
Abstract
Rechargeable magnesium batteries (RMBs) have faced challenges in utilizing oxide cathodes due to the inherently sluggish Mg diffusion and poor compatibility with electrolytes, despite the high redox potential. Herein, we present a prototype RMB that is operational at room temperature, consisting of a nanoparticulate amorphous oxide cathode, fluorinated alkoxyborate (Mg[B(HFIP)4]2) as the electrolyte, and a Mg metal anode. The amorphous MgxTi1/9Mo2/9O cathode contains a considerable free volume formed by ion exchange between monovalent and divalent cations, facilitating Mg diffusion and eventually realizing reversible Mg insertion/extraction at room temperature. The reasonable compatibility of the present cathode with the electrolyte enables full cell operation with a Mg metal anode, and various analyses have demonstrated that Mg intercalation is responsible for the battery performance. The discharging capacity is ~150 mAh g−1, and 70 mAh g−1 is maintained after 200 cycles. These findings demonstrate the feasibility of RMBs with oxide cathodes that are operational at room temperature.
Similar content being viewed by others

Introduction
Rechargeable magnesium batteries (RMBs) are potential alternatives for high-energy-density chemical energy storage due to the abundance of magnesium in the Earth’s crust1; the development of RMBs would mitigate the sole dependence on scarce lithium, which is essential for current lithium-ion batteries (LIBs). The high energy density of RMBs is due to the availability of magnesium metal anodes, which exhibit a specific capacity of 2205 mAh g−1 and a relatively low redox potential of −2.38 V vs. SHE. Additionally, these anodes have a favorable plating morphology in selected electrolytes and conditions1,2,3, unlike Li and Na anodes, which tend to form problematic dendrites4. This makes RMBs a safe and viable choice for applications such as stationary energy storage systems. Despite their promise, however, the development of practical RMBs has faced obstacles since the RMB prototype in 2000, which consisted of a sulfide cathode and a Grignard-based electrolyte5, primarily due to the lack of available cathode materials6,7.
Oxide materials8,9,10,11,12,13,14 are generally beneficial for achieving high energy densities due to their inherently high redox potential (2–3 V vs. Mg2+/Mg), which fully leverages the low potential of magnesium metal anodes compared with other chalcogenides15,16,17,18,19,20, such as sulfides and selenides. Furthermore, the advent of halide-free electrolytes21,22,23,24,25,26 that are capable of relatively reversible Mg deposition/stripping opens up the possibility of utilizing oxide electrodes. Among them, electrolytes with weakly coordinating anions24 such as Mg[B(HFIP)4]225 and Mg[Al(HFIP)4]226 (B(HFIP)4: tetrakis(hexafluoro-iso-propoxy)borate, Al(HFIP)4: tetrakis(hexafluoro-iso-propoxy)aluminate) are among the most promising due to their high oxidative resistance in comparison with that of amine-based/amine-added electrolytes21,22,23 and conventional Grignard-based electrolytes of dichloro complexes and phenyl complexes7. Nevertheless, it is still challenging to build a full cell prototype based on an oxide cathode27 due to the inherently sluggish Mg diffusion in oxide cathodes28, unfavorable rocksalt transformation accompanied by Mg insertion8,11,12, and poor electrode compatibility with the electrolyte29. Indeed, the low diffusivity of Mg drastically narrows the choice of oxide cathodes, such as α-MnO2 and α-V2O56, that are operational at room temperature. α-MnO2 is a promising cathode material11, yielding a capacity of 110 mAh g−1 due to substantial Mg intercalation29 in a half-cell with an electrolyte consisting of Mg[TFSA]2 in triglyme (TFSA: bis(trifluoromethanesulfonyl)amide; triglyme (G3): triethylene glycol dimethyl ether). The reason why the system is defined as a half-cell is that the electrolyte containing Mg[TFSA]2 induces a large overpotential (>2 V) for stripping the Mg metal anode, which poses challenges in full cell operation. A simple solution for this issue is to replace the salt in Mg[TFSA]2 with Mg[B(HFIP)4]2, which is capable of reversible Mg stripping/deposition. However, the drastic change in the solvation structure of Mg cations due to the use of Mg[B(HFIP)4]2 salt results in the decomposition of α-MnO2 during discharge, which is accompanied by significant side reactions with the electrolyte, inhibiting the construction of operational RMB full cells29.
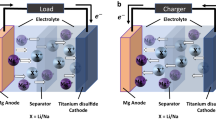
In the present study, we develop a oxide cathode material, Mg0.27Li0.09Ti0.11Mo0.22O (MLTMO), which is a composite of major amorphous and minor rocksalt phases (Fig. 1a). The Mg-containing amorphous oxide is prepared via amorphization of the Li-containing oxides followed by ion exchange between Li and Mg; this process spontaneously introduces a considerable free volume into the resultant amorphous phase as remnants of Li cation vacancies due to the valence state discrepancy between monovalent Li and divalent Mg. This amorphous structure with a large free volume enables facile Mg insertion and extraction. Additionally, the significant difference between the amorphous and rocksalt structures suppresses detrimental rocksalt transformation, in contrast to spinel oxides8, whose oxygen sublattice is similar to that of rocksalt oxide. We selected Mg[B(HFIP)4]2-G3 electrolyte24,25 due to its high oxidative resistance, reversible Mg stripping/plating with relatively low overpotential, compatibility with aluminum cell components, and potential for commercial availability30. We demonstrate that this material is capable of Mg extraction/insertion at room temperature and fairly stable in Mg[B(HFIP)4]2-G3 electrolyte and evaluate its full cell feasibility at room temperature in an orthodox split cell, which consists of a composite cathode electrode, a Mg metal anode, a separator, and a relatively lean electrolyte (Fig. 1b). The discharging capacity remains at 70 mAh g−1 for more than 200 cycles, and rigorous chemical analysis via inductively coupled plasma optical emission spectroscopy (ICP‒OES) confirms that Mg intercalation is the primary contributor to the electrochemically observed capacity during cycling.

a Features of amorphous oxide cathodes. External perturbations cause amorphization of Li-rich layered oxides, and subsequent ion exchange yields a Mg-containing oxide cathode. The valence-state discrepancy between monovalent Li and divalent Mg forms a large free volume. The free volume in the amorphous oxide cathode ensures a percolative Mg migration path and reversible Mg intercalation. The significant difference in structure between the amorphous oxide and rocksalt oxide suppresses the phase transformation into the electrochemically inactive rocksalt structure in the Mg-rich composition. b Cell configuration of present RMBs, in which a Mg metal anode and its compatible electrolyte, Mg[B(HFIP)4]2 in triglyme, is used with an amorphous cathode.
