
Abstract
Comprehensively studying metabolism requires metabolite measurements. Such measurements, however, are often unavailable in large cohorts of tissue samples. To address this basic barrier, we propose a Bayesian framework (‘UnitedMet’) that leverages RNA–metabolite covariation to impute otherwise unmeasured metabolite levels from widely available transcriptomic data. UnitedMet is equally capable of imputing whole pool sizes and outcomes of isotope tracing experiments. We apply UnitedMet to investigate the metabolic impact of driver mutations in kidney cancer, identifying an association between BAP1 and a highly oxidative tumor phenotype. We similarly apply UnitedMet to determine that advanced kidney cancers upregulate oxidative phosphorylation relative to early-stage disease, that oxidative metabolism in kidney cancer is associated with inferior outcomes to anti-angiogenic therapy and that kidney cancer metastases demonstrate elevated oxidative phosphorylation. UnitedMet provides a scalable tool for assessing metabolic phenotypes when direct measurements are infeasible, facilitating unexplored avenues for metabolite-focused hypothesis generation.
Similar content being viewed by others

Main
Changes to metabolite pool sizes and metabolic flux are fundamental to numerous diseases and biological phenomena1; consequently, measurement of metabolites themselves is critical to the discovery of disease biomarkers, therapeutic vulnerabilities and mechanisms of action2,3,4,5,6,7. However, despite the translational value of metabolite measurement, large-scale profiling of metabolite levels in clinical specimens remains scarce because of the technical challenges associated with metabolomic measurements (for example, the need for fresh, snap-frozen tissue and the analytical challenges of measuring chemically diverse compounds)8. Overcoming this data scarcity, therefore, comes with the potential reward of expanded access to the large space of underexplored, metabolite-centered biological hypotheses.
Two simultaneous and recent developments have now poised the metabolism field to overcome the lack of large-scale metabolite measurements. First, recent developments in machine learning have demonstrated the promise of using reference multimodal data (that is, measurements of two or more distinct data modalities) to ultimately impute measurements of interest in single-modality data9,10. For example, multimodal learning methods for single-cell multiomics9,10,11 have been successful at cross-modal prediction for single-modality datasets (for example, protein prediction by jointly modeling with single-cell RNA sequencing (RNA-seq) in TotalVI (ref. 12) and single-cell ATAC prediction through modeling with single-cell RNA-seq in MultiVI (ref. 13)). Second, we and other groups have identified both cancer-type-specific and lineage-agnostic patterns of RNA–metabolite covariation14,15,16,17,18,19. Together, these developments suggest that suitably designed machine learning models may, by leveraging strong covariation between transcripts and metabolite pools, be able to predict otherwise unmeasured metabolite levels from matched single-modality transcriptomic data. Such a joint framework for modeling metabolic and RNA measurements would also produce a unified, low-rank representation of multimodal metabolite and RNA data, enabling downstream sample clustering, visualization and integration in a latent space.
Three key quantitative challenges must be addressed by multimodal models of metabolite and RNA levels. First, mass-spectrometry-derived metabolomics and isotope labeling data are predominantly reported in semiquantitative relative abundances, impeding comparisons of identical metabolites and isotopologs across datasets (and of different metabolites within the same dataset). Second, different metabolomic measurement platforms often detect a subset of metabolites with limited overlap. As a result, each metabolic reference dataset exhibits a varying degree of missing measurements. Third, both metabolic and RNA modalities possess distinct sources of technical errors and noise that need to be suitably modeled. Prior attempts at predicting metabolic profiles from RNA-seq data had limited success, in part because of their inefficacy in addressing the aforementioned challenges. One method, reliant on correlation networks, struggled with missing values, resulting in a limited ability to predict cross-dataset outcomes for only 34 metabolites, with the highest Pearson’s ρ below 0.5 (ref. 20). Similarly, a different approach using multivariate Lasso regression yielded poor performance, with a median R2 value of 0 for within-dataset prediction and an inability to perform cross-dataset prediction21.
Here, we present UnitedMet, a Bayesian probabilistic method for joint modeling of metabolic and RNA-seq data. UnitedMet addresses the above challenges by mapping both RNA and metabolite data onto a shared rank-transformed scale and inferring missing metabolic measurements in reference datasets. UnitedMet operates as a comprehensive framework at two levels. In the latent space, it learns a unified representation for both metabolic and RNA data, facilitating tasks such as sample clustering and dataset integration. At a higher level, UnitedMet seizes on the strength of RNA–metabolite covariation to impute either metabolite pool sizes or isotopolog distributions from isotope labeling experiments directly from RNA abundance. We demonstrate that UnitedMet performs well on both imputation of pool sizes and imputation of isotope tracing experiments. We subsequently apply UnitedMet to identify the metabolite phenotypes of driver mutations in clinical specimens from persons with clear cell renal carcinoma (ccRCC) and study the metabolic phenotypes associated with metastatic disease in ccRCC.
Results
UnitedMet: a Bayesian model for multimodal metabolic data analysis
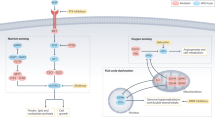
UnitedMet is a Bayesian generative method that jointly models RNA-seq and metabolite data. The input to UnitedMet comprises the paired matrices of RNA counts (X) and total ion counts of metabolites or isotopologs (Y) from samples with both RNA-seq and metabolite data measured (defined as reference datasets) and single-modality matrices with only RNA-seq data available (defined as target datasets) (Fig. 1a). To map metabolite relative abundances and gene expression levels onto a shared measurement scale, we rank-transform the metabolite or isotopolog and gene expression levels across all the samples within each dataset. Such a rank transformation places the distribution of values for metabolite features onto a common, nonparametric scale that naturally accounts for the semiquantitative nature of mass-spectrometry-based metabolomics data. UnitedMet then takes in an aggregate multiple-dataset matrix (R) containing the rankings data from both paired and single-modality samples. UnitedMet assumes observations are generated from a Plackett–Luce ranking distribution of a latent variable Z, which is the matrix product of a latent sample embedding matrix (W) and a latent feature embedding matrix (H) (Fig. 1b). UnitedMet infers posterior distributions of gene expressions and metabolic profiles for all samples in the aggregate matrix and predicts metabolic profiles for single-modality samples using stochastic variational inference (SVI). A hyperparameter λ, the number of latent embedding dimensions, is selected by grid search. The output of UnitedMet is a fully imputed multimodal data matrix, where any missing measurements from single-modality data in the input matrix R are replaced with their posterior estimates.

a, Workflow of a metabolite imputation pipeline with UnitedMet. UnitedMet takes paired matrices of RNA counts (\(X\)) and total ion counts of metabolites or isotopologs (\(Y\)) (defined as reference datasets) and single-modality matrices with only RNA-seq data available (\({X}_{0}\)) (defined as target datasets) as inputs. UnitedMet then normalizes and rank-transforms both RNA-seq and metabolic data. By probabilistic modeling, UnitedMet infers posterior distributions of metabolic profiles for single-modality target samples, which can be used in downstream analysis for biological hypothesis testing. b, Architecture of the UnitedMet model. An aggregate matrix (\(R\)) containing ranking data from both paired and single-modality samples is modeled with a Plackett–Luce ranking distribution based on latent variables derived from embedding matrices \(W\) and \(H\). UnitedMet integrates transcriptomic and metabolic data into a common low-dimensional space for tasks such as clustering and visualization. Next, UnitedMet imputes missing metabolite levels from gene expression data, offering predictions and uncertainty quantification (some icons in the figure were created with BioRender.com).
UnitedMet provides a unified solution for multimodal metabolomic data analysis at two levels. First, UnitedMet learns a shared representation of both transcriptomic and metabolic data, including from samples where one type of measurement is missing, and integrates these data into a common low-dimensional latent space. Such a low-dimensional, integrated representation facilitates downstream tasks such as sample clustering and data visualization (Fig. 1b). Second, by learning a unified representation of metabolomic and transcriptomic features from reference data, UnitedMet enables the imputation of otherwise unmeasured metabolite levels and/or isotopolog distributions from gene expression data alone and delivers these predictions along with a quantification of their uncertainty. Together, these functions of UnitedMet enable the interrogation of metabolism and the evaluation of hypotheses relying on metabolite measurements in large, deeply profiled cohorts of tumors otherwise lacking metabolomic data.
UnitedMet accurately predicts metabolite levels from RNA-seq data in human tumor samples
To evaluate UnitedMet’s capacity to predict metabolite abundances on real-world patient-derived data, we first applied UnitedMet to four datasets of ccRCC samples with fully paired RNA-seq and metabolomics profiles. The aggregated data contained two datasets from the NIH Clinical Proteomic Tumor Analysis Consortium (CPTAC) project, CPTAC (n = 50, no. of metabolites = 183, no. of genes = 60,483)and CPTAC_val (n = 71, no. of metabolites = 130, no. of genes = 60,483), and two in-house datasets RC18 (n = 144, no. of metabolites = 783, no. of genes = 22,937) and RC20 (n = 76, no. of metabolites = 1,012, no. of genes = 22,987) (Supplementary Table 1). These data represented a typical use case for UnitedMet; while 20,171 genes were represented in all four datasets (corresponding largely to protein-coding genes uniformly measured across all data), only 86 (7% of the 1,148 unique metabolites in the entire dataset) were measured in all four datasets.
We designed a benchmarking experiment to evaluate the performance of UnitedMet and comparator methods for the imputation of otherwise unmeasured metabolites. At each iteration of our benchmarking experiment, we treated three of the four ccRCC datasets as ‘reference’ datasets for UnitedMet (in which both metabolomic and transcriptomic data were available) and treated the remaining ccRCC dataset as a ‘target’ dataset (where only transcriptomic data were available). We subsequently trained four distinct UnitedMet models (one for each iteration of the benchmarking experiment, each with different hyperparameters λ) (Extended Data Fig. 1a) and evaluated the accuracy of UnitedMet metabolite predictions in the target dataset. For each metabolite, predicted levels from UnitedMet were compared to their ground-truth values by Spearman correlation (Fig. 2a). We considered a metabolite well predicted if the correlation between ground-truth and imputed abundance for that metabolite was positive and statistically significant (false discovery rate (FDR)-adjusted P value < 0.1). By calculating the percentage of well-predicted metabolites among the total number of available metabolites in the target dataset that were also measured in at least one other training dataset, UnitedMet successfully imputed between 48% and 67% of metabolites in the four target datasets (Fig. 2b and Supplementary Table 2). We compared the performance of UnitedMet to two existing methods for prediction of metabolite abundance from gene expression with the same datasets: multivariate Lasso regression21 and MIRTH22,23 (Methods). We used two metrics to quantify how well each method predicted metabolite abundance: the Spearman ρ among all predicted metabolites and the number of well-predicted metabolites. UnitedMet outperformed the other methods in all four cross-validation datasets by both metrics (Fig. 2c, Extended Data Fig. 1b and Supplementary Table 2). From these experiments, we conclude that UnitedMet can successfully impute a subset of metabolites directly from RNA-seq and that the accuracy of this imputation varies significantly across metabolites.

