
Abstract
Self-supervised learning (SSL) has emerged as a powerful method for extracting meaningful representations from vast, unlabelled datasets, transforming computer vision and natural language processing. In single-cell genomics (SCG), representation learning offers insights into the complex biological data, especially with emerging foundation models. However, identifying scenarios in SCG where SSL outperforms traditional learning methods remains a nuanced challenge. Furthermore, selecting the most effective pretext tasks within the SSL framework for SCG is a critical yet unresolved question. Here we address this gap by adapting and benchmarking SSL methods in SCG, including masked autoencoders with multiple masking strategies and contrastive learning methods. Models trained on over 20 million cells were examined across multiple downstream tasks, including cell-type prediction, gene-expression reconstruction, cross-modality prediction and data integration. Our empirical analyses underscore the nuanced role of SSL, namely, in transfer learning scenarios leveraging auxiliary data or analysing unseen datasets. Masked autoencoders excel over contrastive methods in SCG, diverging from computer vision trends. Moreover, our findings reveal the notable capabilities of SSL in zero-shot settings and its potential in cross-modality prediction and data integration. In summary, we study SSL methods in SCG on fully connected networks and benchmark their utility across key representation learning scenarios.
Similar content being viewed by others

Main
Single-cell genomics (SCG) has rapidly expanded into a big-data domain, primarily driven by advancements in single-cell RNA-sequencing technologies1. This expansion has shifted the focus from analysing data in isolated studies to using machine learning models for interpreting data within the context of existing datasets2. Recent efforts towards comprehensive atlases, such as the Human Cell Atlas3, underscore this development. However, larger datasets introduce additional methodological challenges, such as technical batch effects across studies and the variability in labelling quality4,5. Large-scale models have gained interest and emerged for their potential to address these issues6. Yet a gap remains in understanding their use cases and how to effectively leverage the emerging datasets comprising millions of cells7. The SCG field now not only requires computational power but also strategic use of methods that handle the complexities of big data. In this context, self-supervised learning (SSL) is a promising approach. SSL leverages pairwise relationships within data X for training, setting it them apart from supervised learning, which relies on data X with labels Y to guide the loss, and unsupervised learning, which depends solely on data X (refs. 8,9,10). It has proven powerful in other data-intensive domains, such as computer vision11,12 and natural language processing13,14, leveraging large unlabelled datasets. It is thus often the basis for foundation models15.
SSL has already begun to impact SCG on small and large scales. On small scales, specialized SSL methods have deployed contrastive losses, tailored with techniques such as multimodal learning16, graph-based strategies17 and clustering-based approaches18,19,20 to embed cells. The contrastive methods address unique data challenges in SCG, including batch effects and data sparsity18,19,21,22,23,24,25,26,27. Other specialized SSL methods predict blood cell traits28, identify subpopulations of T cells29, boost active learning30 and classify cell types on the whole mouse brain31, indicating the method’s versatility. However, a common limitation among these approaches is their application to relatively small datasets or specific problems, resulting in limited generalizability across downstream tasks. On large scales, foundation models are trained on large datasets and applied to a broad range of tasks. In SCG, they often deploy transformers trained in a supervised32,33 and self-supervised34,35,36,37 fashion. While foundation models have demonstrated improvements through self-supervised pre-training34,35, disentangling the contributions of SSL, scaling laws or the transformer architecture remains difficult. This ongoing debate underscores the relevance of investigating SSL in non-transformer contexts, which are prevalent in SCG38,39. Recent studies in computer vision40,41 also suggest a nuanced perspective on the dominance of transformer architectures, indicating the value of exploring diverse architectural approaches for model development. The ambiguity mainly arises when comparing the performance of models with and without self-supervised pre-training14,42, suggesting a need for a more in-depth exploration of the role of SSL in SCG. Similar to SSL, semi-supervised learning combines unsupervised pre-training with supervised fine-tuning30, as opposed to self-supervised pre-training with optional fine-tuning in SSL. Both learning techniques are useful in transfer learning settings that are popular in SCG as reference mapping methods for single-cell datasets2,43.
To guide the effective usage of SSL in SCG, we need to address these ambiguities through systematic empirical validation. Such a study helps to determine the scenarios in which SSL can effectively contribute to SCG. First, this requires developing SSL methods based on first principles and tailoring them for single-cell applications. These SSL methods learn representations from data and differing pairwise relationships. To assess their impact on downstream performance, we benchmark our SSL methods and compare their performance with their supervised and unsupervised counterparts. Second, this study requires validation across downstream applications, addressing the method’s objective to learn data representations that are helpful across multiple tasks.
Our study aims to identify specific scenarios in SCG where SSL is helpful and to thoroughly analyse and evaluate SSL approaches in SCG. Utilizing the CELLxGENE44 census of scTab5 (scTab dataset), which comprises over 20 million cells, our study assesses the effectiveness of SSL across multiple downstream tasks. On the basis of well-defined benchmark metrics for SSL in SCG, our empirical analysis primarily focuses on the cell-type prediction application, with validation in gene-expression reconstruction, cross-modality prediction and data integration. We find that SSL improves downstream performance in transfer learning settings, that is, when analysing smaller datasets informed by insights from a larger auxiliary dataset and in scenarios involving unseen datasets. This improvement is especially notable in class-imbalance-sensitive metrics, indicating robustness improvements. However, our findings also reveal that self-supervised pre-training on the same dataset as the fine-tuning does not yield improvement compared with only supervised or unsupervised training. In summary, our study clarifies the roles and benefits of SSL in SCG, demonstrating its strengths in specific contexts while identifying its applicability limits. This research contributes to a more informed and strategic use of SSL in SCG, particularly in advancing our understanding of complex biological datasets.
