–g). By comparing the WT contour plots of Pd foil (Fig. 5d) and PdO (Fig. 5e), the WT contour plots of three samples showed a main peak at wave vector k of ∼6.4 Å−1, which is attributed to Pd-N, and a secondary peak at wave vector k of ∼10.3 Å−1, which is attributed to Pd-Pd scattering58.
Overall, the above results consistently demonstrated the coexistence of atomically dispersed Pd1-N4 structure and Pd Cs (mainly Pd-N coordination) in Pd/APTE-glucose and Pd/APTE-BTMO-glucose. The SMSI of Pd SAs/Cs with various coordination groups, especially imino (C = N) group, in non-enzymatic browning glucose achieved to stabilize the high valent (d electron-deficient) Pd sites.
Activity test
The reaction of N-(2-(methylthio)phenyl)propionamide (1a) and 3-methyl iodobenzene (2a) was chosen as a model C(sp3)-H arylation reaction for screening heterogeneous Pd catalysts (see Fig. 6a). Using K2CO3 as base, t-amyl-OH:H2O (16:1) as solvent and Pd/APTE-BTMO-glucose as heterogeneous catalyst, the desired C(sp3)-H arylation products (mono- 3aa, di- 3aa’) were obtained in excellent yields after 24 h reaction at 115 °C, and the catalyst showed similar catalytic performance and selectivity as Pd(OAc)2 for mono- and di- arylation products. By comparison, the typical Pd NPs catalysts, such as commercial 5 wt.% Pd/C and Pd/APTE-BTMO-cellulose, showed inferior catalytic performance and poor selectivity for limiting homocoupling of 2a. Interestingly, Pd/APTE-glucose showed notably different selectivity from homogeneous catalyst for mono- and di- arylation products. This result proved the achievement of tuning reaction selectivity by changing the structure of support in our catalyst as same as ligand-controlled selectivity in homogeneous catalysis. Interestingly, Pd/APTE-BTMO-glucose showed higher catalytic performance than Pd/APTE-glucose in the C(sp2)-H arylation reaction of pentafluorobenzene (4a) and iodobenzene (2b) (Fig. 6b). Not only that, Pd/APTE-BTMO-glucose showed strong substrate universality in the direct coupling of C(sp3)-H bonds of polyfluorobenzenes with aryl iodides (Supplementary Note 5, Fig. S4). It is probably because that the benzene ring of BTMO in Pd/APTE-BTMO-glucose facilitated the adsorption of substrates possessing similar structure59.

a The catalytic performance of the various catalysts for C(sp3)-H arylation reaction. Reaction conditions: 1a (0.1 mmol), 2a (0.3 mmol), K2CO3 (3.0 equiv.), 24 h, 115 °C, Isolated yield, actual amount of Pd of all catalysts is 7 mol%; b The catalytic performance of the various catalysts for C(sp2)-H arylation reaction. Reaction conditions: 4a (0.2 mmol), 2b (0.2 mmol), KHCO3 (0.4 mmol), isobutyric acid (0.1 mmol), 24 h, 110 °C, Isolated yield, actual amount of Pd of all catalysts is 3.5 mol%; c The recycle performances of three kinds of heterogeneous Pd catalysts for C(sp3)-H arylation reaction; Isolated yield. d The recycle performances of three kinds of heterogeneous Pd catalysts for C(sp2)-H arylation reaction; Isolated yield. e The conversion and selectivity of Pd/APTE-glucose, Pd/APTE-BTMO-glucose, and Pd(OAc)2 for C(sp3)-H arylation reaction changed with time; Isolated yield.
