
Abstract
Bisphenol A (BPA)-based epoxy materials are widely used for their excellent adhesion and strong mechanical properties in applications such as food packaging and medical devices. However, due to BPA’s endocrine-disrupting effects, there is increasing pressure to find safe alternatives for applications involving human exposure. Here, we address the urgent need for a biocompatible, BPA-free monomer with similar physico-mechanical properties to bisphenol A-glycidyl dimethacrylate (BisGMA), a crucial component in dental composites. We demonstrate a safe-and-sustainable-by-design approach with a bio-derived alternative: the lignin-derived bisguaiacol P vinyl ester resin, BGPGMA. Our findings show that BGPGMA is a promising non-estrogenic BisGMA analog, exhibiting excellent biocompatibility and superior physico-mechanical properties both as a monomer and in BGPGMA-based composites. This proof-of-concept highlights BGPGMA’s potential as a viable replacement for BisGMA, particularly in dental composites, and sets the stage for the development of safer, bio-based resins for wider industrial applications, including epoxy and polycarbonate plastics, that require BPA-free alternatives.

Similar content being viewed by others
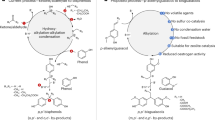

Introduction
Vinyl ester resins (VER) are recognized for their exceptional physico-mechanical properties, including favorable hydrophobicity, a high refractive index, relatively low volumetric shrinkage, and high reactivity, all of which contribute to the excellent durability and mechanical performance of composite materials used in structural applications. The key chemical structure responsible for these superior properties is the diphenylmethane (DPM) core of p-,p’-bisphenol A (BPA), which is characterized by its high rigidity and stiffness1. BPA-glycidyl dimethacrylate (BisGMA), a commercial crosslinking monomer derived from BPA, also named Bowen’s resin, is used in resin composites, thanks to its low polymerization shrinkage, high esthetic quality, and superior mechanical strength2. BisGMA is particularly well-suited for high-value materials like tooth-colored dental resin-based composites (RBCs, Fig. 1). RBCs, composed of silanated inorganic fillers embedded in a methacrylate resin matrix that is polymerized by blue light during tooth repair, are a cornerstone of modern dentistry. These composites are highly valued for their ability to bond effectively with tooth structure and their versatility in a wide range of dental applications. RBCs are most commonly used to esthetically restore teeth affected by decay, trauma, or wear, providing a cost-effective and tooth-preserving solution3.

Both BisGMA and BGPGMA were synthesized through three downstream reactions that follow the upstream (bio)refinery process, including dimerization of the respective fossil-based bisphenol (i.e., phenol, acetone) or bio-based bisguaiacol (i.e., isoeugenol, guaiacol) precursors, epoxidation, and esterification. A composite matrix was produced by mixing BGPGMA (or BisGMA) with TEGDMA and the CQ/EDMAB photo-initiation system. Barium-borosilicate glass filler was vacuum-mixed into the matrix to generate the bio-based composite.
However, support for using BisGMA in RBCs and other applications has diminished due to concerns about human exposure to BPA via its release in the oral environment4. BPA is classified as a substance of very high concern because of its endocrine-disrupting properties and its toxicity5. It interferes with the human endocrine system by mimicking natural oestrogens6, leading to a variety of adverse health effects, including infertility, diabetes, and immune system dysfunction7. Although BPA is not intentionally added to composites, it can be present as an impurity or degradation product8. Despite the low quantities of BPA released from RBC dental composites, the European Scientific Committee on Emerging Newly Identified Health Risks issued a statement in 2015 recommending that medical devices, including RBCs, should be BPA-free where practicable6. The European Food Safety Authority has progressively lowered the tolerable daily intake for BPA5, and in December 2024, they adopted a ban on the use of BPA in food contact materials9,10. Given the increasing public awareness of BPA exposure and the tightening regulatory limits, the use of BPA-based compounds, particularly in dental applications, is expected to be phased out in the near future.
Safer, preferably bio-based alternatives for BisGMA in composites with similar or improved physico-mechanical properties are thus desired. One effective strategy to eliminate the endocrine-disrupting potential of bisphenols is to inhibit their molecular interaction with hormone receptors by introducing steric and electronic hindrance at the ortho position to the phenolic OH11. Accordingly, ortho-methoxy bisphenols (i.e., bisguaiacols) are currently proposed as promising, safer alternatives to BPA, as we previously proved that these bisguaiacols significantly lower the estrogenic responses compared to BPA12. Unlike BPA, which originates from petroleum refineries, bisguaiacols can be derived from biomass. Emerging biorefinery technologies provide the necessary synthetic starting point for producing such bisguaiacols from non-edible, abundant lignocellulose biomass, such as wood13,14. Developing bio-based chemicals and materials is key in transitioning to a more sustainable circular (bio)economy, part of a worldwide strategy to combat climate change and global warming15.
Interestingly, bio-based alternatives for BisGMA have been presented in literature starting from sugars, such as isosorbide- and furan-based methacrylates16,17. These monomers featured mechanical properties similar to BisGMA but were quite water-sensitive due to their high hydrophilicity, making them less suitable for intraoral use16,18. In 2022, Sun et al. successfully improved the mechanical properties of bio-based dental composites, reporting a bio-based composite of a bisguaiacol, m,m’-BGF-4M. This bisguaiacol, derived from creosol, a lignin pyrolysis product, and formaldehyde, is characterized by the DPM core structure19. Their bio-based composite showed promising mechanical properties, surpassing those of the BisGMA composite. However, the synthetic pathway towards m,m’-BGP-4M follows the classic bisphenol hydroxyalkylation-alkylation condensation, which has several sustainable disadvantages, including the use of volatile organic alkylation agents, co-production of water, and low product selectivity due to the formation of oligomers, resulting in a complex purification process20,21. Trullemans et al. recently reported a more sustainable, highly selective pathway towards p,p’-bisguaiacols starting from major lignocellulose biorefinery products12. These p,p’-bisguaiacols consist of bisguaiacol structures, which, in contrast to m,m’-BGF-4M, not only preserve the DPM core structure but also the desired p,p’-isomer orientation of BPA, showing even lower in vitro estrogenic activity than m,m’-BGF-4M. From the presented portfolio on safe p,p’-bisguaiacols, p,p’-bisguaiacol P (BGP) appeared adequate to synthesize high-performance epoxy resins (i.e., the chemical intermediate for the synthesis of VER) and is, therefore, a promising building block for truly safe and sustainable RBCs12.
In this study, we introduce the BPA-free VER developed as a biosafe BisGMA analog. RBCs made from this BPA-free VER, a critical dental application in need of non-regrettable alternatives, demonstrated excellent biocompatibility and superior physico-mechanical properties compared to BisGMA-based composites (Fig. 1).
Results and discussion
BGP as a building block
We synthesized VER of BGP (i.e., BGPGMA) via a two-step reaction involving the synthesis of the epoxy resin followed by esterification with methacrylic acid (Fig. 1). The nucleophilic attack of the carboxylate in the esterification reaction can occur on both carbon atoms of the asymmetric epoxy ring, where the reaction on the, less substituted, more electrophilic terminal carbon is preferred. This induces two different end-groups: the major “normal” (n) end-group and the minor isomeric (i) end-group22,23. Combining both end-groups in VERs, such as BisGMA, led to three BisGMA isomers: BisGMAn,n, BisGMAn,i and BisGMAi,i. In Supplementary Fig. 1, the molecular structure of BisGMAn,i is presented with the characterizing protons assigned to their signal. Although 1H-NMR can be used to define the ratio of both end-groups, it cannot be used to determine their combinations; hence, the BisGMA isomer distribution. Therefore, the isomers and potential side products were quantified by high-performance liquid chromatography (HPLC) in combination with 1H-NMR22,24. Commercial BisGMA showed a purity of 94 wt.% with an isomer distribution of 2% BisGMAi,i, 26% BisGMAn,i, and 72% BisGMAn,n. In addition, bisphenol A diglycidyl ether (BADGE) and side products were observed, compromising 1 mol% of the substrate. Initially, we validated the synthetic procedure starting from the commercial BADGE, reaching full conversion and an isolated yield of 86% synthetic BisGMA with a purity and isomer distribution like commercial BisGMA (Supplementary Fig. 2, 3, Supplementary Table 1). To obtain BGP, the esterification reaction needed to be prolonged by 1 h to achieve full conversion and an isolated yield of 87% BGPGMA with a purity and isomer distribution similar to that of BisGMA. The HPLC, 1H, 2D 1H-13C HSQC, and 13C-NMR analysis of the BGPGMA product are presented in Supplementary Fig. 2–4 and Supplementary Table 1. The lower reactivity of BGP can be attributed to the mesomeric and weak inductive electron-donating effect of the o-methoxy moieties23. We obtained BGPGMA, molecular weight of 572,64 g/mol, as a highly viscous transparent liquid at room temperature. It should be noted that the resins, including synthesized BisGMA, were highly sensitive to colourizing during the experimental work-up; hence, colorless to yellowish resins were obtained.