a, Schematic of the benchmarking experiment to evaluate model performance in a cross-validation scenario. Each time, three of four ccRCC datasets were designated as reference datasets, while the fourth dataset served as the target dataset with only transcriptomic data. The accuracy of UnitedMet’s predictions was then evaluated by comparing predicted metabolite abundances to their ground-truth levels. \(X\), RNA-seq data; \(Y\), metabolomics data. b, The imputation performance for each dataset was assessed by Spearman ρ values between predicted values and their ground truths across all simulated missing features. Metabolites with predicted ranks that showed a significant positive correlation (two-sided FDR-adjusted P < 0.1 and Spearman ρ > 0) with the actual ranks are labeled red. c, Performance of UnitedMet, multivariate Lasso regression and MIRTH based on the Spearman ρ among all predicted metabolites. Significance was assessed using a two-sided Wilcoxon signed-rank test. (CPTAC, n = 156 participant samples, PLasso_UnitedMet = 2.1 × 10−11, PMIRTH_UnitedMet = 2.7 × 10−8; CPTAC_val, n = 129 participant samples, PLasso_UnitedMet = 6.4 × 10−12, PMIRTH_UnitedMet = 6.8 × 10−4; RC18, n = 709 participant samples, PLasso_UnitedMet = 6.6 × 10−6, PMIRTH_UnitedMet = 2.1 × 10−11; RC20, n = 718 participant samples, PLasso_UnitedMet = 1.7 × 10−33, PMIRTH_UnitedMet = 4 × 10−3). In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles) and the whiskers extend to the minima and maxima within 1.5 times the interquartile range. Data points outside this range are shown as individual outliers. d, Correlation between actual and predicted metabolite ranks for two reproducibly well-predicted metabolites: kynurenine (top) and N-acetylneuraminate (bottom). Each point represents one sample in which the metabolite was measured and predicted. e, The imputation performance for each metabolite is summarized across datasets, with average Spearman ρ values plotted. A subset of consistently well-imputed metabolites is labeled and those that are reproducibly well predicted are marked in blue.
As targeted mass spectrometry can only measure a specific class of metabolites, a related challenge is imputing a large panel of metabolites from a subset of measured metabolites and RNA-seq data. To address this, we extended UnitedMet’s capabilities by introducing a weighted loss function to address the imbalanced metabolomics and RNA-seq modalities. To benchmark the imputation accuracy, we randomly selected 50% of all measured metabolites as simulated missing in each dataset. Once again, we found UnitedMet was the top performer on all datasets in terms of the same metrics mentioned above (Extended Data Fig. 1c,d and Supplementary Table 3).
We next investigated the consistency of prediction accuracy across the four ccRCC datasets in UnitedMet. For each pair of two datasets from the four, we observed strong correlation between their metabolite-level prediction performances (Spearman ρ = 0.32–0.83 in all pairwise comparisons, P < 0.001 in all pairwise comparisons; Extended Data Fig. 2a). The highest concordance was observed between the two in-house datasets, likely because of the larger overlap in measured metabolites that could be used for training and the larger sample sizes in these datasets. These results together confirmed that well-predicted metabolites were highly consistent across all four datasets. For instance, kynurenine (average Spearman ρ = 0.64, FDR-adjusted P value < 0.1 in all four datasets) and N-acetylneuraminate (average Spearman ρ = 0.64, FDR-adjusted P value < 0.1 in all four datasets) exhibited robust prediction results across four datasets (Fig. 2d). Combining these prediction results in four datasets, we labeled 59 metabolites as ‘reproducibly’ well-predicted metabolites, indicating that they were well predicted in at least three of four target datasets (Supplementary Table 4). Reproducibly well-predicted metabolites were enriched for amino acids and carbohydrates but depleted of lipids relative to the full panel of metabolites (Fig. 2e). We also explored UnitedMet’s capacity to estimate model uncertainty by evaluating the s.d. of 1,000 draws from the posterior distribution of metabolite levels. We found that prediction uncertainty was negatively correlated with the prediction accuracy (Extended Data Fig. 2b), indicating that posterior uncertainty could guide the selection of reliable predictions for downstream analyses.
To further validate UnitedMet’s reliability, we conducted an external validation using three independent breast cancer cohorts24,25,26. Training on two breast cancer datasets24,25 and testing on a triple-negative breast cancer (TNBC) dataset26,27, UnitedMet successfully predicted 42% of available metabolites in the TNBC dataset (Extended Data Fig. 2c), demonstrating its ability to generalize across different cancer types. Importantly, these predictions preserved well-characterized metabolic alterations of TNBC subtypes, such as elevated lipid metabolism in the C1 subtype and increased carbohydrate and glutathione metabolism in the C2 subtype (Extended Data Fig. 2d), highlighting the biological relevance of the model.
UnitedMet can predict isotopolog distributions from RNA-seq data in vitro and in vivo
Unlike measurements of metabolite pool sizes, isotopolog distributions produced from steady-state isotopic labeling experiments capture the flow of nutrients through cellular metabolism. However, labeling experiments are technically challenging; consequently, there are even less publicly available isotopic labeling data (both in cell lines and in tissue specimens) compared to conventional metabolomic data. Motivated by the ability of UnitedMet to predict metabolite levels by jointly modeling metabolomics and RNA-seq data and the generalizability of our model, we hypothesized that UnitedMet might be able to predict isotopolog distributions from RNA-seq data. To test this hypothesis, we obtained three datasets with paired RNA-seq data and isotopic labeling data (measured by mass spectrometry). Dataset RCC contained RCC tumor samples obtained from 76 participants receiving infusions of [U-13C]glucose before surgery28. A total of 64 isotopologs and 12,300 genes were measured in the RCC dataset28. The other two datasets were composed of human non-small cell lung cancer (NSCLC) cell lines labeled with either [U-13C]glucose or [U-13C]glutamine: NSCLC-G (n = 85, no. of isotopologs = 28, no. of genes = 16,383) and NSCLC-Q (n = 85, no. of isotopologs = 21, no. of genes = 16,383) (Supplementary Table 1)3.
To evaluate UnitedMet’s performance of predicting isotopolog distributions, we conducted a simulation where 50% of the samples in a given dataset were randomly selected and treated as target data for UnitedMet (that is, with isotopolog measurements masked) (Fig. 3a). The remaining 50% of samples were treated as a reference dataset for UnitedMet. We trained three distinct UnitedMet models (one for each dataset) with different hyperparameters λ (Extended Data Fig. 3a). UnitedMet was able to successfully impute 52% (RCC), 56% (NSCLC-G) and 63% (NSCLC-Q) of the held-out isotopologs (Spearman ρ > 0, FDR-adjusted P value < 0.1) (Fig. 3b and Supplementary Table 5). Citrate M + 2, which reflects the contribution of glucose-derived carbon to the tricarboxylic acid (TCA) cycle through pyruvate in [U-13C]glucose-labeled data, was reproducibly predicted with high accuracy in both the in vitro NSCLC dataset (Spearman ρ = 0.44, P = 0.003) and the in vivo RCC dataset (Spearman ρ = 0.39, P = 0.01) (Fig. 3c). In contrast, gene expression scores of either oxidative phosphorylation signature or TCA cycle signature, calculated directly from RNA-seq data, were not correlated to citrate M + 2 labeling in these datasets (oxidative phosphorylation signature: Spearman ρ = 0.16, P = 0.3 (NSCLC) and Spearman ρ = 0.22, P = 0.19 (RCC), Fig. 3c; TCA cycle signature: ρ = 0.06, P = 0.3 (NSCLC) and ρ = 0.25, P = 0.13 (RCC); Extended Data Fig. 3b). Similarly, lactate M + 3, which reflects glucose contribution to glycolysis in [U-13C]glucose-labeled data, was accurately predicted in the RCC dataset (Spearman ρ = 0.43, P = 0.007), while a glycolysis gene expression signature was not correlated to lactate M + 3 (Spearman ρ = 0.05, P = 0.8; Extended Data Fig. 3c). Together, these results demonstrate that UnitedMet can accurately predict isotopologs that characterize specific metabolic phenotypes, an achievement not possible with standard gene set enrichment analysis of RNA-seq data.

a, Schematic of the benchmarking experiment to assess model performance on isotopolog predictions. Here, 50% of the samples in a given dataset were randomly selected and treated as target data for UnitedMet (that is, simulated as unmeasured). The remaining 50% of samples were treated as a reference dataset for UnitedMet. \(X\), RNA-seq data; \(Y\), Isotopolog data. b, Imputation performance for each dataset was evaluated using Spearman ρ values between predicted values and their ground truths across all simulated missing features. Isotopologs with predicted ranks that exhibited a significant positive correlation with the actual ranks are marked in red. c, True ranks of citrate M + 2 were well predicted by UnitedMet but not by the gene expression signature of the Hallmark oxidative phosphorylation pathway. For each sample in the [U-13C]glucose-labeled NSCLC (top) and RCC (bottom) datasets, true ranks of citrate M + 2 were compared to predicted ranks from UnitedMet (left) and oxidative phosphorylation pathway scores calculated from gene expressions in the corresponding Hallmark gene set (right). Significance was assessed using a two-sided Spearman correlation. d, Uniform manifold approximation and projection plots of sample embedding matrix W (posterior means) learned by UnitedMet reveal integration of batches (top) and clustering across renal cell carcinoma subtypes in TCGA KIPAN batch (bottom). Each dot represents a participant sample. RCC and TCGA KIPAN samples overlap in the latent space. e, UnitedMet captures histology-associated differences in metabolism across RCC subtypes. DA of imputed isotopologs across RCC subtypes in TCGA KIPAN were compared to ground-truth differences in the measured RCC cohort. Significance was assessed using a two-sided Spearman correlation. Isotopologs in blue were consistently and significantly enriched (FDR-adjusted P < 0.1, two-sided Wilcoxon rank-sum test) in both measured and predicted cohorts. f, UnitedMet captures mutation-driven metabolic reprogramming in ChRCC. For each sample in the ChRCC cohort (n = 61), predicted levels of lactate M + 3/glucose M + 6 (left) and citrate M + 2/glucose M + 6 (right) are shown. Error bars represent ±1 s.d. The x axis is sorted by predicted abundances of corresponding isotopologs. Samples with complex I insertions or deletions are labeled red. Samples with complex I single-nucleotide variations are labeled green. P values show the results of a two-sided Wilcoxon rank-sum test between complex I indel samples and the other samples.