Hot filtration study using Pd/APTE-glucose was performed to check whether the leached Pd species in reaction process is the actually catalyst. When the reaction was run 4 h after catalyst removal, the conversion rate of 1a almost unchanged, which excludes the possibility of the C(sp3)-H arylation reaction through a homogeneous pathway (Supplementary Note 5, Table S5). In comparison, when the reaction was run 4 h after Pd/APTE-BTMO-cellulose catalyst removal, the conversion rate of 1a increased by 9% (Supplementary Note 5, Table S5). To further compare the stability of our catalysts with Pd NPs catalysts, we conducted the catalyst recycling experiments. The recycling experiment results revealed that the catalytic activity of Pd/APTE-glucose was maintained from the first to the fifth run in the C(sp3)-H arylation reaction (Fig. 6c). In contrast, Pd/C and Pd/APTE-BTMO-cellulose showed particularly poor recyclability. The possible reason is that the oxidative addition of iodobenzene to Pd sites weakens the Pd-Pd bonds and Pd-support coordination bonds, resulting in Pd leaching (Supplementary Note 2, Table S2) and Pd0 aggregate (Supplementary Note 1, Fig. S1f). In the analysis of ICP-OES, Pd loading of 3.27, 2.09 and 1.96 wt.% ( ± 10% error) were detected in fresh Pd/APTE-glucose and after the first and fifth cycles (Supplementary Note 2, Table S1). There was almost no Pd loss was detected, except the first cycle. These results suggested that the antioxidation ability may be attributed to SMSI between Pd species with support in Pd/APTE-glucose. The best stability and recyclability of Pd/APTE-BTMO-glucose in C(sp2)-H arylation reaction further confirmed the conclusion (Fig. 6d and Supplementary Note 2, Table S2 and Note5, S6). Next, the conversion and selectivity of C(sp3)-H arylation reaction using Pd/APTE-glucose, Pd/APTE-BTMO-glucose and Pd(OAc)2 as catalysts changed with time were tested. As shown as Fig. 6e, the di-arylation product (3aa’) was generated synchronously with the mono-arylation product (3aa) and the amount of di-arylation product increased steadily over time in the Pd/APTE-glucose catalyzed reaction. This result provided evidence for the synergistic effect of Pd SAs with Cs (or SAs with SAs), which is distinguished from homogeneous catalytic system. Based on many reported successful synergistic composite catalysts combining SAs with Cs33,34,35,36, the synergistic effect of Pd SAs and Cs may be the most likely reason for the best catalytic performance of Pd/APTE-glucose.
Using Pd/APTE-glucose as catalyst, we next explored the scope of this heterogeneous catalyzed aliphatic C(sp3)-H arylation of 1a with aryl iodides (Fig. 7). We were pleased to see that various aryl iodides bearing diverse functional groups such as -alkyl, -OMe, -CF3, -F, -CN, -COMe and -COOMe reacted smoothly with 1a to afford desired aliphatic C(sp3)-H arylation product in moderate to good yields (3aa-3aj). All of non-, para- and meta- substituted aryl iodides afforded mono- and di- arylation products. Surprisingly, the presence of an ortho- substituent at the aromatic ring of aryl iodide did not affect the reaction activity except the selectivity, as only mono- arylation product (3ad) was obtained in a satisfactory yield. Moreover, heteroaryl iodide was also tolerated in the heterogeneous catalyzed aliphatic C(sp3)-H arylation reaction (3ak). When N-(2-(methylthio)phenyl)-3- phenylpropanamide (1b) was used as substrate, the C(sp3)-H bond of methylene was arylated, but the yields were lower than C(sp3)-H bond arylation of methyl affected by steric hindrance (3ba-3ca). The site-selective β- C(sp3)-H bond (methylene) arylation of N-(2-(methylthio)phenyl)butyramide (1c) confirmed that the reaction proceeds through [5,5]-fused palladacycle intermediate. Apart from N-(2-(methylthio)phenyl)-propanamides, N-(quinolin-8-yl)- propanamides also reacted smoothly with aryl iodides to afford desired aliphatic C(sp3)-H arylation products (3ec-3en). To demonstrate the potential industrial applications of our catalyst, we performed scale-up reaction. The Pd/APTE-glucose catalyst afforded 3aa and 3aa’ in 85% total yield in gram-scale reaction.

1 (0.1 mmol), 2 (0.3 mmol), Pd/APTE-glucose (25 mg), K2CO3 (3.0 equiv.), and t-Amyl-OH: H2O (16:1, 1 mL) for 24 h at 115 °C, Isolated yield.
