
Abstract
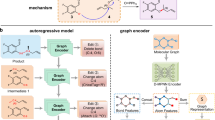
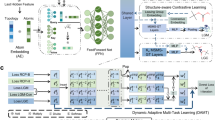
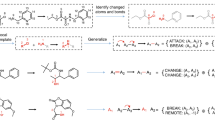
Artificial intelligence has transformed the field of precise organic synthesis. Data-driven methods, including machine learning and deep learning, have shown great promise in predicting reaction performance and synthesis planning. However, the inherent methodological divergence between numerical regression-driven reaction performance prediction and sequence generation-based synthesis planning creates formidable challenges in constructing a unified deep learning architecture. Here we present RXNGraphormer, a framework to jointly address these tasks through a unified pre-training approach. By synergizing graph neural networks for intramolecular pattern recognition with Transformer-based models for intermolecular interaction modelling, and training on 13 million reactions via a carefully designed strategy, RXNGraphormer achieves state-of-the-art performance across eight benchmark datasets for reactivity or selectivity prediction and forward-synthesis or retrosynthesis planning, as well as three external realistic datasets for reactivity and selectivity prediction. Notably, the model generates chemically meaningful embeddings that spontaneously cluster reactions by type without explicit supervision. This work bridges the critical gap between performance prediction and synthesis planning tasks in chemical AI, offering a versatile tool for accurate reaction prediction and synthesis design.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
Data availability
The preprocessed dataset comprising 13 million chemical reactions used for model pre-training is available via GitHub at https://github.com/licheng-xu-echo/RXNGraphormer. A permanently archived version is accessible through Zenodo at https://doi.org/10.5281/zenodo.15770470 (ref. 47) and via figshare at https://doi.org/10.6084/m9.figshare.28356077 (ref. 48). Specific datasets, including the Buchwald–Hartwig reaction dataset, Suzuki–Miyaura reaction dataset, the C–H functionalization dataset, the asymmetric thiol addition dataset, the USPTO-derived series and three external validation datasets are available via GitHub at https://github.com/licheng-xu-echo/RXNGraphormer.
Code availability
Codes for data preprocessing, fictitious product molecule generation, delta-mol graph generation, model training, performance evaluation, as well as scripts for analysing pre-trained and fine-tuned models, are available via GitHub at https://github.com/licheng-xu-echo/RXNGraphormer. A permanently archived version is accessible through Zenodo at https://doi.org/10.5281/zenodo.15770470 (ref. 47).
References
Żurański, A. M., Martinez Alvarado, J. I., Shields, B. J. & Doyle, A. G. Predicting reaction yields via supervised learning. Acc. Chem. Res. 54, 1856–1865 (2021).
Zahrt, A. F., Athavale, S. V. & Denmark, S. E. Quantitative structure–selectivity relationships in enantioselective catalysis: past, present, and future. Chem. Rev. 120, 1620–1689 (2020).
Corey, E. J., Long, A. K. & Rubenstein, S. D. Computer-assisted analysis in organic synthesis. Science 228, 408–418 (1985).
Todd, M. H. Computer-aided organic synthesis. Chem. Soc. Rev. 34, 247 (2005).
Cheong, P. H.-Y., Legault, C. Y., Um, J. M., Çelebi-Ölçüm, N. & Houk, K. N. Quantum Mechanical investigations of organocatalysis: mechanisms, reactivities, and selectivities. Chem. Rev. 111, 5042–5137 (2011).
Klucznik, T. et al. Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 4, 522–532 (2018).
Crawford, J. M., Kingston, C., Toste, F. D. & Sigman, M. S. Data science meets physical organic chemistry. Acc. Chem. Res. 54, 3136–3148 (2021).
Rinehart, N. I., Zahrt, A. F., Henle, J. J. & Denmark, S. E. Dreams, false starts, dead ends, and redemption: a chronicle of the evolution of a chemoinformatic workflow for the optimization of enantioselective catalysts. Acc. Chem. Res. 54, 2041–2054 (2021).
