
Abstract
The evolutionary processes that drive malignant progression of IDH-mutant astrocytomas remain unclear. Here, we performed multiomics on matched initial and recurrent tumor samples from a cohort of 105 patients and overlaid the data with detailed clinical annotation. We identified overlapping features associated with malignant progression that are derived from three molecular mechanisms: cell cycling, tumor cell (de)differentiation and remodeling of the extracellular matrix. Together, they provide a rationale of the underlying biology of tumor malignancy. DNA methylation levels decreased over time, predominantly in tumors with malignant transformation, and co-occurred with poor prognostic genetic events. We identified a DNA methylation-based signature strongly associated with survival, which allows objective, molecular-based grading of IDH-mutant astrocytomas to aid clinical decision making. Our findings were validated on large, independent cohorts of IDH-mutant astrocytoma samples. Lastly, in this retrospective study, we found little effect of radiotherapy or chemotherapy on the molecular features associated with malignant progression.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
GLASS-NL data are available from the European Genome-Phenome Archive (EGA) under accession codes EGAS00001007546 (DNA methylation), EGAS00001007551 (RNA-seq) and EGAS00001007527 (sWGS and WES) and at the Proteomics Identifications Database (PRIDE) under accession code PXD062328 (proteomics). Access to data deposited to the EGA is controlled to comply with the General Data Protection Regulation and patient consent. To request access, a formal application must be submitted to the Data Access Committee (DAC) that is associated with the respective dataset(s) through the EGA. Further information can be found at https://ega-archive.org. The accession of files is limited to research purposes and regulated with a data use agreement. The timeframe for response to requests is as promptly as possible (expected within 10 working days), pending DAC approval. Access to the proteomics data will be made public through the same PRIDE identifier upon obtaining a PMID or DOI. Source data are provided with this paper.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 22, iv1–iv96 (2020).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23, 1231–1251 (2021).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Cancer Genome Atlas Research, N. et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498 (2015).
Barthel, F. P., Wesseling, P. & Verhaak, R. G. W. Reconstructing the molecular life history of gliomas. Acta Neuropathol. 135, 649–670 (2018).
Weller, M. et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 18, 170–186 (2021).
Barthel, F. P. et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576, 112–120 (2019).
Consortium, G. Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the Glioma Longitudinal Analysis Consortium. Neuro Oncol 20, 873–884 (2018).
Varn, F. S. et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199 (2022).
van den Bent, M. J. et al. The biological significance of tumor grade, age, enhancement and extent of resection in IDH mutant gliomas: how should they inform treatment decision in the era of IDH inhibitors? Neuro Oncol. 26, 1805–1822 (2024).
Mellinghoff, I. K. et al. Vorasidenib in IDH1- or IDH2-mutant low-grade glioma. N. Engl. J. Med. 389, 589–601 (2023).
Tesileanu, C. M. S. et al. Prognostic significance of genome-wide DNA methylation profiles within the randomized, phase 3, EORTC CATNON trial on non-1p/19q deleted anaplastic glioma. Neuro Oncol. 23, 1547–1559 (2021).
van den Bent, M. J. et al. Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet 390, 1645–1653 (2017).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol. 24, v1–v95 (2022).
van Dijk, E. et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 12, 3188 (2021).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166 (2018).
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010).
Mazor, T. et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 28, 307–317 (2015).
Tesileanu, C. M. S., Vallentgoed, W. R., French, P. J. & van den Bent, M. J. Molecular markers related to patient outcome in patients with IDH-mutant astrocytomas grade 2 to 4: a systematic review. Eur. J. Cancer 175, 214–223 (2022).
Glowacka, W. K. et al. 5-Hydroxymethylcytosine preferentially targets genes upregulated in isocitrate dehydrogenase 1 mutant high-grade glioma. Acta Neuropathol. 135, 617–634 (2018).
Yuan, J. et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 10, 57 (2018).
Wang, L. et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 9, 1708–1719 (2019).
Johnson, K. C. et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 53, 1456–1468 (2021).
Hoogstrate, Y. et al. Transcriptome analysis reveals tumor microenvironment changes in glioblastoma. Cancer Cell 41, 678–692 (2023).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849 (2019).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169 (2021).
Kelman, Z. PCNA: structure, functions and interactions. Oncogene 14, 629–640 (1997).