Universality of catalyst
Our catalyst also showed higher activities and stability in almost all kinds of arylation reactions using aryl iodides as arylation reagent, confirming the universality of non-enzymatic browning glucose supported Pd SAs/Cs synergistic catalyst (Fig. 8). Based on the best catalytic activity of Pd/APTE-BTMO-glucose in C(sp2)-H arylation reaction, the direct coupling of iodobenzene with C(sp2)-H bond of benzo[d]thiazole, 1-methyl-1H-indole, benzo[h]quinoline and quinoline-8-carbaldehyde were achieved using the same reaction condition of C(sp2)-H arylation of pentafluorobenzene, respectively (6-9). Compared with Pd/APTE-BTMO-glucose, Pd/APTE-glucose showed inferior catalytic activity in C(sp2)-H arylation reactions under same reaction conditions. However, Pd/APTE-glucose showed superior catalytic activity than Pd/APTE-BTMO-glucose in transient-directed C(sp3)-H arylation of o-methyl benzaldehyde (10). These results further demonstrated that Pd/APTE-glucose is more applicable to C(sp3)-H bond activation reactions and Pd/APTE-BTMO-glucose is more applicable to C(sp2)-H bond activation reactions. Targeted to either C(sp3)-H or C(sp2)-H bond activation, designing specific heterogeneous Pd catalyst was achieved by tuning the structure of support in our catalyst. Not only that, our catalyst showed excellent catalytic activity and recyclability in Suzuki, Heck and C-N cross-coupling reaction (11-13).

The universality of catalyst for various cross-coupling reactions.
Conclusion
In summary, non-enzymatic browning glucose was firstly used as support to prepare Pd SAs/Cs cocatalyst, which exhibits promising catalytic activity and stability for chelation-assisted site-selective C(sp3)-H arylation reaction. Multi-technique characterizations illustrated the coexistence of atomically dispersed Pd1-N4 structure and Pd Cs (mainly Pd-N coordination) in Pd/APTE-glucose and Pd/APTE-BTMO-glucose. The SMSI between Pd sites with non-enzymatic browning glucose perfectly achieved to stabilize high valent (d electron-deficient) active Pd sites on the support surface in the reaction process and broke the activity-stability trade-off in chelation-assisted C(sp3)-H arylation reaction via PdII/PdIV catalytic cycle. Moreover, the tuning reaction selectivity to distinguish from homogeneous Pd catalyst and designing specific heterogenous Pd catalyst targeted at either C(sp3)-H or C(sp2)-H bond activation was achieved by tailoring the structure of support in our catalysts. This work not only paves the way to green C-H arylation with non-enzymatic browning glucose supported Pd catalyst, but also provides theoretical guidance for the design and development of more targeted heterogeneous catalyst for either C(sp3)-H or C(sp2)-H bond activation.
Methods
Catalyst preparation
Preparation of non-enzymatic browning glucose supported Pd catalysts
In an oven dried flask, glucose (1.1 g), L-lysine (144 mg) were dispersed in dry toluene (40 mL). Next, 3-aminopropyl triethoxysilane (APTE, 4.3 mL) was added and the mixture was stirred at 82 °C for 48 h. The crude APTE-glucose was removed and washed using Soxhlet extractor with acetone (150 mL). After 16 h, the APTE-glucose was dried under vacuum. APTE-glucose (1.0 g) was suspended in pH-adjusted (9) aqueous solution using 0.1 mol/L NaOH (25 mL) and was allowed to stir at room temperature for 10 min. PdCl2 (67.6 mg) was added and the resulting suspension was stirred for 24 h. And then the suspension was centrifuged (3900 pm, 3 min), and the separate solid was washed with water (3 times) and acetone (3 times). Finally, the solid dried under vacuum for 24 h and denoted as Pd/APTE-glucose. The Pd/APTE-BTMO-glucose was prepared by a similar procedure except adding trimethoxyphenylsilane (BTMO, 3.5 mL).