Results
SSL framework for SCG
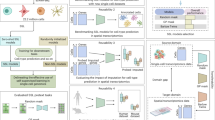
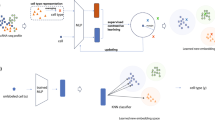
We present an SSL framework to develop self-supervision methods and study different use cases in SCG. Central to our framework is the use of fully connected autoencoder architectures, selected for their ubiquitous application in SCG tasks38,39 and for minimizing architectural influences on our study, yet still large enough to capture underlying biological variations. In this framework, we integrate key SSL pretext tasks based on masked autoencoders45 and contrastive learning46,47 to benchmark their performance. The framework operates in two stages. The first stage is pre-training, also called pretext task, where the model learns from unlabelled data. We call the resulting model ‘zero-shot SSL’ for its zero-shot evaluation. The second stage is the optional fine-tuning. We call the resulting model the ‘SSL’ model, which is further trained to specific downstream tasks such as cell-type annotation (Fig. 1a). The pretext task builds a rich data representation based on a comprehensive dataset. We chose the scTab dataset5 because of its extent and diversity. We used all 19,331 human protein-encoding genes from scTab to maximize generalizability, ensuring gene coverage for analyses of unseen datasets, regardless of their feature selections. Our SSL framework leverages masked autoencoder with random masking and gene programme (GP) masking strategies, along with our isolated masked autoencoder approaches gene programme to gene programme (GP to GP) and gene programme to transcription factor (GP to TF) masking, considering isolated sets of genes (Fig. 1b). The strategies entail leveraging different degrees of biological insight, from random masking with a minimal inductive bias to isolated masking that intensively utilizes known gene functions, emphasizing targeted biological relationships. For contrastive learning, we incorporate the negative-pair-free methods bootstrap your own latent (BYOL)46 and Barlow twins47, known for their effectiveness in computer vision (Fig. 1c), with negative binomial noise and masking as data augmentations. We benchmarked these strategies for their efficacy in improving downstream performance. Our SSL framework, including these strategies, is depicted in Fig. 1a, outlining its architecture and pivotal components. Detailed descriptions of the specific implementations and adaptations of these SSL methods for SCG are further elaborated in Methods.

a, Overview of the SSL framework (Methods). The zero-shot SSL model is trained on scTab RNA-sequencing data using masked autoencoders (MAEs) and contrastive learning (CL). Its weights initialize the SSL model, which is fine-tuned for downstream tasks (for example, cell-type prediction, gene-expression reconstruction). The non-SSL model is initialized randomly and fine-tuned only for downstream tasks. b, Masking strategies. Input features are either zeroed out (black) or left unchanged. The autoencoder (grey) predicts the masked features, and the loss is computed only on those. GP and TF masking is also shown (Methods). c, Contrastive learning. Input is augmented to create views. BYOL and Barlow twins are contrastive methods for data representation (Methods). d, Results from individual datasets. (1) Random model, (2) non-SSL model (for example, supervised for cell-type prediction, unsupervised for gene expression), (3) zero-shot SSL model, and (4) SSL model. Models are tested on PBMC, Tabula Sapiens and HLCA (Methods). Cell-type prediction is evaluated with macro/micro F1 scores (higher is better; see Supplementary Fig. 5 for loss curves); gene-expression reconstruction is evaluated with weighted explained variance (EV W, higher is better). The best performance is in bold. e, Relative cell prediction accuracy for the SSL and supervised models for cell types with the largest performance differences (see Supplementary Fig. 1 for other datasets). f, Macro F1 score differences between SSL and supervised models plotted against cell-type abundance, with the number of cell types for each abundance shown above (see Supplementary Fig. 1 for other datasets). g, Cell-type prediction performance of SSL models pre-trained on random scTab donor subsets and fine-tuned on PBMC, compared with the supervised model trained on only PBMC. Shaded error bands represent 95% confidence intervals (mean ± s.e. × t-value at 95% confidence). Results are from five random seeds (see Supplementary Fig. 6 for other datasets).
Pre-training on auxiliary data boosts cell-type prediction
As a first use case for self-supervision in SCG, we asked whether analyses on cell atlases or smaller datasets can benefit from self-supervised pre-training on auxiliary data. We answered this using three datasets: the Human Lung Cell Atlas (HLCA)4 (2,282,447 cells, 51 cell types), peripheral blood mononuclear cells (PBMCs) after severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection48 (422,220 cells, 30 cell types), and the Tabula Sapiens Atlas (483,152 cells, 161 cell types)49. These datasets vary in size, biological context and complexity, providing a robust test bed for our models. We evaluated cell-type prediction with the macro F1 score, supplemented by the micro F1 score, to compare robustness against class imbalances. We evaluated gene-expression reconstruction with the weighted explained variance. For the PBMC and Tabula Sapiens datasets, the self-supervised pre-training on additional scTab data significantly improved cell-type prediction and gene-expression reconstruction (Fig. 1d and Supplementary Fig. 2): from [0.7013 ± 0.0077] to [0.7466 ± 0.0057] macro F1 in the PBMC dataset and from [0.2722 ± 0.0123] to [0.3085 ± 0.0040] macro F1 in the Tabula Sapiens dataset. In the Tabula Sapiens dataset, this improvement is driven by strongly enhancing the classification of specific cell types, correctly classifying 6,881 of 7,717 type II pneumocytes instead of 2,441 (Fig. 1e; for other datasets, see Supplementary Fig. 1). For the PBMC dataset, this improvement is pronounced for underrepresented cell types (Fig. 1f), also indicated by the stronger macro F1 improvement versus micro F1 improvement. In contrast, the HLCA dataset presented a marginal performance improvement through self-supervised pre-training. Notably, SSL outperforms supervised learning if pre-trained on a large number of donors, highlighting the necessity of a rich pre-training dataset (Fig. 1g and Supplementary Fig. 6).
Tailored pre-training yields strong zero-shot performance
The scenario in which SSL is typically evaluated in computer vision is the zero-shot setting, where the model’s ability to represent and distinguish unobserved classes is assessed using data representations obtained solely through self-supervised pre-training. The labels are predicted, for example, with k-nearest-neighbours (kNN) classification or by training a prediction head while freezing the encoder weights. This perspective is noteworthy in SCG, where datasets’ increasing volume and complexity often come with challenges in obtaining accurate and comprehensive labels4. The ability of zero-shot SSL to achieve up to a 0.6725 macro F1 score on the scTab test set stands out as a strong performance (Fig. 2a). Likewise, in the test cases of HLCA, PBMC and Tabula Sapiens, zero-shot SSL comes close to their fine-tuned counterparts (Fig. 1d and Supplementary Fig. 1). The embedding from the zero-shot model illustrates this implicitly learned distinction of cell types (Fig. 2b). These findings highlight SSL’s potential in SCG to reduce the reliance on curated labels50 and propose adding self-supervised pre-trained model embeddings to biological analyses alongside principal component analysis (PCA), a practice exemplified by platforms such as CELLxGENE44. However, our benchmarking of SSL methods revealed the sensitivity to the choice of pre-training strategy. Contrastive learning has proven effective in domains such as language or vision modelling10,46,47. It has further proved effective on smaller scales18,19,21,22,23,24,25,26,27 in SCG and worked in principle on large scales, as shown in ref. 51 and this benchmark. Still, our study finds that masking outperforms contrastive learning in large SCG tasks. This result highlights the challenges of applying these methods as generalizable pretext tasks for single-cell data. Conversely, masked autoencoders performed better: the random masking strategy consistently ranked among the top performers across different tasks (Fig. 2a). Notably, in the specific context of gene-expression reconstruction, the GP to TF isolated masking showed superior performance compared with other methods (Supplementary Figs. 1 and 2). This finding highlights the potential of tailored masking strategies in capturing the nuanced biological variations inherent in SCG data.