Human kidney cancer arises in a variety of subtypes, including ccRCC, papillary RCC (pRCC) and chromophobe RCC (ChRCC), presenting with functionally distinct metabolic activity. To further benchmark the capacity of UnitedMet to impute isotopolog distributions, we assessed its capacity to capture histology-associated differences in metabolism across RCC subtypes. To do so, we applied UnitedMet, using multimodal RNA-seq and isotopolog data from Bezwada et al.28 as a reference dataset and 1,020 RCC tumor and adjacent normal samples from The Cancer Genome Atlas (TCGA) pan-kidney cohort (KIPAN) (encompassing ccRCC, pRCC and ChRCC) as a target dataset (Extended Data Fig. 3d and Supplementary Table 6). At the low-dimensional latent space learned by UnitedMet, we found that UnitedMet successfully embedded samples in both the reference and the target datasets according to their subtype, despite missing measurements of isotopologs in TCGA KIPAN (Fig. 3d). Furthermore, imputed labeling patterns in TCGA KIPAN dataset preserved ground-truth differences between ChRCC and ccRCC samples (Spearman ρ = 0.85, P = 4.2 × 10−15) and between pRCC and ccRCC samples (Spearman ρ = 0.79, P = 5.0 × 10−12) (Fig. 3e). Consistent with prior findings28, ccRCC samples demonstrated higher glycolytic labeling, such as lactate M + 3/glucose M + 6, while ChRCC and pRCC samples displayed higher ratios of TCA cycle labeling, such as citrate M + 2/glucose M + 6 and succinate M + 2/glucose M + 6 (Fig. 3e and Supplementary Table 7). While ChRCC displays increased use of the TCA cycle, loss-of-function alterations to mitochondrial DNA (mtDNA)-encoded complex I genes can result in loss of oxidative phosphorylation and metabolic reprogramming in favor of glycolysis29. Consistent with these findings, we found that ChRCC samples with complex I alterations demonstrated a shift to an alternative glycolytic metabolic pathway with higher levels of lactate M + 3/glucose M + 6 (P = 0.02) and lower levels of citrate M + 2/glucose M + 6 (P = 0.04), evaluated using the Wilcoxon rank-sum test) (Fig. 3f and Supplementary Table 8). This suggested that UnitedMet captured mutation-driven metabolic reprogramming in ChRCC, which further validated UnitedMet’s capability to generate biologically meaningful predictions.
In total, the analysis presented in Figs. 2 and 3 demonstrates that UnitedMet is capable of accurately imputing both metabolite levels and isotopolog distributions from RNA-seq data through joint, multimodal modeling with reference datasets.
BAP1 mutations are associated with an oxidative metabolic phenotype in ccRCC
Although both oncogenes (such as MYC and PIK3CA) and tumor suppressor genes (PTEN and VHL) are well-recognized regulators of metabolism30,31, the functional consequences of driver alterations on tumor metabolism in vivo are poorly studied32,33. In fact, the lack of population-scale metabolomic profiling in contemporary cohorts of molecularly profiled tumors renders a direct evaluation of the association of either metabolite levels or metabolic flux with the presence of specific driver alterations infeasible. We reasoned that we could apply UnitedMet to impute both metabolite levels and isotope labeling patterns in richly profiled cohorts of tumors, such as those from TCGA, to assess whether genomic alterations were associated with specific metabolite changes.
We focused our efforts on understanding the genome–metabolome covariation in ccRCC, for which we have several reference datasets with both transcriptomic and metabolomic or labeling data. The canonical founder mutation in ccRCC is the biallelic inactivation of the tumor suppressor gene VHL (affecting between 50% (ref. 34) and 80% (ref. 35) of ccRCC cases) and the subsequent activation of a pseudohypoxic transcriptional and metabolic program. The subsequent evolution of ccRCC includes the acquisition of secondary driver mutations in genes (such as PIK3CA, PTEN, MTOR and BAP1), whose functions are (at least in part) metabolic2. To understand the associations between genetic mutations and metabolic variations in ccRCC, we applied UnitedMet to large-scale multiomics TCGA kidney ccRCC (KIRC) cohort (n = 606), which has paired RNA-seq and whole-exome sequencing (WES) data. Training the RNA-seq data from TCGA KIRC with four ccRCC reference datasets (CPTAC, CPTAC_val, RC18 and RC20; n = 341) containing paired RNA-seq and metabolomics data, UnitedMet predicted metabolite levels for TCGA KIRC samples (Fig. 4a and Supplementary Table 9).

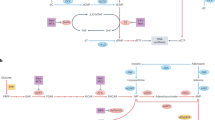
a, Schematic of the metabolite-level prediction and downstream analysis for TCGA KIRC samples with UnitedMet. RNA-seq data (\({X}_{\rm{TCGA}}\)) of TCGA KIRC cohort (target dataset) were trained with four ccRCC reference datasets (CPTAC, CPTAC_val, RC18 and RC20; n = 341) containing paired RNA-seq and metabolomics data. \(X\), RNA-seq data; \(Y\), metabolomics data. Predicted metabolite levels (\({Y}_{\rm{TCGA}}\)) are leveraged for association analysis with ccRCC driver mutations. b, BAP1 mutation demonstrates the strongest association with a broad range of predicted metabolites. Top, distribution of the total number of significantly associated metabolites across 14 key driver mutations in ccRCC. The x axis is sorted by the number of significantly associated metabolites. Bottom, mutation frequency of 14 driver genes. The x axis is sorted by mutation frequency. c, Pathway-based analysis of predicted metabolic changes in BAP1-mutant versus BAP1 wild-type samples in TCGA KIRC cohort. MUT, mutant; WT, wild type. d, Predicted metabolite-level changes in BAP1-mutant versus BAP1 wild-type samples in TCGA KIRC cohort. Left, diagram of glucose metabolism pathways: glycolysis and TCA cycle (created with BioRender.com). α-KG, α-ketoglutarate. Right, box plots comparing predicted unphosphorylated glucose, citrate, fumarate and malate levels in mutant BAP1 (n = 38) versus wild-type BAP1 (n = 330) participant samples. P values were calculated using unpaired two-tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles) and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots. e, Schematic of the isotopolog distribution prediction for TCGA KIRC samples with UnitedMet. RNA-seq data (\({X}_{\rm{TCGA}}\)) of TCGA KIRC cohort (target dataset) were trained with ccRCC samples in the [U-13C]glucose-labeled RCC dataset containing paired RNA-seq and isotope labeling data. \(X\), RNA-seq data; \(Y\), isotopolog data. f, Predicted isotopolog changes in BAP1-mutant versus BAP1 wild-type samples in TCGA KIRC cohort. Box plots compare the predicted citrate M + 2/pyruvate M + 3, succinate M + 2/pyruvate M + 3 and malate M + 2/pyruvate M + 3 ratios in mutant BAP1 (n = 38) versus wild-type BAP1 (n = 330) participant samples. P values were calculated using unpaired two-tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles) and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots.
We first studied associations between the predicted metabolite abundances and genetic mutations in TCGA KIRC cohort. For each of the 14 key driver mutations in ccRCC (VHL, PBRM1, SETD2, BAP1, MTOR, KDM5C, PTEN, TP53, PIK3CA, TSC2, TCEB1, TSC1, PIK3R1 and SDHB), we compared metabolite levels (considering only reproducibly well-predicted metabolites) between mutant and wild-type samples, using an FDR-corrected Wilcoxon test (Fig. 4b). We identified significantly higher or lower mutation-specific abundance of metabolites in BAP1 (n = 38 metabolites), PBRM1 (n = 37), VHL (n = 22), SETD2 (n = 15) and TP53 (n = 3) mutations. The BAP1 mutation showed the strongest association with the largest variety of predicted metabolites despite a relatively low mutation rate (~10%) in participants with ccRCC (Fig. 4b). For example, BAP1-mutant samples exhibited lower levels of β-alanine, glutamine, glutamate and oxidized glutathione, which aligns with the loss of BAP1 impairing cellular redox homeostasis and weakening antioxidant defense mechanisms36,37,38. Additionally, prior studies showed that BAP1 mutations have a role in several aspects of cellular metabolism including glucose metabolism37,38,39. Mass spectrometry measurements demonstrated that germline BAP1 mutations induced the Warburg effect in human fibroblasts, including depleted TCA cycle activity and increased aerobic glycolysis40. Additionally, transcriptome analysis showed that BAP1-mutant ccRCC samples were enriched in glycolytic gene expression41. To gather insight into the interplay between BAP1 mutation and metabolite abundance, we performed a pathway-based differential abundance (DA) analysis of predicted metabolic changes in BAP1-mutant and wild-type samples in TCGA KIRC. BAP1-mutant samples showed significant depletion in the TCA cycle metabolism (DA score = −1), including drops in the levels of citrate (P = 0.03), fumarate (P = 0.007) and malate (P = 0.008) (Fig. 4c,d). BAP1-mutant samples also demonstrated lower levels of free, unphosphorylated glucose (P = 3 × 10−7), suggesting that these tumors may upregulate glucose uptake from the microenvironment (Fig. 4d). Consistent with these findings, similar trends of BAP1 mutation-specific changes were observed in the directly measured metabolite abundances from both CPTAC and CPTAC_val datasets, further validating the metabolite-level differences between BAP1-mutant and wild-type samples (Extended Data Fig. 4a,b).
To more granularly understand the metabolic flux patterns associated with the above-described pool size changes, we trained the RNA-seq data from TCGA KIRC with the [U-13C]glucose-labeled reference dataset RCC (Fig. 4e and Supplementary Table 10) and leveraged imputed [U-13C]glucose-labeled isotopolog distribution data from TCGA KIRC. Relative to BAP1 wild-type tumors, BAP1-mutant tumors demonstrated increased levels of citrate M + 2/pyruvate M + 3 (P = 3 × 10−4), succinate M + 2/pyruvate M + 3 (P = 0.003) and malate M + 2/pyruvate M + 3 (P = 0.03) (Fig. 4f), indicating an elevated contribution of glucose to TCA cycle activity in BAP1-mutant ccRCC. These data indicated that pool size drops in TCA cycle metabolites are not caused by decreased entry of glucose into the TCA cycle. Instead, they suggest that BAP1-mutant tumors undergo reduced entry of other anapleurotic sources of TCA cycle intermediates, such as glutamate, or alternatively increase diversion of TCA cycle intermediates into alternate pathways, such as the use of acetyl-CoA for fatty acid synthesis. Such hypotheses are directly testable by analogous infusion experiments using, for example, labeled glutamine, and suggest that BAP1 tumors may harbor metabolically distinct (and potentially therapeutically targetable) metabolic alterations.