Preparation of Pd/APTE-BTMO-cellulose
The APTE-BTMO-cellulose supported Pd NPs was prepared by a modified impregnation method based on Bäckvall’s work37. In an oven dried flask, cellulose (1.0 g) and tartaric acid (148 mg) were dispersed in dry toluene (40 mL). Next, 3-aminopropyl triethoxysilane (APTE, 4.3 mL) and trimethoxyphenylsilane (BTMO, 3.5 mL) were added and the mixture was stirred at 82 °C for 48 h. The crude APTE-BTMO-cellulose was removed and washed using Soxhlet extractor with acetone (150 mL). After 16 h, the APTE-BTMO-cellulose was dried under vacuum. APTE-BTMO-cellulose (1.0 g) was suspended in pH-adjusted (9) aqueous solution using 0.1 mol/L NaOH (25 mL) and was allowed to stir at room temperature for 10 min. PdCl2 (67.6 mg) was added and the resulting suspension was stirred for 24 h. And then the suspension was centrifuged (3900 pm, 3 min), and the separate solid was washed with water (3 times) and acetone (3 times). Finally, the solid dried under vacuum for 24 h and denoted as Pd/APTE-BTMO-cellulose.
Activity test
General procedure for C(sp3)-H arylation
A mixture of N-(2-(methylthio)phenyl)propionamide 1a (0.1 mmol), 3-methyl iodobenzene 2a (0.3 mmol), catalyst (25 mg), K2CO3 (3.0 equiv.), and t-Amyl-OH: H2O (16:1, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 115 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether/1,2-dichloroethane = 1:8:1 as an eluent) to afford the desired product.
General procedure for C(sp2)-H arylation
A mixture of pentafluorobenzene 4a (0.2 mmol), iodobenzene 2b (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.) and N, N-dimethylformamide (DMF, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:30 as eluent) to afford the desired product.
Synthetic procedure of 2-phenylbenzo[d]thiazole 6
A mixture of iodobenzene (0.2 mmol), benzothiazole (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.) and N, N-dimethylformamide (DMF, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C under nitrogen atmosphere. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 3 as eluent) to afford the desired product.
Synthetic procedure of 1-methyl-2-phenyl-1H-indole 7
A mixture of iodobenzene (0.24 mmol), 1-methylindole (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.), and N, N-dimethylformamide (DMF, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 3 as eluent) to afford the desired product.
Synthetic procedure of 10-phenylbenzo[h]quinoline 8
A mixture of iodobenzene (0.2 mmol), benzo[h]quinoline (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.), and N, N-dimethylformamide (DMF, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 3 as eluent) to afford the desired product.
Synthetic procedure of phenyl(quinolin-8-yl)methanone 9
A mixture of iodobenzene (0.3 mmol), quinoline-8-carboxaldehyde (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.), and N, N-dimethylformamide (DMF, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 3 as eluent) to afford the desired product.
Synthetic procedure of 2-(4-methoxybenzyl)benzaldehyde 10
A mixture of 4-iodoanisole (0.1 mmol), 2-methylbenzaldehyde (0.12 mmol), catalyst (25 mg), acethydrazide (40 mol%), silver acetate (2.0 equiv.), and acetic acid (AcOH, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 24 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 30 as eluent) to afford the desired product.
Synthetic procedure of 4-(tert-butyl)-1,1’-biphenyl 11
A mixture of iodobenzene (0.2 mmol), 4-tert-butylphenylboronic acid (0.24 mmol), catalyst (25 mg), K2CO3 (2.0 equiv.), and solvent (toluene: water = 3:1, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 6 h at 110 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 30 as eluent) to afford the desired product.
Synthetic procedure of methyl cinnamate 12
A mixture of iodobenzene (0.2 mmol), methyl acrylate (0.24 mmol), catalyst (25 mg), triethylamine (1.2 equiv.), and N-methylpyrrolidone (NMP, 1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 8 h at 100 °C. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 30 as eluent) to afford the desired product.