a, Benchmark result of cell-type prediction on the scTab holdout test set using kNN classification (Methods). We compare: (1) baseline methods of kNN classification on a randomly initialized model, on the PCA embeddings, and deploying GeneFormer34 in a zero-shot setting; (2) our zero-shot SSL methods, pre-trained on the scTab training data; (3) our SSL methods; and (4) the supervised model. b, t-distributed stochastic neighbor embedding (t-SNE) visualization of the baseline PCA embedding, and the embedding obtained from the zero-shot SSL model and the supervised model. c, Classification performance on unseen datasets: (I, Human Brain Atlas, tail of hippocampus (HiT) - caudal hippocampus - CA4-DGC52; II, Human Brain Atlas, all non-neuronal cells52; III, single-cell analysis of prenatal and postnatal human cortical development53; IV, circulating immune cells after CV19 infection, vaccination and HC54; V, human, great apes study55) measuring the macro F1 score of a random baseline, a zero-shot SSL model, the supervised model and an SSL model, all trained on scTab without exposure to any unseen dataset. The box plots show the median (centre), 25th and 75th percentiles (box bounds) and whiskers extend to the minima and maxima within 1.5 times the interquartile range (seaborn default).
The efficacy of SSL depends on its context
While the previous evaluations focused on carefully curated and widely used benchmarks, we also set out to investigate SSL’s nuanced behaviour in analysing in-distribution versus unseen data settings. If the supervised and SSL model are provided access to the same data, their performance is remarkably similar (Fig. 2a). This finding is notable across cell-type annotation and gene-expression reconstruction. Extending to unseen datasets, we evaluated the supervised and SSL models on five datasets52,53,54,55 published after the CELLxGENE44 census of scTab (Methods). In this setting, self-supervised pre-training improves performance (Fig. 2c and Supplementary Fig. 2), for example, from [0.0877 ± 0.0215] to [0.1797 ± 0.0450] macro F1 for cell-type prediction in the great apes study55. So, while inside the distribution (Fig. 2a), supervised and self-supervised learning perform similarly, this finding offers another dimension of SSL’s utility: when analysing unseen datasets, where generalization is crucial, SSL shows its advantages.
Help cross-modality prediction with SSL
Having benchmarked the utility of SSL on transcriptomics, we extended our study to multiomics56, asking whether SSL can leverage auxiliary data from one modality to enhance multimodal downstream tasks, here focusing on cross-modality prediction (Fig. 3a). The NeurIPS multiomics dataset57, a rich multi-donor, multi-site and multimodal bone marrow dataset containing coupled gene expression and proteomics counts from CITE-seq58 experiments, provided a suitable test bed. The models obtain RNA-sequencing counts as input and predict protein counts. The SSL models are additionally pre-trained on RNA-sequencing data from the auxiliary scTab and the NeurIPS multiome dataset. When pre-trained on scTab, SSL significantly outperforms its supervised counterpart (P < 0.01) and the baseline method totalVI59 (P < 0.01; Fig. 3b,d). The Pearson correlation between predicted and true protein counts improved from [0.8809 ± 0.0013] for the unsupervised model to [0.8943 ± 0.011] for the self-supervised model. Notably, the improvement is smaller if pre-trained on the same data, to a Pearson correlation of [0.8824 ± 0.0037]. This finding highlights the advantage of self-supervision in cases where one modality is more abundant. This effect is reproducible on other modalities, as verified by predicting the assay for transposase-accessible chromatin with sequencing (ATAC-Seq) from RNA counts in the NeurIPS multiome dataset57 (Fig. 3c), proving the robust advantage of self-supervision on auxiliary data.

a, Scheme of cross-modality prediction training. The SSL models are pre-trained with masked autoencoders on RNA-sequencing data (1) of the downstream NeurIPS multiome dataset or (2) of the auxiliary scTab dataset. The SSL model, initialized with the pre-trained weights, and the unsupervised model, randomly initialized, predict the protein counts from the RNA counts. The baseline totalVI model learns a joint distribution of RNA and protein counts. In inference, all models predict the protein counts from a holdout test set. b,c, Cross-modality prediction performance for two tasks: predicting the normalized counts of 134 proteins (b) and the TF-IDF transformed ATAC-seq counts of 116,490 genes (c); both given coupled RNA counts. Shown is the Pearson correlation between predicted and true counts. The box plots show the median (centre), 25th and 75th percentiles (box bounds), and whiskers extend to the minima and maxima within 1.5 times the interquartile range (seaborn default). Results are from five experiments at random seeds. d, Scatter plot of predicted log counts against true log counts for exemplary proteins with correlation.
Self-supervised pre-training enhances data integration
Integrating single-cell datasets for joint analysis is difficult due to batch effects, for example, experimental conditions or confounding factors, posing unique challenges to atlasing efforts4. Large-scale models in SCG have already been deployed to address this challenge32,34. To clarify the role of SSL in these efforts, we set out to integrate three datasets included in scTab: the molecular cell atlas of the human lung (65,662 cells, 45 cell types)60, the molecular atlas of lung development of LungMap (46,500 cells, 28 cell types)61 and the molecular single-cell lung atlas of lethal coronavirus disease 2019 (COVID-19; 116,313 cells, 30 cell types)62. The datasets vary in cell-type composition and donor health or disease states, providing a challenging environment for this task. The single-cell integration benchmarking (scIB) metrics63 evaluate the data integration performance, indicating how well batch effects are corrected while conserving biological variability (Fig. 4a). The score aggregates five batch correction metrics (PCR batch, batch ASW, graph iLISI, graph connectivity and kBET) and nine biological conservation metrics (NMI cluster/label, ARI cluster/label, cell-type ASW, isolated label F1, isolated label silhouette, graph cLISI, cell cycle conservation, HVG conservation and trajectory conservation) that cover cell identity labels and variance beyond that. We fine-tuned the unsupervised and SSL models using gene-expression reconstruction on these three datasets. To improve data integration performance and model comparison, we added batch covariates to all models56. This led to the SSL-shallow model, which fine-tunes the last encoder layer of the zero-shot SSL model with batch covariates. PCA and scVI38 embeddings serve as baselines for data integration. The scIB metrics indicate that self-supervised pre-training improves the data integration performance (Fig. 4b) with a total scIB score of [0.5638 ± 0.0089] (SSL shallow) and [0.5571 ± 0.0080] (SSL) compared with [0.5354 ± 0.0110] (unsupervised). The SSL-shallow model performed best, hinting at a meaningful data representation learned through the self-supervision algorithms, underscored by the comparable performance of the specialized data integration method scVI38. This finding supports the advantage of leveraging auxiliary data through SSL and showcases the effectiveness of minimal fine-tuning compared with unsupervised learning.