BGPGMA features reactivity and polymerization kinetics similar to BisGMA
Due to its high viscosity induced by intermolecular hydrogen bonding, BisGMA is always combined in RBC formulations with low-viscous VERs, such as triethylene glycol dimethacrylate (TEGDMA)25. Generally, a formulation of an organic matrix including the base monomer (e.g., BisGMA) and diluent (e.g., TEGDMA) is characterized by a viscosity (η) around 1 Pa·s at 21 °C to strike a balance between workability and mechanical properties26. Therefore, viscosity is an important parameter that should be assessed in the quest for safer but functional BisGMA alternatives. We measured η for all resins by rotational rheology at 21 °C with a shear rate of 1 s−1 (Fig. 2a). Furthermore, the impact of TEGDMA on the viscosity of BisGMA and BGPGMA was investigated by determining the viscosity of mixtures with 40–60 mol% base monomer at 21 °C (Fig. 2a, b, Supplementary Table 2–4). The viscosity of BGPGMATEGDMA was twice that of BisGMATEGDMA for all matrix compositions, indicating that TEGDMA’s impact on η was similar for both resins. o-Methoxy groups are known to be hydrogen bond acceptors; therefore, this increase in η is most likely attributed to the formation of additional hydrogen bond interactions27. Noteworthy, certain excellent mechanical properties of BisGMA-based RBCs, such as impact strength, are also associated with intermolecular hydrogen bonding, which acts as additional physical crosslinks28. Hence, despite increasing resin viscosity, which can be tuned by composition optimization, the additional hydrogen bond interactions, here induced by lignin-typical functionalities (e.g., o-methoxy group), might be beneficial for the RBCs’ mechanical properties (vide infra). With η < 1 Pa·s, the 40 mol% resin composition, corresponding to ≈50 wt.% base monomer, resulted in 0.28 Pa·s for BisGMATEGDMA and 0.50 Pa·s for BGPGMATEGDMA, satisfying the viscosity requirements for RBCs (Fig. 2b).

a Viscosity of pure base monomer (VER) at 21 °C and a shear rate of 1 s−1 in Pa·s. b Viscosity at 21 °C and a shear rate of 50 s−1 in Pa·s of the BisGMATEGDMA (gray symbols) and BGPGMATEGDMA (green symbols) formulations with 40–100 mol% base monomer. Storage (G’) and loss (G”) modulus during photopolymerization. Photorheology of BisGMATEGDMA (c) and BGPGMATEGDMA (d) was conducted at an oscillatory strain of 0.1%, frequency of 1 Hz, gap-width of 100 µm, and light intensity of 10 mW cm−2 at 21 °C. After 2.5 min, the lamp was turned on for 10 min, revealing the storage modulus (dark) and loss modulus (light) for the whole-time frame. The lamp symbol indicates the time frame of irradiation; the arrow points to the moduli crossover or gel-time. e–g Conversion/polymerization rate profiles of BisGMATEGDMA (black, dashed line) and BGPGMATEGDMA (green, full line) at 21 °C. Photocalorimetry was conducted with a light intensity of 10 mW cm−2 at 21 °C. Conversion in function of time (e), polymerization rate in function of time (f), and polymerization rate in function of conversion (g) are shown.
The photocurable formulation consisted of the photoinitiator system (0.45 mol% camphorquinone [CQ] and 1.55 mol% ethyl 4-dimethylaminobenzoate [EDMAB]) and photocurable resins with preserved methacrylate groups29. In what follows, we studied the curing kinetics of BGPGMATEGDMA and BisGMATEGDMA, containing 40 mol% base monomer, to assess their potential in dental applications.
A critical parameter for radical polymerization is the time to gelation (i.e., gel-time), which indicates the formation of the polymer network30. This was determined by photorheometry, where the storage (G’), loss modulus (G”), and consequently gel-time were measured, as defined as the crossover point of both moduli (i.e., G’ = G”) during photopolymerization. For BisGMATEGDMA and BGPGMATEGDMA, a crossover after, respectively, 21.8 ± 2.0 s and 21.3 ± 0.3 s of light irradiation was observed, using a light intensity of 10 mW cm−2 and a sample thickness of 10 µm (Fig. 2c, d). The comparison showed that both formulations did not significantly differ in gelation time. Furthermore, the gel-time could be lowered by increasing the light intensity up to 23 mW cm−2, reaching a gel-time of 9 s (Supplementary Fig. 5).
The radical polymerization of VERs is an exothermic reaction where the polymerization heat (∆Hp) of methacrylate is 54.8 kJ mol−1 and can be monitored by photocalorimetry (photo-DSC, pDSC), a differential scanning calorimetry (DSC) equipped with additional blue light irradiation (≈500 nm, 10 mW cm−2)25. This technique has been used since 197931 to study the curing kinetics of photopolymerization, particularly of VERs for dental restorative purposes25,32. Hence, pDSC is an ideal tool to study the photopolymerization of the new bio-based VERs. At 21 °C, BisGMATEGDMA and BGPGMATEGDMA showed (I) similar conversion (α) with time, reaching a maximal conversion (\({\alpha }_{\max }\)) of, respectively, 0.43 and 0.45, (II) similar time to reach the maximum polymerization rate (\({{Rp}}_{\max }\)), being 36.8 s and 39.0 s, respectively, and (III) similar conversion at maximum polymerization rate, being for both 0.16 (Fig. 2e, g). However, at 37 °C and 50 °C, we found different relations (Supplementary Fig. 6, Supplementary Tables 3 and 4, including standard deviations (SD)). BisGMATEGDMA and BGPGMATEGDMA reached \({{Rp}}_{\max }\) at significantly different times, being 52.4 and 69.2 s, and conversions of 0.26 and 0.33 at 50 °C, respectively, resulting in a slightly higher final conversion of BGPGMATEGDMA (0.58) compared to BisGMATEGDMA (0.54).
During curing, the resin viscosity changes rapidly, resulting in diffusion limitations, which complicates the reaction kinetics. When conversion increases, the termination reaction becomes diffusion controlled at early stages of the reaction. This results in an increase of the radical concentration and a consequent increase of polymerization rate (i.e., auto-acceleration). At a certain conversion, the propagating radicals are more mobile by propagation than translational diffusion, and the reaction becomes reaction-diffusion controlled. During this reaction-diffusion mechanism, the termination reaction proceeds via the propagation reactions, and both reactions are proportional, resulting in a constant acceleration of the polymerization reaction. Ultimately, the propagation reaction gets diffusion controlled and the reaction rate starts to decelerate until the glass-transition temperature (Tg) of the system reaches the isothermal curing temperature (Tc) and vitrification takes place25,32. Accordingly, it is hypothesized that at elevated temperatures, diffusion limitation of the propagation reaction is delayed for BGPGMATEGDMA, resulting in a longer reaction-diffusion mechanism compared to BisGMATEGDMA. Dickens et al.25 observed this phenomenon for urethane demethacrylate (UDMA) and attributed this aspect to the higher flexibility of the UDMA molecule compared to BisGMA25,32. All this suggests that the backbone flexibility and its temperature dependence are higher for BGPGMA than for BisGMA, inducing deviation in the curing kinetics at temperatures higher than 21 °C. In general, a higher amount of base monomer (up to 50 mol%) in a formulation with TEGDMA lowers \({{Rp}}_{\max }\) and \({\alpha }_{\max }\) but improves the mechanical properties of the final cured materials25,33. Hence, these results imply that by increasing the base monomer content, a BGPGMATEGDMA formulation with curing kinetics (i.e., \({{Rp}}_{\max }\), \({\alpha }_{\max }\)) at elevated temperatures similar to that of BisGMATEGDMA can be obtained and result in materials with even better mechanical properties.
BGPGMA-based composite revealed superior physico-mechanical properties
BisGMA-based composite possesses good physico-mechanical properties, necessary to withstand daily chewing34. Despite efforts to replace BisGMA, manufacturers still prefer BisGMA as the main monomer in commercial RBCs, since current BisGMA-free RBCs do not guarantee sufficiently strong physico-mechanical properties and are often complicated and (more) costly to synthesize35. We developed two experimental composites that only differ in the BisGMA analog and subjected them to seven physico-mechanical tests to determine properties considered as minimum requirements RBC should meet. Rectangular one-sided mirror-polished samples were prepared for efficient measuring in a material-saving manner (Fig. 3). As a reference, a market-representative commercial composite (MajestyCOMP) was tested.