Cathode preparation and electrochemical properties
The cathode material MLTMO was prepared by a modified solution combustion synthesis (SCS)31 followed by ion exchange between Li and Mg from the ion-exchange precursor Li2Ti1/3Mo2/3O313, which is a pseudobinary oxide of Li2MoO3 and Li2TiO3. The ion-exchange precursor Li2Ti1/3Mo2/3O3 has a Li-rich layered rocksalt structure (Fig. 2a and Supplementary Fig. S1), whose crystalline size was estimated to be 3.65(2) nm from the 104 diffraction peak according to the Scherrer equation. In contrast, the secondary particles (white regions in the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image) were well dispersed and ranged from 7 to 10 nm in diameter, as determined from HAADF-STEM images (Fig. 2b). The dark gray interstitial space corresponds to amorphous carbon, into which the oxide particles were embedded (Supplementary Fig. S2). Fuel-rich combustion31 followed by calcination at 600 °C in a reductive environment of 10% H2-Ar yielded amorphous carbon covering the target oxide, which presumably decreased the oxide particle size by suppressing excessive growth at high temperature (Fig. 2c), in contrast to particles obtained by the conventional Pechini method13. Energy-dispersive X-ray spectroscopy (EDS) images revealed that the composition was virtually homogeneous, while the low fluorescence yield of Ti and O relative to that of Mo obscured the particle outlines in the corresponding EDS images. Constituent Li was exchanged with Mg by immersing the ion-exchange precursor Li2Ti1/3Mo2/3O3 in Mg[TFSA]2-G3 electrolyte at 90 °C for 24 h (Supplementary Fig. S3), which yielded the composition Mg0.269(8)Li0.085(2)Ti0.111Mo0.215(6)O, according to ICP‒OES. X-ray powder diffraction (XRPD, Fig. 2a) indicated that MLTMO consisted of major amorphous and minor crystalline phases with a rocksalt structure, where the ball-milling of the ion-exchange precursor Li2Ti1/3Mo2/3O3 before ion exchange induced amorphization (Supplementary Fig. S1).

a X-ray diffraction profiles of (black broken line) Li2Ti1/3Mo2/3O3 and (blue solid line) ion-exchanged Mg0.27Li0.09Ti0.11Mo0.22O. h and c in the diffraction indexes represent hexagonal and cubic lattices, respectively. The asterisk indicates the halo peak of the amorphous phase. The simulated XRD profiles of Li2MoO3 and MgO are also presented at the bottom of the panel as references to the hexagonal layered rocksalt structure and cubic rocksalt structure, respectively. b STEM-HAADF and EDS images of crystalline Li2Ti1/3Mo2/3O3. The scale bar represents 20 nm. c Schematic of the modified solution combustion synthesis.
The cell performance was evaluated using a three-electrode split cell operated at room temperature. The cell consisted of a composite electrode with MLTMO as the cathode, Mg metal foil as the anode, a glass fiber filter as the separator, and 0.53 M Mg[B(HFIP)4]2 in G3 as the electrolyte. The cell was also equipped with a reference electrode of Li metal immersed in 1 M LiTFSA in G3, which was separated from the main bath by a ceramic filter. Charge/discharge tests were conducted at a constant current density of 10 mA g−1 with cutoff potentials of 1.8 and 4.1 V vs. Li RE (Fig. 3 and Supplementary Fig. S4).

a Potential profiles of Mg0.27Li0.09Ti0.11Mo0.22O at room temperature with a current density of 10 mA g−1 in the potential window of 4.1 V for charging and 1.8 V vs. Li RE for discharging. The inset shows the rate capability for discharging to 1.7 V vs. Li RE, in which the cell was charged to 4.2 V vs. Li RE at a common current density of 10 mA g−1 before each discharging test. b Cell voltage profiles obtained from EWE − ECE in (a), where the counter electrode was pure Mg metal. The vertical axis was, therefore, converted from the electrode potential into net cell voltage. c Capacity retention corresponding to (a). Filled and open circles indicate the discharging and charging capacity, respectively. Green diamonds represent the Coulombic efficiency.
The discharge capacity was highest at ~100 mAh g−1 at the ~6th cycle and remained at ~75 mAh g−1 after 200 cycles. The present cell, therefore, showed much better cyclability than previously reported oxide cathodes operated at 60 °C13 and 90 °C12. ICP‒OES analysis of the material after the 5th cycle demonstrated that reversible Mg insertion/extraction was predominantly responsible for the electrochemically observed capacity, in which the changes in Mg and Li composition were equivalent to electrochemical capacities of 69.3 and 8.2 mAh g−1, respectively (Supplementary Table S1). This contrasts with the case of an α-MnO2 cathode discharged in a similar weakly coordinating electrolyte, in which side reactions rather than Mg insertion contributed the most to the electrochemically observed discharging capacity29. The present cell was equipped with a Li reference electrode separated from the main bath by a ceramic filter due to the high potential stability of Li metal. Nevertheless, the small amount of Li in the main bath derived from the reference electrode hardly affected the electrode properties. Indeed, the amount of Li in the electrolyte, derived from the reference electrode, was confirmed to be negligibly small (Li/Mg <10−3) by the ICP-OES analysis in a similar system13. This was further corroborated by the virtually identical cell performance obtained in the same cell configuration with a Mg reference electrode (Fig. 3a and Supplementary Fig. S5), while a Mg reference tends to be more sensitive to external influences and less stable in potential (Supplementary Fig. S6). The small changes in Li composition at the discharged state of the 5th and 109th cycle observed via ICP‒OES (Supplementary Table S1) also support these observations.
The charging capacity always exceeded the discharging capacity, which was attributed to oxidative decomposition of the electrolyte because excess charging capacity was hardly observed in the high-concentration electrolyte Mg[TFSA]2-G3 even at an elevated temperature of 60 °C13. The excess charging capacity was conspicuous in the first 20 cycles, whereas it decreased in the later cycles, reflecting the corresponding Coulombic efficiency (CE) values: 45–70% in the first 20 cycles and more than 70% in the later cycles. This cycle dependence of CE implies that this parasitic charging capacity can be attributed to the formation of a cathode electrolyte interphase. For instance, the amount of boron on the electrode, derived only from the electrolyte, considerably increased after long-term cycling, indicating that continuous electrolyte decomposition during charging/discharging delivered the parasitic capacities (Supplementary Table S1).
The rate capability of discharging was evaluated by changing the current density from 5 to 500 mA g−1, where the cell was charged at a constant current density of 10 mA g−1 before discharging at various current densities. Only discharging was evaluated because the charging capacity would include the contribution from the oxidative decomposition of the electrolyte. The upper limit of the electrochemical window is estimated to be ~3.5 V vs. Mg2+/Mg on the Pt electrode in the present electrolyte system24. Namely, the electrolyte decomposition is minimal when discharging below 3.5 V vs. Mg2+/Mg, while charging would be accompanied by the electrolyte decomposition, because the electrode potential reaches close the upper limit of the electrochemical window. Note that the upper limit changes depending on the electrode and tends to be lower in the composite electrode relative to the Pt electrode due to the large surface area of the composite electrode and the catalytic activity of the cathode materials32. The slowest discharge of 5 mA g−1 yielded the largest discharging capacity of ~150 mAh g−1. The discharging capacity remained at ~70 mAh g−1 even at the fastest discharge rate of 500 mA g−1, implying the high rate capability of the present cathode material and cell configuration.
The scan rate dependence of the cyclic voltammetry (CV)9,33,34 results (Supplementary Fig. S7) indicated that the diffusion-limited redox reaction, rather than the electrochemical capacitor reaction, was predominant at relatively slow charging/discharging rates of up to 50 mA g−1, which is approximately equivalent to 0.5 mV s−1 in CV, while fast discharge involved a considerable contribution from the electrochemical capacitor reaction as seen in the CV result at 5.0 mV s−1. The relatively good rate capability can, therefore, be attributed to two factors: Mg-ion diffusion and pseudocapacitive contributions33. The small particle size of the present MLTMO matches the diffusion length of Mg at room temperature, thereby enabling battery operation under ambient conditions. Additionally, reducing particle size increases the specific surface area, which in turn enhances the pseudocapacitive contributions, particularly during high-rate operation.