Synthetic procedure of 4-bromo-N,N-diphenylaniline 13
A mixture of 1-bromo-4-iodobenzene (0.36 mmol), diphenylamine (0.43 mmol), catalyst (25 mg), Xantphos (1 mol%), NaOtBu (1.5 eq.), and dioxane (1 mL) was charged in a 25 mL oven dried reaction tube. The reaction mixture was stirred for 12 h at 100 °C under nitrogen atmosphere. After being cooled to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1: 50 as eluent) to afford the desired product.
Hot filtration test
Hot filtration test of C(sp3)-H arylation
N-(2-(methylthio)phenyl)propionamide 1a (0.1 mmol), 3-methyl iodobenzene 2a (0.3 mmol), catalyst (25 mg), K2CO3 (3.0 equiv.), and t-Amyl-OH: H2O (16:1, 1 mL) were added into two reaction tube, respectively. One reaction was ceased after 4 h and the 1a was isolated for calculating TON. Another reaction was applied by filtering the reaction mixture through a pre-heated Celite pad after the reaction for 4 h. Then the filtered reaction solution continued to react for 4 h under normal conditions and the 1a was isolated for calculating TON.
Hot filtration test of C(sp2)-H arylation
Pentafluorobenzene 4a (0.2 mmol), iodobenzene 2b (0.2 mmol), catalyst (25 mg), KHCO3 (2.0 equiv.), isobutyric acid (0.5 equiv.) and N, N-dimethylformamide (DMF, 1 mL) were added into two reaction tube, respectively. One reaction was ceased after 4 h and the 4a was isolated for calculating TON. Another reaction was applied by filtering the reaction mixture through a pre-heated Celite pad after the reaction for 4 h. Then the filtered reaction solution continued to react for 4 h under normal conditions and the 4a was isolated for calculating TON.
Catalyst recycle experiment
After each reaction cycle, the reactants, solvent, and products were removed by centrifugation; the separated catalyst was washed thoroughly with deionized water (twice) and then washed with acetone (once) followed by centrifugal separation and drying at 60 °C overnight. The recovered catalyst was used for the next cycle.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information files. Raw data are available from the corresponding author on reasonable request. Materials and methods, experimental procedures, characterization data, 1H, 13C NMR spectra, and mass spectrometry data are available in the Supplementary Information.
References
He, J., Wasa, M., Chan, K. S. L., Shao, Q. & Yu, J.-Q. Palladium-catalyzed transformations of alkyl C-H bonds. Chem. Rev. 117, 8754–8786 (2017).
Albano, G., Punzi, A., Capozzi, M. A. M. & Farinola, G. M. Sustainable protocols for direct C-H bond arylation of (hetero) arenes. Green. Chem. 24, 1809–1894 (2022).
Ritleng, V., Sirlin, C. & Pfeffer, M. Ru-, Rh-, and Pd-catalyzed C-C bond formation involving C-H activation and addition on unsaturated substrates: Reactions and mechanistic aspects. Chem. Rev. 102, 1731–1770 (2002).
Alberico, D., Scott, M. E. & Lautens, M. Aryl−Aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 107, 174–238 (2007).
Achar, T. K., Maiti, S., Jana, S. & Maiti, D. Transition metal catalyzed enantioselective C(sp2)-H bond functionalization. ACS Catal. 10, 13748–13793 (2020).
Kalyani, D., Deprez, N. R., Desai, L. V. & Sanford, M. S. Oxidative C-H activation/C-C bond forming reactions: Synthetic scope and mechanistic insights. J. Am. Chem. Soc. 127, 7330–7331 (2005).
Zhang, X. et al. C-C coupling on single-atom-based heterogeneous catalyst. J. Am. Chem. Soc. 140, 954–962 (2018).
Collins, K. D., Honeker, R., Vásquez-Céspedes, S., Tang, D.-T. D. & Glorius, F. C-H arylation of triphenylene, naphthalene and related arenes using Pd/C. Chem. Sci. 6, 1816–1824 (2015).