a Degree of conversion (DC in %) measured at time points 30 min, 2 h, 72 h, and 1 week. b Elastic modulus (EM in GPa) measured by the non-destructive impulse excitation technique (IET). c Flexural strength (FS in MPa) is measured by four-point bending. d Curves of the Weibull characteristic strength, which is the strength occurring at a failure probability of 63.2%. e Knoop hardness (KH in kgf/mm2). f KH was measured after water immersion for 1 week, 1 month, 4 months, 6 months, and 1 year. g Water absorption (Wa) and water solubility (Ws) in µg/mm3. h The color comparison was measured through Delta E (note that the commercial product contained a color-stabilizing BHT compound). In (a, f–h), error bars represent the mean ± standard deviation. In (b, c, e), the horizontal lines within each box plot indicate, from top to bottom: the maximum value, upper quartile, median, lower quartile, and minimum value.
Because it directly impacts the mechanical properties and monomer elution36, we first assessed the degree of conversion (DC). The quality of the cross-linked polymeric network after polymerization is a critical determinant in biocompatibility and overall clinical performance37. A significant difference in DC was recorded between the composites at corresponding time points, with the BGPGMA composite (BGPGMACOMP) revealing the highest 1 week DC (Fig. 3a). Aligning with the literature, the composite underwent dark polymerization after initial light-curing38,39. DC significantly increased with time to reach maximum DC at 72 h.
As primary mechanical properties, we next measured elastic modulus (EM), flexural strength (FS), and Knoop hardness (KH). BGPGMACOMP scored the significantly highest EM (Fig. 3b). The significantly higher FS for BGPGMACOMP and BisGMACOMP than MajestyCOMP was unexpected (Fig. 3c) since MajestyCOMP contains 78 wt.% (nano-hybrid) filler compared to 70 wt.% (micro-hybrid) filler within the experimental composites40. However, MajestyCOMP features a more complex formulation than the simplified experimental composites, as it is designed to achieve excellent esthetic properties for optimal tooth color matching41. This requires the incorporation of pigments and other additives that enhance translucency and shade adaptation. However, these pigments can absorb light, potentially interfering with the photoinitiator system and reducing polymerization efficiency, which may, in turn, lower the physico-mechanical properties of the material2,42. The higher Weibull modulus (m) recorded for MajestyCOMP is indicative of a more consistent material (Fig. 3d)43. This must most likely be ascribed to the manufacturer’s fine-tuned production process as compared to our laboratory composite mixing. BGPGMACOMP featured the significantly highest KH, followed by BisGMACOMP (Fig. 3e). As both in-house prepared composites contain the same filler (amount), the matrix monomer must have significantly impacted KH, which indicates that BGPGMACOMP should be more resistant to wear and scratching44. RBCs should withstand the challenging wet oral environment. Upon water sorption, hydrolytic degradation can result in polymer-matrix degradation and filler-matrix decoupling, hereby irreversibly affecting the composite structure and impacting mechanical strength44,45. To evaluate the resistance of the composite to hydrolytic degradation, the essential parameters KH upon water storage, as well as water absorption (Wa) and solubility (Ws), were assessed at specific time points up to 1 year. Despite BGPGMA’s slightly higher hydrophilicity, BGPGMACOMP’s KH remained relatively stable during the 1-year water storage and was always significantly higher than that of the other composites (Fig. 3f). We recorded a significant increase in Wa between 1 week and 1 month for all composites, whereafter Wa leveled off. BGPGMACOMP revealed a slightly but significantly higher Wa at all time points than the other composites, except at 4 months (Fig. 3g). This higher Wa of BGPGMACOMP must be attributed to the o-methoxy moieties, enhancing water sorption as compared to BisGMA46. Ws was similar for all composites (Fig. 3g). Interestingly, negative or almost zero Ws was mostly recorded, suggesting the absorbed water was bound to the hydrophilic groups so that no net solubility was measured.
As the final parameter of clinical relevance concerning esthetic performance, we determined the color stability of the experimental RBCs, having been reported in terms of Delta E (ΔE). Generally, ΔE above 3 can be detected by the human eye47. For all composites, there was a significant increase in ΔE compared to baseline (2 weeks after curing, before water immersion). All composites significantly differed for ΔE at the same time points, with the highest ΔE recorded for BGPGMACOMP at 6 months (Fig. 3h). Despite the somewhat less favorable color stability of the here proposed BGPGMA, much potential remains to optimize the experimental composite formulation, for example, by adding inhibitors such as butylated hydroxytoluene (BHT) or preferably safer antioxidants. We are currently investigating possibilities to optimize the composition accordingly.
Overall, the selected market-representative composite MajestyCOMP exhibited lower values for the tested physico-mechanical properties, while BGPGMACOMP, formulated with the novel BisGMA analog, showed superior physico-mechanical behavior. This underscores the great potential for BGPGMA as a BisGMA alternative. However, future research should focus on refining the experimental composite formulation to optimize not only its mechanical properties but also its esthetic characteristics, ensuring a well-balanced material suitable for clinical applications.
BGPGMA and its composite are biocompatible
Besides excellent physico-mechanical properties, BGPGMA is also biocompatible. This is very important as RBCs may leach residual monomer due to incomplete in situ polymerization48,49,50,51,52. Thorough biocompatibility assessment of novel monomers is thus warranted. Evaluating biocompatibility involves a battery of in vitro tests assessing various endpoints and toxicity mechanisms53. We evaluated cytotoxicity by assessing metabolic activity (XTT assay) and membrane integrity (lactate-dehydrogenase (LDH) assay), as well as genotoxicity and bioaccumulation potential. We tested not only the monomers but also their precursors, which may also leach from the composites and cause biocompatibility issues. This goes beyond the conventional approach outlined in ISO 10993 (“Biological evaluation of medical devices”) that emphasizes evaluating only the end-product (i.e., RBC)54.
Half maximal effective concentration (EC50) for cytotoxicity of the here synthesized bisguaiacol precursor BGP, the bisguaiacol epoxide, and BGPGMA showed no statistically significant differences compared to BPA, BPA-epoxide, and BisGMA, respectively (Fig. 4a). BisGMA’s EC50, derived from LDH measurements, aligns well with previous research findings48,50,55,56. However, a dose-dependent response could not be recorded for BGPGMA in the XTT assay, not allowing EC50 to be calculated (Supplementary Fig. 7). This could be due to cellular properties, compound characteristics, or assay conditions. It is possible that the interaction between cells and compounds caused minimal disruptions in cell membranes, leading to LDH leakage from the cytosol while viable mitochondria remained, enabling the metabolism of XTT salt that may have generated the observed assay results. Furthermore, using light microscopy, we noticed compound darkening for the two highest concentrations (0.5 and 1 mM) that exhibited cytotoxicity in the LDH assay. This can most likely be ascribed to compound agglomeration at the well bottom due to low monomer solubility in the aqueous solution. Such darkening may mask cytotoxic effects in the XTT assay. In the LDH assay, supernatant with leaked cytosol was collected to measure lactate-dehydrogenase activity, and did not affect data recording.

a EC50 of BGP- and BPA-based bisphenols/guaiacols, epoxides, and monomers, as assessed by the LDH and XTT assays. b TailDNA% as assessed by comet assay. Data are presented as mean ± standard deviation (SD) of medians of cells scored per test condition per replicate; *indicates statistical significance at p < 0.05; **indicates statistical significance at p < 0.01 (One-Way ANOVA, post-hoc Tukey’s) (n = 3; in triplicate). c Computed logP. d Relative cell viability (mean% ± SD) after exposure to eluates from BGPGMACOMP, BisGMACOMP, and MajestyCOMP composites.
An initial cell proliferation assay (Incucyte Label-Free Cell Proliferation Assay) revealed significant differences in cell proliferation after 24 h exposure to subcytotoxic concentrations of BGPGMA, when compared to the positive control, methyl methanesulfonate (MMS), a widely used DNA-alkylating agent in genotoxicity testing (Supplementary Fig. 8). Following this, a comet assay involving 3 h exposure to subcytotoxic concentrations of BGPGMA and BisGMA, equivalent to 1/4 EC50LDH, 1/8 EC50LDH, and 1/16 EC50LDH, revealed no statistically significant differences between the BisGMA and BGPGMA for various concentrations when DNA damage was assessed by measuring DNA percentage in the comet tail (TailDNA%). BisGMA at concentrations corresponding to 1/4 EC50LDH and 1/8 EC50LDH caused significantly more DNA damage than the negative control (Fig. 4b).
The octanol-water partition coefficient (logP) was calculated in silico as a surrogate measure of bioaccumulation potential of the rather hydrophobic monomers, since chemicals with logP greater than 5 are considered to have bioaccumulation potential57. Interestingly, BGPGMA exhibited a lower logP (4.11) compared to BisGMA (4.94) (Fig. 4c).
Cytotoxicity of extracts from the experimental composites and MajestyCOMP was assessed by XTT and LDH assays following ISO 10993-5, and we found no cytotoxic effects (Fig. 4d)54. Additionally, cell-cycle analysis of cells exposed to composite extracts to identify potential subcytotoxic effects did not reveal any difference in cell-cycle distribution among the three composites tested (Supplementary Fig. 9).