Lastly, we sought to evaluate whether other diseases may similarly display associations between genotype and metabolic phenotype. To do so, we trained UnitedMet on a reference dataset containing paired RNA-seq and [U-13C]glucose-labeled isotopolog distribution data from 42 NSCLC samples42 and used TCGA lung adenocarcinoma (LUAD) cohort (n = 576) as a target dataset (Supplementary Table 11). Partitioning the reference data into 50% training and 50% testing, we found that UnitedMet was able to successfully impute 7/29 isotopologs. We subsequently studied associations between the well-predicted isotopolog distributions and genetic mutations in TCGA LUAD cohort. Consistent with a prior study3, EGFR-mutant tumors demonstrated decreased levels of citrate M + 2/pyruvate M + 3 (P = 2 × 10−6) and glutamate M + 2/pyruvate M + 3 (P = 2 × 10−4) (Extended Data Fig. 5a) relative to EGFR wild-type tumors, indicating a diminished contribution of glucose to the pool sizes of TCA cycle constituents in EGFR-mutant LUAD. Functionally, this suggests that EGFR mutations in persons with cancer are associated with a less oxidative phenotype, although additional data from tracing of other nutrient sources such as glutamine and acetate are necessary to more completely resolve TCA cycle flux. Together, our findings suggested that, in diverse settings, driver mutations induce specific metabolic phenotypes.
Shift to oxidative metabolism correlates with disease progression and poorer clinical outcome
Recent work suggested that, although ccRCC tumors generally downregulate mitochondrial gene expression and limit entry of glucose-derived carbon into the TCA cycle relative to normal tissue, distant metastases in ccRCC upregulate oxidative phosphorylation and glucose entry into the TCA cycle28. However, there are no large-scale data available on the metabolism of metastatic tumors. We reasoned that high-stage, aggressive ccRCC tumors, which ultimately seed distant metastases, should exhibit signatures of upregulation of oxidative glucose metabolism. To test this hypothesis, we again leveraged predicted isotopolog distribution data from TCGA KIRC and compared isotopolog levels of [U-13C]glucose-labeled TCA cycle intermediates normalized by pyruvate M + 3 in ccRCC tumors from different pathological stages. Aggressive ccRCCs with a higher stage demonstrated higher levels of citrate M + 2/pyruvate M + 3 (P = 3 × 10−4), succinate M + 2/pyruvate M + 3 (P = 7 × 10−6) and malate M + 2/pyruvate M + 3 (P = 2 × 10−4), evaluated using the Kruskal–Wallis test (Fig. 5a), consistent with increased glucose-derived carbon entry into the TCA cycle. Motivated by this finding, we sought to evaluate whether high-stage, aggressive tumors in other cancers also displayed a similar shift to oxidative glucose metabolism. We leveraged predicted isotopolog distribution data from TCGA LUAD and found no significant associations between TCA cycle labelings such as citrate M + 2/pyruvate M + 3 (P = 0.8), glutamate M + 2/pyruvate M + 3 (P = 0.8) and malate M + 2/pyruvate M + 3 (P = 0.3) and pathological stages, evaluated using the Kruskal–Wallis test (Extended Data Fig. 5b). This suggested that metabolic reprogramming in aggressive tumors is cancer-type-specific.

a, Aggressive ccRCCs with higher pathological stage demonstrate higher ratios of predicted citrate M + 2/pyruvate M + 3 (left), succinate M + 2/pyruvate M + 3 (middle) and malate M + 2/pyruvate M + 3 (right) in TCGA KIRC cohort (stage 1, n = 267 participant samples; stage 2, n = 57 participant samples; stage 3, n = 123 participant samples; stage 4, n = 84 participant samples). The significance between any two stages was assessed using a pairwise two-sided t-test. P values are FDR adjusted. Citrate M + 2/pyruvate M + 3, P1_3 = 0.0014 and P1_4 = 0.0014; succinate M + 2/pyruvate M + 3, P1_2 = 0.04, P1_3 = 0.0011 and P1_4 = 3 × 10−5; malate M + 2/pyruvate M + 3, P1_3 = 0.067 and P1_4 = 0.00015. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles) and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots. b, Samples from metastatic sites in participants with ccRCC show higher ratios (compared to samples from primary tumor sites) of predicted citrate M + 2/pyruvate M + 3 (left), succinate M + 2/pyruvate M + 3 (middle) and malate M + 2/pyruvate M + 3 (right) in the IMmotion151 cohort (primary ccRCC, n = 625 participant samples; metastatic ccRCC, n = 198 participant samples). Significance was assessed using two-sided Wilcoxon rank-sum tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles) and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots. c, Kaplan–Meier plot showing PFS of ccRCC participants with a high level of citrate M + 2/pyruvate M + 3 (based on median level) versus a low level of citrate M + 2/pyruvate M + 3 in both the atezolizumab + bevacizumab arm (top) and the sunitinib arm (bottom). Significance was assessed using a log-rank test. d, Same as c but for succinate M + 2/pyruvate M + 3. e, Same as c but for malate M + 2/pyruvate M + 3.
We then applied UnitedMet to predict isotopolog distribution data for 823 primary or metastatic tumor samples from a publicly available advanced ccRCC clinical trial (IMmotion151) (Supplementary Table 12). We trained RNA-seq data from IMmotion151 with the ccRCC samples from the RCC reference dataset. Predicted isotopolog levels of [U-13C]glucose-labeled TCA cycle intermediates normalized by pyruvate M + 3 were compared between primary and metastatic ccRCC tumors in IMmotion151. Metastatic ccRCC tumor samples demonstrated higher levels of citrate M + 2/pyruvate M + 3 (P = 5 × 10−13), succinate M + 2/pyruvate M + 3 (P = 3 × 10−10) and malate M + 2/pyruvate M + 3 (P = 2 × 10−9), evaluated using the Wilcoxon rank-sum test (Fig. 5b). This finding was further validated in a second set of trials (the CheckMate cohort43), where predictions for metastatic ccRCC tumors also showed increased oxidative TCA cycle labeling (Extended Data Fig. 5c). Together, these results indicated that, in ccRCC, increased TCA cycle activity is associated both with (1) high stage or disease progression and (2) the establishment of metastasis itself.
We next interrogated whether this oxidative metabolic phenotype may be linked to poor clinical outcomes. Participants with ccRCC in the IMmotion151 trial were treated with either atezolizumab plus bevacizumab (a combination of tyrosine kinase inhibitor and immunotherapy) or sunitinib (an antiangiogenic tyrosine kinase inhibitor). We evaluated the association between isotopolog levels of TCA cycle intermediates and progression-free survival (PFS) using multivariate Cox proportional hazard models (evaluating different treatment arms separately). In the atezolizumab + bevacizumab arm, participants with high levels of citrate M + 2/pyruvate M + 3, succinate M + 2/pyruvate M + 3 and malate M + 2/pyruvate M + 3 did not exhibit a significant survival difference (Fig. 5c–e). In the sunitinib arm, we observed that participants with high succinate M + 2/pyruvate M + 3 (P = 4.4 × 10−5) Fig. 5d) and malate M + 2/pyruvate M + 3 (P = 1.6 × 10−6) (Fig. 5e) had significantly poorer PFS. These data highlighted oxidative metabolism of glucose as a potential druggable target to diminish cancer progression and metastasis in persons receiving antiangiogenic agents in ccRCC.
Discussion
This work presents an advanced methodology for the joint, probabilistic modeling of multimodal metabolic data. In doing so, it addresses the numerous challenges associated with the analysis of metabolomics data (including but not limited to semiquantitative data and batch effects) and its joint modeling with transcriptomics data. After establishing that UnitedMet accurately imputes metabolite features with estimates of uncertainty in benchmark datasets, we applied UnitedMet to study the metabolic consequences of key driver mutations and the metabolic adaptations associated with aggressive disease and metastatic competency.
The era of cancer genomics has revealed that only a small number of metabolic enzymes (including, for example, IDH1, IDH2, FH and SDH) are recurrently mutated or otherwise lost in cancer. However, a much larger number of recurrently altered genes are so-called regulators of metabolism (for example, PIK3CA, MTOR and MYC) or other proteins indirectly drawing on metabolites as substrates for their action (for example, epigenetic regulators such as DNA methyltransferases). Our observations here suggest that certain molecular subtypes of cancer, associated with the presence or absence of key driver mutations such as BAP1 in ccRCC or EGFR in LUAD, may themselves be associated with unique metabolic features. Whether these associations between genotype and metabolism are gene intrinsic or potentially extend across cancer lineages (for example, to BAP1-mutant mesothelioma) is an open question. While such observations have been made in the past24,44, the growing number of metabolomics datasets in primary tumors and, now, the availability of UnitedMet suggest that the field is now poised to carry out a more comprehensive analysis of the metabolic impact of driver mutations.
Several key limitations underlie UnitedMet and represent important challenges in the development of next-generation methods for joint modeling of multimodal metabolic data. First, the accuracy of UnitedMet varied widely across metabolites (Fig. 2), rendering a large fraction of the metabolite nonimputable. The successive addition of relevant data for training for UnitedMet is the most direct route to addressing this limitation, although the possibility remains that a subset of metabolites will remain nonimputable with even very large training data. Nonetheless, UnitedMet’s ability to estimate the uncertainty of the model for each imputed metabolite enables users to filter reliable predictions, which can help mitigate the variability in performance and ensure robust downstream analyses. Second, while rank transformation has proven useful in both UnitedMet and MIRTH23 for the comparison of semiquantitative metabolite data produced in distinct batches, the process of rank transformation produces a loss of information, where large effect sizes (that is, large fold changes between pairs of samples) in one metabolite feature can be equated to small effect sizes in another metabolite feature in the rank-transformed space. Third, the vast majority of the training data for UnitedMet are derived from single-site tissue biopsies, leaving open the possibility that fluctuations in the global tumor nutrient milieu may be incompletely captured by RNA-seq from a single site. Multiregional sampling of both metabolites and RNA (or coregistered spatial metabolomics and transcriptomics) is likely necessary to address this limitation. Fourth, UnitedMet requires at least one reference dataset of sufficient size to carry out imputation. For the majority of diseases, such a dataset does not exist14,45. One potential avenue to overcoming this challenge is to train disease-agnostic models to impute metabolite features. This seems feasible for at least some metabolite features that demonstrate lineage-agnostic covariation with gene expression, such as IDO1 and kynurenine14,22. However, while our own prior studies focused on analyzing lineage-agnostic covariation between individual genes and individual metabolites, such interactions are rare. Conversely, it remains unknown whether generalized, multivariate patterns of gene–metabolite covariation (for example, a multidisease implementation of UnitedMet) could be used to impute metabolite levels in different contexts. It is reasonable to speculate that certain fundamental and common metabolic phenotypes (such as hypoxia or aerobic glycolysis) may be associated with shared transcriptomic signatures across diseases, whereby cross-disease or cross-tissue imputation would be feasible. Encoding tissue site as an additional factor in future versions of UnitedMet may improve model performance in this setting. Lastly, while UnitedMet demonstrates high accuracy in predicting metabolic phenotypes, we acknowledge that further reference data with paired measurements of RNA-seq and metabolomics are required to expand its predictive capabilities comprehensively. Given the abundance of gene expression data in the field, we advise cautious interpretation to prevent overreliance on imputed metabolite predictions without adequate empirical validation.