a, Scheme of data integration fine-tuning for SSL and SSL-shallow models. The zero-shot SSL model is fine-tuned, along with the batch covariates available for the data integration benchmarking. A complete fine-tuning results in an SSL model, and a shallow fine-tuning of the last encoder layer results in an SSL-shallow model for the data integration task. Uniform manifold approximation and projection (UMAP) plot for good and bad integration. b, The scIB data integration benchmarking results63 of five runs. The benchmarking analysis comprises two sets of bio conservation and batch correction scores aggregated into a total score with a weighted mean. The box plots show the median (centre), 25th and 75th percentiles (box bounds), and whiskers extend to the minima and maxima within 1.5 times the interquartile range (seaborn default). Results are from five experiments at random seeds.
Discussion
We analysed the application of SSL in SGC to guide its effective usage, leading us to adapt and benchmark several SSL techniques tailored for SCG. Our empirical study illuminates the context in which SSL can excel, especially when leveraging insights from vast, auxiliary datasets for smaller dataset tasks and in unseen dataset scenarios. We also demonstrated that SSL shows parity with supervised methods where both access the same data and that the zero-shot SSL model comes close to that performance. Our insights contribute to a more nuanced understanding of SSL’s applications in SCG. By rigorously testing these methods on an expansive dataset encompassing over 20 million cells, we offer a robust, empirically grounded perspective on SSL in SCG, paving the way for more informed, data-driven approaches to studying complex biological systems. In the context of large-scale and foundation models34,35,36, this understanding could help design pre-training and select pretext tasks. For broad applicability within SCG, we address diverse, meaningful tasks, including cell-type prediction, gene-expression reconstruction, cross-modality prediction and data integration. By demonstrating that SSL’s advantages emerge predominantly in scenarios involving transfer learning tasks through auxiliary data or distributional shifts, we offer a pragmatic lens through which the SCG community can view SSL—not as a universal solution but as a strategic tool tailored for specific challenges. This insight is particularly relevant as SCG moves towards larger data analyses, analysing cell atlases and leveraging the consortia of millions of cells in foundation models. The adaptability and robustness of SSL, as evidenced in our empirical analysis, are crucial in this context to leverage large datasets. Our approach thus shows an example in SCG for the contextual application of SSL, guiding researchers to leverage this methodology where it most effectively addresses the field’s unique data challenges.
The benchmark of SSL methods provides a clear recommendation for practitioners regarding which approach is advantageous in the aforementioned settings. As a primary approach, we recommend masked pre-training with a random masking strategy due to its robustness and versatility across various tasks, which is central to foundation models. However, when focusing on specific problems, more tailored techniques might be beneficial. For instance, cell-type-specific tasks such as zero-shot cell-type prediction may benefit from masking gene programmes associated with cell types. Tasks prioritizing cell–cell interactions over subcellular resolutions may prefer contrastive methods, such as Barlow twins, which also showed strong zero-shot performance. These recommendations provide a strategic framework for applying SSL methods in SCG, ensuring researchers can select the most appropriate SSL method.
Future work on SSL in SCG may follow up on our findings. First, we identified several scenarios where SSL can improve performance across downstream tasks. These scenarios can serve as a baseline for future work, such as adding further downstream applications or developing SSL methods. Second, the remarkable performance improvement through SSL pre-training on auxiliary data promises further applications in which the data or data modality are scarce. Our solution can potentially improve analysis performance in applications such as dynamics modelling, where datasets with temporal resolutions are limited in size and availability, or in smaller applications with very small datasets. Third, the findings of this work are in the context of fully connected neural networks. Some conclusions may not generalize to other architectures, such as transformers. Extending the investigation to another base architecture is an interesting direction. Still, practitioners might consider pre-training their chosen model with SSL on auxiliary data, in particular masked autoencoders, as our benchmark suggests. Models natively equipped with self-supervised pre-training on auxiliary datasets, such as scGPT35 or Nicheformer37, can be a good starting point for practical purposes.
Finally, our study clarifies the scenarios in which SSL pre-training can improve performance in SCG. Namely, SSL excels in transfer learning tasks by leveraging auxiliary data and distributional shift scenarios. In the context of foundation models, we illuminate methodological innovations stemming from the SSL pre-training. For the broader computational biology community, we have shown that self-supervised pre-training on atlas-level data can help to improve performance on smaller datasets of biological or medical relevance that are commonly more difficult to scale.
Methods
Data curation
Preprocessing
All datasets used in this study underwent a commonly used preprocessing pipeline in SCG. This involved normalization to 10,000 counts per cell and log1p transformation to mitigate technical variations and facilitate more meaningful biological comparisons. This uniform preprocessing approach ensured that our models were trained and evaluated on data closely reflecting the underlying biological realities while minimizing technical noise.
scTab dataset
The core dataset for our study stems from scTab5 and is derived from the CELLxGENE44 census version 2023-05-15, a long-term supported release hosted by CELLxGENE. This dataset represents a substantial collection of human single-cell RNA-sequencing data, encompassing 22.2 million cells spanning 164 unique cell types, 5,052 unique donors and 56 different tissues. To ensure the reproducibility of dataset creation, scTab applied stringent criteria for inclusion, focusing on primary data from 10x-based sequencing protocols and ensuring a broad representation across cell types and donors. The scTab data are divided into training, validation and test sets based on donors, avoiding label leakage and ensuring each set contains unique donors. This donor-based splitting approach allowed us to maintain a proportional representation of cells across the sets. It ensured that each cell type was represented in the training and testing phases. It further presented a challenging test split with unseen donors. The final split resulted in 15.2 million cells for training, 3.5 million for validation and 3.4 million for testing.