Conclusion
We synthesized bio-based BGPGMA, a safe and sustainable alternative to BisGMA derived from wood. This monomer not only demonstrated excellent biocompatibility but also imparted similar to superior physico-mechanical properties to composites compared to BisGMA, due to the retained p,p’-orientated DPM core. As a first successful example, proof-of-concept was established that BGPGMA can replace BisGMA in dental RBCs, opening the door for the development of a next generation of biosafe, BPA-free dental materials. More broadly, in the context of the global effort to phase out BPA, the knowledge gained from this research is highly relevant for the resin industry as a whole, including epoxy and polycarbonate resins, to produce high-performance, BPA-free alternatives.
Methods
Chemicals and materials
0.5% Trypsin-EDTA, 250 µg/mL amphotericin B, Dulbecco’s Modified Eagle Medium (DMEM), Glutamax cell-culture medium, fetal bovine serum (FBS), Hanks’ Balanced Salt Solution (HBSS), penicillin (10,000 Units/mL)-streptomycin (10,000 µg/mL), and SYBR Gold (10,000x) were supplied by Life Technologies (Merelbeke, Belgium). The chemicals 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)−2H-tetrazolium-5-carboxanilide (XTT), agarose, BisGMA (CAS 1565-94-2), CQ (Cas nr: 10373-78-1), dimethyl sulfoxide (DMSO), ethylenediaminetetraacetic acid (EDTA), EDMAB (Cas nr: 10287-53-3), HCO3Na, low gelling temperature agarose, nicotinamide adenine dinucleotide (NADH), phosphate buffered saline (PBS), pyruvate, TEGDMA, Triton X-100, trizma base, triphenylphosphine (TPP, Cas Nr 603-35-0) were purchased from Sigma-Aldrich (Diegem, Belgium). Bisphenol A (BPA; 80-05-7) was acquired from TCI Europe (Zwijndrecht, Belgium). 2,6-di-tert-butyl-4-methyphenol (BHT, Cas nr: 128-37-0), methacryl acid (MAA, Cas Nr: 79-41-4), and chloroform with 0.03 vol% tetramethylsilane (CDCl3 0.03 vol% TMS; Cas Nr: 865-49-6) were purchased from Fisher Scientific (Brussels, Belgium).
N-methyl dibenzo pyrazine methyl sulfate (PMS) was obtained from AppliChem (Darmstadt, Germany). We also used cavity slides with PTFE coating and sodium chloride, which were supplied by VWR International (Leuven, Belgium). Sodium hydroxide (NaOH) was obtained from Merck KGaA (Darmstadt, Germany). Additionally, Clearfil Majesty Es-2 Classic (Shade A3) was generously provided by Kuraray Noritake (Tokyo, Japan).
Reaction procedures
The gram-scale synthesis of VERs from lignin-derived substrates involves a three-step reaction process. In the first step, BGP was synthesized. This was followed by the synthesis of the intermediate epoxy resin, BGPDGE, which was then used to produce the final VER BGPBMA. The synthesis of BGP- and BGPDGE, its purification, and analysis are well described by Trullemans et al.12 as follows: For the synthesis of BGP, a fed-batch reactor is used. 200 mmol of guaiacol (25.34 g) was added to 0.5 mmol catalytic H+ in a 100 mL round-bottom flask containing a magnetic stirring bar (13 × 3 mm). The flask was sealed with a rubber septum and flushed with N2. The flask was submerged in a preheated, magnetically stirred (stir bar: 25 × 6 mm, 750 rpm), temperature-controlled oil bath at 80 °C. To start the reaction, 10 mmol (1.64 g) of isoeugenol was continuously administered for 3 h via a syringe infusion pump system (B. Braun Perfusor® Space) equipped with a long needle (B. Braun Sterican®, 0.8 × 120 mm) and 20 mL syringe (BD DiscarditTM). After the addition, the reaction was stirred for one extra hour to reach full conversion. After a multi-gram-scale synthesis, the reaction mixture was filtered and excess guaiacol was removed via reduced-pressure distillation (90 °C, 7 mbar). The p,p’-regioisomer was purified and isolated by hot (re)crystallization from n-heptane. The obtained crystals were dried in vacuo, pulverized, and analyzed by GC-FID to assess the purity. Samples were recrystallized in case the regioisomeric purity was below 99.5%. Then the epoxy resin was prepared in a two-step reaction process. In the first step, 4–12 mmol of BGP and 0.2 eq. of the catalyst (TEBAC) were dissolved in an excess of 20 eq. epichlorohydrin in a sealed single-neck round-bottom flask. After purging with N2 gas for 5 min, the reaction was started in a temperature-controlled oil bath (80 °C) and stirred (750 rpm) for 3 h. Subsequently, the reaction mixture was cooled down in an ice bath (0 °C). The second step was initiated by adding dropwise 4 eq. of NaOH in a 40 wt.% aqueous solution to the stirred reaction vessel at RT. After 3 h of reaction, 110 mL of DCM was added, and the reaction mixture was washed until pH 7 was reached. The organic phase was then dried over magnesium sulfate, filtered, and concentrated via reduced-pressure distillation. Prior to analysis with 1H-NMR, HPLC, and GPC, solvent trace amounts were removed in vacuo overnight. BGPDGE was obtained as an off-white solid, m.p. 94 °C, with an absolute yield of 94% and purity higher than 97 mol%.
In this work, the VER was synthesized from the BGPDGE. Here, 60 mmol of MAA was added to 3 mmol epoxy resin, 0.14 mmol triphenylphosphine (PPh3) catalyst, and 0.015 mmol BHT stabilizer in a sealed round-bottom flask. The excess of MAA directed the reaction towards high selectivity and yield. Additionally, no extra solvents were required for producing the high-viscosity resin. After purging with N2 gas for 5 min, the reaction was launched in a preheated oil bath at 100 °C and stirred (750 rpm) for 4 h for BADGE or 5 h for BGP epoxy resin. Subsequently, MAA was removed via distillation under reduced pressure to quench the reaction. Then, residual MAA was removed by extraction in dichloromethane (DCM) using 0.5 wt.% Na2CO3. The organic phase was dried over magnesium sulfate, filtered, and concentrated under reduced pressure. Noteworthy, the VERs were synthesized following conventional synthetic procedures to obtain a product composition comparable to the commercial BisGMA monomer (e.g., isomer distribution). Alternatively, Shah et al. reported the synthesis of glycidyl methacrylate (GMA) from waste glycerin, a side product from biodiesel production58. Here, the use of bio-based BGP and GMA resulted in fully bio-based dimethacrylate resins; therefore, both safer and sustainable alternatives for BisGMA.
Reaction analysis—product characterization
The epoxy resins and VERs were quantitatively analyzed by 1H-NMR, 1H, 13C HSQC-NMR, and 13C-NMR analysis and reversed-phase HPLC. 1H-NMR spectra were acquired on a Bruker Avance III 400-MHz (Bruker, Billerica, MA, USA), the 1H, 13C HSQC-NMR and 13C-NMR spectra on a Bruker Avance III 600-MHz (Bruker, Billerica, MA, USA) NMR spectrometer. All samples were prepared by dissolving 10-35 mg of the dried compound in 500 µl deuterated solvent (CDCl3 0.03 vol% TMS) and transferred to an NMR tube. Quantitative 1H-NMR integrations involved 64 scans with 5 s delay time. The relative integrations of the regions presented in Supplementary Fig. 1 were used to determine end-group selectivity and conversion. The complete NMR analysis of BGPGMA is presented in Supplementary Fig. 4.
To quantify the reaction mixture, gradient-elution reversed-phase HPLC was performed on an HPLC system (Waters TA Instruments, New Castle, DE, USA) equipped with a 280 nm UV detector and C18 column (250 × 4.6 mm, 5 µm packing size). The mobile phase consisted of two solvents: (A) 100 vol.% acetonitrile (ACN); (B) a solution consisting of 95 vol.% distilled water with 5 vol.% ACN. VER analysis was performed at a column temperature of 40 °C, with an injection volume of 15 µl and a flow rate of 1 ml/min. The following gradient elution of A:B was conducted: 0–1 min. 50:50; 1–22 min. linear-gradient to 62.5:37.5; 22–41 min. 100:0; 41–47 min. linear-gradient to 50:50. The samples were prepared by dissolving 2 mg of dried compound in 1 ml 50:50 A:B solvent. Note that for the analysis of all resins, a solution of 95 vol.% and 5 vol.% distilled water was used as solution A; the gradient elution was adapted to simulate the initial gradient, but a small shift in retention time was observed.