UnitedMet harnesses the covariation between the transcriptome and metabolome to impute otherwise unmeasured metabolite features. In doing so, it enables the inference of pool size and tracing patterns (and, consequently, the evaluation of metabolite-centered hypotheses) in valuable clinical samples where metabolite profiling is difficult or otherwise infeasible. Several valuable clinical use cases come to mind as natural applications of the UnitedMet framework where ancillary transcriptomic data are available. For instance, one may seek to infer metabolite levels in archival formalin-fixed, paraffin-embedded samples of inadequate quality for metabolite profiling or in biopsy samples with an inadequate quantity of material for metabolite profiling. Such data are commonly generated in the pursuit of genomic and transcriptomic biomarkers of response to targeted and immunotherapies (and, indeed, form the basis of our analysis in Fig. 5). Separately, in isotope tracing experiments where one is interested in more than one tracer (for example, 13C-glucose and 13C-glutamine tracing, where the infusion of both tracers in the same person is infeasible or does not provide useful data), UnitedMet could be used to impute the outcome of the counterpart tracer as long a common data modality (for example, RNA-seq) was collected. This would overcome fundamental limitations in the ability to resolve tracers using a common isotope in a single person and, in doing so, facilitate a more complete description of metabolic flux patterns in pathways driven by multiple nutrient sources. UnitedMet, therefore, democratizes metabolomics data for scientific discovery.
Methods
Data preprocessing
This study complies with all relevant ethical regulations and was approved by the institutional review board at Memorial Sloan Kettering Cancer Center (MSKCC).
The input to UnitedMet consists of reference datasets with paired measurements of RNA counts and total ion counts of metabolites or isotopologs and a single-modality target dataset with RNA-seq data only (Fig. 1a). We assume that there are N different reference datasets, each with an RNA-seq sample × gene matrix of raw counts \({X}_{n}\in {{\mathbb{R}}}^{{S}_{n}\times {G}_{n}}(n=1,2,\ldots ,N)\) and a paired sample × metabolite or sample × isotopolog ion count matrix \({Y}_{n}\in {{\mathbb{R}}}^{{S}_{n}\times {M}_{n}}(n=1,2,\ldots ,N)\). Let \({X}_{0}\in {{\mathbb{R}}}^{{S}_{0}\times {M}_{0}}\) be the RNA-seq sample × gene matrix in a single-modality target dataset.
Normalization
We first normalized all input data with distinct techniques. We implemented total ion count normalization to raw ion count matrices of metabolomics data (Y) and transcripts per million (TPM) normalization to raw count matrices of RNA-seq data (X). In metabolomics experiments, ion counts below a threshold were not detected by the mass spectrometry. This ended up with missing metabolite measurements in some samples. We treated these left-censored values as half of the minimum value across all metabolite measurements when calculating the total ion count normalizer.
For sample × isotopolog ion count matrices (Y) of isotope labeling data, we first calculated the fractional labeling (namely, the proportion of each isotopolog relative to the sum of all isotopologs in that metabolite). We then divided all fractions by the fraction of pyruvate M + 3 or glucose M + 6. Normalization by pyruvate M + 3 allowed us to establish the labeling ratio of each isotopolog to pyruvate M + 3, providing insights into the contribution of glucose-derived pyruvate to that specific isotopolog.The labeling ratio of citrate M + 2 to pyruvate M + 3, for instance, suggested the contribution of glucose through the pyruvate dehydrogenase reaction. Normalization by glucose M + 6 instead revealed the contribution of glucose carbon to other metabolites.
Rank transformation
As metabolomics and isotope tracing data generated using mass spectrometry are reported as semiquantitative relative abundances, we are only able to compare measurements of the same metabolite or isotopolog from different samples in the same dataset. To map metabolic relative abundances and gene expression levels into a shared measurement scale across all features and datasets, we rank the metabolite or isotopolog and gene expression levels across all the samples within each dataset. Ranks enable the comparison of features across datasets and transfer learning from RNA-seq modality to metabolic modality. Samples exhibiting the maximum level for a specific feature within the provided dataset are assigned the highest rank. Conversely, samples displaying the minimum level for the same feature are allocated the lowest rank. Left-censored samples are tied, sharing the last rank in the ranking hierarchy. While we use unnormalized rankings for modeling, we normalize ranks by their total number of samples S in downstream analyses, mapping them to a comparable scale of ranks [0, 1) in all datasets. Here, we use 𝑆 to refer to the number of samples within a dataset in a general sense, without referring to a specific dataset. For each feature j, the normalized rank of a measurement \({f}_{{ij}}(i=\mathrm{1,2},\ldots ,S\;)\) in that dataset is defined by \({\rm{rank}}_{{ij}}=\frac{{\sum }_{k=1}^{S}P [\;{f}_{{ij}} > {f}_{{kj}}]}{S}\). Importantly, rank transformations are performed separately for each metabolite, ensuring that comparisons are made only within the same metabolite across samples. The ranked values are specific to each metabolite and are not directly comparable between different metabolites.
Data aggregation
Rankings data of RNA-seq matrices Xn and metabolic matrices \({Y}_{n}(n=\mathrm{1,2},\ldots ,N)\) in reference datasets are aggregated into a single data matrix R along with the rankings data of metabolic matrix X0 in the target dataset. While we take in the common genes shared across datasets to save computation costs, we aggregate metabolic modalities by taking the union of relevant features (namely, the aggregated matrix \(R\in {{\mathbb{R}}}^{{S}_{R}\times {F}_{R}}\), where \({S}_{R}={S}_{0}+{\sum }_{i=1}^{N}{S}_{n}\) and \({F}_{R}=|{G}_{R}|+|{M}_{R}|,{G}_{R}={G}_{0}\bigcap{\cap }_{i=1}^{N}{G}_{i},{M}_{R}={\cup}_{i=1}^{N}{M}_{i}\)). In the benchmarking test on four ccRCC datasets, R contained measurements of 1,148 metabolites and 20,171 genes for 341 samples.
The UnitedMet model
UnitedMet is a probabilistic generative method that jointly models RNA-seq and metabolic data. UnitedMet assumes that the rankings in R are generated by a Plackett–Luce ranking distribution of a latent variable matrix Z, where \(Z=WH\) is the product of the latent sample embedding matrix \(W\in {{\mathbb{R}}}^{{S}_{R}\times \lambda }\) and the latent feature embedding matrix \(H\in {{\mathbb{R}}}^{\lambda \times {F}_{R}}\). The hyperparameter λ is the number of embedding dimensions. We suppose all latent variables in both latent embedding matrices are generated by normal prior distributions: \({W}_{{ik}}\overset{\text{iid}}\sim {\rm{Normal}}(\mathrm{0,1}),{H}_{{kj}}\overset{\text{iid}}\sim {\rm{Normal}}(\mathrm{0,1}),\) where Wik is the entry in the ith sample and the kth embedding column in embedding matrix W and Hkj is the entry in the kth embedding row and the jth feature in embedding matrix H.
Plackett–Luce ranking distribution
The Plackett–Luce distribution46,47 models a ranking of T items as an ordered series of choices. It begins by choosing the top-ranked item from the entire set of T options, followed by choosing the second-ranked item from the remaining options and so on48. Given a set of T options \(\{{Q}_{1},\ldots ,{Q}_{T}\}\), the probability of selecting the ith item Qi is defined as \(P(i|\{1,\ldots ,T\})=\frac{{u}_{i}}{{\sum }_{t=1}^{T}{u}_{t}}\) by the Luce choice axiom, where ui represents the utility score of Qi. The probability of a full ordering \(\{{\sigma }_{1},\ldots ,{\sigma }_{T}\}\), where we assume \({Q}_{{\sigma }_{1}} > \ldots > {Q}_{{\sigma }_{T}}\), is then given by recursively applying the Plackett–Luce distribution, choosing σ1 from \(\{1,\ldots ,T\}\), σ2 from \(\{1,\ldots ,T\}\backslash \{{\sigma }_{1}\}\) and σ3 from \(\{1,\ldots ,T\}\backslash \{{\sigma }_{1},{\sigma }_{2}\}\), yielding \(P(\{{\sigma }_{1},\ldots ,{\sigma }_{T}\}|\{1,\ldots ,T\})={\prod }_{i=1}^{T}\frac{{u}_{{\sigma }_{i}}}{{\sum }_{r=i}^{T}{u}_{{\sigma }_{r}}}\). Given the latent variable matrix \(Z=WH\) in UnitedMet, we suppose the utility score of the item in the ith sample and the jth feature is defined as \({\rm{exp}}({Z}_{ij})={\rm{exp}}({{W}_{i}}{H}_{j})\). Extending this to censored rankings in UnitedMet, the likelihood of observing a censored ordering \(\{{\sigma }_{1},{\sigma }_{2},\ldots ,{\sigma }_{K},\{{\sigma }_{K+1},\ldots ,{\sigma }_{S}\}\}\) in the jth feature of a batch is then defined by \(P\left({R}_{\!j}=\left\{{Z}_{{\sigma }_{1},\,j} > {Z}_{{\sigma }_{2},\,j} > \ldots > {Z}_{{\sigma }_{K},\,j} >\right.\right.\) \(\left.\left.\{{Z}_{{\sigma }_{K+1},\,{j}},\ldots ,{Z}_{{\sigma }_{S},\,{j}}\}\right\}|\{{Z}_{1,\,{j}},\ldots ,{Z}_{S,\,{j}}\}\right)\)\(={\prod }_{i=1}^{K}\frac{{\rm{exp}}({Z}_{{\sigma }_{i},\,{j}})}{{\sum }_{r=i}^{S}{\rm{exp}}({Z}_{{\sigma }_{r},\,{j}})}\). Detailed definitions of UnitedMet are described below.