The differential capacity curve (dQ/dE) of the charge/discharge test (Supplementary Fig. S8) was in good agreement with the CV profile at the lowest sweeping rate. The differential capacity increased monotonically during charging and decreased during discharging, without showing a significant peak. This profile suggests that charge compensation of Mo, as discussed in the X-ray absorption spectroscopy section below, occurs over a broad potential range in the operating cathode material. The overpotential of the Mg metal anode was within 0.2 and 0.45 V during stripping (discharge) and deposition (charge), respectively (Supplementary Fig. S9), which is in good agreement with the previous studies24,25,30 on the present Mg[B(HFIP)4]2-G3 electrolyte. Amine-additive21 or amine-based22 electrolytes with Mg[TFSA]2 salt represent an alternative electrolyte option, offering broader availability and reasonably low overpotential. However, enhancing the oxidative resistance of amine-based compounds remains a challenge22, particularly for high-voltage oxide cathodes.
Notably, the present cell configuration is equivalent to what is commonly referred to as a full cell, which consists of a fully operational cathode, anode, and electrolyte. Indeed, the charge/discharge performance measured in the two-electrode coin cell shows good agreement with that obtained from the three-electrode split cells (Supplementary Fig. S10). Nevertheless, in the present study, the working electrode potential was controlled in the charge/discharge tests with the split cell to meticulously evaluate the cell behavior without being disturbed by the unstable potential of the Mg metal anode and electrolyte decomposition. We, therefore, defined the present cell operation as a reference-controlled full cell (RC full cell). Note that the RC full cell is distinct from the conventional half-cells used in previous RMB studies, in which either only the cathode or anode is fully operational, such as in the case of oxide cathodes with Mg[TFSA]2-G3 electrolyte12,13. In such cells, the passivated Mg metal anode hampers substantial cell discharge because of the significant overpotential of Mg stripping. Thus, the present study with the RC full cell contrasts with studies based on conventional half-cells. The present results imply that strictly defined full cell operation is feasible by adjusting the galvanostat setting to control the cutoff voltage based on the net cell voltage (i.e., EWE − ECE) rather than on the electrode potential of the cathode (EWE) measured via the reference electrode. Here, EWE and ECE represent the electrode potentials of the cathode (working electrode) and anode (counter electrode), respectively.
Charge compensation mechanism
X-ray absorption spectroscopy (XAS) was used to clarify the redox mechanism and cyclic evolution of the charge and discharge states. X-ray absorption near edge structure (XANES) demonstrated that the charge during charging and discharging was predominantly compensated by changes in the valence state of Mo between 6+ and lower valence states, while the effect of Ti was minimal, based on the same composition of Ti-Mo oxides13. The superimposed spectra exhibited isosbestic points (indicated by arrows in Fig. 4a), implying that MLTMO charged and discharged via a two-phase reaction. Indeed, STEM-EDS analysis revealed two distinct types of amorphous materials, i.e., Mg-rich and Mg-poor amorphous materials, in a previous study on coarse MLTMO operated at 60 °C13. Figure 4b shows the magnified pre-edge peaks indicated by an asterisk in Fig. 4a, representing the average valence states of Mo in different states of charge and discharge.

a Mo K-edge XANES. Arrows indicated the isosbestic points observed in the various states of charge/discharge in this energy region. The asterisk represents a pre-edge peak. b Magnified spectra of the pre-edge peak indicated in (a) by the asterisk.
The intensity of the pre-edge peak represents the average valence state of Mo based on the reference materials Mo(IV)O2 and Mo(VI)O3; a stronger pre-edge peak corresponds to a higher valence state. The spectra corresponding to the 10th cycle (green solid and dashed lines in Fig. 4b) showed the largest change in pre-edge peak intensity with charging and discharging, in good agreement with the almost largest capacity observed in this cycle (Fig. 3c). The intermediate charging/discharging states and completely charged/discharged states were examined in the 3rd cycle, where the discharging capacity increased with increasing number of cycles. The charged state in the 3rd cycle (pink solid line) was virtually identical to that in the 10th cycle, indicating that repeated cycling minimally affected the charged state. The intermediate charging state (pink broken line) fell somewhat between the charged (pink solid line) and discharged states (blue solid line), indicating that electrochemical charging continuously oxidized the constituent Mo cations even though the electrolyte was also oxidatively decomposed in the high potential region. In contrast to charging, discharging reduced the valence state of Mo up to only ~70% of the electrochemically observed capacity, while further discharging did not reduce Mo, judging from the very similar spectra corresponding to a discharging state of ~70% (blue dashed line in Fig. 4b) and the discharged state (blue solid line). This implies that side reactions of the composite electrode or electrolyte partially consumed the electrochemically observed charge at the end of discharging. Nevertheless, the compatibility of MLTMO with the electrolyte was much better than that of α-MnO2, as ICP‒OES indicated substantial Mg insertion. Indeed, side reactions during discharging hinder Mg insertion into the α-MnO2 cathode material due to the inherent incompatibility between bare α-MnO2 and the electrolyte29. Further cycling improved the electrochemical reduction of Mo, as observed in the most reduced state at the 10th cycle. This behavior contrasts with those of the charged states in the 3rd (red solid line) and 10th (green broken line) cycles, where the valence state of Mo was similar, independent of the number of cycles. Thus, with increasing number of cycles, the active range of Mo valence states expands toward the reduced state rather than the oxidized state after charging. This extended Mo valence change with cycling can be attributed to the increase in the amount of active material accessible through Mg intercalation from the electrolyte, as revealed by STEM-EDS13.
Free volume in the amorphous structure
Free volume is estimated via an amorphous structure simulated by molecular dynamics and ab initio calculations. In the crystal system, cation vacancies are sites where cations are absent, and cations can possibly occupy via an intercalation or elemental step of diffusion. Likewise, in the present study, we define free volume in amorphous oxide as a counterpart of the cation vacancy in the crystal structure, since no vacancies can be defined in the amorphous structure. Namely, the free volume is the region where atoms, especially cations in the present study, can possibly occupy35,36. Note that the free volume defined in the present study is not necessarily equivalent to that generally used for studies of amorphous materials37. The simulated amorphous structure was prepared via a melt-quench method starting from the Li2Ti1/3Mo2/3O3 (Li0.67Ti0.11Mo0.22O) crystal of 1000 atoms, followed by replacing Li atoms with Mg. Half of the original Li atoms were randomly substituted with Mg, and the remaining half were removed, in accordance with the ion-exchange strategy used in the corresponding experiment. The resulting amorphous structure was then fully relaxed using ab initio density functional theory (DFT) calculations (Fig. 5a).

a Atomic structure of amorphous Mg0.33Ti0.11Mo0.22O obtained via molecular dynamics and ab initio calculations. Mo–O and Ti–O coordination polyhedra are shown in purple and light blue, respectively. Orange spheres represent Mg atoms. Green regions indicate free volume regions within the structure where bond valence sum (BVS) values are between 1.6 and 2.4, suggesting favorable sites for Mg occupancy. b Same structure as in a with coordination polyhedra removed to highlight the spatial distribution of Mg atoms and BVS-based free volume.