BGPGMA: Off-white liquid. 572.64 g/mol. HPLC 3% BGPGMAi,i, 24% BGPGMAi,n and 73%BGPGMAn,n 1H-NMR (400 MHz, CDCl3, 25 °C, TMS): δH (ppm) = 0.94 (t, 3H), 2.00 (s, 6H), 2.05 (q, 2H), 3.73 (t, 1H); 3.86 (s, 6H), [4.02 (dd), 4.10 (dd), 4.30 (m), 4.37 (dd), 5.27 (m)−10H], 5.64 (s, 2H), 6.18 (s, 2H), 6.78 (d, 2H), 6.81 (dd, 2H), 6.91 (d, 2H). 13C-NMR (150 MHz, CDCl3, 25 °C, TMS): δC (ppm) = 12.8, 18.3, 28.9, 52.5, 55.9, 62.2, 65.4, 68.4, 71.4, 72.9, 112.0, 115.1, 119.9, 126.1, 135.9, 139.5, 146.2, 149.6, 167.4.
Resin formulation
The photocurable formulations from BGPGMATEGDMA and BisGMATEGDMA contained TEGDMA (≈60 mol%), CQ (≈0.45 mol%), and EDMAB (≈1.55 mol%) in addition to the base monomer. The amounts were accurately measured, first mixed with a stirred bar at 1000 rpm (IKA RCT basic stirring plate, IKA, Staufen, Germany) and then sonicated (Branson 2800 sonication bath, Hilsonic, Wirral, UK) for 5 min to remove air and obtain a homogeneous formulation. The resin’s composition in mol% is provided in Supplementary Table 5.
(Photo)rheology
Rheology measurements were performed with an MCR702 twin drive rheometer (Anton Paar, Gent, Belgium) equipped with a parallel plate UV light-curing system, consisting of a UV mercury lamp (Omnicure S2000, Lumen Dynamics, Ontario, Canada) with a 500 nm external filter. To assure representative viscosity and photorheology measurement, the regions in which the resins (i.e., BGPGMA and BisGMA, and the formulations thereof) show ideally viscous behavior and follow Hooke’s law (Supplementary Fig. 10) were initially determined. The results showed an ideal viscous behavior at a shear rate of 1 s−1 for pure base monomer, a shear rate of 50 s−1 for the formulations, and linear viscoelastic behavior for a shear strain of 0.1 up to 100%.
The viscosities reported were obtained at 1 s−1 for the 100% pure VER samples and at 50 s−1 for the formulations of base monomer and TEGDMA at 21 °C or 37 °C. An 8 mm geometry (MCF702b-PP08) and gap-width of 200 µm was used in a parallel plate modus, using the UV light-curing system setup (Anton Paar). The viscosity experiments were repeated in triplicate. Photopolymerization studies were conducted at an oscillatory strain of 0.1%, frequency of 1 Hz, and gap-width of 100 µm, using a light intensity of 10 mW cm−2. After 2.5 min, the lamp was turned on for 10 min. Photorheology measurements were performed in fivefold. The average value was taken for the viscosity and gel-time results, with associated SD.
(Photo)calorimetry
When the photocurable BGPGMATEGDMA and BisGMATEGDMA formulations are irradiated with a monochromatic light source in the UV-Vis range59, the photoinitiator passes to its excited state to promote a radical or cationic polymerization reaction via a chain-propagation process60. Differential scanning photocalorimetry was performed with the differential scanning calorimeter DSC Q200 (Waters TA Instruments, New Castle, DE, USA) equipped with a photocalorimeter accessory, a UV mercury lamp (Omnicure S2000, Lumen Dynamics) light-source accessory and 500 nm external filter. Samples of 10 mg were placed in uncovered aluminum pans and mounted in the DSC cell together with an empty reference pan. Tc of 21 °C, 37 °C, or 50 °C was reached in 1 min. After 2.5 min, the lamp was turned on for 10 min with a light intensity of 10 mW cm−2, followed by 2.5 min of darkness at Tc. This cycle was repeated twice to (I) verify for further polymerization and (II) obtain an extrapolated baseline. The experiments were performed in triplicate under N2 (50 ml min−1). At all Tc and for both resins, the isothermal curing experiment was followed by a dynamic DSC experiment, from −30 to 300 °C with a heating ramp of 20 °C min−1 to detect heat-induced post-cure. All experiments were performed threefold; no subsequent polymerization upon light exposure was observed. The average value is taken for the maximal rate of polymerization and conversion results, with associated SD.
Composite and sample preparation
Two experimental composite formulations that only differed in the BisGMA analog were prepared following a standardized protocol. First, 49.5 wt.% BisGMA or novel BGPGMA monomer was hand-mixed with 49.5 wt.% TEGDMA, upon which 0.2 wt.% CQ and 0.8 wt.% EDMAB was added as a photoinitiator system, resulting in the composite matrix that comprised 30 wt.% of the final composite. Further, 70 wt.% barium-borosilicate glass filler (1.5% silanated) with an average particle size of 0.7 µm (Esschem Europe, Seaham, UK) was mixed with the 30 wt.% composite matrix in a DAC 400.2 VAC-P Speedmixer (Hauschild SpeedMixer, Hamm, Germany) following a standardized mixing protocol. The last mixing step took place under vacuum to remove air entrapment.
Customized rectangular silicon molds with dimensions of 25 × 3.2 × 2.2 mm were filled with composite. Through vibration and load application, the material was uniformly distributed over the mold while air entrapment was minimized. The whole specimen was homogenously light-cured from the top and bottom for 40 s with a Smartlite Pro (Dentsply Sirona, Konstanz, Germany) light-curing unit with an output of 1250 mW cm−2 and wavelength of 450–480 nm.
Clearfil Majesty ES-2 Classic (Kuraray Noritake) was used as a market-representative reference/control. Before mechanical testing, the composite samples were mirror-polished at one of the 3.2-mm long sides with a grinding/polishing device (Beta Grinder Polisher, Buehler, IL, USA) at a speed of 200 rpm under water spray. A total of 22 samples per composite, with final dimensions of 25 × 3 × 2 mm, were used for physico-mechanical testing. The specimens were stored in dry and dark conditions at 37 °C.
Physico-mechanical properties of experimental composites
DC was measured with micro-Raman spectroscopy (Senterra, Buker Optik, Ettlingen, Germany), which is a widely employed method, while the rectangular bar-shaped specimens used in this study resulted in poor contact with the ATR crystal when using FTIR61. The initial measurement, taken before curing, was used as the reference for calculating DC. Subsequent measurements were taken after curing at time points 30 min, 2 h, 72 h, and 1 week (n = 22). DC was determined by comparing the changes in the C=C stretching band relative to the pre-cure measurement. No internal standard was used. Micro-Raman spectra were acquired consecutively at 5 different points with the following settings: 785 nm Ar-ion laser, 100 mW power, 9–15 cm−1 resolution, 50 µm aperture, and 100× objective. The integration time was 20 s with the recorded spectra averaged over three co-additions. The DC of the composite was measured in the mold when covered with a 100 µm microscope cover glass to avoid oxygen inhibition (Supplementary Fig. 11). The DC was calculated using the following formula:
C aliphatic is the area under the absorption peak at 1638 cm−1 for the cured specimen, while C aromatic is the reference peak at 1608 cm−1 for the cured specimen. U aliphatic is the area under the absorption peak at 1638 cm−1 for the uncured specimen, while U aromatic is the reference peak at 1608 cm−1 for the uncured specimen38,61,62,63 (Supplementary Fig. 12).
EM was measured in the same samples with a non-destructive impulse excitation technique (“IET”; GrindoSonic MK7, GrindoSonic, Leuven, Belgium) (n = 22).
FS was next measured with a universal material tester (Instron 5848 MicroTester, Instron, Norwood, MA, USA), by four-point bending (0.5 mm/min crosshead speed; 20 mm outer and 10 mm inner specimen span between supports) (n = 22), considered being more appropriate than three-point bending64. SEM (JSM-6610, Jeol, Tokyo, Japan) fractographic analysis was conducted to identify the cause of composite-specimen failure, following gold sputtering of the fractured specimen for enhanced surface imaging (Supplementary Fig 13).
The Weibull modulus as an indication of material reliability was determined by the Maximum Likelihood Estimation method with Minitab (Minitab, State College, PA, USA).
The composite specimens were next re-dimensioned into bars of 6 × 3 × 2 mm to measure KH (Falcon 400, Innovatest, Maastricht, The Netherlands) using a 50 gf load and 15 s dwell time (n = 44).
Color stability was evaluated by taking standardized pictures with an Elab greycard (Elab greycard, Emulation S. Hein, Briesgau, Germany) before and after water immersion for 1 week, 1 month, 4 months, 6 months, and 1 year (n = 9). Delta E-values on a scale from 0 to 100 were calculated with Adobe Photoshop (Adobe, San Jose, CA, USA), representing the degree of color difference.