Ibarra, A., Schwob, E. & Mendez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl Acad. Sci. USA 105, 8956–8961 (2008).
Bai, H. et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 48, 59–66 (2016).
van Thuijl, H. F. et al. Spatial and temporal evolution of distal 10q deletion, a prognostically unfavorable event in diffuse low-grade gliomas. Genome Biol. 15, 471 (2014).
Wong, Q. H. et al. Molecular landscape of IDH-mutant primary astrocytoma grade IV/glioblastomas. Mod. Pathol. 34, 1245–1260 (2021).
Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Malta, T. M. et al. The epigenetic evolution of glioma is determined by the IDH1 mutation status and treatment regimen. Cancer Res. 84, 741–756 (2024).
Choi, S. et al. Temozolomide-associated hypermutation in gliomas. Neuro Oncol. 20, 1300–1309 (2018).
Cadieux, B., Ching, T. T., VandenBerg, S. R. & Costello, J. F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 66, 8469–8476 (2006).
Carstam, L. et al. WHO grade loses its prognostic value in molecularly defined diffuse lower-grade gliomas. Front. Oncol. 11, 803975 (2021).
Mellinghoff, I. K. et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin. Cancer Res. 27, 4491–4499 (2021).
Louis, D. N. et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 30, 844–856 (2020).
Sahm, F. et al. Molecular diagnostic tools for the World Health Organization (WHO) 2021 classification of gliomas, glioneuronal and neuronal tumors; an EANO guideline. Neuro Oncol. 25, 1731–1749 (2023).
Boehm, E. M., Gildenberg, M. S. & Washington, M. T. The many roles of PCNA in eukaryotic DNA replication. Enzymes 39, 231–254 (2016).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
He, Y. F. et al. TET-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011).
Goll, M. G. & Bestor, T. H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514 (2005).
Chuang, L. S. et al. Human DNA-(cytosine-5) methyltransferase–PCNA complex as a target for p21WAF1. Science 277, 1996–2000 (1997).
Nomura, M. et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 9, 1903 (2019).
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Sawaya, R. et al. Neurosurgical outcomes in a modern series of 400 craniotomies for treatment of parenchymal tumors. Neurosurgery 42, 1044–1055 (1998).
Kickingereder, P. et al. Automated quantitative tumour response assessment of MRI in neuro-oncology with artificial neural networks: a multicentre, retrospective study. Lancet Oncol. 20, 728–740 (2019).
Yushkevich, P. A. et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 31, 1116–1128 (2006).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Scheinin, I. et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014).
Poell, J. B. et al. ACE: absolute copy number estimation from low-coverage whole-genome sequencing data. Bioinformatics 35, 2847–2849 (2019).
van de Wiel, M. A. et al. CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics 23, 892–894 (2007).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Tesileanu, C. M. S. et al. Non-IDH1-R132H IDH1/2 mutations are associated with increased DNA methylation and improved survival in astrocytomas, compared to IDH1-R132H mutations. Acta Neuropathol. 141, 945–957 (2021).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 8, 6 (2015).
Law, C. W. et al. A guide to creating design matrices for gene expression experiments. F1000Res. 9, 1444 (2020).
Stewart, S. K. et al. oxBS-450K: a method for analysing hydroxymethylation using 450K BeadChips. Methods 72, 9–15 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Germain, P. L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979 (2021).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
McKenzie, A. T. et al. Brain cell type specific gene expression and co-expression network architectures. Sci. Rep. 8, 8868 (2018).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Buehler, M. et al. Quantitative proteomic landscapes of primary and recurrent glioblastoma reveal a protumorigeneic role for FBXO2-dependent glioma-microenvironment interactions. Neuro Oncol. 25, 290–302 (2023).
Meier, F. et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236 (2020).
Giai Gianetto, Q., Wieczorek, L., Couté, Y. & Burger, T. A peptide-level multiple imputation strategy accounting for the different natures of missing values in proteomics data. Preprint at bioRxiv https://doi.org/10.1101/2020.05.29.122770 (2020).
Wang, J. et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 48, 768–776 (2016).
