
Abstract
Ensembles of suspended spinning particles in liquids form a distinct category of active matter systems known as chiral fluids. Recent experimental instances of dense chiral fluids have comprised of spinning colloidal magnets powered by an external rotating magnetic field. These particles interact through both magnetic and hydrodynamic forces, organizing collectively into circulating clusters characterized by unidirectional edge flows. Here, we externally drive the collective behavior of spinning colloids by leveraging diffusiophoretic interactions among the geometrically anisotropic particles. We show that these nanoscale interfacial flows lead to the formation of bound states between spinning colloids that are stabilized through near-field hydrodynamic and chemical interactions. At a collective level, we demonstrate that added diffusiophoretic interactions cause a loss in structural cohesion of the circulating clusters and promote expansion, while preserving global cluster inter-connectivity. The expanded cluster state is characterized by the formation of a dynamic interconnected network promoted by axi-asymmetric interactions around particles with attractive dipolar interactions dominating along the direction of the magnetic moment. This process is observed to be entirely reversible, offering external control over the emergent dynamics in dense chiral fluids, paving the way for new self-organization routes in chiral fluids and broader forms of active matter.
Similar content being viewed by others

Introduction
A modern approach to developing self-organization processes capable of producing structural and functional complexity is to use dissipative active systems whose constituents consume energy from their environment and remain out of equilibrium. A classic example of synthetic systems capable of self-organization are self-propelled active colloids, where individual particles locally exert mechanical forces and stresses on the environment and nearby particles, leading towards large-scale collective phenomenon inaccessible at thermal equilibrium1,2. Self-organization in self-propelled systems arises broadly from two distinct phenomenon: inter-particle interactions and motility induced phase separation (MIPS). Systems with dominant velocity-alignment interactions between particles have been shown to evolve from isotropic gas-like distributions into spatially localized swarms, and eventually into a homogenous polar liquid phase with increasing particle concentration3,4. Other complex spatiotemporal patterns such as chains, clusters, and vortices have been achieved by tuning either individual particle properties, such as shape, or inter-particle interactions via electric, magnetic, chemical, or hydrodynamic fields5,6,7,8,9,10. In contrast to interaction-induced self-organization, dense clusters of low-motility particles can spontaneously form even in the absence of attractive interactions, directly as a result of particle motility - a phenomenon known as MIPS11. In MIPS, cluster formation arises from a combination of self-propulsion and isotropic excluded volume interactions, where particle accumulation leads to attenuation of particle motion, which in turn reinforces further accumulation, a self-trapping mechanism that results in cluster formation.
In comparison to self-propelled active systems, chiral active systems, composed of spinning rotor particles, that utilize their rotation as a route to pattern formation and self-organization, are only beginning to be explored systematically12,13,14,15,16,17,18,19. While in these systems there is no significant propulsion at the level of an individual particle, spinning particles exert local torques, and translational motion emerges as a result of hydrodynamic interactions between spinners at a collective level. Interactions between chiral particles are mediated through the fluid and nearby interfaces and have been shown to result in formation of complex structures such as vortices and dynamic 2-D crystalline lattices20,21,22. Recently, a collection of hematite micro-particles that assemble due to an in-plane rotating magnetic field has been proposed as a model system to investigate chiral fluids and has been shown to form rotating vortices with novel properties including sustained chiral edge modes13,23,24. However, unlike their counterparts in self-propelled systems, there have been no demonstrations of the reconfigurability of collective states in chiral fluids by tuning inter-particle interactions. The potential ability to arbitrarily control emergent properties on-the-fly via external signals is an important feature of synthetic active matter systems that enables programmable self-assembly.
Here, we show collective states in chiral fluids of colloidal spinners can be externally reconfigured by introducing inter-particle chemical interactions. In the absence of this interaction, the spinning particles interact strongly via coupled hydrodynamic fields and magnetic dipolar interactions, leading to the formation of circulating clusters with sustained edge currents in a background of freely rotating individual spinners. The size of the separated clusters can be controlled via the rotational frequency of the applied magnetic field. When the same particle system is suspended in H2O2, in the presence of UV light, each hematite particle acts as a catalyst for local decomposition of H2O2, introducing local diffusiophoretic interactions. We find that these additional interactions lead to a loss in cohesivity of the circulating clusters and deplete the edge currents observed in rigid clusters. Our simulations show that near-field hydrodynamic and diffusiophoretic interactions are responsible for the formation of coupled bound states in the presence of chemical interactions, leading to loosening of chiral clusters without destroying them. The expanded cluster state is characterized by the formation of a dynamic inter-connected structure that results from the interplay between axi-asymmetric magnetic-dipolar attractions and effective diffusiophoretic repulsions and is completely reversible. This allows us to switch between rigid and relaxed cluster states in chiral fluids via an external signal. Additionally, we show that the rotating clusters form inter-connected networks of colloids, once the rotating field is removed, whose structural properties such as porosity and conformation can also be controlled by chemical interactions.