The free volume ratio, \({f}_{V}\), was calculated by \({f}_{V}=1-{\Sigma }_{i}{n}_{i}{V}_{i}/{V}_{{{\rm{cell}}}}\), where \({n}_{i}\) is the number of atoms of element, i, \({V}_{i}\) is the Voronoi volume, and \({V}_{{{\rm{cell}}}}\) is the volume for the simulation cell of the amorphous structure. \({V}_{i}\) for Mg, Ti, and Mo were determined from the simulated amorphous structure, while \({V}_{{{\rm{O}}}}\) from crystalline MgO was used to evaluate the free volume, due to its well-defined coordination environment. \({f}_{V}\) was estimated to be 9.7%, which is equivalent to 52% of the \({V}_{{{\rm{Mg}}}}\). Namely, the chemical formula can be nominally described as Mg0.33□0.17Ti0.11Mo0.22O when explicitly describing the amount of free volume, □, in the structure. Therefore, approximately half of the free volume introduced by the ion exchange is secured in the amorphous structure according to the theoretical prediction. The distribution of the free volume is further visualized based on the bond valence sum (BVS) in the simulated amorphous structure (Fig. 5b). The green region in Fig. 5b represents an isosurface defined by \(\left|{v}_{{{\rm{BVS}}}}-{v}_{{{\rm{ideal}}}}\right| < 0.4\), where \({v}_{{{\rm{BVS}}}}\) is a calculated BVS at each mesh point, and \({v}_{{{\rm{ideal}}}}\) is a nominal valence of divalent Mg2+. The widespread and well-dispersed distribution of the green regions indicates that the free volume is uniformly distributed throughout the amorphous matrix, without forming large voids or aggregated clusters. Such free volume would contribute as diffusion path of Mg in the amorphous structure.
Outlook for full cell realization
The present demonstration of RC full cell operation at room temperature with MLTMO implies the following general requirements for oxide cathodes for RMBs: an atomic structure with considerable vacancy/free volume, a small particle size, a redox potential within the electrochemical window of the electrolyte, and compatible cell components. Interactions between neighboring Mg cations significantly affect the energy barrier in cathode materials38; in addition to strong Coulombic interactions between divalent Mg cations and surrounding O anions, which lead to a relatively high energy barrier for Mg migration. The former factor is easily overlooked since such interactions are virtually negligible in LIB cathodes39. However, for instance, a previous study38 showed that it was extremely challenging to substantially extract/insert Mg from/into Mg-rich disordered rocksalt oxides while introducing cation vacancies to facilitate Mg migration and eventually activate the cathode material12,38. Specifically, substantial Mg migration requires considerable vacancies/free volume in the initial structure. This could also explain why previously proposed oxide cathodes that are operational at room temperature, such as MnO2, V2O5, and TiO2, contain little Mg6. Consequently, candidate materials with adequate space to accommodate Mg are limited to specific structures.
In contrast to conventional material synthesis, the present study employed ion exchange between monovalent alkaline and divalent Mg cations, which simultaneously introduced vacancies/free volume (Fig. 1b). Ideally, the amount of vacancies/free volume is the same as that of exchanged Mg cations, given that the host structure maintains space for movable cations during ion exchange. This vacancy introduction, realized by balancing the electric charge between monovalent and divalent ions, is likely critical for facilitating Mg migration in oxide cathodes by mitigating the interactions between Mg cations. If the cations completely exchange, the relevant chemical reaction can be described as AMxOy + 0.5Mg2+ → Mg0.5□0.5MxOy + A+, where A is an alkaline element, M is a metal element, and □ is a vacancy/free volume in the atomic structure. The present study employed Li as the alkaline metal, A, because Li-containing compounds have been well studied for use in LIBs. However, A is not necessarily limited to Li; Na and K would also be candidates for this procedure. The cations can be exchanged chemically and electrochemically; moreover, the chemical process of simply immersing the Li-containing compound in excess Mg electrolyte should be milder for oxide cathodes because this process circumvents the formation of higher valence states of redox-active transition metals, which are frequently unstable. The present study also implies that Mg-containing cathodes such as MLTMO can ultimately lead to anode-free cells with no Mg metal in the anode, even though a demonstration of this idea is beyond the scope of this work. This approach would be beneficial for the industrial manufacturing of RMBs because Mg metal is much safer than alkaline metals but is prone to oxidization in ambient air.
The residual alkaline cations in the cathode materials may enhance electrochemical properties by facilitating Mg diffusion in the materials12,40,41 or suppressing adverse phase transitions42. This diffusion-enhancement effect was elaborated in the Chevrel compound with Li40 and further extended to oxides incorporating various alkaline cations of Li, Na, and K41, where the extent of enhancement depends on the specific cation species. The small amount of residual Li in the present MLTMO would affect the electrode performances, while the detailed analysis is beyond the scope of this work. These effects can potentially be leveraged in the aforementioned ion-exchange approach to obtain Mg-containing oxide cathodes.
This concept has been intensively explored in so-called “dual-salt batteries”43,44 and also referred to as “hybrid batteries” in later studies45,46. The dual-salt/hybrid batteries employ electrolytes containing both mono- and multi-valent cations (e.g., Li+ and Mg2+), where the primary reactions typically involve Li intercalation at cathode, and Mg plating/stripping at anode. However, co-intercalation of both Li and Mg into the cathode has also been explored as a strategy to increase the overall energy density of the dual-salt/hybrid battery systems44,45. The charge/discharge mechanism of the present MLTMO partially aligns with the dual-salt/hybrid battery concept, particularly in terms of the co-intercalation of Mg2+ and Li+. However, the present system exhibits a dominant contribution from Mg intercalation ( ~ 90%) and does not include any Li salt in the initial electrolyte. These features contrast from the original dual-salt/hybrid battery design, although categorical distinction between dual-salt/hybrid batteries and RMBs employing the cathode containing monovalent cations remains inherently ambiguous.
It is also well known that particle size is an important factor influencing the activity of cathode materials, as it should be comparable to the diffusion length. The diffusion length, ld, is approximated by \({l}_{{{\rm{d}}}} \sim \sqrt{{Dt}}\), where D is the chemical diffusion coefficient and t is the time; cathode materials for RMBs typically have an ld in the range of one nanometer at room temperature27. The particle size of coarse MLTMO in the previous study13 was tens of nm (Supplementary Fig. S11c), as heat treatment inevitably accompanies particle growth. Consequently, the course cathode material could deliver less than 30 mAh g−1 at room temperature (Supplementary Fig. S11a). In contrast to the previous study, we suppressed the particle growth during heat treatment by embedding the nucleus in the carbon matrix in the modified solution combustion synthesis (Fig. 2c). This yielded the cathode materials with a primary particle size of 3.65(2) nm, which was comparable to the typical diffusion length of Mg at room temperature. The smaller particle size increased the particle regions for the substantial Mg extraction/insertion, and therefore, delivered the capacity of up to 150 mAh g−1 at room temperature.
The substantial electrochemical window of the electrolyte, specifically the oxidative decomposition potential, tends to become narrower on cathode composite electrodes because of the large surface area. A well-defined archetypal electrode, such as Pt foil, is typically used to evaluate the electrochemical window using linear sweep voltammetry (LSV) in electrolyte studies. The potential where the current drastically increases is defined as the upper limit of the electrochemical window. However, even a small oxidative current within the defined electrochemical window preceding the drastic current increase effectively limits the maximum potential for charging a composite electrode with a large surface area. The different types of the materials used for these purposes, namely, Pt for LSV and transition metal oxides for the cell, may also affect the catalytic activity of oxidative electrolyte decomposition and, ultimately, the practical electrochemical window. For instance, an excessive charging capacity resulted in a moderate CE of ~70%. This value should be increased to ~100% for practical RMBs in the future by developing more electrochemically stable electrolytes and/or modifying the surface of the cathode materials to suppress the catalytic activity of electrolyte decomposition.