Impact of water on the composite
Wa and Ws were measured following the ISO 4049 standard65, by first drying the specimens until a constant mass (m1) was reached, upon which the volume (V) was calculated. All specimens (n = 9) were randomly assigned into 5 groups for 1 week, 1 month, 4 months, 6 months, and 1 year immersion in distilled water. After the respective time, the specimens were removed from the water and again weighed (m2). After complete drying, the third mass (m3) was measured. Wa and Ws were calculated as follows:
Following water immersion and Wa/Ws measurement, KH was re-measured to assess the difference in KH before and after water immersion.
Cytotoxicity assessment by XTT and LDH assay
Primary human gingival fibroblasts (HGFs) were seeded in 96-well plates at a density of 20,000 cells/well. HGFs culture was established from residual gingival tissue that was collected from healthy adult patients undergoing treatment at the University Hospitals Leuven (UZ Leuven) (approval by UZ Leuven Ethics Committee: S45254). To be used in experiments, HGFs (passages 4–6) were cultured in 175 cm2 flasks at a constant atmosphere of 5% CO2 and 100% relative humidity at 37 °C, and DMEM supplemented with 10% FBS was used. Experiments were performed when cells reached monolayer confluence with a density of 80–90%.
After 24 h incubation, the cell-culture medium was removed, and 50 µL freshly prepared dilutions of the compounds were dosed to the cells. BGP- and BPA-based VERs and their precursors, bisphenols/guaiacols and epoxides, were tested as follows. A series of six dilutions of BPA and BGP, ranging from 6 to 0.19 mM, with a dilution factor of 1/2, and 6 dilutions of BADGE and BGP.DGE, ranging from 0.5 to 0.07 mM, with a dilution factor of 2/3, were prepared in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 1% fungizone (vehicle) from DMSO stock solution. Furthermore, a series of 6 dilutions of BGPGMA, ranging from 1.0 to 0.031 mM, and 6 dilutions of BisGMA, ranging from 0.5 to 0.016 mM, with a dilution factor of 1/2, were prepared using an indirect dosing method. To avoid precipitation, an additional series of six dilutions was prepared in 100% DMSO. Next, 10 µL DMSO dilution was added to the vehicle to obtain a 100-fold dilution that was dosed to the cells.
As negative and positive controls, cells were exposed to the vehicle containing either 1% DMSO or 1% Triton X-100, respectively. The viability of the exposed cells was evaluated 24 h after exposure by the XTT and LDH assay. Three independent assays were performed in triplicate.
The XTT assay evaluated the mitochondrial activity of the exposed cells via the cleavage of the yellowish tetrazolium salt XTT to form an orange formazan dye in the presence of the co-factor PMS.
After 20 h of exposure, 50 µL XTT solution, containing 1 mg/mL XTT and 0.383 mg/mL PMS at a ratio of 50:1, was added to the 96-well plate. After 4 h incubation, absorbance was measured at 450 nm (reference wavelength: 690 nm) with the Thermo Multiskan Ascent plate reader with the Ascent Software Version 2.6 (ThermoFisher, Asse, Belgium). Cell viability (%) was calculated as the ratio of absorbance measured for the treated cells over that of the negative control.
The integrity of the cellular membrane was evaluated with the LDH assay via the leakage of LDH. After 24 h exposure, 50 µL “supernatant” was collected and transferred to an empty 96-well plate. The cells were washed twice with PBS, and subsequently, the cells were incubated for 30 min with 200 µL of 0.2% Triton X-100 in PBS to lyse the cells. After 30 min, 50 µL of the “lysate” was transferred to an empty 96-well plate. Next, 100 µL HBSS was added to the “supernatant” and “lysate.” Right before the measurement, a 100 µL measurement mix (0.458 mg/mL pyruvate, 0.532 mg/mL NADH, and 0.794 mg/mL HCO3Na in PBS) was added. Absorbance was recorded 13 times in 15 s intervals at 340 nm using the Thermo Multiskan Ascent plate reader with the Ascent Software Version 2.6 (ThermoFisher). After determining the slope of the absorbance curves, cell viability was calculated according to the following formula: cell viability (%) = [slope of “lysate”/(slope of “lysate” + slope of “supernatant”)] × 100 and relative cell viability (%) = (viability of sample/viability of negative control) × 100.
Incucyte Label-Free Cell Proliferation Assay
For the Incucyte Label-Free Cell Proliferation Assay, HGFs at passage 4 were seeded in a 96-well plate at a low density of ~2000 cells/well in a volume of 100 µL/well. The plate was left in a laminar flow hood for ~30 min to allow cells to settle and start attaching. The plate was then placed in the IncuCyte ZOOM Live-Cell Analysis System (Essen BioScience, Ann Arbor, MI, USA) for 24 h to allow cells to attach and start dividing. After 24 h of incubation, the medium was removed from the wells, and the cells were exposed to serial dilutions of BGPGMA. The highest concentration was based on the calculated 80% relative viability from the LDH assay. Starting from 0.10 mM, a series of five dilutions were made using the indirect dosing method as described above. DMSO (1%) vehicle and MMS (50 µm) in DMEM served as negative and positive controls, respectively.
The cells were next monitored with IncuCyte ZOOM (Essen BioScience) for an additional 6 days, including images taken every 2 h with a 10× objective. Four images were taken per well at each time point, upon which they were analyzed using the accompanying software (version 2018A).
Cell proliferation was assessed by measuring the confluence of cells in each well. These confluence data were then used to generate growth curves for each treatment condition. Three independent experiments in triplicate were carried out.
Genotoxicity assessment
The genotoxic potential of BGPGMA and BisGMA was investigated in vitro with the comet assay (single-cell alkaline gel electrophoresis). HGFs at passage 4 or 5 were seeded in 96-well plates at a concentration of 20,000 cells/well in 100 µL of cell-culture medium per well. After 24 h of incubation, the medium was removed, and the cells were exposed to a serial dilution of BGPGMA and BisGMA. Serial dilutions were prepared according to the indirect dosing method as described above. Subcytotoxic concentrations corresponding to 1/16, 1/8, and 1/4 EC50LDH were prepared (concentrations of 0.050 mM, 0.025 mM, and 0.013 mM for BGPGMA, and concentrations of 0.023 mM, 0.011 mM, and 0.006 mM for BisGMA). As positive control, the cells were exposed to 50 µM MMS. As a negative control, cells were exposed to 1% DMSO in a vehicle.
After 3 h exposure, the compounds were aspirated, after which the cells were washed with PBS and detached from the wells with 0.25% Trypsin-EDTA. Approximately 10,000 cells in 75 µL PBS were mixed with 75 µL of 1% low-melting point agarose (37 °C), upon which 20 µL of cell suspension was applied per cavity on microscope slides pre-coated with 1% normal-melting agarose. The gels were left in the dark at 4 °C to allow agarose to set. Next, in-gel lysis was performed overnight in the dark at 4 °C in Coplin jars immersed in the lysis buffer containing 2.5 M NaCl, 100 mM EDA, 10 mM Trizma base, and 1 vol% Triton X-100. The slides were washed in PBS and placed in the electrophoresis buffer containing 300 mM NaOH and 1 mM EDTA (pH > 13) for 20 min at 4 °C to allow for alkaline unwinding of DNA. After the power supply was adjusted to ~0.74 V/cm2, electrophoresis was run for 20 min. The slides were rinsed three times for 5 min in neutralization buffer (0.4 mM Tris, pH = 7.5). The experiment was performed three times in triplicate.
The slides were then stained with 1:10000 SYBR Gold nucleic acid gel stain, upon which DNA damage was examined with a fluorescence microscope equipped with a fluorescence camera (Olympus BX61, Olympus Europa, Hamburg, Germany) using a 10× objective. Ideally, 450 cells per condition (150 cells per independent replicate) were analyzed with TriTek CometScore 2.0.0.0 (https://rexhoover.com/index.php?id=cometscore, accessed 23 June 2023) with the percentage of DNA in the comet’s tail (TailDNA%) measured as an indicator of DNA damage. The mean (±SD) DNA damage of medians of cells scored per test condition per replicate (n = 3 in triplicate) was calculated.
Partition coefficient (logP) prediction
In this study, the partition coefficient (logP) was predicted in silico using the EPI Suite-Estimation Program Interface (EPI Suite, US EPA, Washington D.C., USA). Specifically, the KOWWIN model (version 1.69, April 2015) within EPI Suite (US EPA) was used. This model is based on the atom/fragment-contribution method to estimate the octanol-water logP. The input for the model was the Simplified Molecular Input Line Entry System (SMILES) notation of the compounds, which were generated from structures drawn with ACD/ChemSketch 2020.1.0 Freeware (Advanced Chemistry Development, Toronto, ON, Canada). logP was calculated based on the sum of fragment and atom contributions and correction factors.