Results
Clustering of spinning colloidal particles
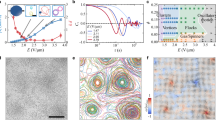
Our experimental system is composed of an aqueous solution with monodisperse ellipsoidal hematite colloids, with long axis (short axis) equal to b = 1.4 µm (a = 1.0 µm) and a permanent magnetic moment oriented perpendicular to b (Fig. 1c). These particles are density mismatched (ρ = 5.26 g/cm3) and sediment close to the bottom glass surface in our experimental cell (two glass slides separated by a h = 120 µm spacer with r = 2.5 cm) and are quasi-confined to their sedimentation plane. In the absence of an external magnetic field, we find that most of the ellipsoidal particles spontaneously arrange into chain structures of finite length due to magnetic dipolar interactions and are aligned in the direction of the earth’s weak magnetic field (Supplementary Fig. 2A). Since the direction of the particles permanent magnetic moment is perpendicular to the long axis, their chaining direction is perpendicular to b. We also observe a dilute concentration of individual particles interspersed with the ellipsoidal particle chains which are subject to thermal fluctuations and participate in exchange of particles at the ends of the chain structures. Due to the small size of the ellipsoids, they are subject to thermal fluctuations and the shape of the chain fluctuates while remaining generally aligned with the direction of the weak magnetic field.

Schematic representation of the distribution of ellipsoidal hematite colloids before (a) and after (b) application of an in-plane rotational magnetic field. c SEM image of an ellipsoidal hematite particle. Arrow indicates the direction of magnetic moment. d In-plane distribution of colloidal particles after magnetic field actuation at f = 20 Hz. Each image is obtained by averaging the positions of colloidal particles over 100 s after application of the rotating magnetic field, showing the formation and growth of clusters over t = 500 s. e Timelapse microscopy images showing the growth of cluster area in a single cluster at f = 20 Hz. f Timelapse microscopy images showing the coalescence of neighboring clusters to form a larger cluster (f = 20 Hz); Supplementary Movie 1. g Time evolution of the normalized fraction of clusters (magenta) and individual particles (brown) over t = 600 s at f = 20 Hz. The plots are normalized to maximum observed values of Nclusters and Nparticles. h Probability density function of cluster areas at different frequencies from f = 20 to 50 Hz over time. i Time evolution of representative contour areas of clusters at different frequencies over t = 0 to t = 600 s. j Time evolution of average cluster area growth at different frequencies from f = 20 to 50 Hz. Scale bar in (c) corresponds to 0.5 µm. All other scale bars correspond to 10 µm.
We begin by applying a static magnetic field in the \(\hat{{\boldsymbol{z}}}\) direction, \({\boldsymbol{B}}={B}_{0}\hat{{\boldsymbol{z}}}\). The ellipsoidal particles immediately break away from the chain structures and orient with their short axis perpendicular to the glass substrate. The particle motion is now dominated by thermal fluctuations leading to a randomly distributed system over time (Supplementary Fig. 2B). We let our system evolve for t = 600 s after application of the static field in the \(\hat{{\boldsymbol{z}}}\) direction to obtain a state of randomly dispersed particles (Fig. 1a, d). From this initial state, we begin to apply a rotating magnetic field, circularly polarized in the \((\hat{{\boldsymbol{x}}}, \, \hat{{\boldsymbol{y}}})\) plane with magnitude \({B}_{0}\) and frequency f, \({\boldsymbol{B}}={B}_{0}(\cos (2\pi {ft})\hat{{\boldsymbol{x}}}+\sin (2\pi {ft})\hat{{\boldsymbol{y}}})\) applying a torque on each particle \(\tau ={\boldsymbol{m}}\times {\boldsymbol{B}}\). While the applied rotating field frequency is lower than the step-out frequency (~70 Hz; Supplementary Fig. 1) the colloids remain phase locked with the direction of the external magnetic field, and we obtain colloidal spinners.
The spinning colloids interact with each other both via hydrodynamic flows and the aforementioned magnetic dipolar forces. When a colloid is externally driven, as in our case, the spinner transfers external torque to the fluid, and at high frequencies, the generated dipolar azimuthal flow can be approximated to be that of a sphere, which decays as \(1/{r}^{2}\), given by \({\boldsymbol{u}}\left({\boldsymbol{r}}\right)=\frac{\tau }{8\pi \eta {r}^{2}}\hat{{\boldsymbol{z}}}\times \hat{{\boldsymbol{r}}}\), where \(\eta\) is the fluid viscosity, leading to transverse pairwise forces between spinning colloids and advection in the generated flow fields25. In the absence of magnetic dipolar attraction, the two colloids would have a static center of mass and rotate about their center of mass with an angular velocity \(\varOmega =\frac{{\rm{\tau }}}{{4\pi \eta r}_{0}^{3}}\), where \({r}_{0}\) is the initial separation between the two colloids. Our simulations show the formation of these orbits for spinning ellipsoidal colloids with hydrodynamic interactions (Supplementary Fig. 3, Methods). However, in the presence of dipolar attractions the orbital trajectories become contractile and collapse into each other (Supplementary Fig. 4)23. The collective behavior of a system of externally torqued spinners is determined by a combination of short-range hydrodynamic repulsion and magnetic dipolar attraction. Upon application of the rotating magnetic field on a randomly dispersed system of magnetic particles, we observe configurational transformation to a system consisting of dense rotating clusters in a background of dilute free spinners (Fig. 1b, d). These clusters grow with time both via accumulation of individual particles from the bulk phase and also by the merging of nearby clusters to form larger clusters (Fig. 1e, f). In order to characterize the emergent behavior, we track the number of individual particles and clusters in the system and find that over time, the fraction of single particles reduces, as the fraction of clusters grows simultaneously, with both values saturating at t = 270 s (Fig. 1g).