The last but critical challenge of developing operational RMBs is compatibility between the cell components. Mg[Al(HFIP)4]2, a derivative of the compound Mg[B(HFIP)4]2 used in the present study, is corrosive to Al metal at high potential26,29,47 and is typically used as the cathode current collector in RMBs as well as LIBs. We confirmed that the present electrolyte, i.e., Mg[B(HFIP)4]2 in G3, was not corrosive to the present cell components, including the Al current collector. The most critical compatibility is that between the electrolyte and the cathode material. For instance, bare α-MnO2 used as a cathode material is not compatible with hexafluoroisopropoxide-derived electrolytes during discharging29. This is because the solvation structure of the solvate-separated ion pair (SSIP) is highly reactive to unstable surface oxygen on the α-MnO2 cathode, whereas the contact ion pair (CIP) in the Mg[TFSA]2-glyme electrolyte can form a stable interface that suppresses the continuous reaction between α-MnO2 and the electrolyte. Such incompatibility results in a considerable discrepancy between the electrochemically observed discharging capacity and that calculated from the composition; Mg insertion into the cathode material is minimal despite the substantial discharging capacity. In contrast to conventional oxide cathodes, MLTMO exhibited reasonable compatibility with the Mg[B(HFIP)4]2-G3 electrolyte, judging from the substantial Mg insertion/extraction demonstrated by chemical composition analysis via ICP‒OES. For instance, the electrochemical discharging capacity of approximately 100 mAh g−1 at the 5th cycle (Fig. 3c) was in good agreement with the capacity of 77 mAh g−1 estimated from the composition change (Supplementary Table S1). This consistency implies the high stability of Ti-Mo oxides in the presence of the Mg[B(HFIP)4]2-G3 electrolyte, which may be attributed to the inherent high chemical resistance of Ti and Mo oxides in general.
Capacity retention is also a crucial factor in realizing practical full cells. Reported degradation mechanisms of cathode materials can be categorized as follows: (1) conversion or semi-topotactic reactions8,11,48,49, (2) electrolyte decomposition12,48,50, (3) surface reactions51,52, and (4) decomposition or dissolution53. Conversion and semi-topotactic reactions do not necessarily degrade capacity retention, but cyclability is compromised when these reactions are not fully reversible. A representative example is the semi-topotactic transition from spinel to rocksalt induced by Mg insertion. The reverse transition from rocksalt back to spinel is often incomplete due to sluggish Mg diffusion within the rocksalt structure, resulting in poor cycle life10. This issue can be mitigated by limiting the Mg composition range to maintain structural integrity during charge/discharge, as demonstrated in defect spinel cathodes9. The present MLTMO material would circumvent this issue by incorporating elements such as Mo and Ti, which are less prone to forming rocksalt structures, and by leveraging the large structural difference between the amorphous and rocksalt phases.
Electrolyte decomposition, particularly at high voltages, also leads to poor cyclability, either through the formation of insulating decomposition products on the cathode surface12 or due to a limited electrochemical window that restricts full cathode utilization. When the electrochemical window is fixed, such limitations stem from the intrinsic redox potential of the cathode material and the overpotential during Mg extraction, which is linked to intercalation kinetics48,50. Therefore, the redox potential must be carefully tuned by selecting appropriate redox-active elements, and the structure should be designed to enable fast Mg-extraction kinetics.
Surface reactions generally result in the formation of Mg-impermeable layers51,52, which are often tied to the chemical compatibility between the cathode and the electrolyte. Decomposition and dissolution are observed in, for example, sulfide cathodes53, which dissolve into the electrolyte during cycling. However, such degradation may also occur in oxide cathodes, particularly when halide-based electrolytes are used. As described above, capacity retention is influenced by multiple interrelated factors, such as redox potential, Mg intercalation kinetics, phase transition behavior, and electrolyte compatibility. At the current stage of RMB development, improving cyclability requires addressing these challenges in an integrated manner during material design, possibly with the aid of computational materials science, rather than relying on a single specific strategy.
The present MLTMO is further compared with the typical oxide and polyanion cathode materials previously reported, considering not only the electrode performance but also the compatibility between cell components and rigor of the cathode characterization (Supplementary Table S2). The following conditions are considered such that each study implies the feasibility of the full cell operation at room temperature, and is comparable to the present study. We believe that essential characterizations to demonstrate reversible Mg insertion/extraction into/from the cathodes are bulk-sensitive analyses of the chemical composition, crystal structure, and chemical state. This is because excessive side reactions, such as electrolyte decomposition29 and proton insertion54, can falsely give seemingly good electrochemical performances and lead to misinterpretation that reversible Mg insertion and extraction are occurring. We also evaluate the electrolyte used in terms of compatibility with the Mg metal anode. Six materials out of the 32 listed materials fulfill the three conditions of cathode characterizations, electrolyte compatibility, and operation at room temperature. It is further narrowed down to two materials when ruling out the use of halide-containing electrolytes of all phenyl complex and dichloro complex, which are corrosive to the oxide materials and whose electrochemical window is limited by halide ions. The screened materials are α-MnO248 and γ-MnO2-based Mg0.15MnO221, both of which show the highest capacity of ~270 mAh g−1 and the average potential/voltage of ~1.0 and ~2.4 V for α-MnO2 and Mg0.15MnO2, respectively. The present MLTMO cathode material delivers the energy density of ~255 Wh kg−1 (=1.7 × 150), which is comparable to α-MnO2 (270 Wh kg−1 (=1.0 × 270)), but less than Mg0.15MnO2 (648 Wh kg−1 (=2.4 × 270)). However, the MLTMO shows the relatively good capacity retention of 80% after 200 cycles (=80/100, Fig. 3c), while α-MnO2 and Mg0.15MnO2 show 19% (=30/160) at 30 cycles and 70% (=190/270) at 25 cycles, respectively.
The feasibility of using the present cell configuration as an RMB operational at room temperature was further examined by using a minimal setup of a 2032 coin cell connected to a commercial blue LED (Fig. 6). The current‒voltage curve (I‒V curve) of the LED (Supplementary Fig. S12) indicated that a substantial current flowed above ~2.5 V with blue light emission. Therefore, cells consisting of conventional sulfide-based cathodes, which have a potential of 1–1.5 V, hardly drive the blue LED. Additionally, even cells with oxide cathodes do not necessarily deliver a voltage higher than 2.5 V when combined with electrolytes that passivate the Mg metal anode. This is the case for, for example, Mg[TFSA]2-based electrolytes, whereas Mg[TFSA]2-based electrolytes are widely used for cathode characterization due to their high stability with respect to oxide cathodes. A single 2032 coin cell charged to 3.5 V after eight charging/discharging cycles continuously activated the blue LED for over 7 min. This finding demonstrates that the present RMB prototype delivers a relatively high voltage due to effective use of the oxide cathode material.

Blue LED emission test using a single prototype coin cell consisting of a Mg0.27Li0.09Ti0.11Mo0.22O cathode, a Mg metal anode, and Mg[B(HFIP)4]2 in G3 as the electrolyte.