Single-cell atlases
We further considered smaller, focused datasets to test whether access to the auxiliary data gives an advantage. These datasets are subsets of the CELLxGENE44 census of scTab5 (scTab dataset), tailored to specific applications, and consist of the Human Lung Cell Atlas (HLCA)4 (available at cellxgene.cziscience.com/e/9f222629-9e39-47d0-b83f-e08d610c7479.cxg; 775,790 cells after filtering, 51 cell types, 540,732 training, 117,541 validation, 117,517 test samples), peripheral blood mononuclear cells (PBMCs) after SARS-CoV-2 infection48 (available at cellxgene.cziscience.com/e/2a498ace-872a-4935-984b-1afa70fd9886.cxg; 78,354 cells after filtering, 30 cell types, 78,354 training, 33,761 validation, 189,756 test samples), and the Tabula Sapiens Atlas (available at cellxgene.cziscience.com/e/53d208b0-2cfd-4366-9866-c3c6114081bc.cxg; 335,861 cells after filtering, 161 cell types, 223,337 training, 54,908 validation, 57,616 test samples)49. The division into training, validation and test sets is derived from their allocation within the scTab dataset to prevent data leakage. Note that the training, validation and test sets of the PBMC, Tabula Sapiens and HLCA datasets are also part of the corresponding splits of the full scTab dataset.
Unseen datasets
To evaluate our models’ performance in unseen data analysis scenarios, we incorporated five unseen datasets published after the CELLxGENE census version of scTab: (1) all non-neuronal cells from the Human Brain Atlas52 (available at cellxgene.cziscience.com/e/b165f033-9dec-468a-9248-802fc6902a74.cxg) (2) dissection, tail of hippocampus (HiT) - caudal hippocampus - CA4-DGC from the Human Brain Atlas52 (available at cellxgene.cziscience.com/e/9f499d32-400d-4c42-ac9a-fb1481844fee.cxg), (3) the single-cell analysis of prenatal and postnatal human cortical development53 (available at cellxgene.cziscience.com/e/1a38e762-2465-418f-b81c-6a4bce261c34.cxg), (4) circulating immune cells—CV19 infection, vaccination and HC54 (available at cellxgene.cziscience.com/e/242c6e7f-9016-4048-af70-d631f5eea188.cxg), and (v) human, great apes study55 (available at cellxgene.cziscience.com/e/2bdd3a2c-2ff4-4314-adf3-8a06b797a33a.cxg). The unseen datasets were filtered for the genes used in scTab; missing genes were zero-padded. The datasets were then normalized to 10,000 counts per cell and log1p transformed. The full datasets were used as the test split, that is, no samples were used for training.
NeurIPS multiome dataset
Our study included the NeurIPS multiome dataset57, a multimodal bone marrow dataset that integrates gene-expression counts with proteomics data. While distinct in its multi-omic nature, this dataset underwent similar preprocessing steps to our other datasets, ensuring consistency across all analyses. We split the dataset into training, validation and test sets using an 80/10/10 random split. We chose 2,000 highly variable genes using Scanpy64 as a standard preprocessing step for this dataset.
Self-supervision methods
Overview
SSL is the concept that data, along with their inherent pairwise relationships, are sufficient for learning meaningful data representations, even in the absence of explicit labels. While supervised learning relies on paired observations and labels (X, Y), SSL thus depends on only the input X and an inter-sample relationship (X, G), where G is constructed through a data augmentation that sustains the semantic information of X8. Thereby, the method distils signal from noise65, a crucial aspect for managing challenges such as class imbalances in large, real-world datasets66. In single-cell data, this means distiling the signal of the cellular omics and removing noise sources such as batch effects or inconsistent labelling.
In the context of SCG, SSL harnesses these capabilities to navigate the complexities of vast, unlabelled datasets replete with intricate biological interdependencies. The framework is structured into two distinct phases: pre-training and fine-tuning. During the pre-training phase, the model employs contrastive learning or denoising methods to learn a data representation. This representation, characterized by its broad applicability, is then utilized in one of two ways. First, as a zero-shot SSL model, it can be directly applied to a downstream task without further label-dependent training. Alternatively, as an SSL model, it undergoes fine-tuning to enhance performance on specific tasks. This fine-tuning capitalizes on the rich data representation acquired during pre-training, adjusting and optimizing it for the desired application. The fine-tuning phase of SSL, therefore, is not only about refining the pre-training but also about strategically leveraging the pre-established data mappings for task-specific optimizations.
Core principles and strategies
The choice of self-supervised pre-training, that is, learning the inter-sample relationship, is critical to obtaining a meaningful data representation as it gives rise to the signal-to-noise distinction in the dataset. Our SSL framework is designed around two primary pre-training strategies: masked autoencoders and contrastive learning, both adapted to meet the unique demands of SCG.
Masked autoencoders
This approach follows the concept of self-prediction, where a significant portion of input features (genes in SCG) are masked (that is, set to zero), and the model is trained to reconstruct these missing parts9,45,67. It thus sets focus on inter-feature dependencies. We implemented various masking strategies. (1) In random masking, 50% of genes are randomly chosen and masked with different choices in each iteration. (2) In GP masking, sets of genes known for biological functions are masked such that n% of genes are masked and reconstructed. The C8 cell-type signature gene sets from the Human MSigDB Collections68,69,70 were used. Next, we introduce isolated masked autoencoders, in which all genes but a defined set are masked, and only this set is reconstructed. (3) For this, we present a GP to TF isolated masking. This masking predicts the expression value of the transcription factor known to correspond to a gene programme. This connection is given in the TFT transcription factor targets subset of C3 regulatory target gene sets from the Human MSigDB Collections71,72. (4) Last, we present a GP to GP isolated masking. In this strategy, a gene programme is kept unmasked and used to predict only itself. The gene programmes for this strategy also stem from the C8 cell-type signature gene sets from the Human MSigDB Collections. These strategies are tailored to capture specific gene interactions and relationships, making them particularly suited for the intricate nature of single-cell data.
Contrastive learning
Unlike self-prediction, contrastive learning focuses on understanding relationships between different samples, thus focusing on inter-sample dependencies. This method minimizes distances between similar samples and maximizes distances between dissimilar ones in the embedded space. Contrastive methods are typically distinguished by their strategy to avoid representation collapse, the trivial solution to contrastive losses of constant representations9,10. BYOL is an example of architectural regularization through its teacher–student network. Barlow twins is an example of an information maximization method that avoids collapse by maximizing the information content of the embedding. We incorporated BYOL and Barlow twins in our framework to benchmark two schools of thought. We used a combination of negative binomial noise and masking as data augmentation, simulating the expected noise profiles in SCG data.