Similar to previous work, we also find that the cluster size has a dependence on the applied frequency, with lower rotating frequencies where attractive interactions dominate over squeezed repulsive flows, leading to larger clusters23,24. We track the size of the clusters by estimating their contour area (see Methods) and plot the probability densities of cluster areas for different applied frequencies ranging from f = 20 Hz to 50 Hz. We observe a clear separation in the probability density function of cluster areas between the different applied frequencies, with the peak for f = 20 Hz at A = 48 \({\mu m}^{2}\), whereas for f = 50 Hz, the peak occurs at A = 17 \({\mu m}^{2}\) (Fig. 1h), indicating the development of smaller clusters for higher frequencies. At higher frequencies, chiral flows generated by the spinning particles dominate over dipolar attractions hindering aggregation and stabilizing smaller clusters. In Fig. 1i we plot the contour of a representative cluster for each driving frequency to show the cluster growth at different frequencies, and in Fig. 1j we show the growth curves. We restrict further experiments to f = 20 Hz which promotes the formation of larger sized spinning clusters.
To understand the motility dynamics of the colloidal spinners within the spinning clusters, we use an optical flow detection method to calculate flow velocities in different regions of the vortex (Fig. 2). This method yields a 2D vector field where each vector is a displacement vector showing the movement of points from each frame to the next. Due to the high density of particles in the clusters we find that optical flow methods provide more reliable analysis than individual particle tracking. We average the optical flow vectors over 1800 frames (30 s) to obtain steady state velocity and vorticity distribution within the formed clusters (Fig. 2). We find that the cluster is composed of a rigid core where very low velocity is detected surrounded by a fast flow at the edges with circulating spinners in the direction of the applied rotational field, as typical for chiral fluids. In Fig. 2a we plot the velocity vector field and in Fig. 2b, c the corresponding velocity and vorticity magnitudes for a vortex with r = 18 µm. In Fig. 2d–f we plot the detected flow velocities along the radial profile of the vortex for three different vortex sizes from r = 8 µm to r = 18 µm and observe consistent behavior in all cases, with clusters containing a rigid core surrounded by an edge current with vedge = 20–25 µm/s. We also calculate the absolute values of mean vorticity magnitude \({\langle {|\langle {\rm{rot}}{\boldsymbol{v}}({\boldsymbol{r}},t) \rangle }_{t}| \rangle }_{r\in {ROI}}\) (Fig. 2c, g–i) which also confirms a rigid core with a circulating edge current of the spinner colloids for all cluster sizes.

a Steady state velocity vectors in a cluster with a rotating in-plane magnetic field (f = 20 Hz, B = 10 mT). The colors correspond to the velocity vector direction. b Velocity magnitude of the cluster represented in (a) with the edge current clearly visible, color bar units are µm/s; Supplementary Movie 2. c Vorticity map of the cluster represented in (a, b); color bar units are 1/s. d–f Velocity magnitude plotted along a radial profile for clusters of 3 different sizes ranging from r = 8 µm to r = 18 µm, with insets on the right showing the velocity map image for each cluster. Error bars represent the standard error of mean. g–i Corresponding vorticity magnitude plotted along a radial profile with insets on the right showing the vorticity map image for each cluster. Error bars represent the standard error of mean.
Influence of diffusiophoretic interactions on spinning clusters
The hematite particles that we use as colloidal spinners are photo-catalytically active when suspended in a solution of H2O2. Briefly, when the hematite colloids are exposed to blue light (wavelength λ = 450–490 nm) they induce a photocatalytic decomposition of H2O2 (2 H2O2 → O2 + 2 H2O) creating a local excess of reaction products. For an individual hematite particle, we observe enhanced diffusion dynamics in the presence of UV illumination (Supplementary Fig. 5). More importantly, it sets up a chemical gradient of the reaction product in the particle vicinity which drives the diffusiophoretic migration of nearby particles26. Since diffusiophoresis is a surface slip phenomenon, the direction of colloidal migration in a chemical gradient is sensitive to the surface properties and depends on the particle surface mobility (µp). Additionally, the reaction also sets up a chemical gradient along the nearby glass substrate, introducing an unbalanced osmotic pressure that drives radial surface flows along the adjacent glass surface, the direction of which is again dependent on the substrate surface mobility (µs). The net effect on a neighboring particle is determined by the balance of both diffusiophoresis and near substrate osmotic flows. In a simple experiment, we affix hematite particles onto a glass substrate by allowing a dilute suspension of the magnetic particles to dry. We then add a suspension of r = 1 µm silica colloids in H2O2 (0.5% v/v) in the area of the affixed hematite particles to study the interaction between the two. The hematite particles remain adhered to the substrate while silica particles display Brownian diffusion in the fluid. In the absence of UV illumination, we only observe excluded volume interactions between the hematite and the silica colloids, but as soon as the UV light is activated, we observe a strong attraction between the two colloids. Over time, we find that diffusing silica colloids in the bulk accumulate in layers around the affixed hematite particle, with inner layers being more rigid than the outer layers, indicating that the net effect of the diffusiophoretic and chemiosmotic interactions is attractive (Supplementary Fig. 3). The number of accumulated passive silica colloids is also dependent on UV intensity, with higher intensity leading to higher number of attracted particles. confirming this to be a osmoto-phoretic effect (Supplementary Fig. 5, Supplementary Movie 3).