Campana, F., Massaccesi, B. M., Santoro, S., Piermatti, O. & Vaccaro, L. Polarclean/water as a safe and recoverable medium for selective C2-arylation of indoles catalyzed by Pd/C. ACS Sustain. Chem. Eng. 8, 16441–16450 (2020).
Tang, D.-T. D., Collins, K. D. & Glorius, F. Completely regioselective direct C-H functionalization of benzo[b]thiophenes using a simple heterogeneous catalyst. J. Am. Chem. Soc. 135, 7450–7453 (2013).
Mao, S., Shi, X., Soulé, J.-F. & Doucet, H. Pd/C as heterogeneous catalyst for the direct arylation of (poly)fluorobenzenes. Chem. Eur. J. 25, 9504–9513 (2019).
Duan, L. et al. An efficient reusable mesoporous solid-based Pd catalyst for selective C2 arylation of indoles in water. ACS Catal. 6, 1062–1074 (2016).
Mandal, T., Mondal, M. & Choudhury, J. Hypercrosslinked polymer platform-anchored single-site heterogeneous Pd−NHC catalysts for diverse C-H functionalization. Organometallics 40, 2443–2449 (2021).
Cao, M., Wu, D., Su, W. & Cao, R. Palladium nanocrystals stabilized by cucurbit[6]uril as efficient heterogeneous catalyst for direct C-H functionalization of polyfluoroarenes. J. Catal. 321, 62–69 (2015).
Limaye, A. S. et al. Greener approach for Pd–NPs synthesis using mangifera indica leaf extract: Heterogeneous nano catalyst for direct C–H arylation of (poly)fluorobenzene, hiyama coupling reaction and hydrogen evolution reaction study. Cat. Lett. 153, 1988–2004 (2023).
Santoro, S., Kozhushkov, S. I., Ackermann, L. & Vaccaro, L. Heterogeneous catalytic approaches in C-H activation reactions. Green. Chem. 18, 3471–3493 (2016).
Luo, Q. et al. Polymer-supported Pd nanoparticles for solvent-free hydrogenation. J. Am. Chem. Soc. 146, 26379–26386 (2024).
Chen, J., Bai, C., Ma, H., Liu, D. & Bao, Y. S. Nano palladium catalyzed C(sp3)-H bonds arylation by a transient directing strategy. Chin. Chem. Let. 32, 465–469 (2021).
Hu, F. et al. Direct synthesis of atomically dispersed palladium atoms supported on graphitic carbon nitride for efficient selective hydrogenation reactions. ACS Appl. Mater. Interfaces 48, 54146–54154 (2020).
Li, Z. et al. Strong electronic interaction of indium oxide with palladium single atoms induced by quenching toward enhanced hydrogenation of nitrobenzene. Appl. Catal. B-environ. 313, 121462 (2022).
Li, Z. et al. Selective hydrogenation on a highly active single-atom catalyst of palladium dispersed on ceria nanorods by defect engineering. ACS Appl. Mater. Interfaces 12, 57569–57577 (2020).
Li, Z. et al. One-step synthesis of single palladium atoms in WO2.72 with high efficiency in chemoselective hydrodeoxygenation of vanillin. Appl. Catal. B-environ. 298, 120535 (2021).
Li, Z. et al. Highly efficient and enhanced sulfur resistance supported bimetallic single-atom palladium–cobalt catalysts for benzene oxidation. Appl. Catal. B-environ. 285, 119844 (2021).
Tiburcio, E. et al. Soluble/MOF-supported palladium single atoms catalyze the ligand-, additive-, and solvent-free aerobic oxidation of benzyl alcohols to benzoic acids. J. Am. Chem. Soc. 143, 2581–2592 (2021).
Liu, J. et al. Molecular engineered palladium single atom catalysts with an M-C1N3 subunit for Suzuki coupling. J. Mater. Chem. A. 9, 11427–11432 (2021).
Ji, S. et al. Construction of a single-atom palladium catalyst by electronic metal-support interaction and interface confinement effect with remarkable performance in Suzuki coupling reaction. Chem. Eng. J. 1, 139205 (2023).