The present study demonstrated the feasibility of RMBs consisting of an oxide cathode and Mg[B(HFIP)4]2-G3 as the electrolyte that are operational at room temperature. An MLTMO cathode material was prepared by a modified SCS followed by ion exchange between Li and Mg, which simultaneously introduces a considerable free volume into the resulting amorphous structure. The small particle size and substantial free volume enable reversible Mg extraction/insertion at room temperature. A full cell consisting of the composite cathode and a Mg metal anode separated by a glass fiber separator and containing a relatively lean Mg[B(HFIP)4]2-G3 electrolyte exhibited the largest capacity of 150 mAh g−1 at a current density of 5 mA g−1 and good capacity retention of 70 mAh g−1 over 200 cycles. Chemical composition analysis via ICP‒OES confirmed that the electrochemically observed capacity was predominantly derived from Mg insertion/extraction. Notably, the current results imply the feasibility of anode-free RMBs since the MLTMO cathode contains Mg, unlike previous cathode materials, such as α-MnO2 and α-V2O5. Furthermore, the present study reveals that ion-exchanged amorphous oxides are viable cathode materials for RMBs, which would be advantageous for Mg diffusion and compatibility with the electrolyte. Importantly, the RC full cell demonstrated in the present study provides opportunities to investigate the charging/discharging mechanism in a whole cell, in which the crosstalk and compatibility of multiple materials and cell components affect cell performance, ultimately leading to the realization of practical RMBs.
Methods
Synthesis
Mg0.27Li0.09Ti0.11Mo0.22O (MLTMO) was produced by a modified solution combustion synthesis (SCS) followed by ion exchange between Li and Mg from the ion exchange precursor Li2Ti1/3Mo2/3O3 (LTMO). Stoichiometric quantities of (NH4)6Mo7O24-4H2O (Wako, purity 99%), titanium tetraisopropoxide ([(CH3)2CHO]4Ti, Wako, purity 95%) and a 5% excess of LiNO3 (Wako) were mixed with propylene glycol C3H8O2 (Wako, purity 98%), citric acid anhydrous (Wako, purity 98%), and ammonium nitrate (Wako, purity 98%) to prepare a precursor gel. The molar ratio of [metal element]:[propylene glycol]:[citric acid]:[ammonium nitrate] was set to 1:2:2:4.5. The molybdenum salt, lithium salt, propylene glycol, and ammonium nitrate were dissolved in deionized water to create a Mo‒Li solution. A citric acid solution (CA solution) was prepared by dissolving citric acid in dehydrated ethanol predried with 3 Å molecular sieves overnight. Titanium tetraisopropoxide was mixed with dehydrated ethanol to create a Ti solution. For batch synthesis of 100 mmol LTMO, these three solutions were prepared with ~250 ml of solvent each.
The CA solution was added to the Ti solution and mixed thoroughly for ~10 min. Then, the Mo–Li solution was carefully dripped into the Ti solution under rapid stirring so that the transparency of the mixed solution was maintained. The mixed solution was heated at 60 °C overnight. The temperature was then changed to 65 °C, and the mixture was heated for a few days until the solution changed to a light orange viscous paste.
The paste was placed on an alumina boat and calcined at 600 °C for 10 h with 10% H2–Ar gas flow. The paste spontaneously combusted at approximately 300 °C during the temperature increase in the furnace. The obtained puff-like black product was transferred to an Ar-filled glove box with minimal exposure to moist air and hand-milled using an agate mortar and pestle. The powder was further ball-milled at 500 rpm for 200 min in total with 0.5 mm-ϕ zirconia balls in dimethyl carbonate in an Ar atmosphere. The pristine LTMO powder contained 24 wt% residual amorphous carbon derived from propylene glycol and citric acid, as determined by infrared absorption spectroscopy after combustion.
Ion exchange and electrochemical tests were performed with composite electrodes fabricated by mixing the synthesized LTMO with carbon black (Super C65, Timcal) and polyvinylidene difluoride (PVDF, Kureha) at a weight ratio of 8:1:3 while maintaining an inert Ar atmosphere in a mixing container. The obtained slurry was uniformly applied onto aluminum foil and vacuum dried at 120 °C for 12 h. The cropped electrode, typically 16 mm ϕ, was immersed in an electrolyte consisting of Mg(TFSA)2 (Wako) and triglyme at a molar ratio of 1:2.22 for 24 h at 90 °C to promote ion exchange between Li and Mg. The ion-exchanged electrodes were washed with ultradry acetonitrile (Wako, >99.8%), dried, and used for electrochemical evaluation and characterization. Ion exchange and subsequent washing were performed in an Ar-filled glove box. The triglyme, which was obtained from Tokyo Chemical Industry, was predried with 3 Å molecular sieves.
Electrochemical testing
Three-electrode split cells (SB9, EC Frontier), were assembled and operated for electrochemical testing in Ar-filled glove boxes. Al foil covered the cell casing on the cathode side to prevent contact between the stainless steel cell body and the electrolyte. Polished pure magnesium foil (Nilaco or Rikazai, 60–100 μm thick, >99.9%) was used as the counter electrode. The reference electrode was lithium foil (Honjo Metal) immersed in 1 M LiTFSA in triglyme as the electrolyte, separated from the main bath by a ceramic filter. The working electrode potential was converted to V vs. Mg2+/Mg by using the experimentally determined potential difference of 1.0 V between Li+/Li and Mg2+/Mg in the system. A glass fiber filter (GF/F, Whatman) was used as a separator. The electrolyte was 0.53 M Mg[B(HFIP)4]2 in triglyme. The electrolyte was pretreated before cell evaluation by applying constant potentials of 1.3 and 4 V vs. Li+/Li for 48 h at each potential to eliminate any redox-active impurities. For electrolyte pretreatment, Pt foil and Mg ribbon were used as working and counter electrodes, and the electrolyte was stirred.
Two-electrode 2032 coin cells (Hosen) were assembled using the same components as the three-electrode split cells except for the absence of a reference electrode. The cathode side of the cell housing was coated with aluminum.
Electrochemical measurements were conducted using VMP3, VSP-300, and VSP apparatuses (Biologic) at room temperature. Galvanostatic charging/discharging tests were conducted at a current density of 10 mA g−1 within a potential window of 1.8–4.1 V versus the Li RE. To evaluate the discharge rate capability, the cell was charged at a common current density of 10 mA g−1 to 4.2 V versus the Li RE before being discharged at various current densities to 1.7 V versus the Li RE.
Sample characterization
After electrochemical testing, all the electrode samples were washed with ultradry acetonitrile for further characterization. X-ray powder diffraction (XRPD) was performed with a SmartLab apparatus (Rigaku) equipped with a molybdenum X-ray source and a 1D D/teX detector (Rigaku) in Debye–Scherrer geometry. The electrode samples were encapsulated within Lindeman glass capillaries (500 μm diameter) in Ar-filled glove boxes to prevent air exposure. The zirconium Kβ filter was placed upstream of the sample to suppress the Mo Kα fluorescence from the Mo-containing samples, which is excited by the high-energy continuous X-ray from the X-ray tube, in addition to the original purpose of Mo Kβ elimination. The background was numerically subtracted from the XRPD profiles using the XRPD profile of a blank capillary.
The cation composition was determined by inductively coupled plasma‒optical emission spectroscopy (ICP‒OES). The carbon content in the pristine sample was evaluated via infrared absorption spectroscopy after combustion.
Transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and EDS were performed utilizing a spherical-aberration-corrected JEM-ARM200F microscope (JEOL) operating at 200 kV. The samples were prepared for STEM observation by suspending them on copper-mesh grids coated with thin perforated carbon.
X-ray absorption spectroscopy (XAS) was performed at the BL11S2 beamline of AichiSR, Japan. Washed electrode samples were mixed with boron nitride, compressed into pellets 3 mm in diameter, and sealed with polyimide film tape to avoid exposure to air during measurement. The Mo K-edge absorption spectra were measured in transmission mode.
Amorphous model and valence-based analysis
To generate an amorphous Mg1/2Ti1/3Mo2/3O3 structure, a 5 × 5 × 5 supercell of a rocksalt-type Li2Ti1/3Mo2/3O3 configuration was constructed, in which Li, Ti, and Mo cations were randomly distributed over the cation sublattice. The 1000-atom system was subjected to classical molecular dynamics simulations using LAMMPS55 with the CHGNet56 machine-learning potential.
The structure was melted at 3000 K using the Langevin thermostat for 10 ps in the NVE ensemble with a damping parameter of 0.02 ps, followed by a linear quench to 300 K over 20 ps. The time step was set to 1 fs. After the quench, energy minimization and isotropic cell relaxation were performed to remove residual stress. Final equilibration was conducted at 300 K for 10 ps using the NVT ensemble.
To achieve the target composition, 50% of the Li atoms were replaced with Mg and the other 50% were with vacancy. The resulting structure was relaxed by density functional theory (DFT) calculations using VASP57,58,59,60, employing the PBE functional and the projector augmented-wave (PAW) method61,62. Ionic relaxation was performed with a force convergence threshold of 0.005 eV/Å and an energy convergence criterion of 5 × 10−4 eV. The cell shape and volume were allowed to change, and Γ-point sampling was used.
Bond valence sum (BVS) analysis was conducted on the amorphous Mg1/2Ti1/3Mo2/3O3 structure to identify energetically favorable ion sites63. A uniform three-dimensional 200 × 200 × 200 grid was generated within the unit cell (a,b,c = 20.38 Å) with a mesh spacing of ~0.1 Å along each axis. Grid points within 1.7 Å of any existing atom were excluded to avoid unphysical overlaps. At each remaining grid point, the BVS was calculated using the bond valence expression: \({v}_{{{\rm{BVS}}}}={\Sigma }_{i}\exp \left\{\left({R}_{0}-{d}_{i}\right)/{b}\right\}\), where \({d}_{i}\) denotes the distance to the ith oxygen atom within 6.0 Å, \({R}_{0}\) = 1.693 Å is the bond valence parameter for the Mg–O ion pair, and b = 0.37 Å is the universal constant commonly applied in bond valence modeling64. The expected valence for Mg was set to 2.0, and the absolute valence mismatch \(\left|{v}_{{{\rm{BVS}}}}-2.0\right|\) was evaluated at each grid point.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Liang, Y., Dong, H., Aurbach, D. & Yao, Y. Current status and future directions of multivalent metal-ion batteries. Nat. Energy 5, 646–656 (2020).
Matsui, M. Study on electrochemically deposited Mg metal. J. Power Sources 196, 7048–7055 (2011).
Leuppert, L. et al. Electrolyte-dependent deposition morphology on magnesium metal utilizing MeMgCl, Mg[B(hfip)4]2 and Mg(HMDS)2–2AlCl3 electrolytes. RSC Appl. Interfaces 1, 1142–1155 (2024).
Liu, W., Liu, P. & Mitlin, D. Review of emerging concepts in SEI analysis and artificial SEI membranes for lithium, sodium, and potassium metal battery anodes. Adv. Energy Mater. 10, 2002297 (2020).
Aurbach, D. et al. Prototype systems for rechargeable magnesium batteries. Nature 407, 724–727 (2000).
Li, Z., Häcker, J., Fichtner, M. & Zhao‐Karger, Z. Cathode materials and chemistries for magnesium batteries: challenges and opportunities. Adv. Energy Mater. 13, 2300682 (2023).
Blázquez, J. A. et al. A practical perspective on the potential of rechargeable Mg batteries. Energy Environ. Sci. 16, 1964–1981 (2023).
Okamoto, S. et al. Intercalation and push‐out process with spinel‐to‐rocksalt transition on Mg insertion into spinel oxides in magnesium batteries. Adv. Sci. 2, 1500072 (2015).
Shimokawa, K. et al. Structure design of long‐life spinel‐oxide cathode materials for magnesium rechargeable batteries. Adv. Mater. 33, 2007539 (2021).
Shimokawa, K. et al. Zinc-based spinel cathode materials for magnesium rechargeable batteries: toward the reversible spinel-rocksalt transition. J. Mater. Chem. A 7, 12225–12235 (2019).
Hatakeyama, T. et al. Accelerated kinetics revealing metastable pathways of magnesiation-induced transformations in MnO2 polymorphs. Chem. Mater. 33, 6983–6996 (2021).
Kawaguchi, T. et al. Securing cation vacancies to enable reversible Mg insertion/extraction in rocksalt oxides. J. Mater. Chem. A 12, 9088–9101 (2024).
Kawaguchi, T., Nemoto, N., Sakurai, H., Okamoto, N. L. & Ichitsubo, T. Utilization of delithiation-induced amorphous oxide as a cathode for rechargeable magnesium batteries. Chem. Mater. 36, 4877–4887 (2024).
Koketsu, T. et al. Reversible magnesium and aluminium ions insertion in cation-deficient anatase TiO2. Nat. Mater. 16, 1142–1148 (2017).
Sun, X. et al. A high capacity thiospinel cathode for Mg batteries. Energy Environ. Sci. 9, 2273–2277 (2016).
Blanc, L. et al. Toward the development of a high-voltage Mg cathode using a chromium sulfide host. ACS Mater. Lett. 3, 1213–1220 (2021).
Mao, M. et al. Tuning anionic chemistry to improve kinetics of Mg intercalation. Chem. Mater. 31, 3183–3191 (2019).
Canepa, P. et al. Odyssey of multivalent cathode materials: open questions and future challenges. Chem. Rev. 117, 4287–4341 (2017).
Li, Z. et al. Multi-electron reactions enabled by anion-based redox chemistry for high-energy multivalent rechargeable batteries. Angew. Chem. Int. Ed. 59, 11483–11490 (2020).
Dey, S. et al. Exploring cation–anion redox processes in one-dimensional linear chain vanadium tetrasulfide rechargeable magnesium ion cathodes. J. Am. Chem. Soc. 142, 19588–19601 (2020).
Hou, S. et al. Solvation sheath reorganization enables divalent metal batteries with fast interfacial charge transfer kinetics. Science 374, 172–178 (2021).
Wang, F. et al. Solvent molecule design enables excellent charge transfer kinetics for a magnesium metal anode. ACS Energy Lett. 8, 780–789 (2023).
Wang, M. et al. Synergy between the coordination and trace ionization of co-solvents enables reversible magnesium electroplating/stripping behavior. Energy Environ. Sci. 17, 630–641 (2024).
Mandai, T., Youn, Y. & Tateyama, Y. Remarkable electrochemical and ion-transport characteristics of magnesium-fluorinated alkoxyaluminate–diglyme electrolytes for magnesium batteries. Mater. Adv. 2, 6283–6296 (2021).