In our system of spinning hematite colloids, when suspended in H2O2, there are several inter-particle interactions that occur in the presence of UV illumination. Magnetic dipolar interactions and external torque induced hydrodynamic flows lead to the formation of circulating clusters as we have described before (Figs. 1, 2). Additionally, the decomposition of H2O2 introduces diffusiophoretic interactions between particles, and osmotic flows in the proximity of the substrate. The overall collective behavior in the presence of UV illumination is determined by the interplay between these different interactions. In our experiments, as soon as UV illumination is switched on, we observe an immediate and rapid expansion of the circulating cluster leading to a loss in cluster cohesion (Fig. 3a–c). To characterize this loss in cohesion, we plot the two-dimensional positional density distribution at different points in time before and after UV exposure in Fig. 3d with darker colors indicating higher values. Before UV exposure we obtain a compact cluster with a dark center and a lighter outer edge where we have the circulating edge velocity. After UV exposure we observe an expansion of the cluster and a decrease in positional density in all regions as a result. The cluster radius increases from r = 7 µm at t = tUV_on to r = 11 µm at t = tUV_on + 18 s, after which the cluster radius saturates. Similar to analysis in Fig. 2, we also employ optical flow detection to estimate velocities in different regions of the circulating cluster and observe a loss in edge velocity (Fig. 3e) upon UV exposure. As the cluster expands, the edge velocity reduces from vedge = 17 µm/s at t = tUV_on to vedge = 12 µm/s at t = tUV_on + 8 s and eventually normalizing across the cluster at t = tUV_on + 18 s to vedge = 8 µm/s (Fig. 3f–h).

Schematic representation of the clusters of ellipsoidal hematite colloids before (a) and after (b) application of an UV illumination. c Optical microscopy images of the colloidal spinner cluster with UV activation turned on and subsequently off, showing the reversible control of cluster cohesiveness; Supplementary Movie 4, 5. d Positional probability distribution of a colloidal spinner cluster under different stages of UV activation (f = 20 Hz, B = 10 mT) over t = 180 s. e Corresponding velocity maps of the cluster with and without UV activation. Velocity magnitude of a colloidal spinner cluster plotted along a radial profile (f) after UV activation (g, h) and UV deactivation (i, j).
The cluster expansion is completely reversible. Once the cluster is in its fully expanded state, we switch off the UV illumination and find that the cluster recovers its cohesive state similar to pre-UV illumination. This reversible process is characterized by the intermediate formation of small local clusters which subsequently merge to form the larger initial cluster. We find that since the expansion radius of the cluster upon UV activation is limited, the recovered clusters tend to be composed of the original particles due to the high local particle density within the expanded cluster compared to bulk density. In Fig. 3d we plot the two-dimensional particle density map which shows after t = tUV_off, the initial formation of small clusters (r = 3–4 µm) in close proximity of each other that subsequently merge to form the larger cluster. Interestingly we recover the edge currents in the intermediate clusters and find that it is has similar vedge to that of the initial (and final) clusters at vedge = 16 µm/s (Fig. 3i, j). The intermediate clusters also have a rigid core (Fig. 3e) and the cluster merging is primarily promoted by the fast-moving spinners at the edges of the clusters.
In order to gain an understanding of the mechanism leading to cluster expansion we record high magnification image sequences of cluster dynamics in the presence of UV light. In our experiments, at t = 0, the cluster is compact with a rigid core and a fast-moving edge layer as described previously (Fig. 2). Immediately after UV light is turned on (t = 8 s), we observe holes that begin to develop within the cluster as it expands. The holes form across the structure simultaneously and we do not observe any obvious directional propagation. While some of the particles at the edges separate out into the bulk during the expansion process, remarkably, we observe that the expanded cluster has a finite radius and is primarily composed of a dynamic network of inter-connected particles. The separated particles continue to rotate in the rotational magnetic field and form transient links with other nearby particles along their magnetic dipole moment creating a dynamic connected network of reconfiguring colloidal chains. To characterize this interconnected structure, we apply a morphological operation where we binarize each frame in our image sequence and reduce all detected binary objects to single-pixel wide representation to obtain the ‘skeleton’ of the expanded cluster structure that preserves topological and connectivity information from the original image. This method allows us to construct the cluster as a network of branches along the formed colloidal chains and track their evolution over time. In Fig. 4a, we plot the number of detected branches (Nbranch) and the average length of branches (\(\left\langle {L}_{{branch}}\right\rangle\)). At the initial state as the cluster is primarily a rigid structure, we see a small number of branches with high Lbranch as the morphological operation reduces to cluster regions around transient gaps that appear in the cluster. However, once the UV light is turned on (t = 8 s; dotted line) we find that Nbranch increases by >66% as the cluster begins to expand and (\(\left\langle {L}_{{branch}}\right\rangle\)) in the system reduces simultaneously indicating the formation of a networked structure.

a Number of detected branches (magenta) and the branch length (brown) obtained by a digital thinning algorithm plotted over time before and after UV activation (dotted line). Branch detection analysis demonstrated on a typical cluster of spinner colloids before (b) and after UV activation (c, d); Supplementary Movie 6. Scale bar corresponds to 10 µm. e Calculated trajectory of spinners with hydrodynamic and magnetic dipole-dipole interactions, but without chemical activity. As they spin, the particles spiral towards each other until they collide. f The particles from (e), but with chemical activity switched on. The particles are stably attracted to a circular orbit with no steric contact.
The expansion of the circulating cluster upon UV illumination is somewhat counter-intuitive considering that the osmoto-phoretic effects result in attractive interactions when tested with tracer silica particles (Supplementary Fig. 5) and that the magnetic dipolar interactions are strongly attractive. However, osmotic and phoretic effects are highly sensitive to the material surface properties and direction of migration changes by switching the sign of the surface mobility parameter (µp). While the value of µp is difficult to directly access experimentally, our observations suggest that for a hematite particle it results in a repulsive particle-particle interaction unlike the hematite-silica case. As we show below, the interplay between the hydrodynamic, magnetic, and phoretic interactions can result in a loosened cluster without destroying it. The axi-asymmetry resulting from the attractive interactions along the direction of magnetic moment (\({\boldsymbol{m}}\)), promotes the dynamic network formation observed in our experiments (Fig. 4).
