
Abstract
With the fast-paced development of artificial intelligence, large language models are increasingly used to tackle various scientific challenges. A critical step in this process is converting domain-specific data into a sequence of tokens for language modelling. In chemistry, molecules are often represented by molecular linear notations, and chemical reactions are depicted as sequence pairs of reactants and products. However, this approach does not capture atomic and bond changes during reactions. Here, we present ReactSeq, a reaction description language that defines molecular editing operations for step-by-step chemical transformation. Based on ReactSeq, language models for retrosynthesis prediction may consistently excel in all benchmark tests, and demonstrate promising emergent abilities in the human-in-the-loop and explainable artificial intelligence. Moreover, ReactSeq has allowed us to obtain universal and reliable representations of chemical reactions, which enable navigation of the reaction space and aid in the recommendation of experimental procedures and prediction of reaction yields. We foresee that ReactSeq can serve as a bridge to narrow the gap between chemistry and artificial intelligence.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
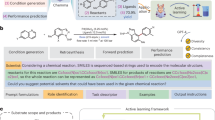

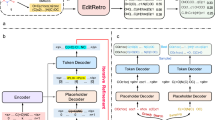
Data availability
All datasets used in this work are available at https://github.com/jiachengxiong/ReactSeq, https://drive.google.com/drive/folders/1a6NL5apcP_7isY3HccLjkSsjJGwp_FwD and via Zenodo at https://doi.org/10.5281/zenodo.13338263 (ref. 51). Source data are provided with this paper.
Code availability
All code for the generating and transforming of ReactSeq, as well as the code for model training and inference, is available at https://github.com/jiachengxiong/ReactSeq and via Zenodo at https://doi.org/10.5281/zenodo.13338263 (ref. 51). The webpage for our prompt learning model is available at https://huggingface.co/spaces/Oopstom/ReactSeq.
References
Thirunavukarasu, A. J. et al. Large language models in medicine. Nat. Med. 29, 1930–1940 (2023).
Boiko, D. A., MacKnight, R., Kline, B. & Gomes, G. Autonomous chemical research with large language models. Nature 624, 570–578 (2023).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023).
Grisoni, F. Chemical language models for de novo drug design: challenges and opportunities. Curr. Opin. Struct. Biol. 79, 102527 (2023).
Skinnider, M. A., Stacey, R. G., Wishart, D. S. & Foster, L. J. Chemical language models enable navigation in sparsely populated chemical space. Nat. Mach. Intell. 3, 759–770 (2021).
Moret, M. et al. Leveraging molecular structure and bioactivity with chemical language models for de novo drug design. Nat. Commun. 14, 114 (2023).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Krenn, M., Häse, F., Nigam, A., Friederich, P. & Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): a 100% robust molecular string representation. Mach. Learn. Sci. Technol. 1, 045024 (2020).
Kuenneth, C. & Ramprasad, R. polyBERT: a chemical language model to enable fully machine-driven ultrafast polymer informatics. Nat. Commun. 14, 4099 (2023).
Krenn, M. et al. SELFIES and the future of molecular string representations. Patterns 3, 100588 (2022).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019).
Liu, B. et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 3, 1103–1113 (2017).
Sun, Y. & Sahinidis, N. V. Computer-aided retrosynthetic design: fundamentals, tools, and outlook. Curr. Opin. Chem. Eng. 35, 100721 (2022).
Wang, X. et al. RetroPrime: a diverse, plausible and transformer-based method for single-step retrosynthesis predictions. Chem. Eng. J. 420, 129845 (2021).
Thakkar, A. et al. Unbiasing retrosynthesis language models with disconnection prompts. ACS Cent. Sci. 9, 1488–1498 (2023).
Huang, T. & Li, Y. Current progress, challenges, and future perspectives of language models for protein representation and protein design. The Innovation 4, 100446 (2023).
Min, B. et al. Recent advances in natural language processing via large pre-trained language models: a survey. ACM Comput. Surv. 56, 1–40 (2023).
Schwaller, P., Hoover, B., Reymond, J.-L., Strobelt, H. & Laino, T. Extraction of organic chemistry grammar from unsupervised learning of chemical reactions. Sci. Adv. 7, eabe4166 (2021).
Schwaller, P. et al. Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach. Intell. 3, 144–152 (2021).