Biocompatibility of experimental composites
The experimental composites and the reference composite Clearfil Majesty ES-2 Classic (Kuraray Noritake) were assessed in vitro for biocompatibility according to the ISO 10993-5:2009 standard54.
Cylindrical composite specimens were prepared in custom-made Teflon molds (10-mm internal diameter, 1-mm thickness). Once placed, the composite was covered with a microscope glass slide to displace excess material, ensure a smooth surface, and prevent oxygen inhibition. The specimens were cured for 20 s from the top using the SmartLite Pro (Dentsply Sirona) light-curing unit, upon which sample extracts were prepared. Each composite specimen was briefly immersed in 70% ethanol and air-dried inside a laminar flow hood to avoid composition changes through sterilization. Eight specimens of each composite were next placed in a 24-well plate, of which each well received 500 μL DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% fungizone. All samples were incubated for 24 h at 37 °C.
All 8 extract samples were pooled and processed as one sample (4 mL extract sample in total) to be dosed to a well plate containing primary HGFs. A series of six dilutions was made from the extract samples with a dilution factor of 1/2. HGFs were seeded in 96-well plates at a density of 20,000 cells/well. After 24 h incubation, the cell-culture medium was removed, and 50 µL of the extracted sample was dosed to cells in triplicate and kept incubated for 24 h.
After 24 h of exposure, cell viability was determined by XTT and LDH assays. Four independent assays were performed as described above. Possible subcytotoxic effects on HGFs were evaluated by flow cytometry. During DNA synthesis (S phase), cells replicate their genetic material before entering the mitotic phase (M phase). The relative number of cells in the gap G0/G1 phase, S phase, and G2/M phase can be determined by analyzing the DNA content of the cells. HGFs were seeded in a 48-well plate (80,000 cells/well). After 24 h of attachment, the cell-culture medium was aspirated, and HGFs were exposed to 300 µL of undiluted extract samples. After 24 h incubation, the cells were collected, fixed in 70% ethanol, and frozen at −80 °C. DNA in the cells was next stained with DNA dye SYBR Gold, upon which 20,000 events per sample were analyzed with a flow cytometer (BD FACSVerse, Becton Dickinson, Erembodegem, Belgium) to determine the cell-cycle distribution of the cell population.
Statistical analysis
The statistical analysis of the physico-mechanical properties of experimental composites involved two-way ANOVA and Tukey’s multiple comparison test, except for EM, for which the Kruskal–Wallis test was used due to non-normal data distribution (α = 0.05). The EC50 of bisphenols/guaiacols, epoxides, and VERs was calculated and statistically compared with one-way ANOVA and post-hoc Tukey’s test (α = 0.05). The statistical analysis of the Incucyte Label-Free Cell Proliferation Assay involved multiple t-tests to compare each test condition with the negative control at each data point, aiming to identify statistically significant differences in cell proliferation. To correct for multiple testing, the obtained p values were plotted, and locally weighted scatterplot smoothing was applied, allowing continuous observation of differences in proliferation rates throughout the experiment across different test conditions compared to the negative control. Data from the genotoxicity assessment were compared by one-way ANOVA and Tukey’s test (α = 0.05) to determine statistical significance between groups.
Statistical analysis of the biocompatibility of experimental composites was performed only with the relative cell viability of the undiluted extracts, since no dose-response relationship was observed. To compare relative cell viability after exposure to the undiluted samples, separate one-way ANOVA tests (α = 0.05) were used for each assay to evaluate relative cell viability between different experimental groups. Two-way ANOVA was used to compare cell-cycle distribution between the different experimental groups. The significance level α was set at 0.05.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. The raw datasets generated during and/or analyzed during the current study are available in the RDR KU Leuven repository, «link will be shared as soon as possible».
References
Ferracane, J. L. Resin composite—state of the art. Dent. Mater. 27, 29–38 (2011).
Cho, K., Rajan, G., Farrar, P., Prentice, L. & Prusty, B. G. Dental resin composites: a review on materials to product realizations. Compos. B Eng. 230, 109495 (2022).
Pratap, B., Gupta, R. K., Bhardwaj, B. & Nag, M. Resin based restorative dental materials: characteristics and future perspectives. Jpn. Dent. Sci. Rev. 55, 126–138 (2019).
Seo, H. et al. Comprehensive assessment of the estrogenic activity of resin composites. Chemosphere 343, 140104 (2023).
European Food Safety Authority (EFSA). Bisphenol A in food is a health risk. https://www.efsa.europa.eu/en/news/bisphenol-food-health-risk (2023).
Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR). Opinion on the Safety of the Use of Bisphenol A in Medical Devices (European Commisson, 2015).
Rochester, J. R. Bisphenol A and human health: a review of the literature. Reprod. Toxicol. 42, 132–155 (2013).
Vandenberg, L. N., Hauser, R., Marcus, M., Olea, N. & Welshons, W. V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 24, 139–177 (2007).
Lambré, C. et al. Re-evaluation of the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J. 21, 6857 (2023).
Food Safety, European Commission. Commission adopts ban of Bisphenol A in food contact materials. https://food.ec.europa.eu/food-safety-news-0/commission-adopts-ban-bisphenol-food-contact-materials-2024-12-19_en (2024).
Koelewijn, S. F. et al. Regioselective synthesis, isomerisation, in vitro oestrogenic activity, and copolymerisation of bisguaiacol F (BGF) isomers. Green Chem. 21, 6622–6633 (2019).
Trullemans, L. et al. Renewable and safer bisphenol A substitutes enabled by selective zeolite alkylation. Nat. Sustain. https://doi.org/10.1038/s41893-023-01201-w (2023).
Sun, Z., Fridrich, B., De Santi, A., Elangovan, S. & Barta, K. Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 118, 614–678 (2018).
Schutyser, W. et al. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 47, 852–908 (2018).
Wagner, M. et al. State of the science on plastic chemicals—identifying and addressing chemicals and polymers of concern. https://doi.org/10.5281/zenodo.10701706 (2024).
Łukaszczyk, J., Janicki, B. & Frick, A. Investigation on synthesis and properties of isosorbide based bis-GMA analogue. J. Mater. Sci. Mater. Med. 23, 1149–1155 (2012).
Kainulainen, T. P., Erkkilä, P., Hukka, T. I., Sirviö, J. A. & Heiskanen, J. P. Application of furan-based dicarboxylic acids in bio-derived dimethacrylate resins. ACS Appl. Polym. Mater. 2, 3215–3225 (2020).
Saxon, D. J., Luke, A. M., Sajjad, H., Tolman, W. B. & Reineke, T. M. Next-generation polymers: Isosorbide as a renewable alternative. Prog. Polym. Sci. 101, 101196 (2020).
Sun, Y. et al. Preparation and evaluation of novel bio-based Bis-GMA-free dental composites with low estrogenic activity. Dent. Mater. 38, 281–293 (2022).
Farnham, A. & Klosek, F. P. Manufacture of diphenylol methanes. https://patents.google.com/patent/US2812364A/en (1957).
Koelewijn, S. F. et al. Sustainable bisphenols from renewable softwood lignin feedstock for polycarbonates and cyanate ester resins. Green Chem. 19, 2561–2570 (2017).
Sheela, M. S., Selvy, K. T., Krishnan, V. K. & Pal, S. N. Studies on the synthesis of a methacrylate‐based dental restorative resin. J. Appl. Polym. Sci. 42, 561–573 (1991).
Pal, N., Srivastava, A. & Rai, J. S. P. Synthesis of vinyl ester resins in the presence of monoepoxies: a kinetic study. Polym. Plast. Technol. 42, 105–122 (2003).
Munteanu, D., Işfan, A. & Bratu, D. High-performance liquid chromatographic separation of BisGMA oligomers and isomers in dental restorative materials. Chromatographia 23, 412–418 (1987).
Dickens, S. H., Stansbury, J. W., Choi, K. M. & Floyd, C. J. E. Photopolymerization kinetics of methacrylate dental resins. Macromolecules 36, 6043–6053 (2003).
Beun, S. et al. Rheological properties of experimental Bis-GMA/TEGDMA flowable resin composites with various macrofiller/microfiller ratio. Dent. Mater. 25, 198–205 (2009).
Stansbury, J. W. Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent. Mater. 28, 13–22 (2012).
Barszczewska-Rybarek, I. & Jurczyk, S. Comparative study of structure-property relationships in polymer networks based on Bis-GMA, TEGDMA and various urethane-dimethacrylates. Materials 8, 1230–1248 (2015).
Crivello, J. V. & Reichmanis, E. Photopolymer materials and processes for advanced technologies. Chem. Mater. 26, 533–548 (2014).
Bassett, A. W. et al. Vanillin-based resin for additive manufacturing. ACS Sustain. Chem. Eng. 8, 5626–5635 (2020).
Tryson, G. R. & Shuktz, A. R. A calorimetric study of acrylate photopolymerization. J. Polym. Sci. 17, 2059–2075 (1979).