To further understand the interplay of chemical activity, hydrodynamics, and magnetic forces, we perform numerical simulations of the trajectories of two interacting magnetic spinners. Our simulations resolve the hydrodynamic and chemical fields sourced by the particles in microscopic detail, through solution of the governing partial differential equations (see Methods). Furthermore, we note that they resolve the ellipsoidal shape of the particles and their rapid spinning motion, as driven by the rotating magnetic field. In the absence of chemical activity, we recover the observation that a pair of rapidly spinning particles will slowly spiral towards each other until making steric contact, due to a net magnetic attraction between their dipole moments (Fig. 4e, Supplementary Movie 8). Keeping the same magnetic parameters, we switch on chemical activity, with the particles’ phoretic surface mobility chosen to give a repulsive diffusiophoretic interaction. We do not consider the effect of charge, whether in the form of charged molecular species (ions) diffusing in solution, or charge on the surface of the particle (i.e., the zeta potential.) as previous experimental observations with tracers in hematite-H2O2 systems could be quantitatively captured by a model assuming production solely of an uncharged molecular species by the hematite particle5,27,28. We find that the chemical interaction can actually stabilize bound states of magnetic spinners that have a finite-sized, liquid-filled gap between the spinners (Fig. 4f, Supplementary Movie 9). This result is particularly surprising when one considers the far-field decay of the magnetic and diffusiophoretic interactions. In the far-field, the magnetic interaction decays as 1/r4, while the diffusiophoretic interaction decays as 1/r2, where r is the distance between the particles. (We note that the interaction through chemi-osmotic flows would also have a 1/r2 decay.) For this combination of long-ranged repulsion and short-ranged attraction, the expectation is that bound states would be unstable. Therefore, the observation of stable bound states demonstrates the importance of near-field hydrodynamic and chemical interactions. To test this hypothesis, we also consider magnetically and chemically interacting particles with a large initial separation. We find that they move away from each other (see Supplementary Fig. 7), consistent with the form taken by the interactions in the far-field regime. Overall, these results from two-body microscopic simulations suggest that diffusiophoretic repulsion can loosen clusters - without destroying them - by promoting formation of non-contact bound states. Moreover, near-field chemical and hydrodynamic interactions are a necessary ingredient for stabilization of the bound states.
Control of colloidal aggregation through magnetic and chemical interactions
Finally, we show that the clustering and expansion phenomenon can be exploited to control static colloidal aggregation behavior via a sequence of applied magnetic and chemical activation signals. Initially, after suspending hematite colloids in an aqueous H2O2 solution, we observe that the density mismatched colloids are arranged as finite length chains, aligned along earth’s weak magnetic field, surrounded by a sparse population of individual Brownian particles as described earlier (Fig. 5a). We then apply a rotating magnetic field (f = 20 Hz) for a period of t = 300 s. During this time the system transitions into large chiral clusters composed of rotating hematite spinners that we have previously described (Fig. 5b). The size of the clusters can be controlled both via applied frequency and the particle density in the system. At t = 300 s we switch off the rotating magnetic field. We observe that this creates a network of inter-connected chain structures that somewhat resembles a colloidal gel, composed of the particles in the rotating clusters collectively aligned along the weak magnetic field (Fig. 5c). Effectively, the rotational magnetic field allows us to transition from a system of sparse chains of colloids through circulating clusters and eventually (when the magnetic field is switched off) to dense interconnected networks of colloidal chains whose extent is dependent on the size of the rotating cluster (controllable via particle density and applied frequency). Further, we can use the axi-asymmetric interactions resulting from magnetic dipolar attractions and diffusiophoretic repulsions to control structural properties of the dense networked structure such as porosity and size. Upon introduction of UV light, we observe that the networked structure expands only in the lateral direction. The colloidal chains remain intact through dipolar attractive interactions but are laterally repelled from each other leading to an expanded porous state (Fig. 5d, e) and the expansion continues to occur until interactions with other neighboring chains stabilizes it (Fig. 5f). Similar to chiral clusters, the process with the static networks is also reversible and the structures revert to their initial configuration over time when chemical interactions are quenched.

a Distribution of colloidal chains composed of ellipsoidal hematite particles formed through magnetic dipolar interactions. b Cluster of spinning colloidal particles obtained t = 300 s after application of a rotating magnetic field. c Colloidal-gel like networked structure composed of inter-connected chains of ellipsoidal hematite particles obtained when in-plane rotating magnetic field is turned off. d, e Expansion of the colloidal network structure after UV activation, over time; Supplementary Movie 7. Scale bar corresponds to 10 µm. f Evolution of cluster width after UV activation.