Acknowledgements
This project was funded by the Dutch Cancer Society (KWF Kankerbestrijding, project number 11026/2017-1, to W.R.V., K.A.v.G., B.A.W., M.Sm., M.J.v.d.B., P.W. and P.J.F.). We acknowledge the contribution of the PALGA foundation in the Netherlands and FGCZ. The CATNON study was funded by Merck, Sharp and Dohme (formerly Schering Plough) by an educational grant and by the provision of TMZ (to M.J.v.d.B.).
Author information
Authors and Affiliations
Contributions
Study design, P.A.R., M.Sm., M.v.d.W., R.G.W.V., M.J.v.d.B., B.A.W., P.W. and P.J.F. Conceptualization, W.R.V., Y.H., K.A.v.G., J.M.K., M.Sm., M.v.d.W., B.Y., R.G.W.V., M.J.v.d.B., B.A.W., P.W. and P.J.F. Methodology, W.R.V., Y.H., K.A.v.G., L.v.H., E.v.D., B.Y., W.W.J.d.L., M.J.v.d.B. and P.J.F. Formal analysis and investigation, W.R.V., I.d.H., Y.H., K.A.v.G., L.v.H., E.v.D., C.M.S.T., A.L., M.B., M.D., I.M. and P.P.E. Resources, W.R.V., K.A.v.G., L.v.H., M.C.M.K., J.M.N., K.D., W.W.J.d.L., C.M.S.T., A.L., W.W., P.M.C., M.Sm., E.F., T.G., M.W., P.A.R., J.M.K., T.W., V.G., M.Sa., M.J.v.d.B., P.W. and P.J.F. Validation, W.R.V., L.v.H., Y.H., C.M.S.T. Writing—original draft, W.R.V., M.J.v.d.B., P.J.F., B.A.W. and P.W. Writing—review and editing, all authors. Supervision and funding, P.W., M.J.v.d.B. and P.J.F.
Corresponding authors
Ethics declarations
Competing interests
P.J.F. declares research support from Servier. E.F. declares research support from Incyte Steering committee (uncompensated) and is on the Genenta Science advisory board. M.W. has received research grants from Novartis, Quercis and Versameb and honoraria for lectures or advisory board participation or consulting from Anheart, Bayer, Curevac, Medac, Neurosense, Novartis, Novocure, Orbus, Pfizer, Philogen, Roche and Servier. R.G.W.V. is cofounder of and holds equity in Boundless Bio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Jiayi Huang, Itay Tirosh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Longitudinal genomic changes.
a, Difference in tumor purity between initial and recurrent tumor samples, calculated by using two-sided Wilcoxon rank sum tests for the ACE estimated cellularity based on sWGS data (left, P = 0.33, 96 initial vs 113 recurrent tumor samples) and the RFpurify Absolute estimated purity based on DNA-methylation array data (right, P = 0.79, 103 initial vs 128 recurrent tumor samples). b, Differential CNA analysis between matched initial and recurrent tumor pairs (n = 93). Each panel represents a chromosome, with on the x-axis the chromosomal position of each segment breakpoint and on the y-axis the log2(read counts), centromere is allocated with a vertical grey line. Segments mean with error bars (SE = SD/sqrt(n)) are separately visualized for initial (orange) and recurrent tumor samples (purple). Significant differential segments between initial and recurrent tumors (in red) were identified using a paired two-sided Wilcoxon signed-rank test (BH-corrected, FDR < 0.05). Chromosomal regions chr1p12-q23.3, chr3p24.3-p11.1, chr9p24.3-p21.2, chr10q21.3-q26.3, chr14q11.2-q32.33, and chr22q11.21-q13.33 had significantly lower log2 read counts in recurrent tumors compared to initial tumors. c, Kaplan-Meier curves for PRS when stratifying patients on the CNA status (HD < -0.4 and LOH < -0.2 mean log2(read counts)) of differential regions (identified in Extended Data Fig. 1b) of the recurrent tumor. Compared to cases without an alteration, loss of chr3p24.3.p11.1 (LOH (n = 16), P = 7.74×10−4, HR = 2.900 (1.559-5.395); HD (n = 4), P = 0.075, HR = 2.564 (0.9101-7.223)), chr9p24.3-p21.2 (LOH (n = 25), P = 0.024, HR = 2.035 (1.098-3.770); HD (n = 18), P = 6.57×10−4, HR = 3.106 (1.618-5.963)) and chr10q21.3-q26.3 (LOH (n = 26), P = 8.62×10−5, HR = 2.968 (1.724-5.109); HD (n = 2), P = 0.004, HR = 8.888 (2.048-38.582), two-sided Wald test) were negatively associated with PRS. d, Hypermutant cut-off value for the GLASS-NL cohort. Upper panel shows the log scale of the TMB (muts/Mb) of each tumor sample, ordered based on the log(TMB). The elbow of the curve is located at 6 mutations per Mb and marked with a horizontal red dashed line. Samples located on the left side of the vertical red dashed line are considered to be hypermutated. Lower panel depicts the TMB per sample with the cut-off of 6 muts/Mb marked with a red dashed line. Hypermutant samples are colored in green. e, Regression coefficients plot of linear regression model testing the effect and pair-wise interaction of TMZ treatment and MMR/POLE mutations to the TMB (TMB ~ TMZ*MMR), with MMR/POLE wildtype tumor samples (15 TMZ treated vs 68 without CT) in the left, and MMR/POLE mutated tumor samples (6 TMZ treated vs 4 without CT) in the right panel. Estimates, 95% CI, and residuals are plotted and coefficients table is included (p-values are obtained using a two-sided Wald test).