Zero-shot SSL concept
A key concept in our study is the differentiation between the zero-shot SSL and SSL models. The zero-shot SSL model represents the initial phase of pre-training, where the model learns from the data without any label guidance through self-supervision algorithms. This model, even without fine-tuning, can provide meaningful insights into data, as demonstrated in various downstream tasks. The SSL model, in contrast, undergoes an additional fine-tuning phase tailored to specific downstream applications. This distinction allows us to explore the full spectrum of SSL’s capabilities, from a generalized understanding of data to specialized, task-specific optimizations.
In summary, our self-supervision methods in SCG are defined by a nuanced application of masked autoencoders and contrastive learning adapted to the field’s specific challenges. The zero-shot SSL concept plays a central role in our approach, highlighting the potential of SSL to derive meaningful insights from large-scale, unlabelled datasets. This methodological framework sets the stage for a detailed exploration and benchmarking of SSL’s impact on various SCG tasks, as detailed in the following sections of our study.
Downstream applications in SCG
Cell-type annotation
Cell-type annotation in SCG is a classification task where data samples, represented as vectors of RNA-sequencing counts, are assigned to distinct cellular identities. Although seemingly straightforward, this task is complicated by the noise and heterogeneity inherent in large-scale datasets. We utilize the scTab dataset as the primary basis for our cell-type annotation analysis. We employ various SSL methods and compare their effectiveness against supervised approaches. We train the classifier using a cross-entropy loss. We evaluate cell-type annotation performance by kNN (k = 5) classification using the scTab validation set as neighbours of the test sample. The validation set is sufficiently large and diverse, making it a simple and scalable alternative to the training set for this purpose. This choice is driven to have the same evaluation, including for the zero-shot SSL model that does not have a prediction head. Our evaluation metrics focus on the macro F1 score, reflecting the models’ ability to handle class imbalances, supplemented by the micro F1 score, offering an additional comparative perspective to class imbalances. Exemplary loss curves for this training are shown in Supplementary Fig. 5 and a list of hyperparameters is shown in Supplementary Table 1.
Gene-expression reconstruction
Gene-expression reconstruction, the process of reconstructing counts from the transcriptome, still presents challenges due to the inherent noise and dispersion in RNA-sequencing data. The popular scVI model38 inspires our approach and diverges in its use of input data. While scVI uses raw counts as input and models them as a negative binomial distribution, our method employs normalized data for consistency with other downstream tasks. Nonetheless, similar to scVI, we predict the parameters of the negative binomial distribution. This strategy of modelling distribution parameters rather than direct RNA-sequencing count prediction enhanced reconstruction accuracy in our experiments. We opt for a non-variational, fully connected autoencoder framework consistent with our cell-type prediction approach. Performance evaluation encompasses MSE and uniform and weighted explained variance. We reported the weighted explained variance to best reflect the actual reconstruction efficacy, accounting for class imbalances. We include the MSE and uniform explained variance in our framework as supplementary evaluation, and they were used in our experiments. The hyperparameters used are shown in Supplementary Table 1.
Cross-modality prediction
Cross-modality prediction is the task of predicting one modality from another. Such a task could potentially augment cellular data by a different modality, offering another perspective. For pre-training, we used masking (1) on the auxiliary scTab dataset and (2) on the downstream task dataset. For fine-tuning, we included two studies, both using normalized and log1p transformed RNA-sequencing counts as originating modalities. First, we predicted all 134 normalized and log1p transformed protein counts (proteomics) available in the NeurIPS CITE-seq dataset57. We trained the models in a random training, validation and test split using coupled RNA and proteomics counts. Second, we predicted all 116,490 TF-IDF (term frequency-inverse document frequency)73-normalized ATAC counts available in the NeurIPS multiome dataset57. Again, we trained the models in a random training, validation and test split using the coupled RNA and ATAC counts. Hyperparameters are shown in Supplementary Table 1.
Data integration
Data integration is an effort to study a set of related SCG datasets, possibly curated from various donors with different pipelines and in different settings that create batch effects and technical artefacts. The scIB63 integration benchmarking is a well-established analysis to determine how well the relevant and meaningful biological signals are preserved in any model data representation while removing the unwanted batch effects resulting in a mixed representation of various datasets. Accordingly, the scIB pipeline measures two metrics, including the bio conservation and batch correction metrics, each consisting of several evaluations through different methods. The hyperparameters for data integration are shown in Supplementary Table 1.
Contrastive method choice
This benchmark developed contrastive methods based on BYOL and Barlow twins, two well-performing negative-pair-free methods. This choice is motivated by their reliance solely on data augmentations rather than sampling negative pairs in a large and heterogeneous dataset and their proven performance46,47. Other reasonable choices include simple Siamese networks74, which were excluded due to repeatedly observed training instability in our setting, and SimCLR12, which was not pursued further as BYOL and Barlow twins showed superior performance in previous benchmarks. While VICReg11 is promising by design, we focused on BYOL and Barlow twins due to their robustness. As contrastive learning methods generally performed worse than masking approaches, we prioritized them for thorough investigation.
Batch effect
Batch effects were not explicitly corrected when working with large datasets, such as scTab, covering 249 datasets. Including many datasets seems to reduce the relative impact of such effects on the overall variation. When working with fewer datasets, such as in the data integration experiments covering three datasets, a batch covariate needs to be included to avoid strong batch effects.
Computational resources
The experiments for this work were conducted on a graphics processing unit (GPU) server with the following specifications:
-
GPU: 16x Tesla V100 GPUs with 32 GB random access memory (RAM) per card
-
GPU: 2x Tesla V100 GPUs with 16 GB RAM per card
-
GPU: 8x A100-SXM4 GPUs with 40 GB RAM per card
All pre-training methods were trained on a single GPU for 2 days with early stopping, using up to 160 GB of system memory at a batch size of 8,192. For practitioners with limited GPU memory, smaller batch sizes can reduce memory usage. For example, a batch size of 2 uses under 1 GB of VRAM but greatly increases training time (>200 h per epoch on scTab). All fine-tuning methods were trained on a single GPU for 1 day with early stopping. All models were checked for convergence in the validation metrics.