Ding, G. et al. Atomically dispersed palladium catalyses Suzuki–Miyaura reactions under phosphine-free conditions. Commun. Chem. 3, 43 (2020).
Chen, Z. et al. A heterogeneous single-atom palladium catalyst surpassing homogeneous systems for Suzuki coupling. Nat. Nanotech 13, 702–707 (2018).
Wei, H. et al. Rapid synthesis of Pd single-atom/cluster as highly active catalysts for Suzuki coupling reactions. Chin. J. Catal. 43, 1058–1065 (2022).
Hu, B. et al. Distinct crystal-facet-dependent behaviors for single-atom palladium-on-ceria catalysts: Enhanced stabilization and catalytic properties. Adv. Mater. 34, 2107721 (2022).
Niu, F. et al. Single Pd–Sx sites in situ coordinated on CdS surface as efficient hydrogen autotransfer shuttles for highly selective visible-light-driven C–N coupling. ACS Catal. 12, 4481–4490 (2022).
Luo, G. et al. Advances of synergistic electrocatalysis between single atoms and nanoparticles/clusters. Nano-Micro Lett. 16, 241 (2024).
Kuai, L. et al. Titania supported synergistic palladium single atoms and nanoparticles for room temperature ketone and aldehydes hydrogenation. Nat. Commun. 11, 48 (2020).
Mo, F. et al. The optimized Fenton- like activity of Fe single- atom sites by Fe atomic clusters–mediated electronic configuration modulation. PNAS 120, e2300281120 (2023).
Ren, X. et al. The fabrication of Pd single atoms/clusters on COF layers as co-catalysts for photocatalytic H2 evolution. ACS Appl. Mater. Interfaces 14, 6885–6893 (2022).
Wang, T. et al. Synergistic Pd single atoms, clusters, and oxygen vacancies on TiO2 for photocatalytic hydrogen evolution coupled with selective organic oxidation. Small 2, 2006255 (2020).
Naidu, V. R., Rafi, A. A., Tai, C.-W., Bäckvall, J.-E. & Córdova, A. Regio- and stereoselective carbon-boron bond formation via heterogeneous palladium-catalyzed hydroboration of enallenes. Chem. Eur. J. 29, e202203950 (2023).
Rizzi, G. P. Electrochemical study of the maillard reaction. J. Agric. Food Chem. 51, 1728–1731 (2003).
Hemmler, D. et al. Insights into the chemistry of non-enzymatic browning reactions in different ribose-amino acid model systems. Sci. Rep. 8, 16879 (2018).
Mohsin, G. F., Schmitt, F. J., Kanzler, C., Alzubaidi, A. K. & Hornemann, A. How alanine catalyzes melanoidin formation and dehydration during synthesis from glucose. Eur. Food Res. Technol. 248, 1615–1624 (2022).
Rufián-Henares, J. A. & De La Cueva, S. P. Antimicrobial activity of coffee melanoidins-A study of their metal-chelating properties. J. Agric. Food Chem. 57, 432–438 (2009).
Morales, F. J., Fernández-Fraguas, C. & Jiménez-Pérez, S. Iron-binding ability of melanoidins from food and model systems. Food Chem. 90, 821–827 (2005).
Wen, X. et al. Effect of roasting on properties of the zinc-chelating substance in coffee brews. J. Agric. Food Chem. 53, 2684–2689 (2005).
Bekedam, E. K., De Laat, M. P. F. C., Schols, H. A., Van Boekel, M. A. J. S. & Smit, G. Arabinogalactan proteins are incorporate in negatively charged coffee brew melanoidins. J. Agric. Food Chem. 55, 761–768 (2007).
Morales, F. J. Application of capillary zone electrophoresis to the study of food and food-model melanoidins. Food Chem. 76, 363–369 (2002).
Hashiba, H. Oxidative browning of Amadori compounds. Color formation by iron with Maillard reaction products. In Amino Carbonyl Reactions in Food and Biological Systems; Fujimaki, M., Kato, H., Namiki, M., Eds.; Kodansha: Amsterdam, The Netherlands pp 155-164 (1986).