Conclusions
In summary, we have demonstrated the ability to control and reconfigure circulating clusters that form in chiral fluids of spinning colloids. The clusters form as a result of both hydrodynamic and magnetic dipolar interactions between spinning colloids and are therefore sensitive to the applied rotational frequency. The activity in chiral fluids arises solely from the rotational motion of the spinners in contrast to self-propelled units in conventional active matter systems and requires new methods of control. Here we have shown that introducing additional chemical interactions between particles gives us the ability to reversibly control the cohesiveness of spinning clusters. Since the hematite particles in the chiral fluid are only catalytically active in the presence of UV illumination, we can switch these interactions on/off at will, giving us external control over the observed collective states in chiral fluids. Our simulations of two particle interactions show that the presence of chemical interactions can stabilize bound states of magnetic spinners that have a finite-sized, liquid-filled gap between the spinners leading to loosening of chiral clusters without destroying them. Our preliminary experimental results also indicate the importance of particle shape in the observed collective states, as we achieve circulating clusters using ellipsoidal and cuboidal hematite particles but not rod-shaped particles under the same magnetic actuation conditions (Supplementary Fig. 8). A detailed investigation on the role of particle shape in chiral clusters will be the subject of a future study. Finally, we have shown that the rotating clusters result in inter-connected colloidal gel-like structures when the external field is switched off and the properties of this structure can be controlled via chemical interactions leading to lateral expansion when the UV illumination is switched on. We hope that the introduction of chemical activity-based interactions in chiral fluids paves the way for developing new routes to control self-organization in these systems. Since osmoto-phoretic interactions are sensitive to surface properties we expect that surface modifications could be an additional control parameter for collective behavior. Additionally these interactions enhance the role of the bottom substrate as the osmotic flows are mediated along the surface and enable potential means to control collective effects via surface-patterning29.
Methods
Experimental details
Iron (III) chloride hexahydride, sodium hydroxide, sodium sulfate, required for ellipsoidal hematite particle synthesis were obtained from Sigma Aldrich (USA). Hematite microparticles were synthesized with sol-gel method as previously reported30. The shape of the particles was controlled by the addition of sodium sulfate. For microparticle synthesis, 5.4 M NaOH was slowly added to the 2 M FeCl3 solution on stirring. Further, solution was shaken vigorously to homogenize the gel. Subsequently 0.2 M Na2SO4 was added to the homogenized gel before aging in an oven at 100 °C for 8 days. After 8 days, particles were centrifuged at 3000 rpm for 10 min, and washed with water thrice to remove unwanted salt from the particles. The obtained particles were suspended in highly deionized H2O to obtain the desired particle concentration and stabilized by adding 1.375 mg/mL sodium dodecyl sulfate (SDS).
For clustering experiments, a droplet (10 µL) of an ellipsoidal hematite particle suspension was sandwiched between two glass slides separated by a spacer (h = 120 µm) and imaged with an inverted microscope at room temperature using a \(\times \!\)10X objective for capturing system dynamics and \(\times \!\)60X objective for capturing individual cluster dynamics. The external magnetic field was applied using a field generator (MagnebotiX, MFG-100i) consisting of 8 stationary electromagnets allowing us to apply arbitrary magnetic fields in a spherical workspace of approximately 10 mm. The MFG system was controlled using custom MATLAB scripts. Blue light was applied via a fluorescent filter cube with an excitation wavelength of 491 nm. The dynamics of the particles were captured via a CMOS camera at 30-60 fps. Image analysis was carried out using custom Python scripts. Images containing hematite particles are binarized with a threshold such that only particles and particle clusters are selected. Their contours are identified using Python’s OpenCV library and their centers and areas are extracted. The optical flow is calculated on the bright-field image sequence using the cv2 implementation of the Gunnar Farneback algorithm. The branching analysis is carried out by applying a thinning algorithm (zhang-suen thinning) implemented in OpenCV. The function iteratively reduces binary objects to single pixel representations while preserving structural information. Average branch length and number are calculated from the structure obtained from the final structure.
Numerical model
We consider two oblate spheroidal particles immersed in an incompressible Newtonian fluid near a solid planar wall. The aspect ratio of the particles is \({r}_{e}=2\), where \({r}_{e}\) is the ratio of the major axis length to the minor axis length, and we assume that the minor axis of each particle lies within the plane of the wall. The wall is located at z = 0, and we assume the particles maintain a constant height \(z/a=2.1\), where \(a\) is the length of the semi-minor axis. Each particle \(i\) has an intrinsic magnetic moment \({\vec{m}}_{i}=m{\hat{d}}_{i}\), where \({\hat{d}}_{i}\) is aligned with the minor axis. The system is driven by a rotating magnetic field described by \(\vec{B}(t)={B}_{0}[\sin (\omega t)\hat{x}+\cos (\omega t)\hat{y}]\). Each particle experiences a magnetic torque \(\vec{\tau }={\mathop{m}\limits^{ \rightharpoonup }}_{i}\times \vec{B}\). In addition, the two particles interact with each other through a dipole-dipole force \({ \vec{F}}_{{ij}}=\frac{3{\mu }_{0}{m}^{2}}{4 \pi {r}^{4}}[{\hat{r}}_{{ij}}(\hat{d}_{i} \cdot {\hat{d}}_{j})+{\vec{m}}_{i}({\hat{r}}_{{ij}} \cdot {\vec{m}}_{j})+{\vec{m}}_{j}({\hat{r}}_{{ij}} \cdot {\vec{m}}_{i})-5{\hat{r}}_{{ij}}({\hat{r}}_{{ij}} \cdot {\vec{m}}_{i})({\hat{r}}_{{ij}} \cdot {\vec{m}}_{j})]\) and torque \({\vec{\tau }}_{{ij}}=\frac{3{\mu }_{0}{m}^{2}}{4\pi {r}^{3}}\)[3(\({\vec{m}}_{i}\cdot {\hat{r}}_{{ij}}\))(\({\vec{m}}_{j}\times {\hat{r}}_{{ij}}\)) + (\({\vec{m}}_{i}\times {\vec{m}}_{j}\))], where \({\vec{F}}_{{ij}}\) is the force on particle \(j\) from particle \(i\), \({\vec{\tau }}_{{ij}}\) is the torque on particle \(j\) due to particle \(i\), \({\hat{r}}_{{ij}}\) is the unit vector pointing from the center of particle \(i\) to the center of particle \(j\), \(r\) is the distance between the particle centers, and \({\mu }_{0}\) is the permeability of free space31. The suspending fluid obeys the Stokes equation \(-\nabla p+\mu {\nabla }^{2}\vec{u}=0\), where \(\vec{u}(\mathop{x}\limits^{ \rightharpoonup })\) is the velocity of the fluid and \(p(\mathop{x}\limits^{ \rightharpoonup })\) is the pressure. The velocity is subject to the boundary conditions \(\vec{u}=\) 0 on the solid wall, \(|\vec{u}|\to 0\) as \(\left|\vec{x}\right|\to \infty\), and \(\vec{u}={\vec{U}}_{i}+{\vec{\Omega }}_{i}\times \left(\vec{x}-{\vec{x}}_{i}\right)+{\vec{v}}_{s,i}(\vec{x})\) on particle \(i\), where \({\vec{U}}_{i}\) and \({\vec{\Omega }}_{i}\) are the (unknown) translational and rotational velocities of particle \(i\), and \({\vec{x}}_{i}\) is the position of particle \(i\). The quantity \({\vec{v}}_{s,i}(\vec{x})\) is the interfacial slip velocity, which results from chemical activity, and is discussed in detail below. Incompressibility imposes the condition \(\nabla \cdot \vec{u}=0\) on the velocity field. In addition, we assume the following model for chemical activity 26,29. Each particle generates a product molecule (“solute”) at a uniform and constant rate over its surface. The number density of solute \(c(\vec{x})\) is assumed to be quasi-steady, i.e., diffusion of solute is much faster than advection. Therefore, it is governed by the Laplace equation \({\nabla }^{2}c=0\). The boundary conditions for the concentration are \(-D[\hat{n}\cdot \nabla c]=\kappa\) on the surface of each particle, \({|c}(\vec{x})|\to 0\) as \(\left|\vec{x}\right|\to \infty\), and \(\hat{n}\cdot \nabla c=0\) on the solid wall (i.e., the wall is impenetrable to solute), where the surface norm \(\hat{n}\) is defined to point into the fluid. Here, \(D\) is the diffusion coefficient of the solute molecule, and \(\kappa\) is the rate of production of solute per unit surface area. The slip velocity is determined by \({\vec{v}}_{s,i}\left(\vec{x}\right)=-b{\nabla }_{{||}}c\). The surface slip represents flow in a thin interfacial layer (relative to the length scale \(a\)), driven by osmotic pressure gradients that originate in the effective interaction between the solute and the surface26. The surface mobility \(b\) encodes the details of the solute/surface interaction, and \(b\) is positive (negative) for an attractive (repulsive) interaction. We choose \(b \, < \, 0\) to obtain diffusiophoretic repulsion between the particles. The quantity \({\nabla }_{{||}}=(\mathop{I}\limits^{\leftrightarrow}-\hat{n}\hat{n})\cdot \nabla\) is the surface gradient operator. We non-dimensionalize the equations by choosing \({\omega }^{-1}\) as the characteristic timescale, and the semi-minor axis length \(a\) as the characteristic length scale. As a result, the chemical activity is parameterized by the dimensionless quantity \(A\equiv \left|b\right|\kappa /{Da}\omega\) and the dipole-dipole force by the dimensionless quantity \(B=\frac{3{\mu }_{0}{m}^{2}}{4\pi \mu \omega {a}^{5}}\). We choose \(B=500\) and (when activity is switched on) \(A=0.025\). This choice of \(B\) gives the slow decay of orbital trajectories, as discussed in the main text, and likewise this choice of \(A\) gives bound states when the activity is on. Sufficiently small variations of \(A\) and \(B\) do not change the pair behavior. In each simulation time step, we first solve for the concentration field numerically, if chemical activity is present, using the boundary element method (BEM)32. Next, we calculate the surface slip \({\vec{v}}_{s,i}\left(\vec{x}\right)\). We then solve the Stokes equation for the particle velocities, also using the BEM. Next, we update the particles’ positions and orientations using a rigid body dynamics engine, assuming that the minor axes remain in the plane of the wall (i.e., \({\vec{\Omega }}_{i}={\Omega }_{z,i}\hat{z}\)) and that the particles do not move in the vertical direction, \({U}_{z,i}=0\). We terminate the simulation if particles come into steric contact.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials
References
Ramaswamy, S. The mechanics and statistics of active matter. Annu. Rev. Condens. Matter Phys. 1, 323–345 (2010).
Bechinger, C. et al. Active particles in complex and crowded environments. Rev. Mod. Phys. 88, 045006 (2016).
Kaiser, A., Snezhko, A. & Aranson, I. S. Flocking ferromagnetic colloids. Sci. Adv. 3, e1601469 (2017).
Bricard, A., Caussin, J.-B., Desreumaux, N., Dauchot, O. & Bartolo, D. Emergence of macroscopic directed motion in populations of motile colloids. Nature 503, 95–98 (2013).
Palacci, J., Sacanna, S., Steinberg, A. P., Pine, D. J. & Chaikin, P. M. Living crystals of light-activated colloidal surfers. Science 339, 936–940 (2013).
Yan, J. et al. Reconfiguring active particles by electrostatic imbalance. Nat. Mater. 15, 1095–1099 (2016).
Katuri, J., Poehnl, R., Sokolov, A., Uspal, W. & Snezhko, A. Arrested-motility states in populations of shape-anisotropic active Janus particles. Sci. Adv. 8, eabo3604 (2022).
Buttinoni, I. et al. Dynamical clustering and phase separation in suspensions of self-propelled colloidal particles. Phys. Rev. Lett. 110, 238301 (2013).