Cook, W. D. Thermal aspects of the kinetics of dimethacrylate photopolymerization. Polymer. 33, 2152 (1992).
Gonçalves, F., Kawano, Y., Pfeifer, C., Stansbury, J. W. & Braga, R. R. Influence of BisGMA, TEGDMA, and BisEMA contents on viscosity, conversion, and flexural strength of experimental resins and composites. Eur. J. Oral. Sci. 117, 442–446 (2009).
Low, I. M., Duraman, N. & Mahmood, U. Mapping the structure, composition and mechanical properties of human teeth. Mater. Sci. Eng. C 28, 243–247 (2008).
He, J., Lassila, L., Garoushi, S. & Vallittu, P. Tailoring the monomers to overcome the shortcomings of current dental resin composites—review. Biomater. Investig. Dent. 10, 2191621 (2023).
Sarcev, B. P. et al. Effect of the degree of conversion on mechanical properties and monomer elution from self-, dual-and light-cured core composites. Materials 14, 5642 (2021).
Ferracane, J. L. et al. Academy of dental materials guidance—resin composites: Part II—technique sensitivity (handling, polymerization, dimensional changes). Dent. Mater. 33, 1171–1191 (2017).
Pongprueksa, P., De Munck, J., Inokoshi, M. & Van Meerbeek, B. Polymerization efficiency affects interfacial fracture toughness of adhesives. Dent. Mater. 34, 684–692 (2018).
Decker, C. Photoinitiated crosslinking polymerisation. Prog. Polym. Sci. 21, 593–650 (1996).
Kim, K.-H., Ong, J. L. & Okuno, O. The effect of filler loading and morphology on the mechanical properties of contemporary composites. J. Prosthet. Dent. 87, 642–649 (2002).
CLEARFIL MAJESTY ES-2. Kuraray Noritake Dental Inc. https://www.kuraraynoritake.com/world/product/composites/clearfil_majesty_es2.html.
Leyva del Rio, D. & Johnston, W. M. Optical characteristics of experimental dental composite resin materials. J. Dent. 118, 103949 (2022).
Zhao, D., Botsis, J. & Drum, J. L. Fracture studies of selected dental restorative composites. Dent. Mater. 13, 198–207 (1997).
De Moraes, R. R. et al. Effects of 6 months of aging in water on hardness and surface roughness of two microhybrid dental composites. J. Prosthodont. 17, 323–326 (2008).
Szczesio-Wlodarczyk, A., Sokolowski, J., Kleczewska, J. & Bociong, K. Ageing of dental composites based on methacrylate resins—a critical review of the causes and method of assessment. Polymers 12, 882 (2020).
Gilormini, P. & Verdu, J. On the role of hydrogen bonding on water absorption in polymers. Polymer 142, 164–169 (2018).
Ruyter, I. E., Nilner, K., Eystein Ruyter, I. & -Scandinavian, N. Color stability of dental composite resin materials for crown and bridge veneers. Dent Mater. 3, 246–251 (1987).
Geurtsen, W., Lehmann, F., Spahl, W. & Leyhausen, G. Cytotoxicity of 35 dental resin composite monomers/additives in permanent 3T3 and three human primary fibroblast cultures. J. Biomed. Mater. Res. 41, 474–480 (1998).
Durner, J., Wellner, P., Hickel, R. & Reichl, F. X. Synergistic interaction caused to human gingival fibroblasts from dental monomers. Dent. Mater. 28, 818–823 (2012).
Styllou, M. et al. Dental composite components induce DNA-damage and altered nuclear morphology in gingiva fibroblasts. Dent. Mater. 31, 1335–1344 (2015).
Putzeys, E. et al. Long-term elution of monomers from resin-based dental composites. Dent. Mater. 35, 477–485 (2019).
De Nys, S. et al. Bisphenol A as degradation product of monomers used in resin-based dental materials. Dent. Mater. 37, 1020–1029 (2021).
Schmalz, G. & Galler, K. M. Biocompatibility of biomaterials—lessons learned and considerations for the design of novel materials. Dent. Mater. 33, 382–393 (2017).
International Organization for Standardization (ISO). Biological evaluation of medical devices (ISO 10993-1:2018). https://www.iso.org/standard/68936.html (2018).
Issa, Y., Watts, D. C., Brunton, P. A., Waters, C. M. & Duxbury, A. J. Resin composite monomers alter MTT and LDH activity of human gingival fibroblasts in vitro. Dent. Mater. 20, 12–20 (2004).
Moharamzadeh, K., Van Noort, R., Brook, I. M. & Scutt, A. M. Cytotoxicity of resin monomers on human gingival fibroblasts and HaCaT keratinocytes. Dent. Mater. 23, 40–44 (2007).
Arnot, J. A. & Gobas, F. A. P. C. A generic QSAR for assessing the bioaccumulation potential of organic chemicals in aquatic food webs. QSAR Comb. Sci. 22, 337–345 (2003).
Shah, P. N. et al. Environmentally benign synthesis of vinyl ester resin from biowaste glycerin. RSC Adv. 5, 38673–38679 (2015).
Ahn, D., Stevens, L. M., Zhou, K. & Page, Z. A. Rapid high-resolution visible light 3D printing. ACS Cent. Sci. 6, 1555–1563 (2020).
Bagheri, A. & Jin, J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 1, 593–611 (2019).
Miletic, V. & Santini, A. Micro-Raman spectroscopic analysis of the degree of conversion of composite resins containing different initiators cured by polywave or monowave LED units. J. Dent. 40, 106–113 (2012).
Gatin, E. et al. Raman spectroscopy as spectral tool for assessing the degree of conversion after curing of two resin-based materials used in restorative dentistry. Diagnostics 12, 1993 (2022).
Pianelli, C., Devaux, J., Bebelman, S. & Leloup, G. The micro-Raman spectroscopy, a useful tool to determine the degree of conversion of light-activated composite resins. J Biomed Mater Res.48, 657-81 (1999).
Ilie, N. et al. Academy of dental materials guidance—resin composites: Part I—mechanical properties. Dent. Mater. 33, 880–894 (2017).
International Organization for Standardization (ISO). Dentistry—polymer-based restorative materials (ISO 4049:2019). https://www.iso.org/standard/67596.html (2019).
Acknowledgements
We gratefully acknowledge Steven-Friso Koelewijn (CSCE, Centre for Sustainable Catalysis and Engineering, KU Leuven) for initiating the project, Stijn Anthonissen (CSCE, Centre for Sustainable Catalysis and Engineering, KU Leuven) for his contribution to upscale the production of the novel monomer, Ben Mercelis (BIOMAT, Department of Oral Health Sciences, KU Leuven) for his technical support with the biocompatibility and physico-mechanical property testing, and Evita Willems (MTM, Department of Materials Engineering, KU Leuven) for the development of the IET protocol. This study was supported by the Research Foundation—Flanders (FWO) grants G0B2618N and G0C7320N, the KU Leuven funds C32/18/021, C24E/19/078, and the Industrial Research Fund ZKC6712.
Author information
Authors and Affiliations
Contributions
Line Etiennot: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—review & editing, and visualization. Laura Trullemans: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—review & editing, and visualization. Marko Turkalj: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—review & editing, and visualization. Fei Zhang: methodology, validation, resources, data curation, writing—review & editing, and supervision. Rashmi Singh: methodology and writing—review & editing. Siemon De Nys: conceptualization, methodology, formal analysis, investigation, and data curation. Fatima Rammal: methodology, validation, formal analysis, and data curation. Xin Li: conceptualization, methodology, writing—review & editing, and funding acquisition. Marleen Peumans: conceptualization, resources, writing—review & editing, and supervision. Manosij Ghosh: methodology, validation and supervision. Bert Lagrain: conceptualization, formal analysis, resources, data curation, writing—review & editing, supervision, project administration, and funding acquisition. Peter Van Puyvelde: methodology, resources, writing—review & editing, supervision, project administration, and funding acquisition. Jeroen Vanoirbeek: resources, writing—review & editing, supervision, project administration, and funding acquisition. Peter Hoet: conceptualization, methodology, resources, writing—review & editing, and supervision. Bart Van Meerbeek: conceptualization, methodology, resources, writing—review & editing, visualization, supervision, project administration, and funding acquisition. Bert Sels: conceptualization, methodology, resources, writing—review & editing, visualization, supervision, project administration, and funding acquisition. Kirsten L. Van Landuyt: conceptualization, methodology, resources, writing—review & editing, visualization, supervision, project administration, and funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Steven Caliari and Jet-Sings.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Etiennot, L., Trullemans, L., Turkalj, M. et al. Lignin-derived bisguaiacol P vinyl ester as a bisphenol A-free resin alternative. Commun Mater 6, 105 (2025). https://doi.org/10.1038/s43246-025-00826-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-025-00826-y