Zhao-Karger, Z., Gil Bardaji, M. E., Fuhr, O. & Fichtner, M. A new class of non-corrosive, highly efficient electrolytes for rechargeable magnesium batteries. J. Mater. Chem. A 5, 10815–10820 (2017).
Herb, J. T., Nist-Lund, C. A. & Arnold, C. B. A fluorinated alkoxyaluminate electrolyte for magnesium-ion batteries. ACS Energy Lett. 1, 1227–1232 (2016).
Kobayashi, H. et al. Ultraporous, ultrasmall MgMn2O4 spinel cathode for a room-temperature magnesium rechargeable battery. ACS Nano 17, 3135–3142 (2023).
Rong, Z. et al. Materials design rules for multivalent ion mobility in intercalation structures. Chem. Mater. 27, 6016–6021 (2015).
Ye, X. et al. Examining electrolyte compatibility on polymorphic MnO2 cathodes for room-temperature rechargeable magnesium batteries. ACS Appl. Mater. Interfaces 14, 56685–56696 (2022).
Mandai, T. Critical issues of fluorinated alkoxyborate-based electrolytes in magnesium battery applications. ACS Appl. Mater. Interfaces 12, 39135–39144 (2020).
Varma, A., Mukasyan, A. S., Rogachev, A. S. & Manukyan, K. V. Solution combustion synthesis of nanoscale materials. Chem. Rev. 116, 14493–14586 (2016).
Han, J., Yagi, S., Takeuchi, H., Nakayama, M. & Ichitsubo, T. Catalytic mechanism of spinel oxides for oxidative electrolyte decomposition in Mg rechargeable batteries. J. Mater. Chem. A 9, 26401–26409 (2021).
Wang, J., Polleux, J., Lim, J. & Dunn, B. Pseudocapacitive contributions to electrochemical energy storage in TiO2 (anatase) nanoparticles. J. Phys. Chem. C 111, 14925–14931 (2007).
Wang, L. et al. Solvent-controlled charge storage mechanisms of spinel oxide electrodes in Mg organohaloaluminate electrolytes. Nano Lett. 18, 763–772 (2018).
Doweidar, H. Volume of ionic sites in silicate glasses. J. Phys. Condens. Matter 19, 156102 (2007).
Nhan, N. T., Hung, P. K., Nghiep, D. M. & Kim, H. S. Molecular dynamics investigation on microstructure and void in amorphous SiO2. Mater. Trans. 49, 1212–1218 (2008).
Turnbull, D. & Cohen, M. H. Free‐volume model of the amorphous phase: glass transition. J. Chem. Phys. 34, 120–125 (1961).
Robert, H. et al. Mg-rich disordered rocksalt oxide cathodes for Mg-ion batteries. J. Mater. Chem. A 12, 27303–27310 (2024).
Lee, J. et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 343, 519–522 (2014).
Li, H. et al. Fast diffusion of multivalent ions facilitated by concerted interactions in dual‐ion battery systems. Adv. Energy Mater. 8, 1801475 (2018).
Qi, Y. et al. Alkali cation additives assisting magnesium cation intercalation in hollandite-type manganese dioxide cathodes. J. Phys. Chem. C 127, 21271–21278 (2023).
Roy, A. et al. Improving rechargeable magnesium batteries through dual cation co-intercalation strategy. Nat. Commun. 15, 492 (2024).
Yagi, S. et al. A concept of dual-salt polyvalent-metal storage battery. J. Mater. Chem. A 2, 1144–1149 (2014).
Ichitsubo, T. et al. Toward “rocking-chair type” Mg–Li dual-salt batteries. J. Mater. Chem. A 3, 10188–10194 (2015).
Gao, T. et al. Hybrid Mg2+/Li+ battery with long cycle life and high rate capability. Adv. Energy Mater. 5, 1401507 (2015).
Zhang, J. et al. Current design strategies for rechargeable magnesium-based batteries. ACS Nano 15, 15594–15624 (2021).
Mandai, T. et al. Toward improved anodic stability of ether-based electrolytes for rechargeable magnesium batteries. J. Phys. Chem. C 127, 10419–10433 (2023).
Iimura, R. et al. Ultrasmall α ‐MnO2 with low aspect ratio: applications to electrochemical multivalent‐ion intercalation hosts and aerobic oxidation catalysts. Small 21, 2411493 (2025).
Ling, C., Zhang, R., Arthur, T. S. & Mizuno, F. How general is the conversion reaction in Mg battery cathode: a case study of the magnesiation of α-MnO2. Chem. Mater. 27, 5799–5807 (2015).
Sun, X., Bonnick, P. & Nazar, L. F. Layered TiS2 positive electrode for Mg batteries. ACS Energy Lett. 1, 297–301 (2016).
Arthur, T. S. et al. Understanding the electrochemical mechanism of K-αMnO2 for magnesium battery cathodes. ACS Appl. Mater. Interfaces 6, 7004–7008 (2014).
Zhang, R. & Ling, C. Unveil the chemistry of olivine FePO4 as magnesium battery cathode. ACS Appl. Mater. Interfaces 8, 18018–18026 (2016).
Shimokawa, K. et al. Electrochemically synthesized liquid-sulfur/sulfide composite materials for high-rate magnesium battery cathodes. J. Mater. Chem. A 9, 16585–16593 (2021).
Verrelli, R. et al. On the strange case of divalent ions intercalation in V2O5. J. Power Sources 407, 162–172 (2018).
Thompson, A. P. et al. LAMMPS—a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Hafner, J. Norm-conserving and ultrasoft pseudopotentials for first-row and transition elements. J. Phys. Condens. Matter 6, 8245–8257 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Mori, K. et al. Visualization of conduction pathways in lithium superionic conductors: Li2S-P2S5 glasses and Li7P3S11 glass–ceramic. Chem. Phys. Lett. 584, 113–118 (2013).
Brown, I. D. & Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. B 41, 244–247 (1985).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 41, 653–658 (2008).
Acknowledgements
The authors thank Dr. Hiroshi Tanimura for fruitful discussions and technical support. The work of T.K. was supported by JSPS KAKENHI (Grant Numbers 21H01646, 22KK0068, and 24K01188). This work was partially supported by the GteX Program of Japan (Grant Number JPMJGX23S1). The work of TI was partially supported by JSPS KAKENHI (Grant Number 23H05452). The XAS measurements were performed with the approval of AichSR (Proposal No. 2024D4004). The ICP analyses were supported by F. Sakamoto, K. Nakayama, and Y. Kabasawa at the Analytical Research Core for Advanced Materials, IMR, Tohoku University. The atomic structure was drawn using Vesta65.
Author information
Authors and Affiliations
Contributions
T.K. and T.I. designed the experiments. T.K. and H.S. synthesized the oxide materials. T.K., H.S. and S.F. performed the electrochemical measurements. T.M. synthesized the electrolyte. T.K. and H.S. characterized the samples via XRPD, XAS, and SEM/EDS. N.L.O. performed the TEM/STEM/EDS observations. T.K., H.S., X.Y. and H.L. developed the operational full cells. T.K. wrote the manuscript, and all the authors discussed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Qinyou An and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kawaguchi, T., Sakurai, H., Fukui, S. et al. Amorphous oxide cathode enabling room-temperature rechargeable magnesium batteries. Commun Mater 6, 203 (2025). https://doi.org/10.1038/s43246-025-00921-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43246-025-00921-0