Terminology
In this paper, we use the following terminology:
We use the above table’s terminology throughout the paper to distinguish between architecture, method and model. This distinction clarifies how different methods impact models that share similar architectures. For example, scGPT trains a transformer architecture using SSL.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scTab data are available with instructions in the corresponding publication5. The smaller datasets are publicly available on CELLxGENE44 and subsets of the scTab datasets (HLCA, Dataset ID 148; PBMC, Dataset ID 87; Tabula Sapiens, Dataset ID 41). The unseen datasets are sourced from CELLxGENE44 with instructions in the corresponding publications52,53,54,55. The NeurIPS multiome dataset is publicly available from NCBI GEO under accession GSE194122 with instructions in the corresponding publication57.
Code availability
The code is available at github.com/theislab/ssl_in_scg and on Zenodo at https://doi.org/10.5281/zenodo.13358872 (ref. 75). A lean version for masked pre-training on adatas fitting into memory is available at github.com/theislab/sc_mae.
References
Angerer, P. et al. Single cells make big data: new challenges and opportunities in transcriptomics. Curr. Opin. Syst. Biol. 4, 85–91 (2017).
Lotfollahi, M. et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 40, 121–130 (2022).
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
Sikkema, L. et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 29, 1563–1577 (2023).
Fischer, F. et al. scTab: scaling cross-tissue single-cell annotation models. Nat. Commun. 15, 6611 (2024).
Consens, M. E. et al. To transformers and beyond: large language models for the genome. Preprint at https://arxiv.org/abs/2311.07621 (2023).
Boiarsky, R., Singh, N., Buendia, A., Getz, G. & Sontag, D. A deep dive into single-cell RNA sequencing foundation models. Preprint at bioRxiv https://doi.org/10.1101/2023.10.19.563100 (2023).
Balestriero, R. et al. Contrastive and non-contrastive self-supervised learning recover global and local spectral embedding methods. Adv. Neural Inf. Process. Syst. 35, 26671–26685 (2022).
Weng, L. et al. Self-supervised learning: self-prediction and contrastive learning. Adv. Neural Inf. Process. Syst. https://nips.cc/media/neurips-2021/Slides/21895.pdf (2021).
Uelwer, T. et al. A survey on self-supervised representation learning. Preprint at https://arxiv.org/abs/2308.11455 (2023).
Bardes, A. et al. Y. VICReg: Variance-Invariance-Covariance regularization for self-supervised learning. Int. Conf. Learn. Represent. https://openreview.net/forum?id=xm6YD62D1Ub (2022).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In Proc. 37th International Conference on Machine Learning Vol. 119 (eds Iii, H. D. & Singh, A.) 1597–1607 (PMLR, 2020).
Radford, A., Narasimhan, K., Salimans, T. & Sutskever, I. Improving language understanding by generative pre-training (2018); https://cdn.openai.com/research-covers/language-unsupervised/language_understanding_paper.pdf
Devlin, J. et al. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2019).
Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at https://arxiv.org/abs/2108.07258 (2021).
Yang, M. et al. Contrastive learning enables rapid mapping to multimodal single-cell atlas of multimillion scale. Nat. Mach. Intell. 4, 696–709 (2022).
Xiong, Z. et al. scGCL: an imputation method for scRNA-seq data based on graph contrastive learning. Bioinformatics 39, btad098 (2023).
Yan, X., Zheng, R., Wu, F. & Li, M. CLAIRE: contrastive learning-based batch correction framework for better balance between batch mixing and preservation of cellular heterogeneity. Bioinformatics 39, btad099 (2023).
Chen, L., Zhai, Y., He, Q., Wang, W. & Deng, M. Integrating deep supervised, self-supervised and unsupervised learning for single-cell RNA-seq clustering and annotation. Genes 11, 792 (2020).
Zhang, R., Luo, Y., Ma, J., Zhang, M. & Wang, S. scPretrain: multi-task self-supervised learning for cell-type classification. Bioinformatics 38, 1607–1614 (2022).
Shen, H. et al. Miscell: an efficient self-supervised learning approach for dissecting single-cell transcriptome. iScience 24, 103200 (2021).
Wan, H., Chen, L. & Deng, M. scNAME: neighborhood contrastive clustering with ancillary mask estimation for scRNA-seq data. Bioinformatics 38, 1575–1583 (2022).
Ciortan, M. & Defrance, M. Contrastive self-supervised clustering of scRNA-seq data. BMC Bioinform. 22, 280 (2021).
Han, W. et al. Self-supervised contrastive learning for integrative single cell RNA-seq data analysis. Brief. Bioinform. 23, bbac377 (2022).
Du, L., Han, R., Liu, B., Wang, Y. & Li, J. ScCCL: single-cell data clustering based on self-supervised contrastive learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 2233–2241 (2023).
Peng, W. et al. Multi-network graph contrastive learning for cancer driver gene identification. IEEE Trans. Netw. Sci. Eng. 11, 3430–3440 (2024).
Zhang, W., Jiang, R., Chen, S. & Wang, Y. scIBD: a self-supervised iterative-optimizing model for boosting the detection of heterotypic doublets in single-cell chromatin accessibility data. Genome Biol. 24, 225 (2023).
Vime: extending the success of self-and semi-supervised learning to tabular domain. https://proceedings.neurips.cc/paper/2020/hash/7d97667a3e056acab9aaf653807b4a03-Abstract.html
Lee, C. et al. Self-supervision enhanced feature selection with correlated gates. In Proc. 10th International Conference on Learning Representations https://openreview.net/forum?id=oDFvtxzPOx (OpenReview.net, 2022).
Geuenich, M. J., Gong, D.-W. & Campbell, K. R. The impacts of active and self-supervised learning on efficient annotation of single-cell expression data. Nat. Commun. 15, 1014 (2024).
Richter, T. et al. SpatialSSL: whole-brain spatial transcriptomics in the mouse brain with self-supervised learning. (2023).
Chen, J. et al. Transformer for one stop interpretable cell type annotation. Nat. Commun. 14, 223 (2023).
Tang, W. et al. Single-cell multimodal prediction via transformers. In Proc. 32nd ACM International Conference on Information and Knowledge Management 2422–2431 (CIKM, 2023).
Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023).
Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 21, 1470–1480 (2024).
Yang, F. et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 4, 852–866 (2022).