Rinehart, N. I. et al. A machine-learning tool to predict substrate-adaptive conditions for Pd-catalyzed C–N couplings. Science 381, 965–972 (2023).
Zahrt, A. F. et al. Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning. Science 363, eaau5631 (2019).
Reid, J. P. & Sigman, M. S. Holistic prediction of enantioselectivity in asymmetric catalysis. Nature 571, 343–348 (2019).
Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. & Doyle, A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 360, 186–190 (2018).
Sandfort, F., Strieth-Kalthoff, F., Kühnemund, M., Beecks, C. & Glorius, F. A structure-based platform for predicting chemical reactivity. Chem 6, 1379–1390 (2020).
Schwaller, P., Vaucher, A. C., Laino, T. & Reymond, J.-L. Prediction of chemical reaction yields using deep learning. Mach. Learn. Sci. Technol. 2, 015016 (2021).
Li, B. et al. A deep learning framework for accurate reaction prediction and its application on high-throughput experimentation data. J. Cheminformatics 15, 72 (2023).
Coley, C. W., Green, W. H. & Jensen, K. F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 51, 1281–1289 (2018).
Segler, M. H. S., Preuss, M. & Waller, M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018).
Coley, C. W., Barzilay, R., Jaakkola, T. S., Green, W. H. & Jensen, K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 3, 434–443 (2017).
Tu, Z. & Coley, C. W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. J. Chem. Inf. Model. 62, 3503–3513 (2022).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019).
Lin, K., Xu, Y., Pei, J. & Lai, L. Automatic retrosynthetic route planning using template-free models. Chem. Sci. 11, 3355–3364 (2020).
Zheng, S., Rao, J., Zhang, Z., Xu, J. & Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. J. Chem. Inf. Model. 60, 47–55 (2020).
Kim, E., Lee, D., Kwon, Y., Park, M. S. & Choi, Y.-S. Valid, plausible, and diverse retrosynthesis using tied two-way transformers with latent variables. J. Chem. Inf. Model. 61, 123–133 (2021).
Sacha, M. et al. Molecule edit graph attention network: modeling chemical reactions as sequences of graph edits. J. Chem. Inf. Model. 61, 3273–3284 (2021).
Mao, K. et al. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 457, 193–202 (2021).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. J. Chemformer: a pre-trained transformer for computational chemistry. Mach. Learn. Sci. Technol. 3, 015022 (2022).
Zhu, J. et al. Dual-view molecular pre-training. In Proc. 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (eds Singh, A. K. et al.) 3615–3627 (ACM, 2023).
Lu, J. & Zhang, Y. Unified deep learning model for multitask reaction predictions with explanation. J. Chem. Inf. Model. 62, 1376–1387 (2022).
Li, S.-W., Xu, L.-C., Zhang, C., Zhang, S.-Q. & Hong, X. Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledge. Nat. Commun. 14, 3569 (2023).
Shi, R., Yu, G., Huo, X. & Yang, Y. Prediction of chemical reaction yields with large-scale multi-view pre-training. J. Cheminformatics 16, 22 (2024).
Xia, J. et al. Mole-BERT: rethinking pre-training graph neural networks for molecules. In Proc. Eleventh International Conference on Learning Representations (eds Yan, L. et al.) https://openreview.net/pdf?id=jevY-DtiZTR (ICLR 2023).
Ying, C. et al. Do transformers really perform badly for graph representation? In 35th Conference on Neural Information Processing Systems (NeurIPS 2021) https://openreview.net/pdf?id=OeWooOxFwDa (NeurIPS, 2021).
Shi, Y. et al. Benchmarking Graphormer on large-scale molecular modeling datasets. Preprint at https://arxiv.org/abs/2203.04810 (2023).
Vaswani, A. et al. Attention is all you need. In 31st Conference on Neural Information Processing Systems (NIPS 2017) https://papers.nips.cc/paper_files/paper/2017/file/3f5ee243547dee91fbd053c1c4a845aa-Paper.pdf (2017).
Perera, D. et al. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 359, 429–434 (2018).