Strieth-Kalthoff, F. et al. Artificial intelligence for retrosynthetic planning needs both data and expert knowledge. J. Am. Chem. Soc. 146, 11005–11017 (2024).
Nugmanov, R. I. et al. CGRtools: Python library for molecule, reaction, and condensed graph of reaction processing. J. Chem. Inf. Model. 59, 2516–2521 (2019).
Shi, C., Xu, M., Guo, H., Zhang, M. & Tang, J. A graph to graphs framework for retrosynthesis prediction. In Proc. 37th International Conference on Machine Learning (eds Blei, D. et al.) 8818–8827 (PMLR, 2020).
Yan, C. et al. Retroxpert: decompose retrosynthesis prediction like a chemist. Adv. Neural Inf. Process. Syst. 33, 11248–11258 (2020).
Somnath, V. R., Bunne, C., Coley, C., Krause, A. & Barzilay, R. Learning graph models for retrosynthesis prediction. Adv. Neural Inf. Process. Syst. 34, 9405–9415 (2021).
Zhong, W., Yang, Z. & Chen, C. Y.-C. Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing. Nat. Commun. 14, 3009 (2023).
Wang, Y. et al. Retrosynthesis prediction with an interpretable deep-learning framework based on molecular assembly tasks. Nat. Commun. 14, 6155 (2023).
Saebi, M. et al. On the use of real-world datasets for reaction yield prediction. Chem. Sci. 14, 4997–5005 (2023).
Lu, J. & Zhang, Y. Unified deep learning model for multitask reaction predictions with explanation. J. Chem. Inf. Model. 62, 1376–1387 (2022).
Wan, Y., Hsieh, C.-Y., Liao, B. & Zhang, S. Retroformer: pushing the limits of end-to-end retrosynthesis transformer. In Proc. 39th International Conference on Machine Learning (eds Chaudhuri, K. et al.) 22475–22490 (PMLR, 2022).
Dong, J. et al. Ketones and aldehydes as alkyl radical equivalents for C-H functionalization of heteroarenes. Sci. Adv. 5, eaax9955 (2019).
Peltzer, R. M., Gauss, J., Eisenstein, O. & Cascella, M. The Grignard reaction–unraveling a chemical puzzle. J. Am. Chem. Soc. 142, 2984–2994 (2020).
Heravi, M. M., Hashemi, E. & Nazari, N. Negishi coupling: an easy progress for C–C bond construction in total synthesis. Mol. Divers. 18, 441–472 (2014).
Kotha, S., Lahiri, K. & Kashinath, D. Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 58, 9633–9695 (2002).
Zhou, J., Zhao, Z. & Shibata, N. Transition-metal-free silylboronate-mediated cross-couplings of organic fluorides with amines. Nat. Commun. 14, 1847 (2023).
Vulovic, B., Cinderella, A. P. & Watson, D. A. Palladium-catalyzed cross-coupling of monochlorosilanes and Grignard reagents. ACS Catal. 7, 8113–8117 (2017).
Xu, W. Q., Xu, X. H. & Qing, F. L. Synthesis and properties of CF3 (OCF3) CH‐substituted arenes and alkenes. Chin. J. Chem. 38, 847–854 (2020).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Probst, D., Schwaller, P. & Reymond, J.-L. Reaction classification and yield prediction using the differential reaction fingerprint DRFP. Digit. Discov. 1, 91–97 (2022).
Schneider, N., Lowe, D. M., Sayle, R. A. & Landrum, G. A. Development of a novel fingerprint for chemical reactions and its application to large-scale reaction classification and similarity. J. Chem. Inf. Model. 55, 39–53 (2015).
Kajino, M., Hasuoka, A. & Nishida, H. 1-heterocyclylsulfonyl, 2-aminomethyl, 5- (hetero-) aryl substituted 1-H-pyrrole derivatives as acid secretion inhibitors. Patent WO2007026916A1 (2007).
Yu, Q.-Y., Zeng, H., Yao, K., Li, J.-Q. & Liu, Y. Novel and practical synthesis of vonoprazan fumarate. Synth. Commun. 47, 1169–1174 (2017).
Chen, S. & Jung, Y. Deep retrosynthetic reaction prediction using local reactivity and global attention. JACS Au. 1, 1612–1620 (2021).