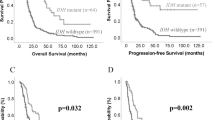
Extended Data Fig. 2 Glioma Methylation Signature identification.
a, DMRs on chromosomal region chr6p22.2-21.33 and overlapping genes of top DMRs (P < 1×10−40, n = 37). b, DMRs on chromosomal region chr11q12.2-13.5 and overlapping genes of top DMRs (P < 1×10−21, n = 41). c, GO enrichment analysis of genes overlapping with on chr11q12.2-13.5. Top 10 pathways are shown, the -log10(Pvalues) for overrepresentation of the gene-set are obtained using Wallenius’ noncentral hypergeometric distribution and FDR adjusted using BH correction. Xaxis depicts the percentage of genes from the gene-set that were differentially methylated, GO subontologies are indicated at the end of the bar for each gene-set. d, Regression coefficients plot of multiple linear regression models testing the effect and pair-wise interaction of known molecular markers of poor prognosis to the GMS-score (GMS ~ (markers)^2) in matched initial (circles) and recurrent (squares) tumor samples (n = 103). Estimates and 95% CI are plotted, the n for each molecular marker is indicated, p-values were calculated using a two-sided Wald test. e, Histogram depicting the distribution of GMS-scores of all GLASS-NL DNA-methylation samples. Bars are colored based on the assigned GMS-class (‘hyper’: >1 M; ‘intermediate’: 0 – 1 M; ‘hypo’: <0 M). f, Kaplan-Meier curve for OS when stratifying patients on the GMS-class of the initial tumor (‘hypo’ vs ‘hyper’, P = 0.699, HR = 1.326 (0.3162-5.564); ‘intermediate’ vs ‘hyper’: P = 0.105, HR = 1.929 (0.8720-4.266), two-sided Wald test). g,h, Multivariate CoxPH models on TTR and PRS with patient and tumor sample characteristics related to initial and recurrent surgery, respectively, including the M-value of all CpGs (DNAm Score) (g) or the GMS-score (h) as a continuous variable. Hazard ratios and 95% CI are plotted, P values are indicated. i, Kaplan-Meier curve for PRS when stratifying patients on the GMS-class and WHO grade of the recurrent tumor. WHO grade 2 and 3, and GMS-class hyper and intermediate are grouped together. Compared to the ‘WHO23/GMShigh’ group, all other groups of recurrent tumors were negatively associated with PRS (‘WHO23/GMSlow’, P = 0.004, HR = 6.306 (1.804-22.042); ‘WHO4/GMShigh’, P = 5.6×10−4, HR = 3.462 (1.711-7.005); ‘WHO4/GMSlow’: P = 1.35×10−10, HR = 8.031 (4.252-15.167), two-sided Wald test). j, Volcano plot depicting the CpG-level temporal changes in DNA-methylation of patients with CNS WHO grade 4 initial and recurrent tumors (n = 10). Differentially methylated CpGs, from a top 10.000 CpG selection, were identified using a paired two-sided Wilcoxon signed-rank test (BH-corrected, FDR < 0.05 & ΔM > 1).