Li, X., Zhang, S., Xu, L. & Hong, X. Predicting regioselectivity in radical C−H functionalization of heterocycles through machine learning. Angew. Chem. Int. Ed. 59, 13253–13259 (2020).
Schneider, N., Stiefl, N. & Landrum, G. A. What’s what: the (nearly) definitive guide to reaction role assignment. J. Chem. Inf. Model. 56, 2336–2346 (2016).
Dai, H., Li, C., Coley, C., Dai, B. & Song, L. Retrosynthesis prediction with conditional graph logic network. In 33rd Conference on Neural Information Processing Systems (NeurIPS 2019) https://proceedings.neurips.cc/paper_files/paper/2019/file/0d2b2061826a5df3221116a5085a6052-Paper.pdf (NeurIPS, 2019).
Schwaller, P., Gaudin, T., Lányi, D., Bekas, C. & Laino, T. ‘Found in translation’: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 9, 6091–6098 (2018).
Jin, W., Coley, C., Barzilay, R. & Jaakkola, T. Predicting organic reaction outcomes with Weisfeiler-Lehman network. In 31st Conference on Neural Information Processing Systems (NIPS 2017) https://papers.nips.cc/paper_files/paper/2017/file/ced556cd9f9c0c8315cfbe0744a3baf0-Paper.pdf (2017).
Arjovsky, M., Bottou, L., Gulrajani, I. & Lopez-Paz, D. Invariant risk minimization. Preprint at https://arxiv.org/abs/1907.02893 (2020).
Schwaller, P. et al. Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach. Intell. 3, 144–152 (2021).
Schneider, N., Lowe, D. M., Sayle, R. A. & Landrum, G. A. Development of a novel fingerprint for chemical reactions and its application to large-scale reaction classification and similarity. J. Chem. Inf. Model. 55, 39–53 (2015).
Schleinitz, J. et al. Machine learning yield prediction from NiCOlit, a small-size literature data set of nickel catalyzed C–O couplings. J. Am. Chem. Soc. 144, 14722–14730 (2022).
Xu, L. et al. Towards data‐driven design of asymmetric hydrogenation of olefins: database and hierarchical learning. Angew. Chem. Int. Ed. 60, 22804–22811 (2021).
Xu, L.-C. et al. Enantioselectivity prediction of pallada-electrocatalysed C–H activation using transition state knowledge in machine learning. Nat. Synth. 2, 321–330 (2023).
Xu, L.-C., Tang, M.-J., An, J., Cao, F. & Qi, Y. A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planning. Zenodo https://doi.org/10.5281/zenodo.15770470 (2025).
Xu, L.-C., Tang, M.-J., An, J., Cao, F. & Qi, Y. A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planning. figshare https://doi.org/10.6084/m9.figshare.28356077 (2025).
Acknowledgements
Generous support by the National Natural Science Foundation of China (grant nos. 82394432 and 92249302, Y.Q.) and the Shanghai Municipal Science and Technology Major Project (grant no. 2023SHZDZX02, Y.Q.). The computations in this research were performed using the CFFF platform at Fudan University. The Data Platform Department at the Shanghai Academy of Artificial Intelligence for Science is acknowledged for their contributions in the open-source reaction dataset collection process.
Author information
Authors and Affiliations
Contributions
L.-C.X., F.C. and Y.Q. conceived the initial idea for the project. F.C. and Y.Q. supervised the project. L.-C.X. designed the RXNGraphormer model framework and the pre-training strategy, wrote the codes, trained the models and analysed the results. M.-J.T. contributed to implement the algorithm for fictitious reaction generation. J.A. contributed to design the model encoder. All authors took part in discussions. L.-C.X. wrote the paper with input from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Kuangbiao Liao, Jolene Reid and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Details of model framework, comparison of results with other models and t-SNE embedding analysis results, together with Supplementary Figs. 1–23 and Tables 1–8.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, LC., Tang, MJ., An, J. et al. A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planning. Nat Mach Intell 7, 1561–1571 (2025). https://doi.org/10.1038/s42256-025-01098-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01098-4