Firuzabadi, F. D. & Asadi, Z. Palladium-catalyzed O-arylation reaction using different heterogeneous catalyst systems: The role of support. ChemistrySelect 3, 9857–9864 (2018).
Ivanovski, V., Bukleski, M., Madalska, M. & Hey-Hawkins, E. Vibrational spectra of a ferrocenyl phosphine derivative chemisorbed on 3-aminopropylsilyl modified silica gel. Vibrational Spectrosc. 69, 57–64 (2013).
Lin, R. et al. In situ immobilization of palladium nanoparticles in microfluidic reactors and assessment of their catalytic activity. Nanotechnology 21, 325605 (2010).
Zhao, W., Liu, S., Wang, H., Yang, J. & Chen, X. Ultrasmall Pd nanoparticles supported on TiO2 for catalytic debenzylation via hydrogenative C-N bond cleavage. ACS Appl. Nano Mater. 4, 159–166 (2021).
Fernandes, S. C. et al. Bioinspired antimicrobial and biocompatible bacterial cellulose membranes obtained by surface functionalization with aminoalkyl groups. ACS Appl. Mater. Interfaces 5, 3290–3297 (2013).
Zinovyeva, V. A., Vorotyntsev, M. A., Bezverkhyy, I., Chaumont, D. & Hierso, J. C. Highly dispersed palladium-polypyrrole nanocomposites: in-water synthesis and application for catalytic arylation of heteroaromatics by direct C-H bond activation. Adv. Funct. Mater. 21, 1064–1075 (2011).
Kumari, S., Maddipoti, K., Das, B. & Ray, S. Palladium–Schiff base complexes encapsulated in zeolite-Y host: Functionality controlled by the structure of a guest complex. Inorg. Chem. 58, 1527–1540 (2019).
Zhao, X. et al. Palladium catalyzed radical relay for the oxidative cross-coupling of quinolines. Nat. Commun. 13, 4180 (2022).
Wu, Y. et al. Cooperative atomic palladium site and island-distributed S-scheme heterostructure for photocatalytic C2H6 production. ACS Cat. 15, 3558–3569 (2025).
Li, Z. et al. Highly active and stable palladium single-atom catalyst achieved by a thermal atomization strategy on an SBA-15 molecular sieve for semi-hydrogenation reactions. ACS Appl. Mater. Interfaces 13, 2530–2537 (2021).
Ouyang, Z. et al. Palladium single atom-supported covalent organic frameworks for aqueous-phase hydrogenative hydrogenolysis of aromatic aldehydes via hydrogen heterolysis. Angew. Chem. Int. Ed. 6, e202418790 (2024).
Li, X. et al. Constructing a highly active Pd atomically dispersed catalyst for cinnamaldehyde hydrogenation: Synergistic catalysis between Pd-N3 single atoms and fully exposed Pd clusters. ACS Cat. 14, 2369–2379 (2024).
Varadwaj, G. B. B., Rana, S. & Parida, K. Pd(0) nanoparticles supported organofunctionalized clay drivingC−C coupling reactions under benign conditions through a Pd(0)/Pd(II) redox interplay. J. Phys. Chem. C. 118, 1640–1651 (2014).
Acknowledgements
Financial support from the National Nature Science Foundation of China (22261044), Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (NMGIRT2302), and Science and technology planning project of Inner Mongolia (2025KYPT0075).
Author information
Authors and Affiliations
Contributions
Y.-S.B. conceived and supervised the whole project and wrote the paper with input from all authors. X.H., W.Z., C.B., and Y.-S.B. designed and discussed the experiments. D.L., A.B., and T.M. performed and analyzed the experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Yong Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, X., Zhang, W., Bai, C. et al. Palladium single-atom/cluster cocatalyst supported on non-enzymatic browning glucose breaks the activity-selectivity trade-off in C(sp3)-H arylation. Commun Chem 8, 307 (2025). https://doi.org/10.1038/s42004-025-01693-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-025-01693-x