Cross-validation
To determine the optimal number of embedding dimensions (\(\lambda\)) of latent matrices \(W\) and \(H\), we use tenfold cross-validation. The range of \(\lambda\) to be tested is contingent on the total number of samples \({S}_{R}\). For instance, performance evaluation spans a \(\lambda\) range of [1, 351] with a step of 10 in the benchmarking test on ccRCC datasets. For each batch, cross-validation features that are used to test model performance are selected separately. Only metabolic features (metabolites or isotopologs) that are measured in at least one other batch are included. These features are then randomly distributed into ten folds. We treat one fold at a time as unmeasured and hold out the fold’s features in the corresponding batch. Masked features are then predicted by UnitedMet. In the end, we calculate the mean absolute error (MAE) between the true ranks of held-out features in the fold and their predicted ranks. The MAE scores across all folds are averaged to obtain a final performance score. We evaluate the MAE scores for all \(\lambda\) values and the one resulting in the elbow of the MAE score curve is chosen as the optimal number of embedding dimensions for the factorization.
Inference
The likelihood is computed from observed rankings in both paired modalities of reference datasets and only in the respective RNA-seq modality of the target dataset. We use SVI within the Pyro49 package for inference. Variational distributions are generated using the AutoNormal function. Optimization is executed through the Adam optimizer, with a default learning rate set to 0.001. Convergence is ascertained when the relative change in evidence lower bound (ELBO) falls below 0.01. To address the inherent imbalance between the RNA-seq and metabolomics modalities in scenarios where we want to impute a large panel of metabolites from a subset of measured metabolites and RNA-seq data, we introduced a weighted loss function in UnitedMet. Typically, RNA-seq data contains measurements for approximately 20,000 genes, whereas metabolomics data comprises around 1,000 measured metabolites. This large difference in the number of features between the two modalities can disproportionately affect the likelihood computation, leading to biases in the inferred rankings. To mitigate this issue, we applied modality-specific weighting when computing the log-likelihood using the Plackett–Luce distribution. Specifically, we assigned equal weight to the metabolomics features, while down-weighting the gene features proportionally by the ratio of the number of metabolites to genes. Given a log-likelihood matrix L of shape (ngenes + nmetabolites) × S, where S is the number of samples. We defined a weight matrix C of the same shape such that: C:,j = 1, if j corresponds to a metabolite; C:,j = nmetabolites/ngenes, if j corresponds to a gene. The weighted log-likelihood matrix was computed as: Lweighted = C ⊙ L. The final total log-likelihood was obtained by summing over all observed values in Lweighted.
Posterior prediction
UnitedMet estimates the joint posterior distribution of the latent embedding matrix \(W\) and \(H\). For every latent variable in \(W\) and \(H\), we draw 1,000 samples from their estimated posterior distribution. Given posterior samples of the latent matrix \(Z(=WH)\), posterior rankings are then generated by the Plackett–Luce ranking distribution. To sample in a computation-efficient way, we implemented the Gumbel–Max trick50, which generates ordered samples from the Plackett–Luce ranking distribution by sorting the perturbed log probability through the addition of independent variables from the Gumbel distribution51 (\({G}_{1,\,j},\ldots ,{G}_{S,\,j}\sim {\rm{Gumbel}}(0),{\rm{iid}}\)). Let \({Z}_{,\;j}\) be the \({j}{\rm{th}}\) column of the latent matrix \(Z\). Set perturbed log probability \({U}_{i,j}\) = \({Z}_{i,j}+{G}_{i,j}\). The ordered indices of the \({j}{\rm{th}}\) column returned by sorting the perturbed log probabilities \(\{{U}_{1,\,j},\ldots ,{U}_{S,\,j}\}\) are equivalent to the orderings generated by the Plackett–Luce model given probabilities (utility scores) \(\{{Z}_{1,\;j},\ldots ,{Z}_{S,\;j}\}\). Specifically, if \(\{{U}_{{\sigma }_{1},\,j} > {U}_{{\sigma }_{2},\,j} > \ldots > {U}_{{\sigma }_{K},\,j} > \{{U}_{{\sigma }_{K+1},\,j},\ldots ,{U}_{{\sigma }_{S},\,j}\}\}\), then we observe \(\{{Z}_{{\sigma }_{1},\;j} > {Z}_{{\sigma }_{2},\;j} > \ldots > {Z}_{{\sigma }_{K},\;j} > \{{Z}_{{\sigma }_{K+1},\;j},\ldots ,{Z}_{{\sigma }_{S},\;j}\}\}\).
Estimates of the rankings can be found as the mean of the 1,000 posterior draws, while the s.d. of posterior samples represents a quantification of the prediction uncertainty.
Benchmarking
Multivariate Lasso regression
We implemented multivariate Lasso regression on four ccRCC datasets according to Li et al.21. In each dataset, metabolomics data were preprocessed by total ion count normalization, while transcript levels were converted into TPM units. At each time in the benchmarking experiments, one ccRCC dataset was treated as the testing set while the other three were training sets. All RNA-seq data were scaled before training or testing. For every metabolite (y), we used gene expressions (x) to predict it in the training set. LassoCV in Python package scikit-learn was used to select the best penalizer α by fivefold cross-validation. The maximum number of iterations fitting along the regularization path was set to default 1,000. After selecting the best model for each metabolite, we assessed model accuracy by calculating Spearman correlation coefficients between predicted metabolite levels and their ground truths.
MIRTH
MIRTH is a matrix factorization approach aimed at predicting the levels of unmeasured metabolites by collectively analyzing the covariation of metabolites across multiple datasets23. We extended MIRTH to the cross-modality prediction problem as previously described22. Metabolomics and RNA-seq data were preprocessed in the same way mentioned above.
MSKCC ccRCC datasets
We obtained two datasets, RC18 (n = 144) and RC20 (n = 76), each with matched RNA-seq and mass spectrometry metabolomics measurements from fresh frozen high-quality tumor or adjacent normal specimens of persons with ccRCC that underwent partial or radical nephrectomies at MSKCC22. Samples were collected under the approval of MSKCC’s institutional review board. The alignment of RNA-seq reads was performed using STAR two-pass alignment against human genome assembly hg19. Metabolites were identified on the basis of the criteria according to Benedetti et al.14. RC18 had measurements for 783 metabolites and 22,937 genes. RC12 had measurements for 1,012 metabolites and 22,987 genes.
CPTAC ccRCC datasets
Metabolite raw count matrices of CPTAC (n = 50) and CPTAC_val (n = 71) were downloaded from Li et al.1. Transcriptomic and WES data were downloaded from Genomic Data Commons (https://portal.gdc.cancer.gov/projects/CPTAC-3; project: CPTAC-3, primary site: kidney). CPTAC contained only ccRCC tumor samples, while CPTAC_val contained tumor and adjacent normal samples of persons with ccRCC. Mass spectrometry peaks were quantified using Thermo Scientific Compound Discoverer software to generate raw counts. HTSeq version 0.11.2 was implemented to calculate the gene-level stranded read count. We then performed total ion count normalization and TPM normalization on metabolite and gene expression count matrices, respectively. CPTAC had measurements for 183 metabolites and 60,483 genes. CPTAC_val had measurements for 130 metabolites and 60,483 genes.
Breast cancer datasets
Matched TPM-normalized RNA-seq and bulk metabolomic data (raw count matrices) of two breast cancer datasets BrCa1 (ref. 24) (n = 108, no. of metabolites = 533, no. of genes = 20,032), BrCa225 (n = 18, no. of metabolites = 397, no. of genes = 21,773) were downloaded from Benedetti et al.14. RNA-seq data of primary tumor tissues from the TNBC cohort (n = 360, no. of genes = 23,211) were downloaded from the National Omics Data Encyclopedia (https://www.biosino.org/node/analysis/detail/OEZ00000398) according to Jiang et al.27. Bulk metabolomics data of the TNBC cohort (n = 479, no. of metabolites = 594) were downloaded from Xiao et al.26. There were 258 tumor samples with paired RNA-seq and metabolomics data after matching.
Human RCC RNA-seq and isotopic labeling data infused with [U-13C]glucose in vivo
Paired RNA-seq and isotopic labeling data from 76 primary tumor or adjacent normal kidney samples of persons with RCC were downloaded from Bezwada et al.28. The RCC dataset had measurements for 64 isotopologs and 12,300 genes. Because small fluctuations of isotopolog levels that are not biologically interpretable can be quantified as signals in mass spectrometry, we set a criterion to filter out isotopologs whose average fraction over all samples was less than 10%. This ended with a total of 23 isotopologs including biologically meaningful isotopologs such as citrate M + 2 and malate M + 2.
Human NSCLC cell line RNA-seq and isotopic labeling data
We downloaded two human NSCLC cell line datasets with paired RNA-seq and isotopic labeling data from Chen et al.3: NSCLC-G (n = 85, no. of isotopologs = 28, no. of genes = 16,383) and NSCLC-Q (n = 85, no. of isotopologs = 21, no. of genes = 16,383). A total of 85 NSCLC cell lines were cultured with medium containing the isotopically enriched nutrient under identical conditions. The isotopic data in NSCLC-G were labeled with [U-13C]glucose, while the isotopic data in NSCLC-Q were labeled with [U-13C]glutamine. After filtering out isotopologs whose average fraction over all samples was less than 10%, there were nine and eight isotopologs in the NSCLC-G and NSCLC-Q datasets, respectively.
TCGA datasets
We downloaded paired RNA-seq, WES and clinical data of 1,020 RCC tumor and adjacent normal samples in TCGA KIPAN from the Genome Data Analysis Center (GDAC) at Broad Institute. A total of 606 TCGA KIRC samples were included in TCGA KIPAN. mtDNA mutation calls using a PCR-based amplification approach for 61 ChRCC cases in TCGA KICH were downloaded from Davis et al.29. Paired RNA-seq, WES and clinical data of TCGA LUAD (n = 576) were also downloaded from GDAC.
Annotation of MAF files from WES data
We downloaded MAF files of WES data for CPTAC, CPTAC_val, TCGA KIPAN and TCGA KIRC from the corresponding websites mentioned above. We annotated all molecular variations to 0 or 1 in a gene-wise way, where 0 represented wild-type or silent variations and 1 represented nonsilent variations. Missense mutation, nonsense mutation, frame-shift deletion, splice site mutation, frame-shift insertion, in-frame deletion, splice-region variant, translation start site mutation, in-frame insertion and nonstop mutation were considered as nonsilent molecular variations. Silent mutations, intron mutation, 3′ UTR mutation and 5′ UTR mutation were considered as silent variations, because they were not able to change gene functions.