Extended Data Fig. 3 Glioma Methylation Signature validation.
a, Histogram depicting the distribution of GMS-scores of CATNON DNA-methylation samples. Bars are colored based on the assigned GMS-class (‘hyper’: >1 M; ‘intermediate’: 0 – 1 M; ‘hypo’: <0 M). b, Kaplan-Meier curve for OS when stratifying patients of the TCGA cohort on the GMS-class. The hypo GMS-class was negatively associated with OS (‘hypo’, P < 0.001, HR = 4.0 (2.21-7.39); ‘intermediate’, P = 0.21, HR = 1.4 (0.81-2.51)). c-d, Kaplan-Meier curves for OS when stratifying based on the GMS-class of WHO grade 2 and grade 3 patients of the TCGA cohort, respectively. As compared to the hyper GMS-class, the hypo GMS-class was negatively associated with OS in WHO grade 2 tumors (c) (‘hypo’, P = 0.002, HR = 3.7495 (1.6344-8.601); ‘intermediate’, P = 0.316, HR = 1.4868 (0.6843-3.230)), and WHO grade 3 tumors (d) (‘hypo’, P = 0.004, HR = 3.9954 (1.55631-10.257); ‘intermediate’, P = 0.765, HR = 1.1407 (0.4812-2.704), two-sided Wald test). e, Output of multivariate CoxPH model on OS using data of the TCGA cohort, with the hyper GMS-class and WHO grade 2 as reference. f, Histogram depicting the distribution of GMS-scores of 5-hmc DNA-methylation samples of Glowaka et al.20. Cut-point for high and low GMS-scores was set at the median (red dotted line). g, Volcanoplot depicting the differential hydroxy-methylation analysis between GMS high (n = 21) and low (n = 21) samples of Glowaka et al. None exceeded the significance threshold of <0.05.
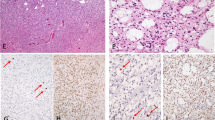
Extended Data Fig. 4 snRNA validation of gene expression clusters.
a, The integrated snRNA dataset of all snRNA samples (n = 6) from three IDH-mutant astrocytoma patients from the GLASS-NL cohort. b, UMAP projection of single nuclei of integrated dataset, colored by the enrichment score of the cell type clusters. c, Density plots of expression of gene expression cluster C2 (left) and C4 (right) by single nuclei. Dashed line indicates cutoff for cells to be marked as ‘positive’ cluster expression. d, Distributions of CNS WHO grades within the expression cluster value classes (‘high’: PC1 > 0; ‘low’: PC1 < 0) of C1 and C3 (graphs of C2 and C4 are found in Fig. 5d).
Supplementary information
Supplementary Tables 1–5
Supplementary Table 1: Identified DMRs between initial and recurrent tumor samples (n = 44,623), with their chromosomal location, number of CpGs and the overlapping genes. DMRs on the chromosome 6p locus are indicated. Supplementary Table 2. Output of differential methylation analysis between initial and recurrent tumor samples. The table includes all probes used for the analysis (n = 656,517), with their chromosomal position and relation to CpG island. Change (up and down) is indicated for all CpGs with FDR < 0.05, CpGs with FDR < 10−9 and absolute logFC > 1, which are included in the GMS. Supplementary Table 3. Output of differential expression analysis between initial and recurrent tumor samples. The table includes all genes used for the analysis (n = 22,346). Genes with FDR < 0.01 and absolute log2FC > 0.75 are differentially expressed (n = 604). The gene expression cluster (C1–C4) to which DEGs belong is indicated. Coefficients of covariates from multivariate analysis are also included for each gene. Supplementary Table 4. Output of differential protein expression analysis. The table includes all proteins used for the analysis (n = 3216). Change (up and down) is indicated for all DEPs with FDR < 0.05 and absolute logFC > 0.75 (n = 80). Supplementary Table 5. Output of mixed-effects model on differential methylation between treated and untreated patients. The table includes all probes used for the analysis (n = 656,517), with their chromosomal position and relation to CpG island. Change (up and down) is indicated for all CpGs with FDR < 0.05 (n = 13).
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vallentgoed, W.R., Hoogstrate, Y., van Garderen, K.A. et al. Evolutionary trajectories of IDH-mutant astrocytoma identify molecular grading markers related to cell cycling. Nat Cancer (2025). https://doi.org/10.1038/s43018-025-01023-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43018-025-01023-z