Schaar, A. C. et al. Nicheformer: a foundation model for single-cell and spatial omics. Preprint at bioRxiv https://doi.org/10.1101/2024.04.15.589472 (2024).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Lotfollahi, M. et al. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
Goldblum, M. et al. Battle of the backbones: a large-scale comparison of pretrained models across computer vision tasks. In Proc. 37th Conference on Neural Information Processing Systems, Datasets and Benchmarks Track https://openreview.net/forum?id=1yOnfDpkVe (NeurIPS, 2023).
Smith, S. L., Brock, A., Berrada, L. & De, S. ConvNets match vision transformers at scale. Preprint at https://arxiv.org/abs/2310.19909 (2023).
Radford, A. et al. Robust speech recognition via large-scale weak supervision. In Proc. 40th International Conference on Machine Learning Vol. 202 (eds Krause, A. et al.) 28492–28518 (PMLR, 2023).
Dann, E. et al. Precise identification of cell states altered in disease using healthy single-cell references. Nat. Genet. 55, 1998–2008 (2023).
CZI Single-Cell Biology Program et al. CZ CELL×GENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Preprint at bioRxiv https://doi.org/10.1101/2023.10.30.563174 (2023).
He, K. et al. Masked autoencoders are scalable vision learners. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition 15979–15988 (IEEE, 2022).
Grill, J.-B. et al. Bootstrap your own latent—a new approach to self-supervised learning. In Advances in Neural Information Processing Systems 21271–21284 (Curran Associates, 2020).
Zbontar, J., Jing, L., Misra, I., LeCun, Y. & Deny, S. Barlow twins: self-supervised learning via redundancy reduction. in Proc. 38th International Conference on Machine Learning 12310–12320 (PMLR, 2021).
Yoshida, M. et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 602, 321–327 (2022).
Tabula Sapiens Consortium et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Fleck, J. S., Camp, J. G. & Treutlein, B. What is a cell type? Science 381, 733–734 (2023).
Heimberg, G. et al. Scalable querying of human cell atlases via a foundational model reveals commonalities across fibrosis-associated macrophages. Preprint at bioRxiv https://doi.org/10.1101/2023.07.18.549537 (2023).
Siletti, K. et al. Transcriptomic diversity of cell types across the adult human brain. Science 382, eadd7046 (2023).
Velmeshev, D. et al. Single-cell analysis of prenatal and postnatal human cortical development. Science 382, eadf0834 (2023).
Ivanova, E. et al. mRNA COVID-19 vaccine elicits potent adaptive immune response without the acute inflammation of SARS-CoV-2 infection. iScience 26, 108572 (2023).
Jorstad, N. L. et al. Comparative transcriptomics reveals human-specific cortical features. Science 382, eade9516 (2023).
Heumos, L. et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 24, 550–572 (2023).
Luecken, M. et al. A sandbox for prediction and integration of DNA, RNA, and proteins in single cells. In Proc. 35th Conference on Neural Information Processing Systems Track on Datasets and Benchmarks (eds Vanschoren, J. & Yeung, S.) https://datasets-benchmarks-proceedings.neurips.cc/paper_files/paper/2021/file/158f3069a435b314a80bdcb024f8e422-Paper-round2.pdf (2021).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Wang, A. et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. eLife 9, e62522 (2020).
Melms, J. C. et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018).
von Kügelgen, J. et al. Self-supervised learning with data augmentations provably isolates content from style. In Advances in Neural Information Processing Systems 16451–16467 (Curran Associates, 2021).
Liu, H., et al. Self-supervised learning is more robust to dataset imbalance. In NeurIPS 2021 Workshop on Distribution Shifts: Connecting Methods and Applications https://openreview.net/forum?id=vUz4JPRLpGx (2021).
Cao, S., Xu, P. & Clifton, D. A. How to understand masked autoencoders. Preprint at https://arxiv.org/abs/2202.03670 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Kolmykov, S. et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 49, D104–D111 (2021).
Xie, X. et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434, 338–345 (2005).
Bredikhin, D., Kats, I. & Stegle, O. MUON: multimodal omics analysis framework. Genome Biol. 23, 42 (2022).
Chen, X. & He, K. Exploring simple Siamese representation learning. Proc. IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit. 15745, 15753 (2020).
Richter, T. & Bahrami, M. Theislab/ssl_in_scg: first release. Zenodo https://doi.org/10.5281/zenodo.13358873 (2024).
Acknowledgements
We thank F. Fischer for his valuable assistance with scTab and his constructive comments, which improved our work’s narrative. For the cross-modality prediction task, we thank A. Litinetskaya for her valuable feedback. We also thank M. Stahl, X. and A. Chernysheva for their contributions during their master practical course, which laid the groundwork for the multiomics task (together with Y. Xia, who continued afterwards). We are particularly grateful to A. Palma for his feedback on the paper’s storyline and to A. Palma, A. Szałata and E. Roellin for their valuable input on the paper, greatly enhancing its quality. We thank F. Curion for her input, sparking our exploration of isolated masked autoencoders, and her feedback on the multiomics application. T.R. and M.B. are supported by the Helmholtz Association under the joint research school ‘Munich School For Data Science’. T.R. and F.J.T. acknowledge support by the Helmholtz Association’s Initiative and Networking Fund through CausalCellDynamics (grant number Interlabs-0029), F.J.T. acknowledges support by the European Union (ERC, DeepCell - 101054957). Views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The language of this paper was refined using ChatGPT by OpenAI and Grammarly by Grammarly Inc.
Funding
Open access funding provided by Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH).
Author information
Authors and Affiliations
Contributions
T.R., D.S.F. and F.J.T. conceptualized the project. T.R. led pilot analyses, method development and implementation. T.R. and F.J.T. outlined the downstream analyses. T.R. performed the cell-type prediction and gene-expression reconstruction studies. T.R. and Y.X. undertook the cross-modality prediction analysis, and M.B. performed the data integration study. The paper was written by T.R., M.B., D.S.F. and F.J.T., with all authors contributing to discussions and providing comments on the paper.
Corresponding author
Ethics declarations
Competing interests
F.J.T. consults for Immunai, CytoReason, Cellarity and Omniscope and has an ownership interest in Dermagnostix GmbH and Cellarity. The other authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Qi Liu, Qing Nie and Zhiyuan Yuan for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–6 and Table 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Richter, T., Bahrami, M., Xia, Y. et al. Delineating the effective use of self-supervised learning in single-cell genomics. Nat Mach Intell 7, 68–78 (2025). https://doi.org/10.1038/s42256-024-00934-3
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s42256-024-00934-3