DA score
The DA score assesses the distinct regulation of a metabolic pathway between two groups. Calculated through a Wilcoxon rank-sum test applied to all pathway metabolites, the score undergoes P-value correction using the Benjamini–Hochberg method (FDR-corrected P value < 0.1). For each pathway, the DA score is derived as follows: (no. of significantly enriched metabolites − no. of significantly depleted metabolites)/no. of total metabolites. Scoring is exclusively applied to pathways exhibiting three or more significantly altered metabolites.
Survival analysis
We collected RNA-seq data and participant-level clinical information from IMmotion151 (refs. 35,52) (n = 823), a published trial exploring immunotherapeutic versus systemic agents in advanced ccRCC. To account for diverse drug effects in clinical trials, we conducted separate statistical analyses for the immunotherapy arm (atezolizumab + bevacizumab) and the sunitinib arm. The survival regression analysis was performed using the Python package lifelines.
Statistical and reproducibility
Statistical analyses were conducted using either R or Python. Differential distribution comparisons were implemented with the Wilcoxon rank-sum test or t-test. All statistical tests were two-sided by default, unless specified otherwise, with P values corrected using the Benjamini–Hochberg method53. No statistical method was used to predetermine sample size and no data were excluded from the analyses. Blinding and randomization were not relevant because this was an observational study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the conclusions of this paper are publicly available through the links provided in the Methods or in the Supplementary Information. The four ccRCC reference datasets, containing paired metabolomics and RNA-seq data, can be accessed from Zenodo (https://doi.org/10.5281/zenodo.11286535)54. Paired RNA-seq and isotopic labeling data from 76 primary tumor or adjacent normal kidney samples of persons with RCC were downloaded from Bezwada et al.28. Paired RNA-seq and isotopolog data from 42 primary tumor or adjacent normal lung samples of persons with NSCLC were downloaded from Cai et al.42. Paired RNA-seq and WES data of 1,020 RCC tumor and adjacent normal samples in TCGA KIPAN were obtained from the GDAC at the Broad Institute (http://firebrowse.org/). mtDNA mutation calls using a PCR-based amplification approach for 61 ChRCC cases in TCGA KICH were sourced from Davis et al.29. RNA-seq data and participant-level clinical information from the IMmotion151 trial (n = 823), exploring immunotherapeutic versus systemic agents in advanced ccRCC, were retrieved from published sources35,52. Source data are provided with this paper.
Code availability
All original code to run UnitedMet and regenerate figures was deposited to GitHub (https://github.com/reznik-lab/UnitedMet) and is publicly available as of the date of publication.
References
Li, Y. et al. Histopathologic and proteogenomic heterogeneity reveals features of clear cell renal cell carcinoma aggressiveness. Cancer Cell 41, 139–163 (2023).
DiNatale, R. G., Sanchez, A., Hakimi, A. A. & Reznik, E. Metabolomics informs common patterns of molecular dysfunction across histologies of renal cell carcinoma. Urol. Oncol. 38, 755–762 (2020).
Chen, P.-H. et al. Metabolic diversity in human non-small cell lung cancer cells. Mol. Cell 76, 838–851 (2019).
Kodama, M. et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 11, 1320 (2020).
Lima, A. R., Bastos, M., de, L., Carvalho, M. & Guedes de Pinho, P. Biomarker discovery in human prostate cancer: an update in metabolomics studies. Transl. Oncol. 9, 357–370 (2016).
Johnson, C. H., Ivanisevic, J. & Siuzdak, G. Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 17, 451–459 (2016).
Kilgour, M. K. et al. 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv. 7, eabe1174 (2021).
Beger, R. D. A review of applications of metabolomics in cancer. Metabolites 3, 552–574 (2013).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Zhu, C., Preissl, S. & Ren, B. Single-cell multimodal omics: the power of many. Nat. Methods 17, 11–14 (2020).
Gong, B., Zhou, Y. & Purdom, E. Cobolt: integrative analysis of multimodal single-cell sequencing data. Genome Biol. 22, 351 (2021).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Ashuach, T. et al. MultiVI: deep generative model for the integration of multimodal data. Nat. Methods 20, 1222–1231 (2023).
Benedetti, E. et al. A multimodal atlas of tumour metabolism reveals the architecture of gene–metabolite covariation. Nat. Metab. 5, 1029–1044 (2023).
Siddiqui, J. K. et al. IntLIM: integration using linear models of metabolomics and gene expression data. BMC Bioinformatics 19, 81 (2018).
Beuchel, C. et al. An atlas of genome-wide gene expression and metabolite associations and possible mediation effects towards body mass index. J. Mol. Med. 101, 1305–1321 (2023).
Yang, M. & Vousden, K. H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16, 650–662 (2016).
Labuschagne, C. F., van den Broek, N. J. F., Mackay, G. M., Vousden, K. H. & Maddocks, O. D. K. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 (2014).
Martínez-Reyes, I. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020).
Cavicchioli, M. V., Santorsola, M., Balboni, N., Mercatelli, D. & Giorgi, F. M. Prediction of metabolic profiles from transcriptomics data in human cancer cell lines. Int. J. Mol. Sci. 23, 3867 (2022).
Li, H., Barbour, J. A., Zhu, X. & Wong, J. W. H. Gene expression is a poor predictor of steady-state metabolite abundance in cancer cells. FASEB J. 36, e22296 (2022).
Tang, C. et al. Immunometabolic coevolution defines unique microenvironmental niches in ccRCC. Cell Metab. 35, 1424–1440 (2023).
Freeman, B. A. et al. MIRTH: metabolite imputation via rank-transformation and harmonization. Genome Biol. 23, 184 (2022).
Terunuma, A. et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 124, 398–412 (2014).
Tang, X. et al. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 16, 415 (2014).
Xiao, Y. et al. Comprehensive metabolomics expands precision medicine for triple-negative breast cancer. Cell Res. 32, 477–490 (2022).
Jiang, Y.-Z. et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell 35, 428–440 (2019).
Bezwada, D. et al. Mitochondrial complex I promotes kidney cancer metastasis. Nature 633, 923–931 (2024).
Davis, C. F. et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330 (2014).
Chen, C.-Y., Chen, J., He, L. & Stiles, B. L. PTEN: tumor suppressor and metabolic regulator. Front. Endocrinol. (Lausanne) 9, 338 (2018).
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L. & Dang, C. V. MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015).
Vander Heiden, M. G. & DeBerardinis, R. J. Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 (2017).
Jones, R. G. & Thompson, C. B. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 23, 537–548 (2009).
Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 (2013).
Motzer, R. J. et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 38, 803–817 (2020).
Carbone, M. et al. Biological mechanisms and clinical significance of BAP1 mutations in human cancer. Cancer Discov. 10, 1103–1120 (2020).
Urso, L. et al. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. Br. J. Cancer 122, 52–61 (2020).
Rohatgi, N. et al. BAP1 promotes osteoclast function by metabolic reprogramming. Nat. Commun. 14, 5923 (2023).
Han, A., Purwin, T. J. & Aplin, A. E. Roles of the BAP1 tumor suppressor in cell metabolism. Cancer Res. 81, 2807–2814 (2021).
Bononi, A. et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 24, 1694–1704 (2017).
Nguyen, G. K., Mellnick, V. M., Yim, A. K.-Y., Salter, A. & Ippolito, J. E. Synergy of sex differences in visceral fat measured with CT and tumor metabolism helps predict overall survival in patients with renal cell carcinoma. Radiology 287, 884–892 (2018).
Cai, L. et al. High glucose contribution to the TCA cycle is a feature of aggressive non-small cell lung cancer in patients. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-23-1319 (2025).
Braun, D. A. et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 26, 909–918 (2020).
Kerr, E. M. & Martins, C. P. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 285, 28–41 (2018).
Reznik, E. et al. A landscape of metabolic variation across tumor types. Cell Syst. 6, 301–313.e3 (2018).
Plackett, R. L. The analysis of permutations. J. R. Stat. Soc. Ser. C Appl. Stat. 24, 193–202 (1975).
Luce, R. D. Individual Choice Behavior: A Theoretical Analysis (Wiley, 1959).
Guiver, J. & Snelson, E. Bayesian inference for Plackett–Luce ranking models. In Proceedings of the 26th Annual International Conference on Machine Learning (eds Danyluk, A., Bottou, L. & Littman, M.) (ACM, 2009).
Bingham, E. et al. Pyro: deep universal probabilistic programming. J. Mach. Learn. Res. 20, 973–978 (2019).
Kool, W., van Hoof, H. & Welling, M. Stochastic beams and where to find them: the Gumbel-Top-k tTrick for sampling sequences without replacement. Preprint at https://arxiv.org/abs/1903.06059 (2019).
Yellott, J. I. The relationship between Luce’s choice axiom, Thurstone’s theory of comparative judgment, and the double exponential distribution. J. Math. Psychol. 15, 109–144 (1977).
Motzer, R. J. et al. Final overall survival and molecular analysis in IMmotion151, a phase 3 trial comparing atezolizumab plus bevacizumab vs sunitinib in patients with previously untreated metastatic renal cell carcinoma. JAMA Oncol. 8, 275–280 (2022).
Glueck, D. H., Mandel, J., Karimpour-Fard, A., Hunter, L. & Muller, K. E. Exact calculations of average power for the Benjamini–Hochberg procedure. Int. J. Biostat. 4, 11 (2008).
Xie, A. ccRCC reference datasets to benchmark UnitedMet. Zenodo https://doi.org/10.5281/zenodo.11286535 (2024).
Acknowledgements
We thank R. Deberardinis (Children’s Medical Center Research Institute, University of Texas Southwestern Medical Center), B. Faubert (Section of Hematology and Oncology, Department of Medicine, University of Chicago) and D. Bezwada (Children’s Medical Center Research Institute, University of Texas Southwestern Medical Center) for thoughtful feedback. Graphics were created with BioRender.com. This project was generously supported by Cycle for Survival, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute Cancer Center Core (grant P30 CA008748) supporting MSKCC. E.R. was supported by the Department of Defense Kidney Cancer Research Program (W81XWH-18-1-0318, HT9425-23-1-0995 and HT9425-24-1-0652), Cycle For Survival Equinox Innovation Award, Kidney Cancer Association Young Investigator Award, Brown Performance Group Innovation in Cancer Informatics Fund and the National Institutes of Health (NIH; R37 CA276200). E.R. was also supported by a grant from the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center. W.T. is supported by the NIH National Cancer Institute (R37 CA271186, U54 CA274492 and P30 CA008748), Break Through Cancer and the Maurice Campbell Initiative at Memorial Sloan Kettering Cancer Center.