Ginot, F., Theurkauff, I., Detcheverry, F., Ybert, C. & Cottin-Bizonne, C. Aggregation-fragmentation and individual dynamics of active clusters. Nat. Commun. 9, 696 (2018).
Bricard, A. et al. Emergent vortices in populations of colloidal rollers. Nat. Commun. 6, 7470 (2015).
Cates, M. E. & Tailleur, J. Motility-induced phase separation. Annu. Rev. Condens. Matter Phys. 6, 219–244 (2015).
van Zuiden, B. C., Paulose, J., Irvine, W. T. M., Bartolo, D. & Vitelli, V. Spatiotemporal order and emergent edge currents in active spinner materials. Proc. Natl Acad. Sci. 113, 12919–12924 (2016).
Soni, V. et al. The odd free surface flows of a colloidal chiral fluid. Nat. Phys. 15, 1188–1194 (2019).
Zhang, B., Sokolov, A. & Snezhko, A. Reconfigurable emergent patterns in active chiral fluids. Nat. Commun. 11, 4401 (2020).
Scholz, C., Engel, M. & Pöschel, T. Rotating robots move collectively and self-organize. Nat. Commun. 9, 931 (2018).
Nguyen, N. H. P., Klotsa, D., Engel, M. & Glotzer, S. C. Emergent collective phenomena in a mixture of hard shapes through active rotation. Phys. Rev. Lett. 112, 075701 (2014).
Snezhko, A. Complex collective dynamics of active torque-driven colloids at interfaces. Curr. Opin. Colloid Interface Sci. 21, 65–75 (2016).
Shen, Z. & Lintuvuori, J. S. Hydrodynamic clustering and emergent phase separation of spherical spinners. Phys. Rev. Res. 2, 013358 (2020).
Shen, Z. & Lintuvuori, J. S. Collective flows drive cavitation in spinner monolayers. Phys. Rev. Lett. 130, 188202 (2023).
Grzybowski, B. A., Stone, H. A. & Whitesides, G. M. Dynamic self-assembly of magnetized, millimetre-sized objects rotating at a liquid–air interface. Nature 405, 1033–1036 (2000).
Kokot, G., Piet, D., Whitesides, G. M., Aranson, I. S. & Snezhko, A. Emergence of reconfigurable wires and spinners via dynamic self-assembly. Sci. Rep. 5, 9528 (2015).
Shen, Z., Würger, A. & Lintuvuori, J. S. Hydrodynamic self-assembly of active colloids: chiral spinners and dynamic crystals. Soft Matter 15, 1508–1521 (2019).
Massana-Cid, H., Levis, D., Hernández, R. J. H., Pagonabarraga, I. & Tierno, P. Arrested phase separation in chiral fluids of colloidal spinners. Phys. Rev. Res. 3, L042021 (2021).
Petrichenko, O. et al. Swarming of micron-sized hematite cubes in a rotating magnetic field – Experiments. J. Magn. Magn. Mater. 500, 166404 (2020).
Fily, Y., Baskaran, A. & Marchetti, M. C. Cooperative self-propulsion of active and passive rotors. Soft Matter 8, 3002–3009 (2012).
Anderson, J. L. Colloid transport by interfacial forces. Annu. Rev. Fluid Mech. 21, 61–99 (1989).
Aubret, A. & Palacci, J. Diffusiophoretic design of self-spinning microgears from colloidal microswimmers. Soft Matter 14, 9577–9588 (2018).
Palacci, J. et al. Light-activated self-propelled colloids. Philos. Trans. R. Soc. A 372, 20130372 (2014).
Uspal, W. E., Popescu, M. N., Dietrich, S. & Tasinkevych, M. Guiding catalytically active particles with chemically patterned surfaces. Phys. Rev. Lett. 117, 048002 (2016).
Meijer, J. M., Rossi, L. & Preparation properties, and applications of magnetic hematite microparticles. Soft Matter 17, 2354–2368 (2021).
Landecker, P. B., Villani, D. D. & Yung, K. W. An analytic solution for the torque between two magnetic dipoles. Phys. Sep. Sci. Eng. 10, 29–33 (1999).
Pozrikidis, C. A Practical Guide to Boundary Element Methods with the Software Library BEMLIB (CRC Press, Boca Raton, 2002).
Acknowledgements
This work was funded by the National Science Foundation (No. EES-2306449, EES-2219558, EES-2000202) and supported by the NSF FAMU CREST Center award (No. EES-1735968). Acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund for support (or partial support) of this research. This material is also based upon work supported by the Air Force Office of Scientific Research under award number FA9550-22-1-0247. Some of the work was performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida. W. E. U. gratefully acknowledges support from the Army Research Office under Grant Number W911NF-23-1-0190. The technical support and advanced computing resources from University of Hawaii Information Technology Services – Cyberinfrastructure, funded in part by the National Science Foundation CC* awards #2201428 and #2232862 are gratefully acknowledged. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the United States Air Force or the United States Army.
Author information
Authors and Affiliations
Contributions
J.K. and J.A. designed research; J.K., A.C., D.Q.J. performed research; N.K. carried out particle synthesis; J.K. analyzed data; W.U. developed theoretical models and performed simulations; J.K., W.U., and J.A. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Katuri, J., Kaur, N., Uspal, W. et al. Control of colloidal cohesive states in active chiral fluids. Commun Phys 7, 291 (2024). https://doi.org/10.1038/s42005-024-01787-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42005-024-01787-3
This article is cited by
-
Transient chirality in the gelation of adhesive spinner monolayers
Communications Physics (2025)