Tetko, I. V., Karpov, P., Van Deursen, R. & Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 11, 5575 (2020).
Zhong, Z. et al. Root-aligned SMILES: a tight representation for chemical reaction prediction. Chem. Sci. 13, 9023–9034 (2022).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. J. Chemformer: a pre-trained transformer for computational chemistry. Mach. Learn. Sci. Technol. 3, 015022 (2022).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2024).
Tingle, B. I. et al. ZINC-22—a free multi-billion-scale database of tangible compounds for ligand discovery. J. Chem. Inf. Model. 63, 1166–1176 (2023).
Chilingaryan, G. et al. BartSmiles: generative masked language models for molecular representations. J. Chem. Inf. Model. 64, 5832–5843 (2024).
zw-SIMM & Xiong, J. jiachengxiong/ReactSeq: ReactSeq (1.0). Zenodo https://doi.org/10.5281/zenodo.13338263 (2024).
Coley, C. W., Rogers, L., Green, W. H. & Jensen, K. F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 3, 1237–1245 (2017).
Segler, M. H. & Waller, M. P. Neural‐symbolic machine learning for retrosynthesis and reaction prediction. Chem-Eur. J. 23, 5966–5971 (2017).
Dai, H., Li, C., Coley, C., Dai, B. & Song, L. Retrosynthesis prediction with conditional graph logic network. Adv. Neural Inf. Process. Syst. 32, 8872–8882 (2019).
Sacha, M. et al. Molecule edit graph attention network: modeling chemical reactions as sequences of graph edits. J. Chem. Inf. Model. 61, 3273–3284 (2021).
Chen, Z., Ayinde, O. R., Fuchs, J. R., Sun, H. & Ning, X. G2Retro as a two-step graph generative models for retrosynthesis prediction. Commun. Chem. 6, 102 (2023).
Yao, L. et al. Node-aligned graph-to-graph: elevating template-free deep learning approaches in single-step retrosynthesis. JACS Au. 4, 992–1003 (2024).
Liu, X. et al. RetroCaptioner: beyond attention in end-to-end retrosynthesis transformer via contrastively captioned learnable graph representation. Bioinformatics 40, btae561 (2024).
Zheng, S., Rao, J., Zhang, Z., Xu, J. & Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. J. Chem. Inf. Model. 60, 47–55 (2019).
Sun, R., Dai, H., Li, L., Kearnes, S. & Dai, B. Towards understanding retrosynthesis by energy-based models. Adv. Neural Inf. Process. Syst. 34, 10186–10194 (2021).
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (grant nos. T2225002 and 82273855, M.Z.), the National Key Research and Development Program of China (grant nos. 2022YFC3400504 and 2023YFC2305904, M.Z.), the Strategic Priority Research Program of the Chinese Academy of sciences (grant no. XDB0830200, M.Z.), the open fund of state key laboratory of Pharmaceutical Biotechnology, Nanjing University, China (grant no. KF-202301, M.Z.) and Shanghai Post-doctoral Excellence Program (grant no. 2024707, J.X.).
Author information
Authors and Affiliations
Contributions
J.X. proposed the idea, conducted computational experiments and drafted the initial paper together with W.Z. Yinquan W., J.H., W.Z., M.X. and M.L. carried out the synthetic experiments. Y.S. developed the web application. Z.F. and J.H. contributed to the case analysis. Z.F., X.K. and M.Z. helped check and improve the paper. Yitian W. and Z.X. participated in the analysis of results. M.Z. led the project and designed the study. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Jannis Born, Xiangliang Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Multistep retrosynthesis predictions by our model for (a) Vonoprazan, (b) Mitapivat, (c) Daridorexant.
The reaction centers and leaving groups are highlighted in different colors at different reaction steps.
Supplementary information
Supplementary Information
Supplementary Figs 1–26, Tables 1–12, Additional Results and Discussion, and Methods.
Source data
Source Data Fig. 3
Raw data for Fig. 3.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiong, J., Zhang, W., Wang, Y. et al. Bridging chemistry and artificial intelligence by a reaction description language. Nat Mach Intell 7, 782–793 (2025). https://doi.org/10.1038/s42256-025-01032-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01032-8
This article is cited by
-
Enzyformer: a two-stage pretrained model for enzymatic retrosynthesis
Journal of Cheminformatics (2026)