Author information
Authors and Affiliations
Contributions
A.X.X., E.R. and W.T. conceptualized the study. A.X.X. designed and implemented the UnitedMet computational framework, conducted the benchmarks and led all the data analysis. A.X.X. and E.R. wrote the manuscript. W.T. edited the manuscript. W.T. and E.R. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Eric Deutsch, Thomas Mitchell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 UnitedMet outperforms Lasso and MIRTH at predicting metabolite levels from RNA-seq data.
a) UnitedMet’s hyperparameter λ, the number of embedding dimensions, is determined by 10-fold cross-validation in the benchmarking experiments of 4 ccRCC datasets. Average mean absolute error between predicted ranks and true ranks across 10 folds changes with different numbers of embedding dimensions. Data are presented as mean values ± SEM (standard error of the mean). Optimal dimension is picked by the elbow point of the curve. Performance evaluation spans a λ range of [1,351] with a step of 10. b) The imputation performance for each dataset is assessed by the number of well-predicted metabolites. Metabolites with predicted ranks that show significant positive correlation (two sided FDR-adjusted p < 0.1 and spearman rho > 0) are defined as well-predicted. c) Performance to impute 50% held-out metabolites from the remaining 50% measured metabolites and RNA-seq data. Performance evaluated by the spearman rho among all predicted metabolites. Extended UnitedMet with a weighted loss function is used. Significant difference was assessed by the two-sided Wilcoxon signed-rank test. (CPTAC, n = 78 patient samples, PLasso_UnitedMet = 4.2 × 10−20, PMIRTH_UnitedMet = 2.6 × 10−20; CPTAC_val, n = 64 patient samples, PLasso_UnitedMet = 3.8 × 10−12, PMIRTH_UnitedMet = 1.3 × 10−6; RC18, n = 354 patient samples, PLasso_UnitedMet = 3.4 × 10−101, PMIRTH_UnitedMet = 2.0 × 10−107; RC20, n = 359 patient samples, PLasso_UnitedMet = 6.0 × 10−105, PMIRTH_UnitedMet = 1.7 × 10−103). In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to the minima and maxima within 1.5 times the interquartile range. Data points outside this range are shown as individual outliers. d) Performance to impute 50% held-out metabolites from the remaining 50% measured metabolites and RNA-seq data. Performance evaluated by the number of well-predicted metabolites. Extended UnitedMet with a weighted loss function is used.
Extended Data Fig. 2 UnitedMet’s metabolite level predictions are consistent across 4 ccRCC datasets.
a) Correlation plots of metabolite-level prediction performances, characterized by their spearman correlation between true ranks and predicted ranks, in all pairwise comparisons of 4 ccRCC datasets. Each dot represents a metabolite. Significant difference was assessed by the two-sided spearman correlation. b) Prediction uncertainty is negatively correlated with the prediction accuracy. Prediction uncertainty is estimated by quantifying the standard deviation of 1000 posterior draws of metabolite levels. For each metabolite, average standard deviation across all samples is associated with its prediction accuracy, characterized by spearman rho values between true ranks and predicted ranks. Each dot, colored by the proportion of censored measurements, represents a metabolite. Significant difference was assessed by the two-sided spearman correlation. c) The imputation performance for the TNBC dataset is assessed by spearman rho values between predicted values and their ground-truths across all simulated missing metabolites. Well-predicted metabolites with predicted ranks that show significant positive correlation (two-sided FDR-adjusted p < 0.1 and spearman rho > 0) with the actual ranks are labeled red. d) Predicted metabolite level changes in metabolic subtype C1 (n = 52 patient samples) v.s. C2 samples (n = 119 patient samples) in the TNBC dataset. Top: Boxplots showing predicted metabolite levels in lipid metabolism. Bottom left: Boxplots showing predicted metabolite levels in glutathione metabolism. Bottom right: Boxplots showing predicted metabolite levels in sugar metabolism. p values are calculated by unpaired two tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots.
Extended Data Fig. 3 UnitedMet’s performance of predicting isotopologues from RNA-seq data.
a) UnitedMet’s hyperparameter λ, the number of embedding dimensions, is determined by 10-fold cross-validation in the benchmarking experiments of 3 isotope labeling datasets respectively. Performance evaluation spans a λ range of [1,81] with a step of 10. Average mean absolute error between predicted ranks and true ranks across 10 folds changes with different numbers of embedding dimensions. Optimal dimension is picked by the elbow point of the curve. b) True ranks of citrate m + 2 were not correlated with gene expression signature of TCA cycle. For each sample in [U-13C]glucose labeled NSCLC dataset (left) and RCC right), true ranks of citrate m + 2 were compared with TCA cycle signature scores calculated from gene expressions in the corresponding Hallmark gene set. Significant difference was assessed by the two-sided spearman correlation. c) True ranks of Lactate m + 3 were well-predicted by UnitedMet but not by gene expression signature of Hallmark glycolysis pathway. For each sample in [U-13C]glucose labeled RCC, true ranks of lactate m + 3 were compared with predicted ranks from UnitedMet (left) and glycolysis pathway scores calculated from gene expressions in the corresponding Hallmark gene set (right). Significant difference was assessed by the two-sided spearman correlation. d) Schematic of the isotopologue distribution prediction for TCGA KIPAN samples with UnitedMet. RNA-seq data (X_TCGA-KIPAN) of the TCGA KIPAN cohort (target dataset) are trained with the [U-13C]glucose labeled RCC dataset containing paired RNA-seq and isotope labeling data. X: RNA-seq data; Y: Isotopologue data. Both target and reference datasets contain different subtypes of RCC.
Extended Data Fig. 4 Validation of BAP1 mutation-specific metabolite changes in CPTAC and CPTAC_val datasets.
a) UnitedMet’s predicted metabolite differences between BAP1-mutant and wildtype samples align with measured data in CPTAC and CPTAC_val. Differential abundances of imputed metabolites between BAP1-mutant and wildtype samples in TCGA KIRC were compared to ground-truth differences in the measured CPTAC (left) and CPTAC_val (right) cohorts. Metabolites in dark blue are consistently and significantly enriched/depleted (FDR-adjusted p < 0.1, two-sided Wilcoxon rank-sum test) in both measured and predicted cohorts. b) Measured metabolite level changes in BAP1 mutant v.s. BAP1 wildtype samples in CPTAC (top, MUT, n = 8 patient samples; WT, n = 42 patient samples) and CPTAC_val (bottom, MUT, n = 6 patient samples; WT, n = 50 patient samples) dataset. Boxplots comparing measured unphosphorylated glucose, citrate, fumarate, and malate levels in MUT BAP1 v.s. WT BAP1 samples. p values are calculated by unpaired two tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots.
Extended Data Fig. 5 TCA cycle labelings were associated with EGFR mutations but not with pathological stages in predicted TCGA-LUAD data.
a) EGFR mutations in LUAD are associated with decreased glucose contribution to the TCA cycle. Predicted citrate m + 2/pyruvate m + 3 levels (left) and glutamate m + 2/pyruvate m + 3 levels (right) in EGFR-mutant (n = 68 patient samples) v.s. EGFR-wildtype samples (n = 445 patient samples) in TCGA LUAD cohort. P values are calculated by unpaired two tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots. b) Oxidative metabolism does not correlate with disease progression in LUAD. Predicted citrate m + 2/pyruvate m + 3 levels (left), glutamate m + 2/pyruvate m + 3 levels (middle), and malate m + 2/pyruvate m + 3 levels (right) are not associated with pathological stages in TCGA LUAD cohort (stage i, n = 275 patient samples; stage ii, n = 122 patient samples, stage iii, n = 84 patient samples; stage iv, n = 26 patient samples. Kruskal–Wallis tests were performed to assess the significance. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots. c) Samples from metastatic sites in ccRCC patients show higher ratios (compared to samples from primary tumor sites) of predicted citrate m + 2/pyruvate m + 3 (left), succinate m + 2/pyruvate m + 3 (middle) and malate m + 2/pyruvate m + 3 (right) in CheckMate cohort (primary ccRCC, n = 225 patient samples; metastatic ccRCC, n = 84 patient samples). Significances are assessed by unpaired two tailed parametric t-tests. In the box plots, the center line represents the median, the bounds of the box indicate the interquartile range (25th to 75th percentiles), and the whiskers extend to 1.5 times the interquartile range. Individual data points are shown as dots.
Supplementary information
Supplementary Tables 1–12
Supplementary Table 1: Overview of datasets used to train UnitedMet. Supplementary Table 2: Performance of UnitedMet, Lasso and MIRTH when 100% metabolites masked. Supplementary Table 3: Performance of UnitedMet, Lasso and MIRTH when 50% metabolites masked. Supplementary Table 4: A list of reproducibly well-predicted metabolites. Supplementary Table 5: Performance of UnitedMet to impute isotopolog distributions. Supplementary Table 6: Imputed [U-13C]glucose-labeled isotopolog distributions for TCGA-KIPAN. Supplementary Table 7: Imputed isotopolog level differences in ccRCC versus pRCC and ChRCC. Supplementary Table 8: Imputed [U-13C]glucose-labeled isotopolog distributions for TCGA KICH. Supplementary Table 9: Imputed metabolite pool sizes for TCGA KIRC. Supplementary Table 10: Imputed [U-13C]glucose-labeled isotopolog distributions for TCGA-KIRC. Supplementary Table 11: Imputed [U-13C]glucose-labeled isotopolog distributions for TCGA LUAD. Supplementary Table 12: Imputed [U-13C]glucose-labeled isotopolog distributions for IMmotion151.
Source data
Source Data Fig. 2
Statistical source data table.
Source Data Fig. 3
Statistical source data table.
Source Data Fig. 4
Statistical source data table.
Source Data Fig. 5
Statistical source data table.
Source Data Extended Data Fig. 1
Statistical source data table.
Source Data Extended Data Fig. 2
Statistical source data table.
Source Data Extended Data Fig. 3
Statistical source data table.
Source Data Extended Data Fig. 4
Statistical source data table.
Source Data Extended Data Fig. 5
Statistical source data table.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, A.X., Tansey, W. & Reznik, E. UnitedMet harnesses RNA–metabolite covariation to impute metabolite levels in clinical samples. Nat Cancer 6, 892–906 (2025). https://doi.org/10.1038/s43018-025-00943-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43018-025-00943-0
This article is cited by
-
Heterogeneity of glucose metabolism and uptake identifies distinct cancer cell and cancer stem cell phenotypes
Cellular & Molecular Biology Letters (2026)
-
Metabolomics and metabolites in cancer diagnosis and treatment
Molecular Biomedicine (2025)
